
Simu-D: A Simulator-Descriptor Suite for Polymer-Based Systems under Extreme Conditions

 , ,
, ,  and
and 
Abstract
:
1. Introduction
2. Molecular Model/Systems Studied
3. Simulator-Descriptor Suite
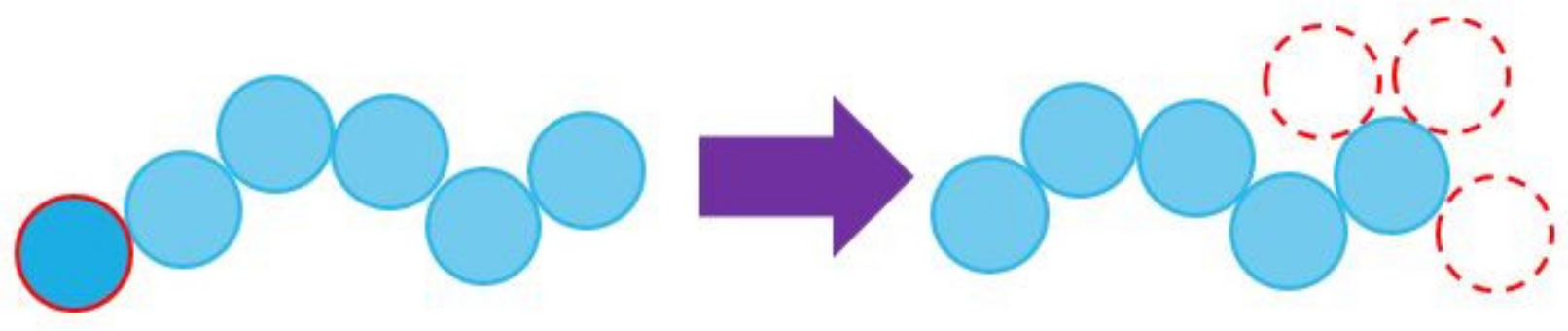
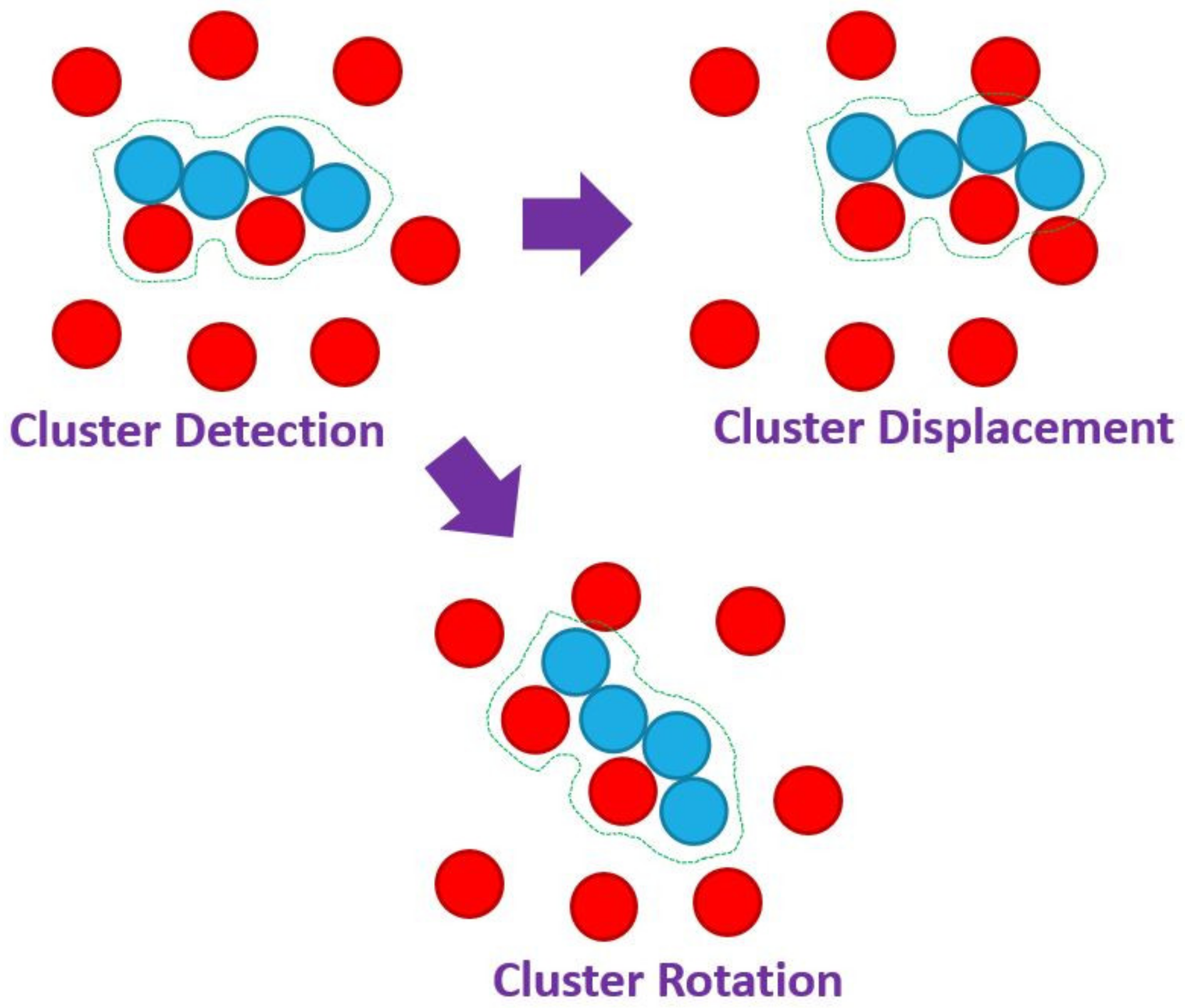
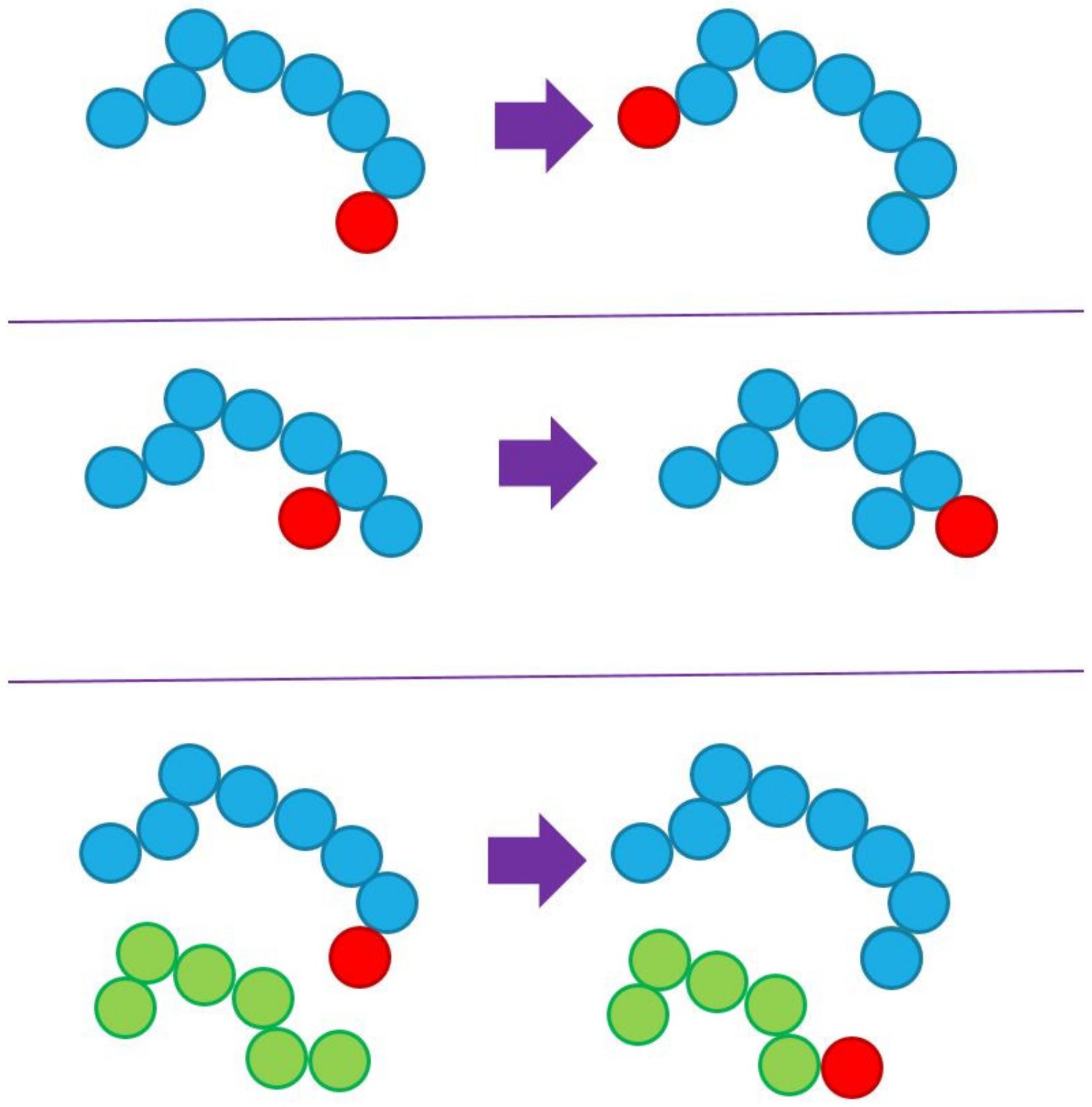
3.1. Simulator
3.2. Descriptor
4. Simu-D: Applications
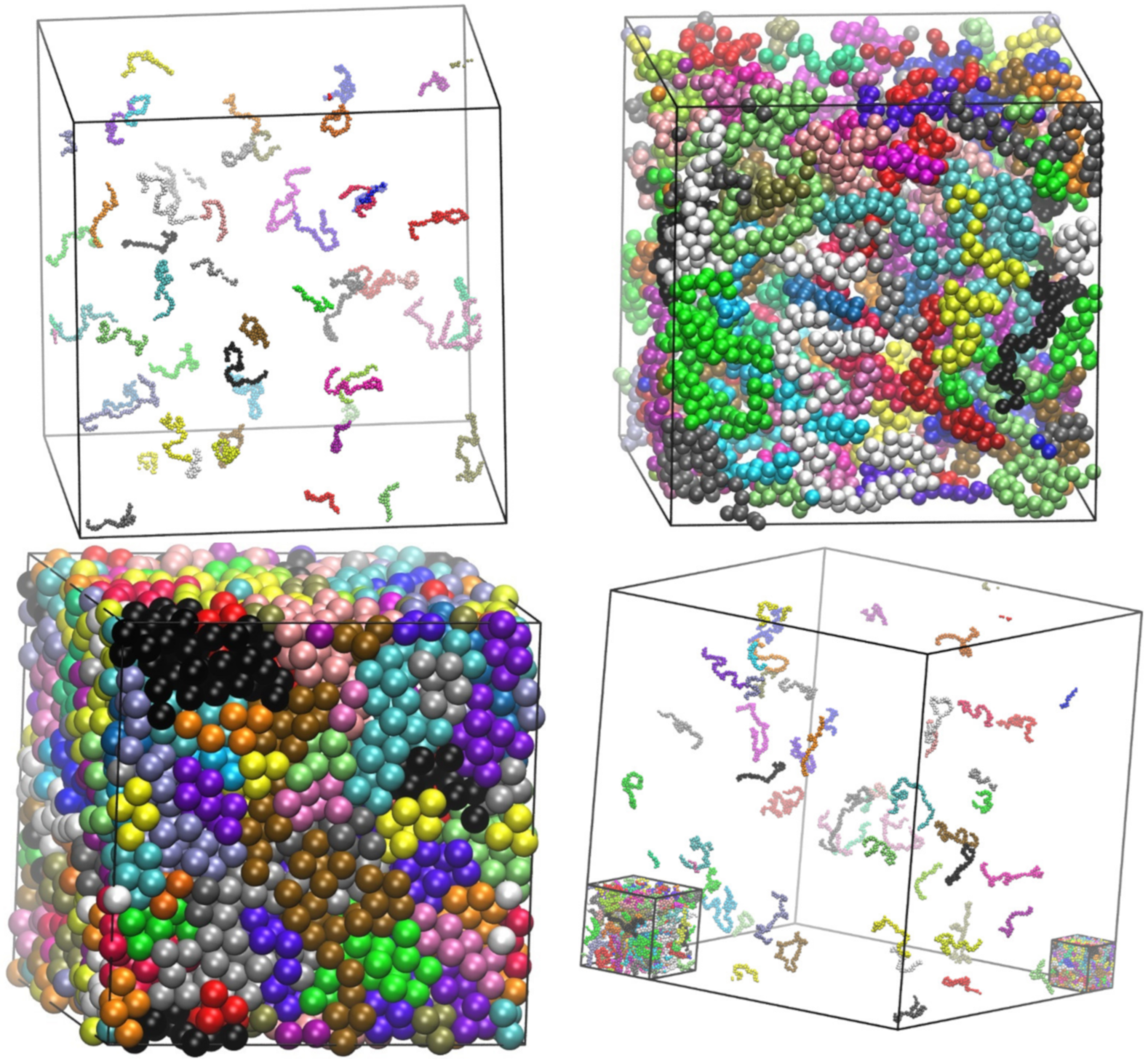
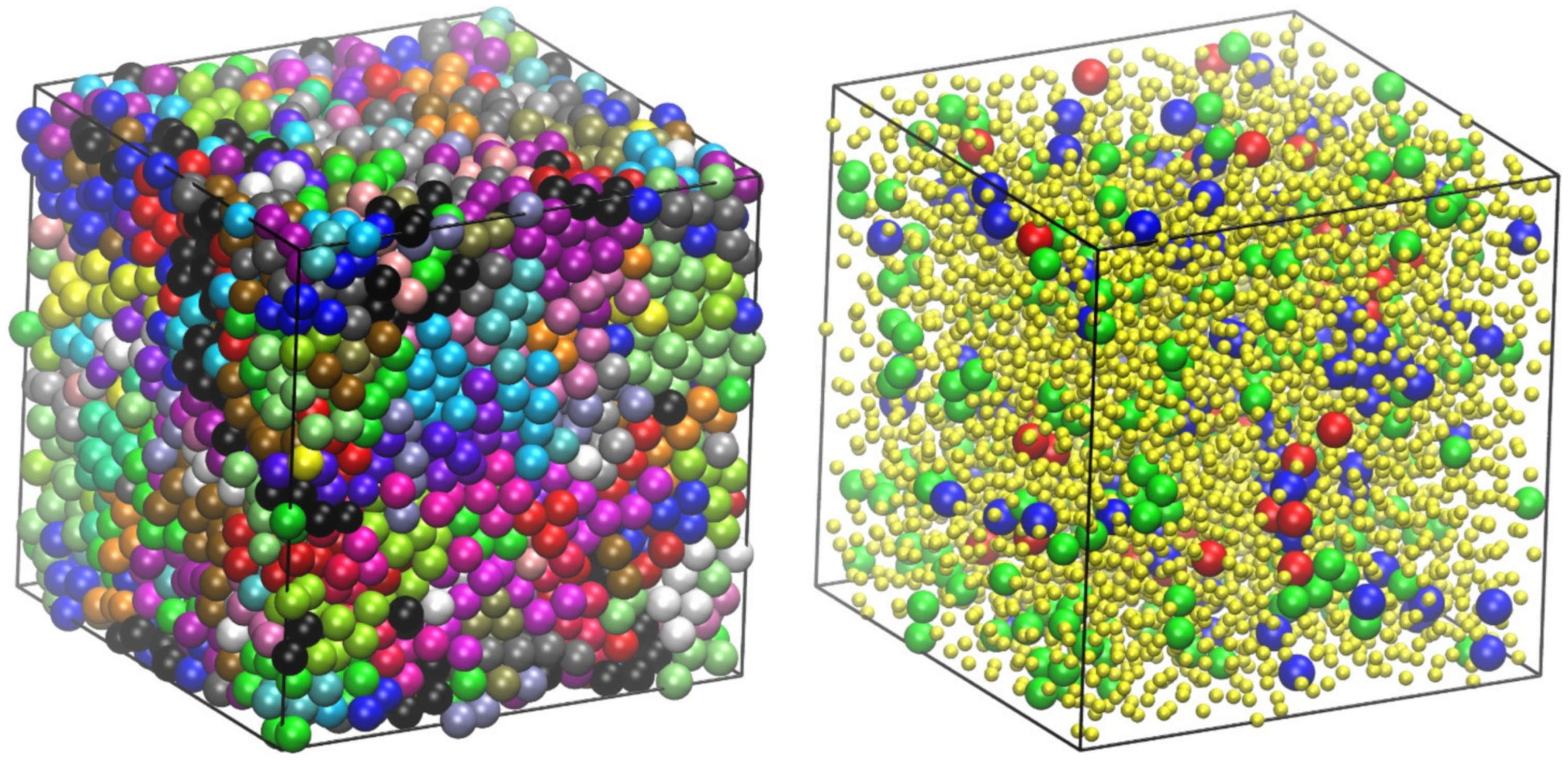
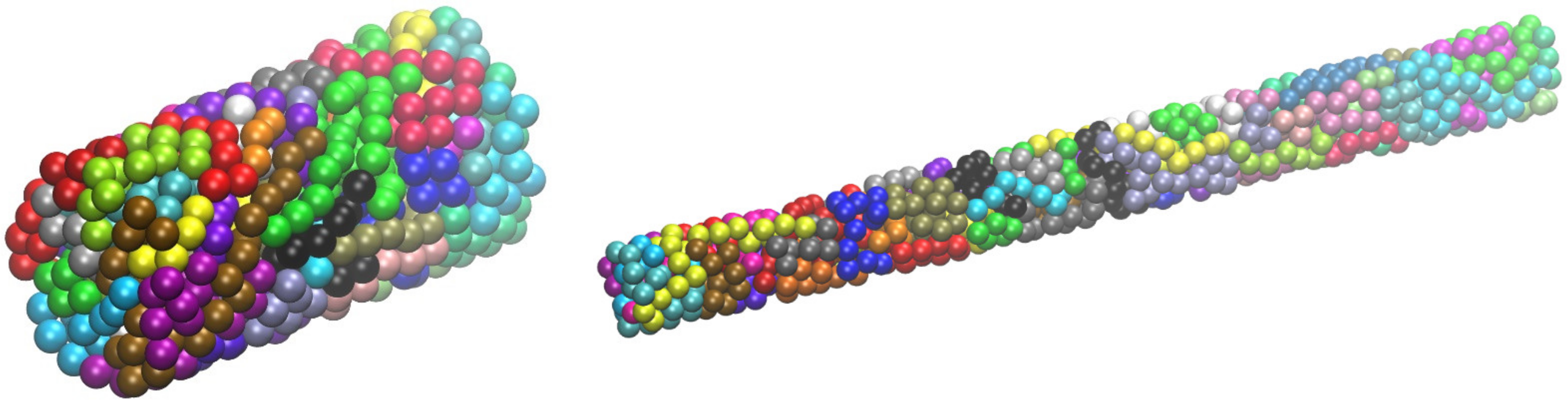
4.1. Packing Efficiency of Semi-Flexible Athermal Polymers (3-D)
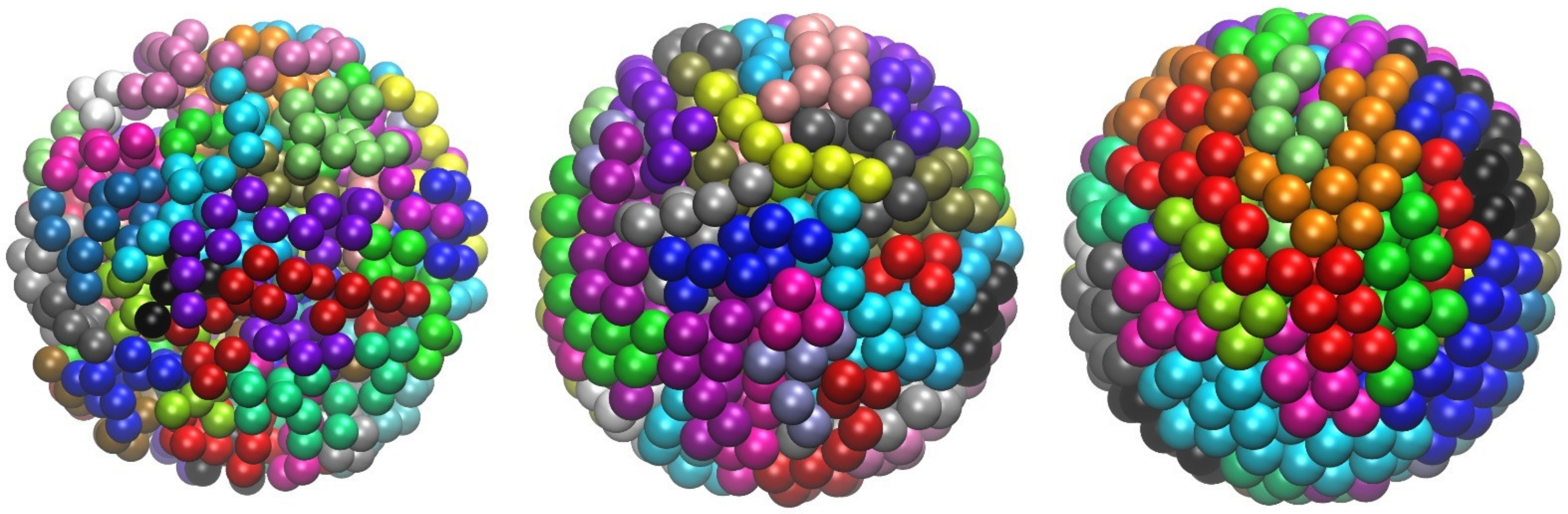
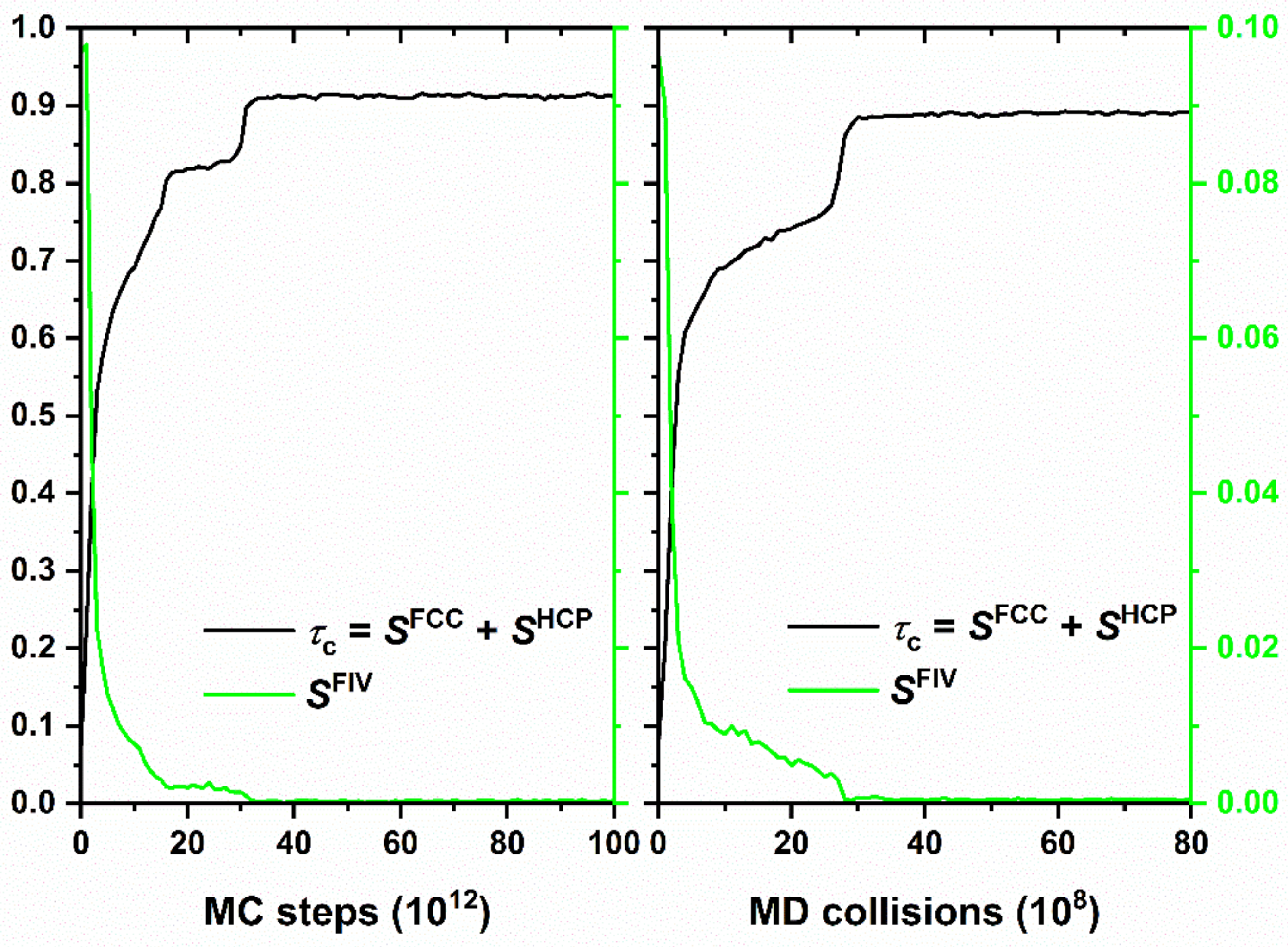
4.2. Entropy-Driven Crystallization of Semi-Flexible Athermal Polymers
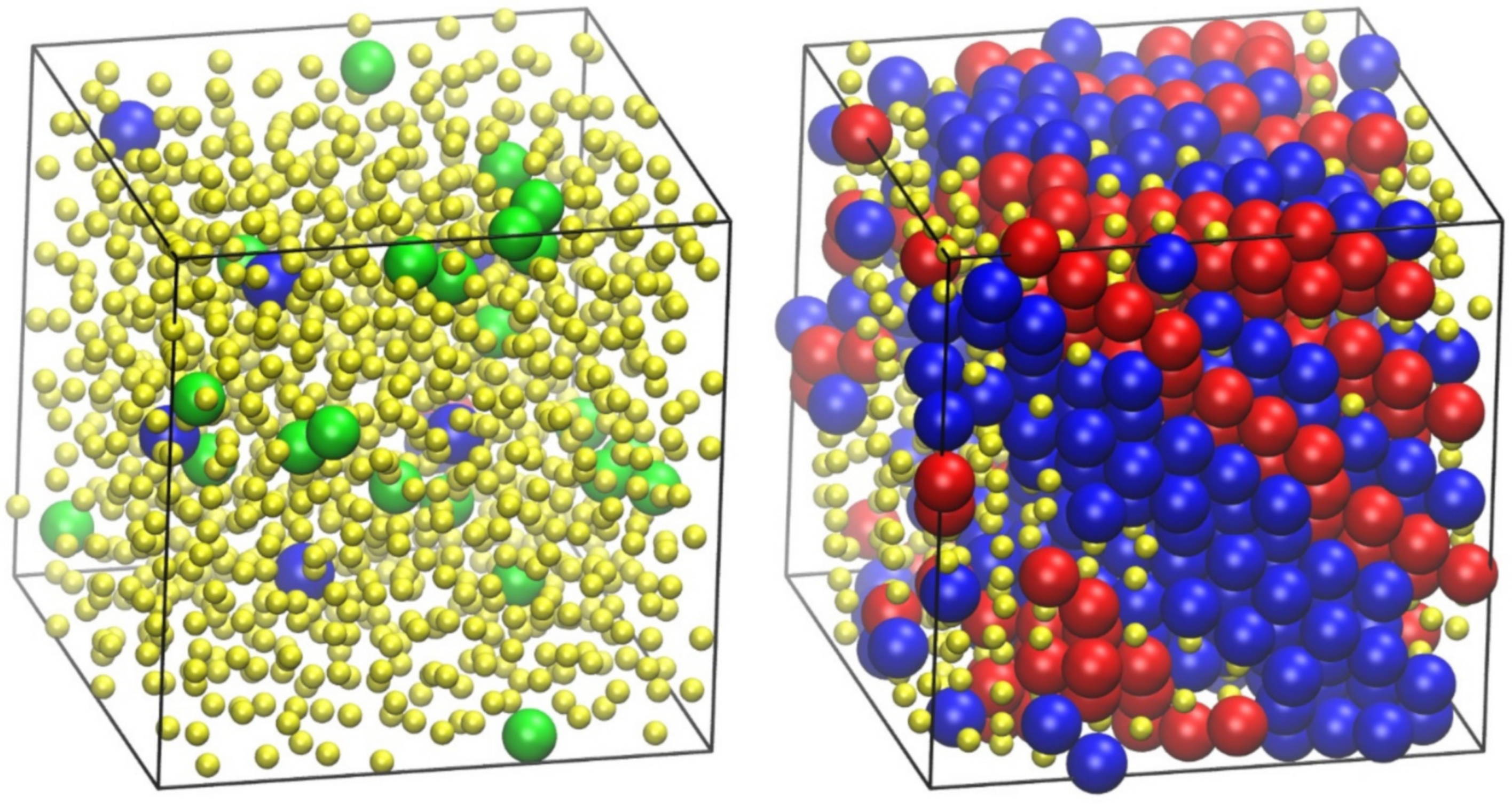
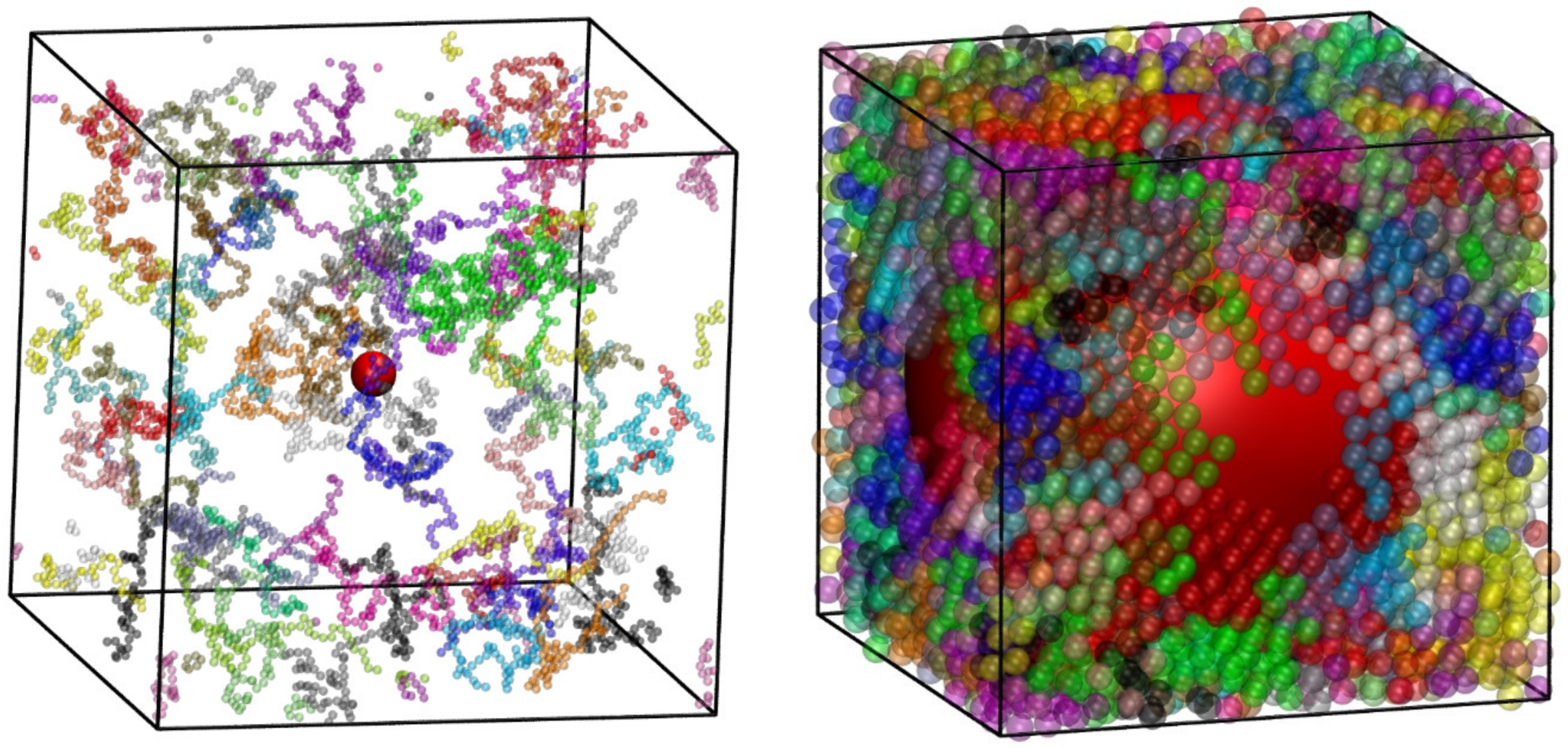
4.3. Phase Behavior of Athermal Blends (Polymers and Monomers)
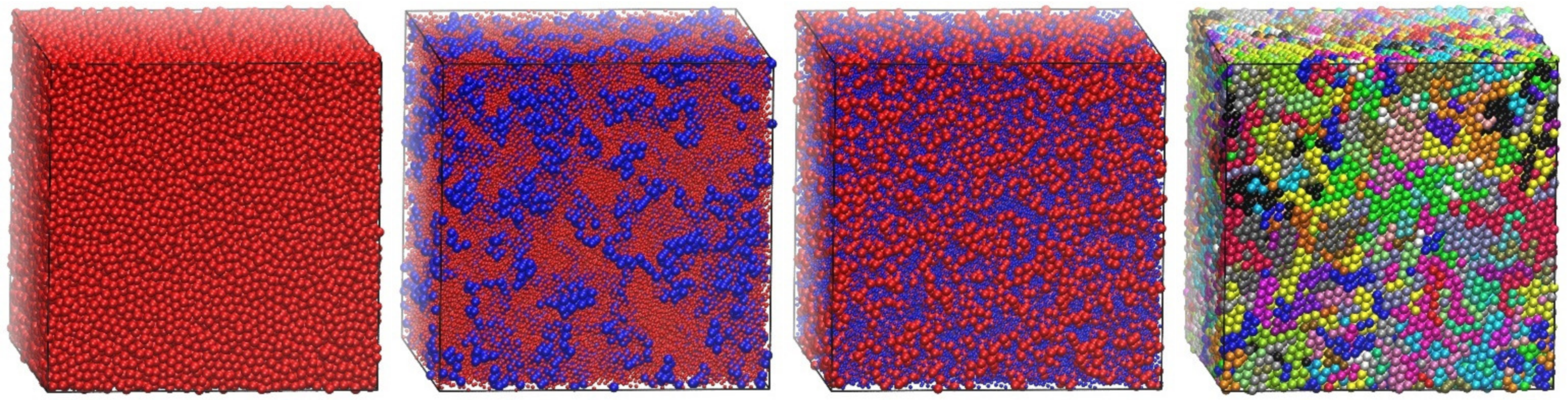
4.4. Energy-Driven Cluster and Crystal Formation of Attractive Chains
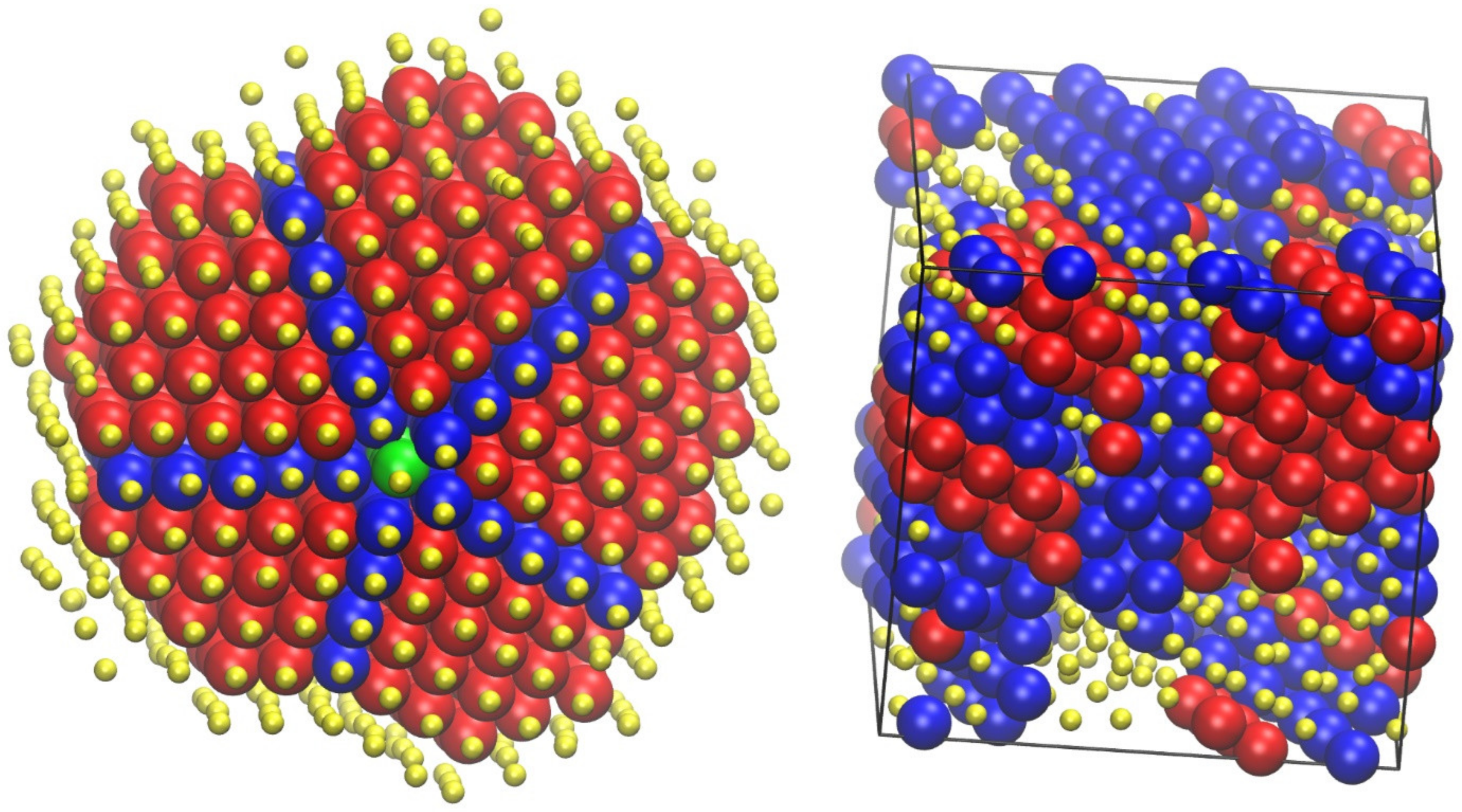
4.5. Polymers under Confinement
4.6. Polymer Nanocomposites
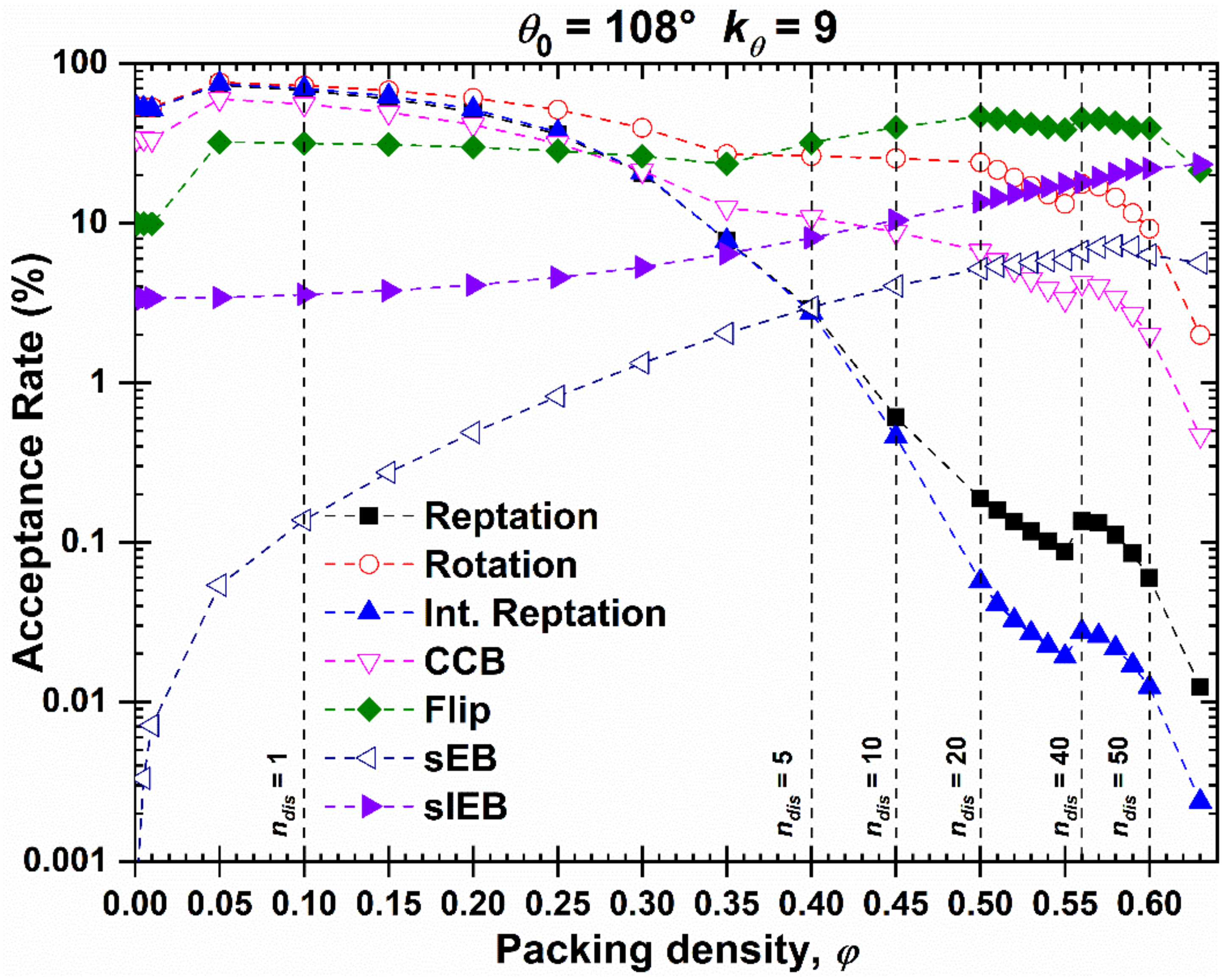
4.7. Comparison with Independent Algorithms
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Type | Description |
|---|---|---|
| D | Categorical | Number of dimensions |
| dconf | Categorical | Number of confined dimensions |
| Nch | Categorical | Number of chains |
| Nat | Numerical | Total number of atoms |
| Nhigh | Numerical | Maximum number of monomers per chain |
| Nlow | Numerical | Minimum number of monomers per chain |
| N | Categorical | Average number of monomers per chain |
| Nmon | Numerical | Number of single monomers |
| Ntrials | Numerical | Number of trials per move in configurational bias scheme |
| Opttrials | Flag | Flag to select the density-dependence of Ntrials |
| ccbcut | Numerical | Maximum number of monomers moved in a CCB move |
| disp | Numerical | Maximum displacement of monomer moves |
| φ | Numerical | Packing density |
| dl | Numerical | Bond gap for chains |
| Nanocomp | Flag | Inclusion of nanoparticles |
| Ncyl | Numerical | Number of nanocylinders |
| Nsph | Numerical | Number of nanospheres |
| dcyl | Numerical | Diameter of nanocylinders |
| dsph | Numerical | Diameter of nanospheres |
| dircyl | Array | Direction of nanocylinders |
| Numerical | Diameter designation | |
| Numerical | Collision diameter for Square-Well/shoulder model | |
| Numerical | Range of interaction for Square-Well/shoulder model | |
| Numerical | Intensity of interaction for Square-Well/shoulder model | |
| OptSW | Flag | Creation of a second cell grid to improve SW performance. |
| Numerical | Intensity of interaction for Square-Well/shoulder of Walls | |
| Numerical | Range of interaction for Square-Well/shoulder model of Walls | |
| Numerical | Intensity of interaction for Square-Well/shoulder of Nanoparticles | |
| Numerical | Range of interaction for Square-Well/shoulder model of Nanoparticles | |
| θeq | Numerical | Supplement of the equilibrium bending angle in radians |
| kbend | Numerical | Energy constant for bending angle potential |
| NPT | Flag | True: Enables NPT ensemble. False: Enables NVT ensemble |
| T | Numerical | Temperature |
| P | Numerical | Pressure |
| Shrink | Flag | True: Runs shrinkage production until a target density. False: Runs normal simulation |
| flucvol | Numerical | Maximum box length reduction when attempting shrinkage |
| Numerical | Target density for the shrinkage production | |
| Isotropic | Flag | True: Volume changes are equal in all direction. False: Volume change is anisotropic |
| Cluster | Flag | Flag to enable cluster moves when there are more than one |
| rclust | Numerical | Radius to detect clusters |
| Vec | Flag | Storage of vectors for crystallographic elements |
| Kiss | Numerical | Coordination number of reference crystal |
| Geom | Flag | Check polymer geometry |
| Neighs | Numerical | Maximum number of Voronoi neighbors |
| HCP | Flag | CCE analysis for HCP crystal |
| FCC | Flag | CCE analysis for FCC crystal |
| BCC | Flag | CCE analysis for BCC crystal |
| HEX | Flag | CCE analysis for HEX crystal |
| FIV | Flag | CCE analysis for FIV symmetry |
| HON | Flag | CCE analysis for HON crystal |
| SQU | Flag | CCE analysis for SQU crystal |
| TRI | Flag | CCE analysis for TRI crystal |
| PEN | Flag | CCE analysis for PEN symmetry |
| Thres | Numerical | CCE threshold of similarity |
| Step | Numerical | Step of the mesh discretization (azimuthal and polar angles) |
| Fast | Flag | No full optimization if norm less than threshold |
References
- Allen, M.P.; Tildesley, D.J. Computer Simulation of Liquids; Oxford University Press: New York, NY, USA, 1987. [Google Scholar]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Landau, D.P.; Binder, K. A Guide to Monte Carlo Simulations in Statistical Physics, 4th ed.; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- Rapaport, D.C. The Art of Molecular Dynamics Simulation, 2nd ed.; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Leach, A. Molecular Modelling: Principles and Applications, 2nd ed.; Pearson: London, UK, 2001. [Google Scholar]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular-Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Abbott, L.J.; Hart, K.E.; Colina, C.M. Polymatic: A generalized simulated polymerization algorithm for amorphous polymers. Theor. Chem. Acc. 2013, 132, 1334. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Glass, C.W.; Reiser, S.; Rutkai, G.; Deublein, S.; Köster, A.; Guevara-Carrion, G.; Wafai, A.; Horsch, M.; Bernreuther, M.; Windmann, T.; et al. ms2: A molecular simulation tool for thermodynamic properties, new version release. Comput. Phys. Commun. 2014, 185, 3302–3306. [Google Scholar] [CrossRef]
- Gezelter, J.D. OpenMD-Molecular Dynamics in the Open. Available online: https:/openmd.org/ (accessed on 15 November 2021).
- Refson, K. Moldy: A portable molecular dynamics simulation program for serial and parallel computers. Comput. Phys. Commun. 2000, 126, 310–329. [Google Scholar] [CrossRef]
- Gale, J.D. GULP: A computer program for the symmetry-adapted simulation of solids. J. Chem. Soc. Faraday Trans. 1997, 93, 629–637. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E.; DeBolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Hypercube. HyperChem. Available online: http://www.hypercubeusa.com (accessed on 15 November 2021).
- Veld, P.J. EMC: Enhanced Monte Carlo. A Multi-Purpose Modular and Easily Extendable Solution to Molecular and Mesoscale Simulations. Available online: http://montecarlo.sourceforge.net (accessed on 15 November 2021).
- In’t Veld, P.J.; Rutledge, G.C. Temperature-Dependent Elasticity of a Semicrystalline Interphase Composed of Freely Rotating Chains. Macromolecules 2003, 36, 7358–7365. [Google Scholar] [CrossRef]
- Yeh, I.-C.; Andzelm, J.W.; Rutledge, G.C. Mechanical and Structural Characterization of Semicrystalline Polyethylene under Tensile Deformation by Molecular Dynamics Simulations. Macromolecules 2015, 48, 4228–4239. [Google Scholar] [CrossRef]
- Kim, J.M.; Locker, R.; Rutledge, G.C. Plastic Deformation of Semicrystalline Polyethylene under Extension, Compression, and Shear Using Molecular Dynamics Simulation. Macromolecules 2014, 47, 2515–2528. [Google Scholar] [CrossRef]
- Kumar, V.; Locker, C.R.; Veld, P.J.; Rutledge, G.C. Effect of Short Chain Branching on the Interlamellar Structure of Semicrystalline Polyethylene. Macromolecules 2017, 50, 1206–1214. [Google Scholar] [CrossRef]
- Martin, M.G. MCCCS Towhee: A tool for Monte Carlo molecular simulation. Mol. Simulat. 2013, 39, 1212–1222. [Google Scholar] [CrossRef]
- Tsimpanogiannis, I.N.; Costandy, J.; Kastanidis, P.; El Meragawi, S.; Michalis, V.K.; Papadimitriou, N.I.; Karozis, S.N.; Diamantonis, N.I.; Moultos, O.A.; Romanos, G.E.; et al. Using clathrate hydrates for gas storage and gas-mixture separations: Experimental and computational studies at multiple length scales. Mol. Phys. 2018, 116, 2041–2060. [Google Scholar] [CrossRef]
- Brukhno, A.V.; Grant, J.; Underwood, T.L.; Stratford, K.; Parker, S.C.; Purton, J.A.; Wilding, N.B. DL_MONTE: A multipurpose code for Monte Carlo simulation. Mol. Simulat. 2021, 47, 131–151. [Google Scholar] [CrossRef]
- Wang, F.; Landau, D.P. Efficient, Multiple-Range Random Walk Algorithm to Calculate the Density of States. Phys. Rev. Lett. 2001, 86, 2050–2053. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.K.; Marin-Rimoldi, E.; Mullen, R.G.; Keene, B.P.; Khan, S.; Paluch, A.S.; Rai, N.; Romanielo, L.L.; Rosch, T.W.; Yoo, B.; et al. Cassandra: An open source Monte Carlo package for molecular simulation. J. Comput. Chem. 2017, 38, 1727–1739. [Google Scholar] [CrossRef]
- Dubbeldam, D.; Calero, S.; Ellis, D.E.; Snurr, R.Q. RASPA: Molecular simulation software for adsorption and diffusion in flexible nanoporous materials. Mol. Simulat. 2016, 42, 81–101. [Google Scholar] [CrossRef]
- Nejahi, Y.; Barhaghi, M.S.; Mick, J.; Jackman, B.; Rushaidat, K.; Li, Y.; Schwiebert, L.; Potoff, J. GOMC: GPU Optimized Monte Carlo for the simulation of phase equilibria and physical properties of complex fluids. SoftwareX 2019, 9, 20–27. [Google Scholar] [CrossRef]
- Mick, J.; Hailat, E.; Russo, V.; Rushaidat, K.; Schwiebert, L.; Potoff, J. GPU-accelerated Gibbs ensemble Monte Carlo simulations of Lennard-Jonesium. Comput. Phys. Commun. 2013, 184, 2662–2669. [Google Scholar] [CrossRef]
- Cezar, H.M.; Canuto, S.; Coutinho, K. DICE: A Monte Carlo Code for Molecular Simulation Including the Configurational Bias Monte Carlo Method. J. Chem. Inf. Model. 2020, 60, 3472–3488. [Google Scholar] [CrossRef] [PubMed]
- Gowers, R.J.; Farmahini, A.H.; Friedrich, D.; Sarkisov, L. Automated analysis and benchmarking of GCMC simulation programs in application to gas adsorption. Mol. Simulat. 2018, 44, 309–321. [Google Scholar] [CrossRef]
- Alexiadis, O.; Cheimarios, N.; Peristeras, L.D.; Bick, A.; Mavrantzas, V.G.; Theodorou, D.N.; Hill, J.-R.; Krokidis, X. Chameleon: A generalized, connectivity altering software for tackling properties of realistic polymer systems. WIREs Comput. Mol. Sci. 2019, 9, e1414. [Google Scholar] [CrossRef]
- Ghobadpour, E.; Kolb, M.; Ejtehadi, M.R.; Everaers, R. Monte Carlo simulation of a lattice model for the dynamics of randomly branching double-folded ring polymers. Phys. Rev. E 2021, 104, 014501. [Google Scholar] [CrossRef] [PubMed]
- Theodorou, D.N. A reversible minimum-to-minimum mapping method for the calculation of free-energy differences. J. Chem. Phys. 2006, 124, 034109. [Google Scholar] [CrossRef] [PubMed]
- Uhlherr, A.; Theodorou, D.N. Accelerating molecular simulations by reversible mapping between local minima. J. Chem. Phys. 2006, 125, 84107. [Google Scholar] [CrossRef]
- Shi, W.; Maginn, E.J. Improvement in Molecule Exchange Efficiency in Gibbs Ensemble Monte Carlo: Development and Implementation of the Continuous Fractional Component Move. J. Comput. Chem. 2008, 29, 2520–2530. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Maginn, E.J. Continuous fractional component Monte Carlo: An adaptive biasing method for open system atomistic simulations. J. Chem. Theory Comput. 2007, 3, 1451–1463. [Google Scholar] [CrossRef]
- Weismantel, O.; Galata, A.A.; Sadeghi, M.; Kroger, A.; Kroger, M. Efficient generation of self-avoiding, semiflexible rotational isomeric chain ensembles in bulk, confined geometries, and on surfaces. Comput. Phys. Commun. 2021, 270, 108176. [Google Scholar] [CrossRef]
- Kroger, M.; Muller, M.; Nievergelt, J. A geometric embedding algorithm for efficiently generating semiflexible chains in the molten state. Cmes-Comput. Modeling Eng. Sci. 2003, 4, 559–569. [Google Scholar]
- Kampmann, T.A.; Muller, D.; Weise, L.P.; Vorsmann, C.F.; Kierfeld, J. Event-Chain Monte-Carlo Simulations of Dense Soft Matter Systems. arXiv 2021, arXiv:2102.05461. [Google Scholar]
- Krauth, W. Event-Chain Monte Carlo: Foundations, Applications, and Prospects. Front. Phys. 2021, 9, 229. [Google Scholar] [CrossRef]
- Klement, M.; Lee, S.; Anderson, J.A.; Engel, M. Newtonian Event-Chain Monte Carlo and Collision Prediction with Polyhedral Particles. J. Chem. Theory Comput. 2021, 17, 4686–4696. [Google Scholar] [CrossRef]
- Kriuchevskyi, I.; Palyulin, V.V.; Milkus, R.; Elder, R.M.; Sirk, T.W.; Zaccone, A. Scaling up the lattice dynamics of amorphous materials by orders of magnitude. Phys. Rev. B 2020, 102, 024108. [Google Scholar] [CrossRef]
- Auhl, R.; Everaers, R.; Grest, G.S.; Kremer, K.; Plimpton, S.J. Equilibration of long chain polymer melts in computer simulations. J. Chem. Phys. 2003, 119, 12718–12728. [Google Scholar] [CrossRef]
- Doshi, U.; Hamelberg, D. Achieving Rigorous Accelerated Conformational Sampling in Explicit Solvent. J. Phys. Chem. Lett. 2014, 5, 1217–1224. [Google Scholar] [CrossRef]
- Subramanian, G. A topology preserving method for generating equilibrated polymer melts in computer simulations. J. Chem. Phys. 2010, 133, 164902. [Google Scholar] [CrossRef]
- Zhang, G.J.; Chazirakis, A.; Harmandaris, V.A.; Stuehn, T.; Daoulas, K.C.; Kremer, K. Hierarchical modelling of polystyrene melts: From soft blobs to atomistic resolution. Soft Matter 2019, 15, 289–302. [Google Scholar] [CrossRef]
- Milchev, A.; Binder, K. Cylindrical confinement of solutions containing semiflexible macromolecules: Surface-induced nematic order versus phase separation. Soft Matter 2021, 17, 3443–3454. [Google Scholar] [CrossRef]
- Zhou, X.L.; Wu, J.X.; Zhang, L.X. Ordered aggregation of semiflexible ring-linear blends in ellipsoidal confinement. Polymer 2020, 197, 122494. [Google Scholar] [CrossRef]
- Milchev, A.; Nikoubashman, A.; Binder, K. The smectic phase in semiflexible polymer materials: A large scale molecular dynamics study. Comput. Mater. Sci. 2019, 166, 230–239. [Google Scholar] [CrossRef]
- Moghimikheirabadi, A.; Kroger, M.; Karatrantos, A.V. Insights from modeling into structure, entanglements, and dynamics in attractive polymer nanocomposites. Soft Matter 2021, 17, 6362–6373. [Google Scholar] [CrossRef] [PubMed]
- Kroger, M. Efficient hybrid algorithm for the dynamic creation of wormlike chains in solutions, brushes, melts and glasses. Comput. Phys. Commun. 2019, 241, 178–179. [Google Scholar] [CrossRef]
- Ahrens, J.; Geveci, B.; Law, C. ParaView: An End-User Tool for Large Data Visualization; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bumstead, M.; Liang, K.Y.; Hanta, G.; Hui, L.S.; Turak, A. disLocate: Tools to rapidly quantify local intermolecular structure to assess two-dimensional order in self-assembled systems. Sci. Rep. 2018, 8, 1–15. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO-the Open Visualization Tool. Modell. Simul. Mater. Sci. Eng. 2010, 18, 015012. [Google Scholar] [CrossRef]
- Tassieri, M.; Ramirez, J.; Karayiannis, N.C.; Sukumaran, S.K.; Masubuchi, Y. i-Rheo GT: Transforming from Time to Frequency Domain without Artifacts. Macromolecules 2018, 51, 5055–5068. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Laso, M. Monte Carlo scheme for generation and relaxation of dense and nearly jammed random structures of freely jointed hard-sphere chains. Macromolecules 2008, 41, 1537–1551. [Google Scholar] [CrossRef]
- Ramos, P.M.; Herranz, M.; Foteinopoulou, K.; Karayiannis, N.C.; Laso, M. Identification of Local Structure in 2-D and 3-D Atomic Systems through Crystallographic Analysis. Crystals 2020, 10, 1008. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Foteinopoulou, K.; Laso, M. The characteristic crystallographic element norm: A descriptor of local structure in atomistic and particulate systems. J. Chem. Phys. 2009, 130, 074704. [Google Scholar] [CrossRef]
- Foteinopoulou, K.; Karayiannis, N.C.; Laso, M. Monte Carlo simulations of densely-packed athermal polymers in the bulk and under confinement. Chem. Eng. Sci. 2015, 121, 118–132. [Google Scholar] [CrossRef]
- Herranz, M.; Santiago, M.; Foteinopoulou, K.; Karayiannis, N.C.; Laso, M. Crystal, Fivefold and Glass Formation in Clusters of Polymers Interacting with the Square Well Potential. Polymers 2020, 12, 1111. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Foteinopoulou, K.; Laso, M. The structure of random packings of freely jointed chains of tangent hard spheres. J. Chem. Phys. 2009, 130, 164908. [Google Scholar] [CrossRef] [PubMed]
- Karayiannis, N.C.; Foteinopoulou, K.; Laso, M. Contact network in nearly jammed disordered packings of hard-sphere chains. Phys. Rev. E 2009, 80, 011307. [Google Scholar] [CrossRef] [PubMed]
- Laso, M.; Karayiannis, N.C.; Foteinopoulou, K.; Mansfield, M.L.; Kroger, M. Random packing of model polymers: Local structure, topological hindrance and universal scaling. Soft Matter 2009, 5, 1762–1770. [Google Scholar] [CrossRef]
- Foteinopoulou, K.; Karayiannis, N.C.; Laso, M.; Kroger, M.; Mansfield, M.L. Universal Scaling, Entanglements, and Knots of Model Chain Molecules. Phys. Rev. Lett. 2008, 101, 265702. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Kroger, M. Combined Molecular Algorithms for the Generation, Equilibration and Topological Analysis of Entangled Polymers: Methodology and Performance. Int. J. Mol. Sci. 2009, 10, 5054–5089. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Foteinopoulou, K.; Laso, M. The role of bond tangency and bond gap in hard sphere crystallization of chains. Soft Matter 2015, 11, 1688–1700. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Foteinopoulou, K.; Laso, M. Jamming and crystallization in athermal polymer packings. Philos. Mag. 2013, 93, 4108–4131. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Foteinopoulou, K.; Abrams, C.F.; Laso, M. Modeling of crystal nucleation and growth in athermal polymers: Self-assembly of layered nano-morphologies. Soft Matter 2010, 6, 2160–2173. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Foteinopoulou, K.; Laso, M. Entropy-Driven Crystallization in Dense Systems of Athermal Chain Molecules. Phys. Rev. Lett. 2009, 103, 045703. [Google Scholar] [CrossRef]
- Ramos, P.M.; Herranz, M.; Foteinopoulou, K.; Karayiannis, N.C.; Laso, M. Entropy-Driven Heterogeneous Crystallization of Hard-Sphere Chains under Unidimensional Confinement. Polymers 2021, 13, 1352. [Google Scholar] [CrossRef] [PubMed]
- Pant, P.V.K.; Theodorou, D.N. Variable Connectivity Method For The Atomistic Monte-Carlo Simulation Of Polydisperse Polymer Melts. Macromolecules 1995, 28, 7224–7234. [Google Scholar] [CrossRef]
- Ramos, P.M.; Karayiannis, N.C.; Laso, M. Off-lattice simulation algorithms for athermal chain molecules under extreme confinement. J. Comput. Phys. 2018, 375, 918–934. [Google Scholar] [CrossRef]
- Siepmann, J.I.; Frenkel, D. Configurational Bias Monte-Carlo—A New Sampling Scheme for Flexible Chains. Mol. Phys. 1992, 75, 59–70. [Google Scholar] [CrossRef]
- Laso, M.; de Pablo, J.J.; Suter, U.W. Simulation of Phase-Equilibria for Chain Molecules. J. Chem. Phys. 1992, 97, 2817–2819. [Google Scholar] [CrossRef]
- Mavrantzas, V.G.; Boone, T.D.; Zervopoulou, E.; Theodorou, D.N. End-bridging Monte Carlo: A fast algorithm for atomistic simulation of condensed phases of long polymer chains. Macromolecules 1999, 32, 5072–5096. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Mavrantzas, V.G.; Theodorou, D.N. A novel Monte Carlo scheme for the rapid equilibration of atomistic model polymer systems of precisely defined molecular architecture. Phys. Rev. Lett. 2002, 88, 105503. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Giannousaki, A.E.; Mavrantzas, V.G.; Theodorou, D.N. Atomistic Monte Carlo simulation of strictly monodisperse long polyethylene melts through a generalized chain bridging algorithm. J. Chem. Phys. 2002, 117, 5465–5479. [Google Scholar] [CrossRef]
- Faken, D.; Jónsson, H. Systematic analysis of local atomic structure combined with 3D computer graphics. Comput. Mater. Sci. 1994, 2, 279–286. [Google Scholar] [CrossRef]
- Martin, A.V.; Kozlov, A.; Berntsen, P.; Roque, F.G.; Flueckiger, L.; Saha, S.; Greaves, T.L.; Conn, C.E.; Hawley, A.M.; Ryan, T.M.; et al. Fluctuation X-ray diffraction reveals three-dimensional nanostructure and disorder in self-assembled lipid phases. Commun. Mater. 2020, 1, 40. [Google Scholar] [CrossRef]
- Steinhardt, P.J.; Nelson, D.R.; Ronchetti, M. Bond-orientational order in liquids and glasses. Phys. Rev. B 1983, 28, 784–805. [Google Scholar] [CrossRef]
- Larsen, P.M.; Schmidt, S.; Schiotz, J. Robust structural identification via polyhedral template matching. Modell. Simul. Mater. Sci. Eng. 2016, 24. [Google Scholar] [CrossRef]
- Tanemura, M.; Hiwatari, Y.; Matsuda, H.; Ogawa, T.; Ogita, N.; Ueda, A. Geometrical analysis of crystallization of the soft-core model. Prog. Theor. Phys. 1977, 58, 1079–1095. [Google Scholar] [CrossRef]
- Anikeenko, A.V.; Medvedev, N.N.; Aste, T. Structural and entropic insights into the nature of the random-close-packing limit. Phys. Rev. E 2008, 77, 031101. [Google Scholar] [CrossRef] [PubMed]
- Ackland, G.J.; Jones, A.P. Applications of local crystal structure measures in experiment and simulation. Phys. Rev. B 2006, 73, 054104. [Google Scholar] [CrossRef]
- Cohen, M.H.; Grest, G.S. Liquid-glass transition, a free-volume approach. Phys. Rev. B 1979, 20, 1077–1098. [Google Scholar] [CrossRef]
- Egami, T.; Maeda, K.; Vitek, V. Structural Defects In Amorphous Solids—A Computer-Simulation Study. Philos. Mag. A 1980, 41, 883–901. [Google Scholar] [CrossRef]
- Ding, J.; Cheng, Y.Q.; Sheng, H.; Asta, M.; Ritchie, R.O.; Ma, E. Universal structural parameter to quantitatively predict metallic glass properties. Nat. Commun. 2016, 7, 13733. [Google Scholar] [CrossRef] [PubMed]
- Malins, A.; Williams, S.R.; Eggers, J.; Royall, C.P. Identification of structure in condensed matter with the topological cluster classification. J. Chem. Phys. 2013, 139, 234506. [Google Scholar] [CrossRef] [PubMed]
- Stukowski, A. Structure identification methods for atomistic simulations of crystalline materials. Modell. Simul. Mater. Sci. Eng. 2012, 20, 045021. [Google Scholar] [CrossRef]
- Paret, J.; Jack, R.L.; Coslovich, D. Assessing the structural heterogeneity of supercooled liquids through community inference. J. Chem. Phys. 2020, 152, 144502. [Google Scholar] [CrossRef] [PubMed]
- Nye, J.F. Physical Properties of Crystals: Their Representation by Tensors and Matrices; Oxford Science Publications: Oxford, UK, 2010. [Google Scholar]
- Malgrange, C.; Ricolleau, C.; Schlenker, M. Symmetry and Physical Properties of Crystals; Springer: Dordrecht, The Netherlands, 2014. [Google Scholar] [CrossRef]
- Giacovazzo, C.; Monaco, H.L.; Artioli, G.; Viterbo, D.; Ferraris, G.; Gilli, G.; Zanotti, G.; Gatti, M. Fundamentals of Crystallography; Oxford Science: Oxford, UK, 2005. [Google Scholar]
- Laso, M.; Jimeno, N. Representation Surfaces for Physical Properties of Materials: A Visual Approach to Understanding Anisotropic Materials; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Hales, T.C. A proof of the Kepler conjecture. Ann. Math. 2005, 162, 1065–1185. [Google Scholar] [CrossRef]
- Hales, T.C.; Harrison, J.; McLaughlin, S.; Nipkow, T.; Obua, S.; Zumkeller, R. A Revision of the Proof of the Kepler Conjecture. Discret. Comput. Geom. 2010, 44, 1–34. [Google Scholar] [CrossRef]
- Bernal, J.D.; Finney, J.L. Random close-packed hard-sphere model. 2. Geometry of random packing of hard spheres. Discuss. Faraday Soc. 1967, 43, 62–69. [Google Scholar] [CrossRef]
- Bernal, J.D. Geometry of The Structure of Monatomic Liquids. Nature 1960, 185, 68–70. [Google Scholar] [CrossRef]
- Torquato, S.; Truskett, T.M.; Debenedetti, P.G. Is random close packing of spheres well defined? Phys. Rev. Lett. 2000, 84, 2064–2067. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Laso, M. Dense and nearly jammed random packings of freely jointed chains of tangent hard spheres. Phys. Rev. Lett. 2008, 100, 050602. [Google Scholar] [CrossRef]
- Hoy, R.S. Jamming of Semiflexible Polymers. Phys. Rev. Lett. 2017, 118, 068002. [Google Scholar] [CrossRef]
- Shakirov, T.; Paul, W. Crystallization in melts of short, semiflexible hard polymer chains: An interplay of entropies and dimensions. Phys. Rev. E 2018, 97, 042501. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, B.; Snook, I. Crystal nucleation in the hard sphere system. Phys. Rev. Lett. 2003, 90, 085702. [Google Scholar] [CrossRef] [PubMed]
- Karayiannis, N.C.; Malshe, R.; de Pablo, J.J.; Laso, M. Fivefold symmetry as an inhibitor to hard-sphere crystallization. Phys. Rev. E 2011, 83, 061505. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Malshe, R.; Kroger, M.; de Pablo, J.J.; Laso, M. Evolution of fivefold local symmetry during crystal nucleation and growth in dense hard-sphere packings. Soft Matter 2012, 8, 844–858. [Google Scholar] [CrossRef]
- Karayiannis, N.C.; Foteinopoulou, K.; Laso, M. Spontaneous Crystallization in Athermal Polymer Packings. Int. J. Mol. Sci. 2013, 14, 332–358. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Yao, Z.W.; Tang, S.X.; Tong, H.; Yanagishima, T.; Tanaka, H.; Tan, P. Morphology selection kinetics of crystallization in a sphere. Nat. Phys. 2021, 17, 121–127. [Google Scholar] [CrossRef]
- Arai, S.; Tanaka, H. Surface-assisted single-crystal formation of charged colloids. Nat. Phys. 2017, 13, 503–509. [Google Scholar] [CrossRef]
- Torquato, S.; Stillinger, F.H. Jammed hard-particle packings: From Kepler to Bernal and beyond. Rev. Mod. Phys. 2010, 82, 2633–2672. [Google Scholar] [CrossRef]
- Wilken, S.; Guerra, R.E.; Levine, D.; Chaikin, P.M. Random Close Packing as a Dynamical Phase Transition. Phys. Rev. Lett. 2021, 127, 038002. [Google Scholar] [CrossRef]
- Rissone, P.; Corwin, E.I.; Parisi, G. Long-Range Anomalous Decay of the Correlation in Jammed Packings. Phys. Rev. Lett. 2021, 127, 038001. [Google Scholar] [CrossRef]
- Alder, B.J.; Wainwright, T.E. Phase Transition For A Hard Sphere System. J. Chem. Phys. 1957, 27, 1208–1209. [Google Scholar] [CrossRef]
- Ni, R.; Dijkstra, M. Effect of bond length fluctuations on crystal nucleation of hard bead chains. Soft Matter 2013, 9, 365–369. [Google Scholar] [CrossRef]
- Johnson, J.K.; Panagiotopoulos, A.Z.; Gubbins, K.E. Reactive canonical monte-carlo—A new simulation technique for reacting or associating fluids. Mol. Phys. 1994, 81, 717–733. [Google Scholar] [CrossRef]
- Laso, M.; Karayiannis, N.C.; Muller, M. Min-map bias Monte Carlo for chain molecules: Biased Monte Carlo sampling based on bijective minimum-to-minimum mapping. J. Chem. Phys. 2006, 125, 164108. [Google Scholar] [CrossRef] [PubMed]
- Uhlherr, A.; Doxastakis, M.; Mavrantzas, V.G.; Theodorou, D.N.; Leak, S.J.; Adam, N.E.; Nyberg, P.E. Atomic structure of a high polymer melt. Europhys. Lett. 2002, 57, 506–511. [Google Scholar] [CrossRef]
- Uhlherr, A.; Leak, S.J.; Adam, N.E.; Nyberg, P.E.; Doxastakis, M.; Mavrantzas, V.G.; Theodorou, D.N. Large scale atomistic polymer simulations using Monte Carlo methods for parallel vector processors. Comput. Phys. Commun. 2002, 144, 1–22. [Google Scholar] [CrossRef]
- Kroger, M. Shortest multiple disconnected path for the analysis of entanglements in two- and three-dimensional polymeric systems. Comput. Phys. Commun. 2005, 168, 209–232. [Google Scholar] [CrossRef]
- Caraglio, M.; Enzo, C.M.; Orlandini, E. Physical Links: Defining and detecting inter-chain entanglement. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Tzoumanekas, C.; Theodorou, D.N. Topological analysis of linear polymer melts: A statistical approach. Macromolecules 2006, 39, 4592–4604. [Google Scholar] [CrossRef]
- Everaers, R.; Sukumaran, S.K.; Grest, G.S.; Svaneborg, C.; Sivasubramanian, A.; Kremer, K. Rheology and microscopic topology of entangled polymeric liquids. Science 2004, 303, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, S.; Larson, R.G. Identification of Topological Constraints in Entangled Polymer Melts Using the Bond-Fluctuation Model. Macromolecules 2006, 39, 2413–2417. [Google Scholar] [CrossRef]















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herranz, M.; Martínez-Fernández, D.; Ramos, P.M.; Foteinopoulou, K.; Karayiannis, N.C.; Laso, M. Simu-D: A Simulator-Descriptor Suite for Polymer-Based Systems under Extreme Conditions. Int. J. Mol. Sci. 2021, 22, 12464. https://doi.org/10.3390/ijms222212464
Herranz M, Martínez-Fernández D, Ramos PM, Foteinopoulou K, Karayiannis NC, Laso M. Simu-D: A Simulator-Descriptor Suite for Polymer-Based Systems under Extreme Conditions. International Journal of Molecular Sciences. 2021; 22(22):12464. https://doi.org/10.3390/ijms222212464
Chicago/Turabian StyleHerranz, Miguel, Daniel Martínez-Fernández, Pablo Miguel Ramos, Katerina Foteinopoulou, Nikos Ch. Karayiannis, and Manuel Laso. 2021. "Simu-D: A Simulator-Descriptor Suite for Polymer-Based Systems under Extreme Conditions" International Journal of Molecular Sciences 22, no. 22: 12464. https://doi.org/10.3390/ijms222212464
APA StyleHerranz, M., Martínez-Fernández, D., Ramos, P. M., Foteinopoulou, K., Karayiannis, N. C., & Laso, M. (2021). Simu-D: A Simulator-Descriptor Suite for Polymer-Based Systems under Extreme Conditions. International Journal of Molecular Sciences, 22(22), 12464. https://doi.org/10.3390/ijms222212464