
Molecular Analysis Uncovers the Mechanism of Fertility Restoration in Temperature-Sensitive Polima Cytoplasmic Male-Sterile Brassica napus

 , ,
, ,
Abstract
:1. Introduction
2. Results
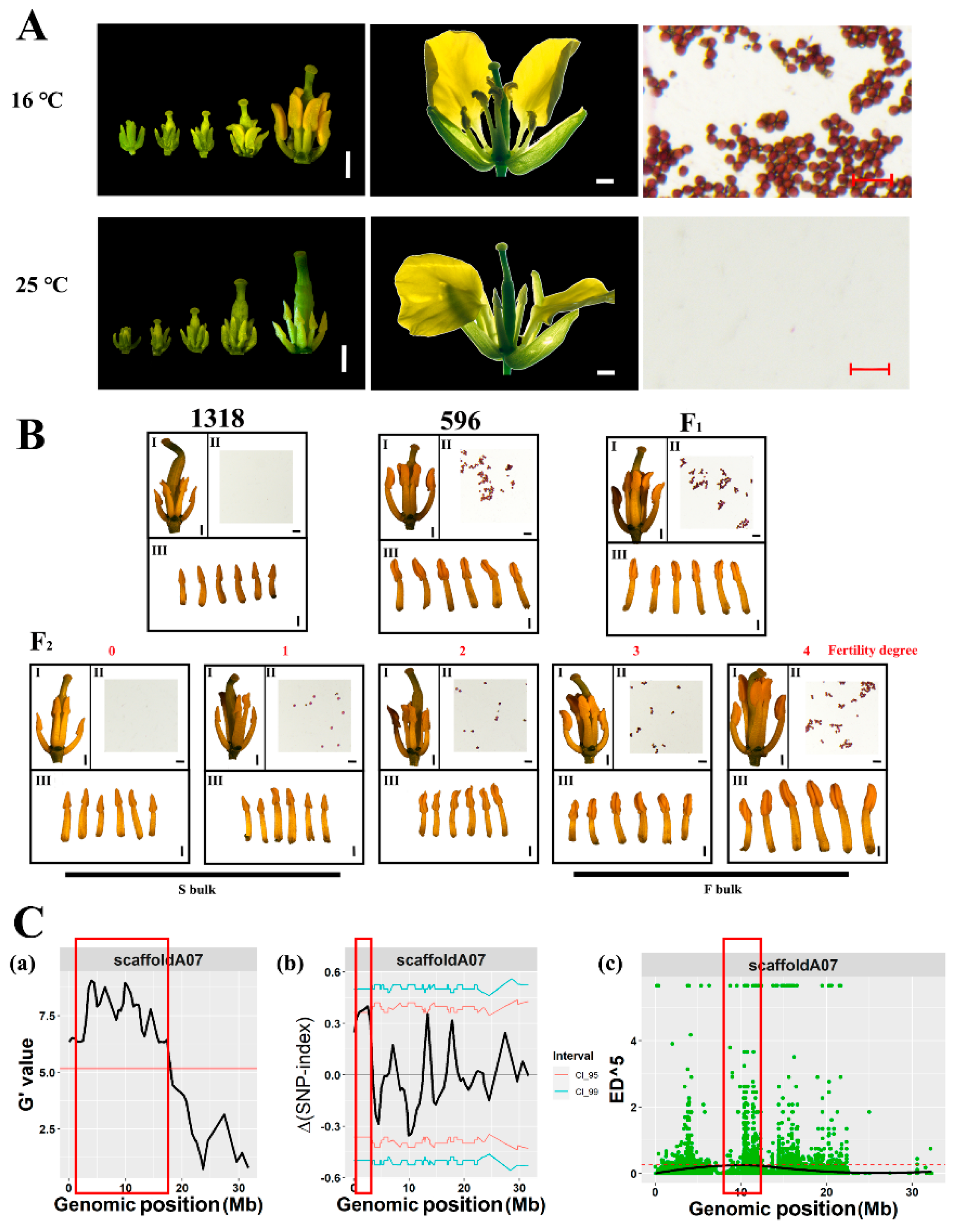
2.1. Flower Morphology
2.2. Identification of Temperature-Sensitive Restorer Locus
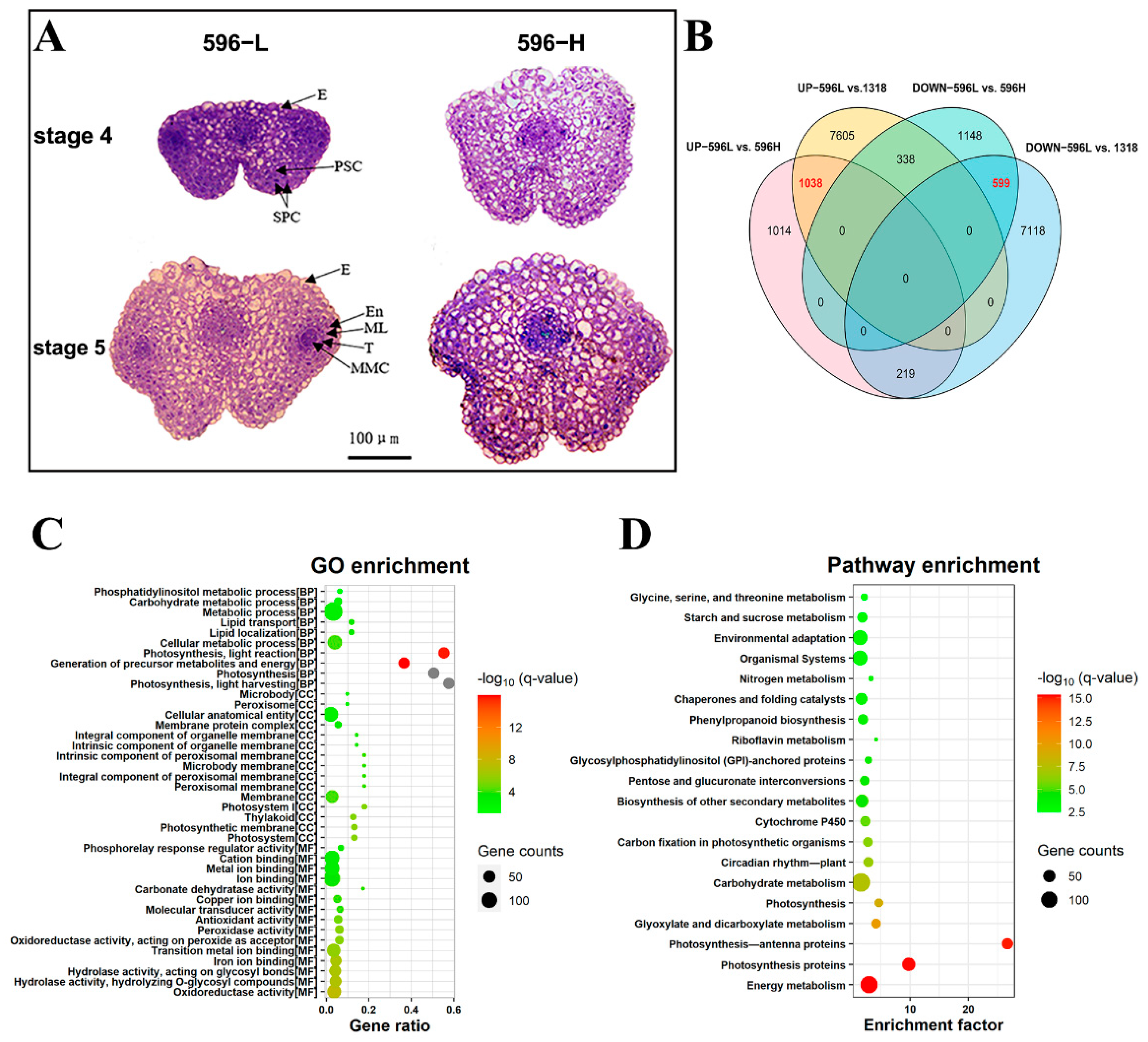
2.3. RNA-Seq and Functional Annotation of Differentially Expressed Genes (DEGs)
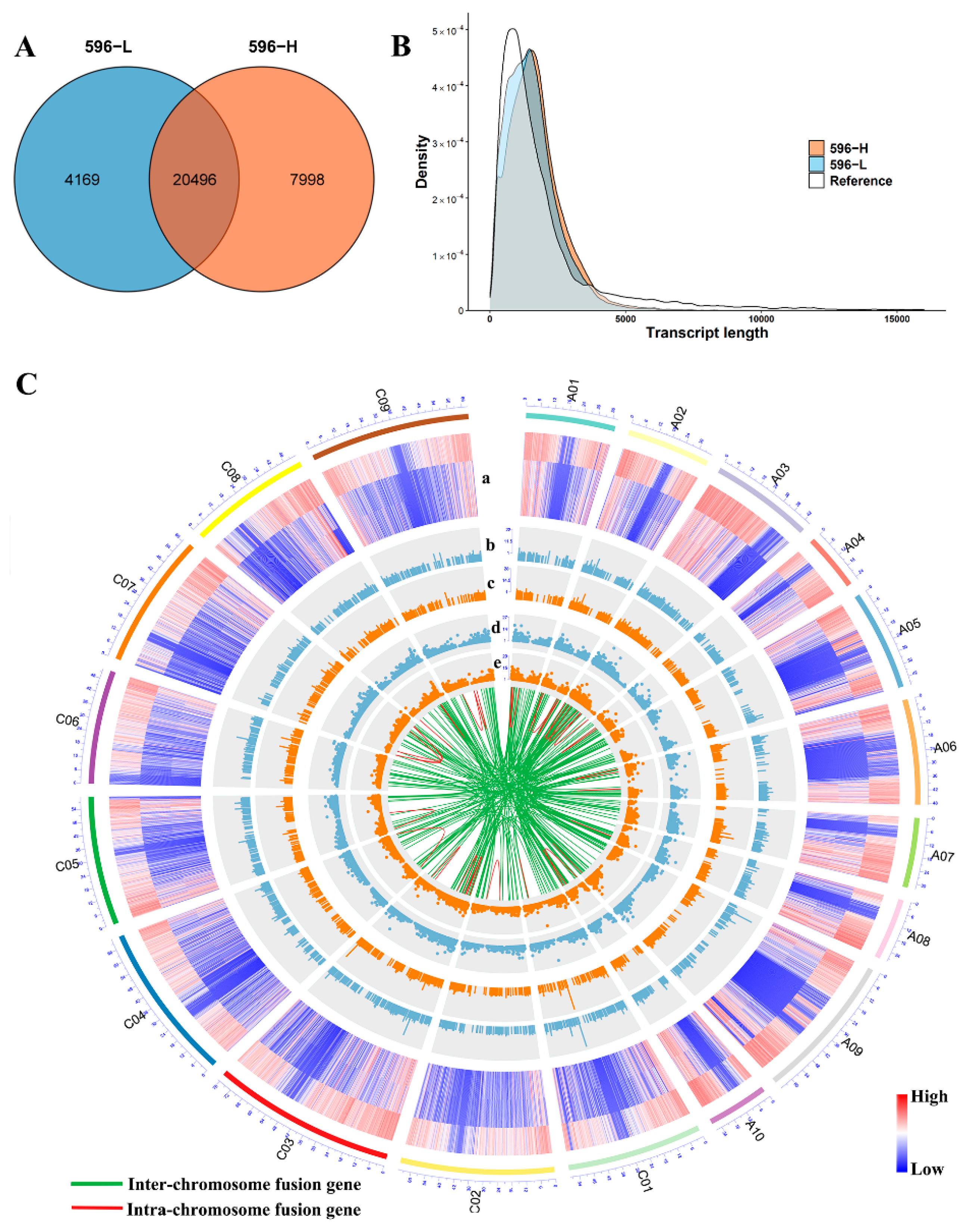
2.4. PacBio Iso-Seq
2.5. AS of Transcripts at Different Ambient Temperatures
2.6. Effect of Ambient Temperature on Mitochondrial Function
2.7. Analysis of the Genes Related to Mitochondrial Retrograde Signal Regulation
2.8. Archesporial Specification Needs Appropriate ROS Levels
2.9. Identification of Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Plant Material, Growth Conditions, and Phenotyping
4.2. Anatomic Analysis
4.3. BSA-Seq and Data Analysis
4.4. RNA Preparation and Sequencing
4.5. Illumina Data Analysis
4.6. PacBio Data Analysis
4.7. Validation by RT-PCR, Quantitative RT-PCR, and Cyclisation RT-PCR
4.8. ROS Analysis of Plant Leaves
4.9. Respiratory Chain Decoupling Treatments
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Linke, B.; Börner, T. Mitochondrial effects on flower and pollen development. Mitochondrion 2005, 5, 389–402. [Google Scholar] [CrossRef]
- Kim, Y.J.; Zhang, D. Molecular Control of Male Fertility for Crop Hybrid Breeding. Trends Plant Sci. 2018, 23, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, H.; Bhat, S.R. Cytoplasmic male sterility in brassicaceae crops. Breed. Sci. 2014, 64, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Guangsheng, Y.; Tingdong, F. The Inheritance of Polima Cytoplasmic Male Sterility in Brassica napus L. Plant Breed. 1990, 104, 121–124. [Google Scholar] [CrossRef]
- Fan, Z.X.; Lei, W.X.; Hong, D.F.; He, J.P.; Wan, L.L.; Xu, Z.H.; Liu, P.W.; Yang, G.S. Development and primary genetic analysis of a fertility temperature-sensitive polima cytoplasmic male sterility restorer in Brassica napus. Plant Breed. 2007, 136, 297–301. [Google Scholar] [CrossRef]
- Handa, H.; Gualberto, J.M.; Grienenberger, J.M. Characterization of the mitochondrial orfB gene and its derivative, orf224, a chimeric open reading frame specific to one mitochondrial genome of the “Polima” male-sterile cytoplasm in rapeseed (Brassica napus L.). Curr. Genet. 1995, 28, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Menassa, R.; L’Homme, Y.; Brown, G.G. Post-transcriptional and developmental regulation of a CMS-associated mitochondrial gene region by a nuclear restorer gene. Plant J. 1999, 17, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Yang, G.S.; Fu, T.D.; Li, Y. Transcriptional control of orf224/atp6 by the pol CMS restorer Rfp gene in Brassica napus L. Acta Genet. Sin. 2003, 30, 469–473. [Google Scholar] [PubMed]
- Liu, Z.; Yang, Z.H.; Wang, X.; Li, K.D.; An, H.; Liu, J.; Yang, G.S.; Fu, T.D.; Yi, B.; Hong, D.F. A Mitochondria-Targeted PPR Protein Restores pol Cytoplasmic Male Sterility by Reducing orf224 Transcript Levels in Oilseed Rape. Mol. Plant 2016, 9, 1082–1084. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Yang, Z.H.; Yi, B.; Wen, J.; Shen, J.X.; Tu, J.X.; Ma, C.Z.; Fu, T.D. Comparative transcript profiling of the fertile and sterile flower buds of pol CMS in B. napus. BMC Genomics 2014, 15, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Farooq, Z.; Chu, L.; Liu, J.; Wang, H.; Guo, J.; Tu, J.; Ma, C.; Dai, C.; Wen, J.; et al. High-generation near-isogenic lines combined with multi-omics to study the mechanism of polima cytoplasmic male sterility. BMC Plant Biol. 2021, 21, 130. [Google Scholar] [CrossRef]
- Barnabás, B.; Jäger, K.; Fehér, A. The effect of drought and heat stress on reproductive processes in cereals. Plant Cell Environ. 2008, 31, 11–38. [Google Scholar] [CrossRef]
- Jagadish, K.S.V.; Craufurd, P.; Shi, W.; Oane, R. A phenotypic marker for quantifying heat stress impact during microsporogenesis in rice (Oryza sativa L.). Funct. Plant Biol. 2014, 41, 48–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, J.P.; Sharkey, T.D. Pollen development at high temperature and role of carbon and nitrogen metabolites. Plant Cell Environ. 2019, 42, 2759–2775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelliher, T.; Walbot, V. Hypoxia triggers meiotic fate acquisition in maize. Science 2012, 337, 345–348. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhou, M.; Yang, Y.; Li, J.; Zhu, L.; Jiang, D.; Dong, J.; Liu, Q.; Gu, L.; Zhou, L.; et al. RNase Z S1 processes Ub L40 mRNAs and controls thermosensitive genic male sterility in rice. Nat. Commun. 2014, 5, 4884. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Liu, Q.; Zhang, L.; Mao, B.; Yan, D.; Jin, Q.; He, Z. Fine mapping and candidate gene analysis of the novel thermo-sensitive genic male sterility tms9-1 gene in rice. Theor. Appl. Genet. 2014, 127, 1173–1182. [Google Scholar] [CrossRef]
- Yu, J.; Han, J.; Kim, Y.J.; Song, M.; Yang, Z.; He, Y.; Fu, R.; Luo, Z.; Hu, J.; Liang, W.; et al. Two rice receptor-like kinases maintain male fertility under changing temperatures. Proc. Natl. Acad. Sci. USA 2017, 114, 12327–12332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Liu, Q.; Li, J.; Jiang, D.; Zhou, L.; Wu, P.; Lu, S.; Li, F.; Zhu, L.; Liu, Z.; et al. Photoperiod- and thermo-sensitive genic male sterility in rice are caused by a point mutation in a novel noncoding RNA that produces a small RNA. Cell Res. 2012, 22, 649–660. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Yang, J.; Mathioni, S.M.; Yu, J.; Shen, J.; Yang, X.; Wang, L.; Zhang, Q.; Cai, Z.; Xu, C.; et al. PMS1T, producing Phased small-interfering RNAs, regulates photoperiod-sensitive male sterility in rice. Proc. Natl. Acad. Sci. USA 2016, 113, 15144–15149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Gao, K.; Ren, H.; Tang, W. Molecular mechanisms governing plant responses to high temperatures. J. Integr. Plant Biol. 2018, 60, 757–779. [Google Scholar] [CrossRef] [PubMed]
- Kotak, S.; Larkindale, J.; Lee, U.; von Koskull-Döring, P.; Vierling, E.; Scharf, K.D. Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 2007, 10, 310–316. [Google Scholar] [CrossRef]
- Li, X.M.; Chao, D.Y.; Wu, Y.; Huang, X.; Chen, K.; Cui, L.G.; Su, L.; Ye, W.W.; Chen, H.; Chen, H.C.; et al. Natural alleles of a proteasome α2 subunit gene contribute to thermotolerance and adaptation of African rice. Nat. Genet. 2015, 47, 827–833. [Google Scholar] [CrossRef]
- Liu, J.X.; Srivastava, R.; Che, P.; Howell, S.H. An endoplasmic reticulum stress response in Arabidopsis is mediated by proteolytic processing and nuclear relocation of a membrane-associated transcription factor, bZIP28. Plant Cell 2007, 19, 4111–4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, S.H. Endoplasmic reticulum stress responses in plants. Annu. Rev. Plant Biol. 2013, 64, 477–499. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Humbert, S.; Liu, J.X.; Srivastava, R.; Rothstein, S.J.; Howell, S.H. Heat induces the splicing by IRE1 of a mRNA encoding a transcription factor involved in the unfolded protein response in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Shi, Y.; Yang, S. Molecular Regulation of Plant Responses to Environmental Temperatures. Mol. Plant 2020, 13, 544–564. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.Y.; Chen, M.X.; Ye, N.H.; Shi, L.; Ma, K.L.; Yang, J.F.; Cao, Y.Y.; Zhang, Y.; Yoshida, T.; Fernie, A.R.; et al. Proteogenomic analysis reveals alternative splicing and translation as part of the abscisic acid response in Arabidopsis seedlings. Plant J. 2017, 91, 518–533. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Sun, M.; Wang, J.; Lei, M.; Li, C.; Zhao, D.; Huang, J.; Li, W.; Li, S.; Li, J.; et al. PacBio full-length cDNA sequencing integrated with RNA-seq reads drastically improves the discovery of splicing transcripts in rice. Plant J. 2019, 97, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Wang, D.; Zheng, X.; Qin, A.; Zhou, J.; Guo, B.; Chen, Y.; Wen, X.; Ye, W.; Zhou, Y.; et al. Multi-strategic RNA-seq analysis reveals a high-resolution transcriptional landscape in cotton. Nat. Commun. 2019, 10, 4714. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chen, S.; Shi, X.; Liu, D.; Zhao, P.; Lu, Y.; Cheng, Y.; Liu, Z.; Nie, X.; Song, W.; et al. Hybrid sequencing reveals insight into heat sensing and signaling of bread wheat. Plant J. 2019, 98, 1015–1032. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.L.; Liang, F.; Gill, R.A.; Huang, J.Y.; Cheng, X.H.; Liu, Y.Y.; Tong, C.B.; Liu, S.Y. A global survey of the transcriptome of allopolyploid Brassica napus based on single-molecule long-read isoform sequencing and Illumina-based RNA sequencing data. Plant J. 2020, 103, 384–857. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.V.S.K.; Gu, L.; Reddy, A.S.N. Analysis of transcriptome and epitranscriptome in plants using pacbio iso-seq and nanopore-based direct RNA sequencing. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilut, D.C.; Coate, J.E.; Luciano, A.K.; Owens, T.G.; May, G.D.; Farmer, A.; Doyle, J.J. A comparative transcriptomic study of an allotetraploid and its diploid progenitors illustrates the unique advantages and challenges of RNA-Seq in plant species. Am. J. Bot. 2012, 99, 383–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickler, S.R.; Bombarely, A.; Mueller, L.A. Designing a transcriptome next-generation sequencing project for a nonmodel plant species. Am. J. Bot. 2012, 99, 257–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, H.; Xiao, Y.; Conklin, P.A.; Govindarajulu, R.; Kelly, J.A.; Scanlon, M.J.; Whipple, C.J.; Bartlett, M. Bulked-segregant analysis coupled to whole genome sequencing (BSA-Seq) for rapid gene cloning in maize. G3 Genes Genomes Genet. 2018, 8, 3583–3592. [Google Scholar] [CrossRef] [Green Version]
- Xu, K.; Wu, Y.; Song, J.; Hu, K.; Wu, Z.; Wen, J.; Yi, B.; Ma, C.; Shen, J.; Fu, T.; et al. Fine mapping and identification of bnac06.Ftsh1, a lethal gene that regulates the psii repair cycle in brassica napus. Int. J. Mol. Sci. 2021, 22, 2087. [Google Scholar] [CrossRef]
- Sanders, P.M.; Bui, A.Q.; Weterings, K.; McIntire, K.N.; Hsu, Y.C.; Lee, P.Y.; Truong, M.T.; Beals, T.P.; Goldberg, R.B. Anther developmental defects in Arabidopsis thaliana male-sterile mutants. Sex. Plant Reprod. 1999, 11, 297–322. [Google Scholar] [CrossRef]
- De Clercq, I.; Vermeirssen, V.; Van Aken, O.; Vandepoele, K.; Murcha, M.W.; Law, S.R.; Inzé, A.; Ng, S.; Ivanova, A.; Rombaut, D.; et al. The membrane-bound NAC transcription factor ANAC013 functions in mitochondrial retrograde regulation of the oxidative stress response in Arabidopsis. Plant Cell 2013, 25, 3472–3490. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xiao, Q.; Wei, C.; Chen, H.; Chen, X.; Dai, C.; Wen, J.; Ma, C.; Tu, J.; Fu, T.; et al. A mitochondria-localized pentatricopeptide repeat protein is required to restore hau cytoplasmic male sterility in Brassica napus. Theor. Appl. Genet. 2021, 134, 1377–1386. [Google Scholar] [CrossRef]
- Lee, K.; Kang, H. Roles of organellar rna-binding proteins in plant growth, development, and abiotic stress responses. Int. J. Mol. Sci. 2020, 21, 4548. [Google Scholar] [CrossRef]
- Wang, X.; An, Y.; Xu, P.; Xiao, J. Functioning of PPR Proteins in Organelle RNA Metabolism and Chloroplast Biogenesis. Front. Plant Sci. 2021, 12, 627501. [Google Scholar] [CrossRef]
- Hu, J.; Wang, K.; Huang, W.; Liu, G.; Gao, Y.; Wang, J.; Huang, Q.; Ji, Y.; Qin, X.; Wan, L.; et al. The rice pentatricopeptide repeat protein RF5 restores fertility in Hong-Lian cytoplasmic male-sterile lines via a complex with the glycine-rich protein GRP162. Plant Cell 2012, 24, 109–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Yu, C.; Hu, J.; Wang, L.; Dan, Z.; Zhou, W.; He, C.; Zeng, Y.; Yao, G.; Qi, J.; et al. Pentatricopeptide-repeat family protein RF6 functions with hexokinase 6 to rescue rice cytoplasmic male sterility. PNAS 2015, 112, 14984–14989. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Dong, F.; Wang, X.; Wang, T.; Su, R.; Hong, D.; Yang, G. A pentatricopeptide repeat protein restores nap cytoplasmic male sterility in Brassica napus. J. Exp. Bot. 2017, 68, 4115–4123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Lezhneva, L.; Arnal, N.; Quadrado, M.; Mireau, H. The radish Ogura fertility restorer impedes translation elongation along its cognate CMS-causing mRNA. Proc. Natl. Acad. Sci. USA 2021, 118, e2105274118. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, Q.; Yin, P. RNA editing machinery in plant organelles. Sci. China Life Sci. 2018, 61, 162–169. [Google Scholar] [CrossRef]
- Takenaka, M.; Zehrmann, A.; Verbitskiy, D.; Kugelmann, M.; Härtel, B.; Brennicke, A. Multiple organellar RNA editing factor (MORF) family proteins are required for RNA editing in mitochondria and plastids of plants. Proc. Natl. Acad. Sci. USA 2012, 109, 5104–5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, S.; Suzuki, T.; Giegé, P.; Higashiyama, T.; Koizuka, N.; Shikanai, T. The Restorer-of-fertility-like 2 pentatricopeptide repeat protein and RNase P are required for the processing of mitochondrial orf291 RNA in Arabidopsis. Plant J. 2016, 86, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Gillman, J.D.; Bentolila, S.; Hanson, M.R. The petunia restorer of fertility protein is part of a large mitochondrial complex that interacts with transcripts of the CMS-associated locus. Plant J. 2007, 49, 217–227. [Google Scholar] [CrossRef]
- Liang, T.; Chi, W.; Huang, L.; Qu, M.; Zhang, S.; Chen, Z.Q.; Chen, Z.J.; Tian, D.; Gui, Y.; Chen, X.; et al. Bulked segregant analysis coupled with whole-genome sequencing (BSA-Seq) mapping identifies a novel pi21 haplotype conferring basal resistance to rice blast disease. Int. J. Mol. Sci. 2020, 21, 2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Yan, C.; Liu, Y.; Liu, Y.; Jia, Y.; Lavelle, D.; An, G.; Zhang, W.; Zhang, L.; Han, R.; et al. Upregulation of a KN1 homolog by transposon insertion promotes leafy head development in lettuce. Proc. Natl. Acad. Sci. USA 2020, 117, 33668–33678. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, X.; Cao, R.; Jiao, G.; Hu, S.; Shao, G.; Sheng, Z.; Xie, L.; Tang, S.; Wei, X.; et al. CDE4 encodes a pentatricopeptide repeat protein involved in chloroplast RNA splicing and affects chloroplast development under low-temperature conditions in rice. J. Integr. Plant Biol. 2021, 63, 1724–1739. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Shi, X.; Li, S.H.A.; Zhang, L.; Song, X. Oxidative stress and aberrant programmed cell death are associated with pollen abortion in isonuclear alloplasmic male-sterile wheat. Front. Plant Sci. 2018, 9, 595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedro Gonçalves, A.; Videira, A. Mitochondrial type II NAD(P)H dehydrogenases in fungal cell death. Microb. Cell 2015, 2, 68–73. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.; De Clercq, I.; Van Aken, O.; Law, S.R.; Ivanova, A.; Willems, P.; Giraud, E.; Van Breusegem, F.; Whelan, J. Anterograde and retrograde regulation of nuclear genes encoding mitochondrial proteins during growth, development, and stress. Mol. Plant 2014, 7, 1075–1093. [Google Scholar] [CrossRef] [Green Version]
- Vanlerberghe, G.C. Alternative oxidase: A mitochondrial respiratory pathway to maintain metabolic and signaling homeostasis during abiotic and biotic stress in plants. Int. J. Mol. Sci. 2013, 14, 6805–6847. [Google Scholar] [CrossRef]
- Clifton, R.; Millar, A.H.; Whelan, J. Alternative oxidases in Arabidopsis: A comparative analysis of differential expression in the gene family provides new insights into function of non-phosphorylating bypasses. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 730–741. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Selinski, J.; Mao, C.; Zhu, Y.; Berkowitz, O.; Whelan, J. Linking mitochondrial and chloroplast retrograde signalling in plants. Philos. Trans. R. Soc. B Biol. Sci. 2020, 375, 20190410. [Google Scholar] [CrossRef]
- Li, Z.; Howell, S.H. Heat stress responses and thermotolerance in Maize. Int. J. Mol. Sci. 2021, 22, 948. [Google Scholar] [CrossRef]
- Wan, S.; Jiang, L. Endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) in plants. Protoplasma 2016, 253, 753–764. [Google Scholar] [CrossRef]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Iwata, Y.; Koizumi, N. Plant transducers of the endoplasmic reticulum unfolded protein response. Trends Plant Sci. 2012, 17, 720–727. [Google Scholar] [CrossRef]
- Liu, R.; Xia, R.; Xie, Q.; Wu, Y. Endoplasmic reticulum-related E3 ubiquitin ligases: Key regulators of plant growth and stress responses. Plant Commun. 2021, 2, 100186. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansfeld, B.N.; Grumet, R. QTLseqr: An R Package for Bulk Segregant Analysis with Next-Generation Sequencing. Plant Genome 2018, 11, 180006. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Landmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Anders, S. Analysing RNA-Seq data with the DESeq package. Mol. Biol. 2012, 43, 1–17. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, Q.; Gao, Z.F.; Zhang, D.; Zhao, B.G.; Dong, F.Q.; Fu, C.X.; Liu, L.J.; Wang, B.C. The developmental dynamics of the Populus stem transcriptome. Plant Biotechnol. J. 2019, 17, 206–219. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Description |
|---|---|
| BnaA07G0014700ZS | Mitochondrial outer membrane import complex protein METAXIN |
| BnaA07G0016300ZS | Pentatricopeptide repeat-containing protein |
| BnaA07G0019600ZS | 60S ribosomal protein L6 |
| BnaA07G0027800ZS | Pentatricopeptide repeat-containing protein |
| BnaA07G0028600ZS | Rhodanese-like domain-containing protein 19, mitochondrial |
| BnaA07G0029700ZS | Mitochondrial phosphate carrier protein 1 |
| BnaA07G0031500ZS | Pentatricopeptide repeat-containing protein |
| BnaA07G0031700ZS | Isocitrate dehydrogenase (NAD) regulatory subunit 2 |
| BnaA07G0033700ZS | Arabidopsis phospholipase-like protein (PEARLI 4) |
| BnaA07G0034100ZS | Pentatricopeptide repeat-containing protein |
| BnaA07G0055400ZS | Pentatricopeptide repeat-containing protein |
| BnaA07G0056400ZS | Aconitate hydratase 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, Q.; Wang, H.; Chen, H.; Chen, X.; Wen, J.; Dai, C.; Ma, C.; Tu, J.; Shen, J.; Fu, T.; et al. Molecular Analysis Uncovers the Mechanism of Fertility Restoration in Temperature-Sensitive Polima Cytoplasmic Male-Sterile Brassica napus. Int. J. Mol. Sci. 2021, 22, 12450. https://doi.org/10.3390/ijms222212450
Xiao Q, Wang H, Chen H, Chen X, Wen J, Dai C, Ma C, Tu J, Shen J, Fu T, et al. Molecular Analysis Uncovers the Mechanism of Fertility Restoration in Temperature-Sensitive Polima Cytoplasmic Male-Sterile Brassica napus. International Journal of Molecular Sciences. 2021; 22(22):12450. https://doi.org/10.3390/ijms222212450
Chicago/Turabian StyleXiao, Qing, Huadong Wang, Hui Chen, Xiaohan Chen, Jing Wen, Cheng Dai, Chaozhi Ma, Jinxing Tu, Jinxiong Shen, Tingdong Fu, and et al. 2021. "Molecular Analysis Uncovers the Mechanism of Fertility Restoration in Temperature-Sensitive Polima Cytoplasmic Male-Sterile Brassica napus" International Journal of Molecular Sciences 22, no. 22: 12450. https://doi.org/10.3390/ijms222212450
APA StyleXiao, Q., Wang, H., Chen, H., Chen, X., Wen, J., Dai, C., Ma, C., Tu, J., Shen, J., Fu, T., & Yi, B. (2021). Molecular Analysis Uncovers the Mechanism of Fertility Restoration in Temperature-Sensitive Polima Cytoplasmic Male-Sterile Brassica napus. International Journal of Molecular Sciences, 22(22), 12450. https://doi.org/10.3390/ijms222212450