3. Discussion
Elevated levels of serum calcium and phosphate are associated with cardiovascular events and mortality in the general population [
1,
2,
3,
4] and subjects with pre-dialysis CKD [
32,
33,
34] or ESRD [
35]. However, hyperphosphatemia accounts for medial arterial calcification [
6], which is also common in patients with CKD and ESRD [
36], but does not provoke atherosclerosis [
6] or intimal calcification [
37]. The mechanism of accelerated coronary artery disease or cerebrovascular disease development in patients with altered mineral homeostasis was previously unclear. Therefore, a need remained to identify mineral-related factors capable of triggering endothelial dysfunction and adventitial/perivascular inflammation, the two major drivers of atherosclerosis [
11,
26,
38].
Insights into the molecular basis of extraskeletal calcification inhibition pinpointed the central role of fetuin-A in the maintenance of mineral homeostasis [
7,
8]. Interactions of fetuin-A with calcium and phosphate result in crystalline nanosized CPPs, which represent a vehicle for the clearance of excessive serum calcium and phosphate [
21]. In vitro and in vivo experiments demonstrated that CPPs are unable to cause cardiovascular calcification per se, attesting their protective significance [
10]. Nevertheless, CPPs exerted significant cytotoxic effects on ECs [
10]. These findings imply an ambiguous role of CPPs in vascular physiology. We hypothesised that while CPPs guard blood vessels from medial calcification, which rapidly causes mechanical incompetence of the arteries, these mineral complexes may also have pathogenic effects ultimately disrupting vascular homeostasis by promoting endothelial dysfunction and adventitial or perivascular inflammation.
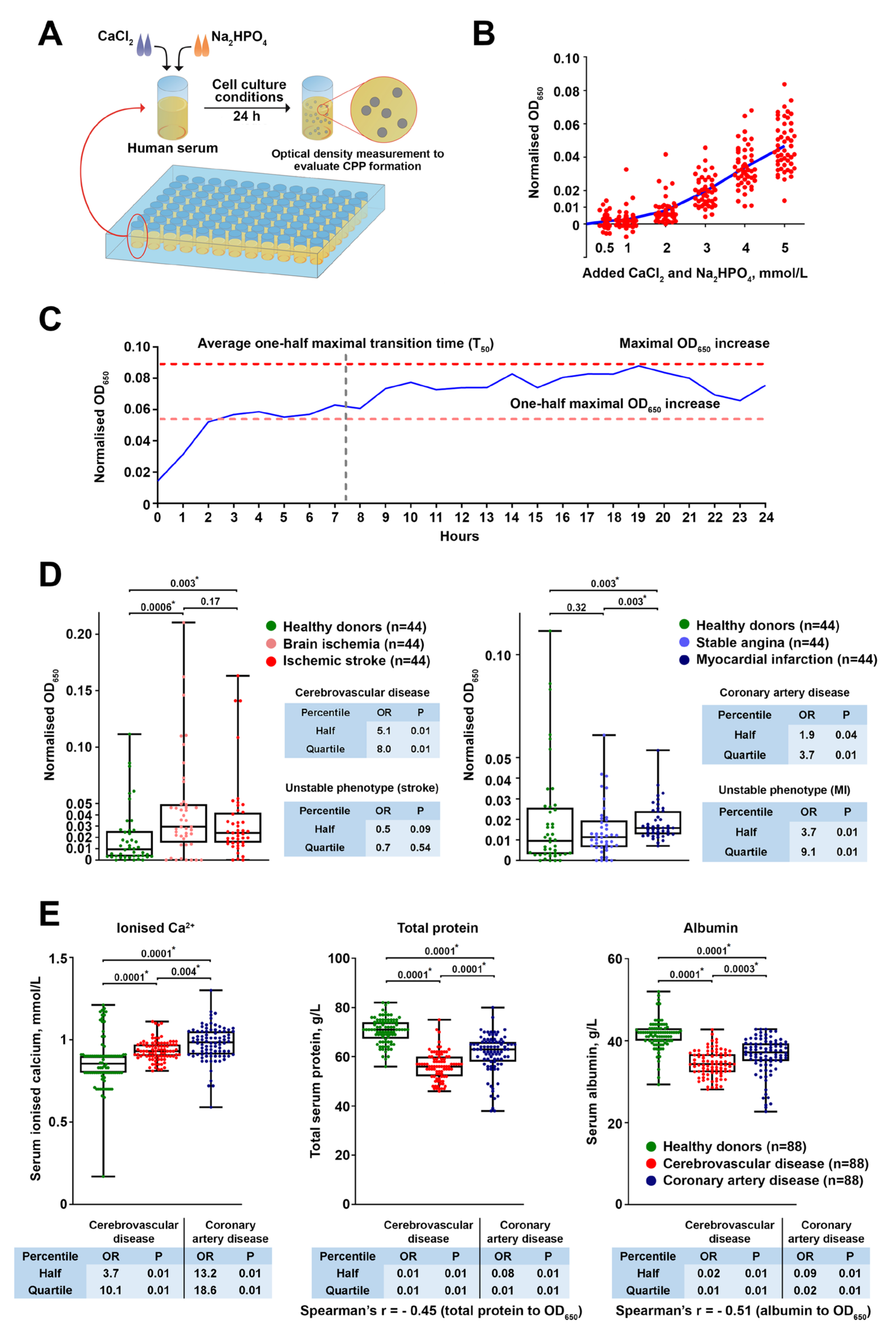
We first sought to explore the clinical relevance of CPP formation in a CVD setting. To this end, we applied a simple assay including a single measurement of optical density increase after 24 h of serum incubation in cell culture conditions following the addition of calcium salts and phosphates, thereby evaluating CPP generation propensity. Patients with coronary artery disease or cerebrovascular disease were characterised by considerably higher CPP yield than healthy blood donors because of increased ionised calcium and reduced albumin, a protein playing a pivotal role in preventing extraskeletal mineralisation by Ca
2+ binding. Notably, the CVD risk pattern in relation to CPP, ionised calcium, and albumin levels was reminiscent of that observed for serum calcium and phosphate in population studies [
1,
2,
3,
4], whereas phosphate and fetuin-A concentrations did not affect cardiovascular risk in our investigation. Taken together, these findings suggest insufficient Ca
2+-binding capacity as a leading mechanism disturbing mineral homeostasis in patients with CVD and promoting CPP formation. As elevated CPP production was also associated with unstable coronary plaque phenotype, we propose that our approach can be implemented in clinical trials both for CVD risk assessment and for higher-accuracy prognostication in patients with coronary artery disease. Intriguingly, serum CPP generation propensity was significantly higher in patients with cerebrovascular disease in comparison with those suffering from coronary artery disease (including those with myocardial infarction). The reason for this finding may include the greater reduction in total protein and albumin, probably because of the lower eGFR (average 74.00 in cerebrovascular disease patients vs. 86.00 in coronary artery disease patients), though these values still did not reach those defining clinically significant CKD. Although reduced eGFR might be partially responsible for the higher CPP generation propensity in patients with cerebrovascular disease or myocardial infarction, the restriction of patient and control cohorts to the subjects having an eGFR >90 mL/min/1.73 m
2 did not change the results, as cardiovascular disease patient cohorts still demonstrated higher CPP generation propensity, increased serum ionised calcium, and reduced total protein and albumin levels. Hence, we found no conclusive evidence that eGFR considerably influences CPP formation in the serum. Nevertheless, these findings require replication in subsequent epidemiological studies, ideally in a comprehensive multicentre research trial aimed at the evaluation of mineral homeostasis parameters in patients with cardiovascular disease and healthy volunteers.
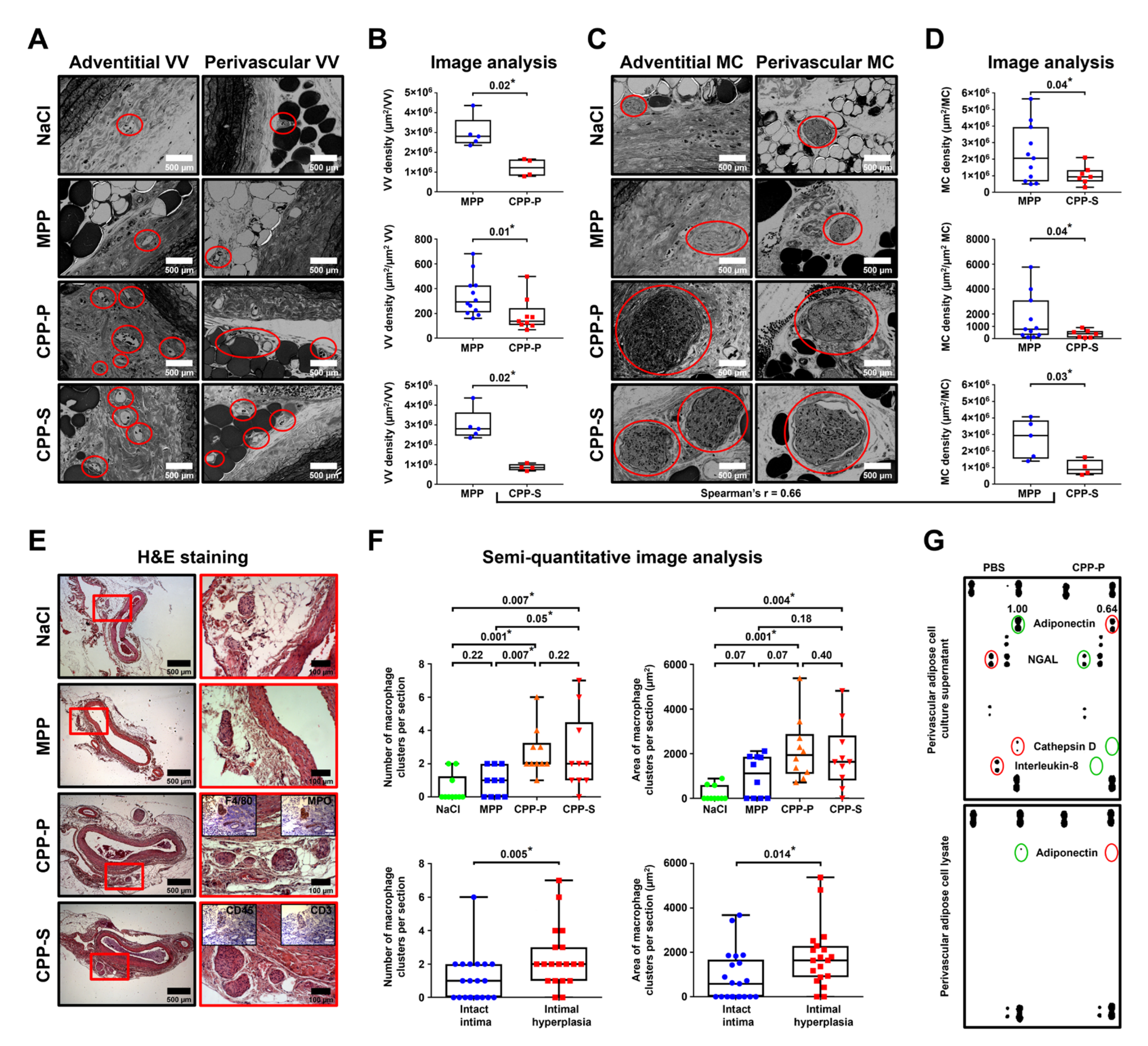
To exclude the possible confounding impacts of cardiovascular risk factors, we intravenously injected CPPs to normolipidemic and normotensive Wistar rats employing various protocols, some of which included artificial vascular injury. Regardless of CPP administration regimen or endothelial integrity, these mineral particles provoked an intimal hyperplasia and adventitial/perivascular inflammation, reflected by increased VV and MCs. These results provide a proof of concept that aggregation of calcium and phosphate into the CPPs disrupts vascular homeostasis, although the details of CPP fate starting from their formation in the blood until their internalisation by ECs are yet to be elucidated.
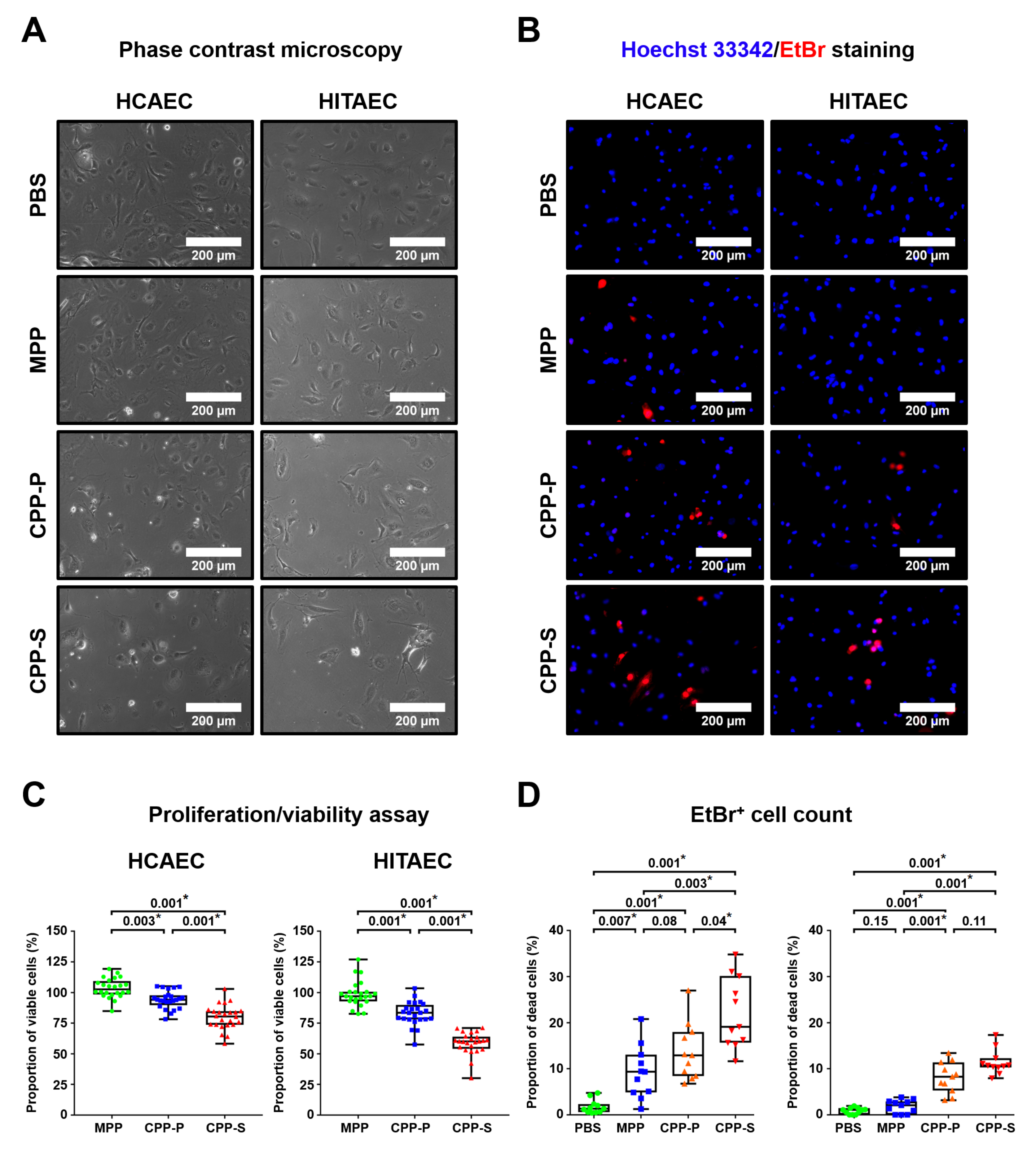
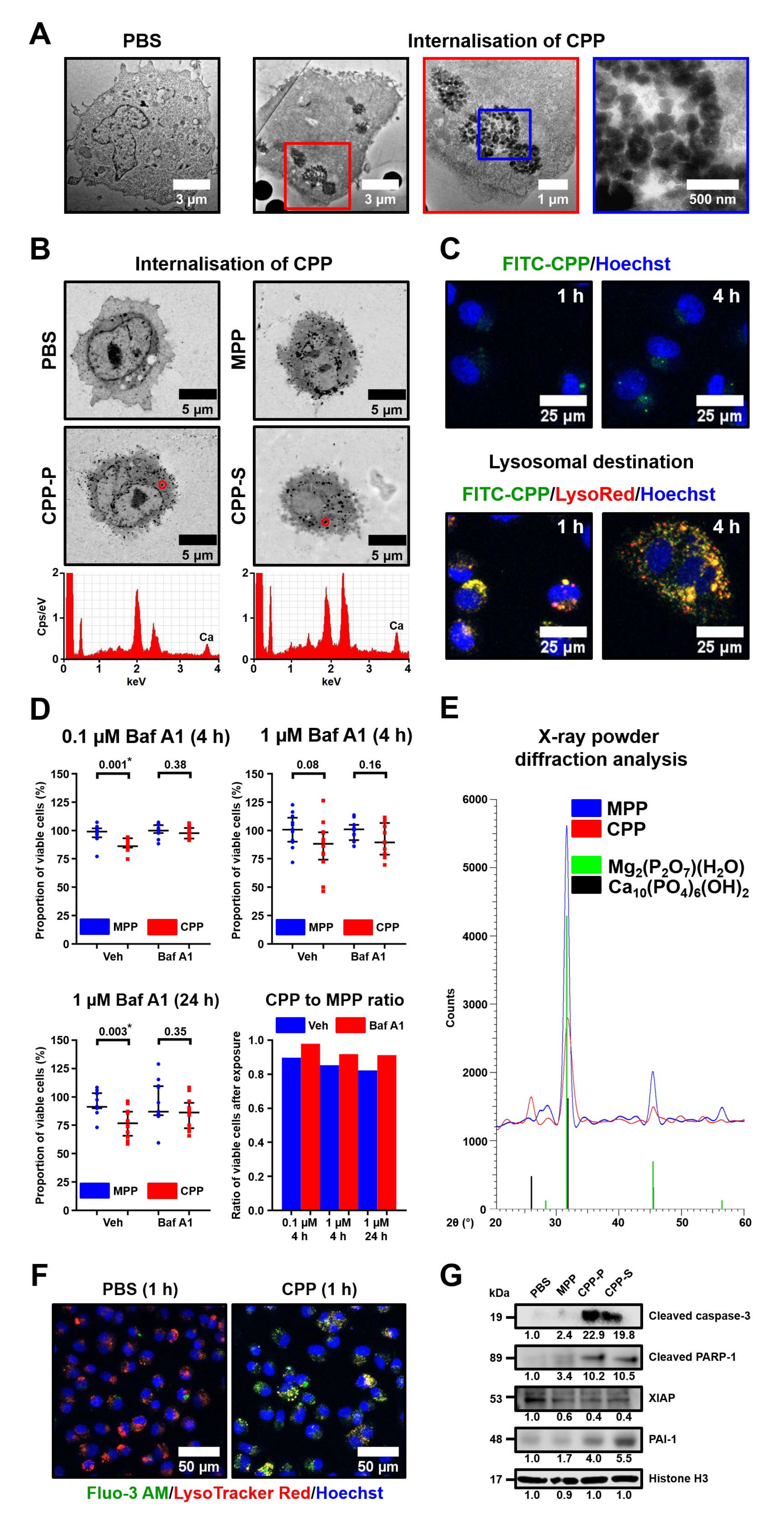
Subsequent experiments on human arterial endothelial cell cultures confirmed the pronounced endothelial toxicity of CPPs (but not MPPs) upon their dissolution in lysosomes, suggesting a mechanism for the deleterious consequences of CPP formation. In agreement with recently acknowledged molecularly oriented definitions of cell death subroutines [
17], we defined the CPP-induced mode of cellular demise as lysosome-dependent cell death, demarcated by primary lysosomal membrane permeabilisation and mediated via executioner caspases. As lysosomal inhibitor bafilomycin A1 abrogated cell death in CPP-treated cells while not endowing MPPs with cytotoxicity, we inferred and showed that the translocation of Ca
2+ ions from the lysosomes to the cytosol triggered mitochondrial outer membrane permeabilisation, evidenced by XIAP downregulation and followed by cleavage of caspase-3, further implementing the regulated cell death. Our prior results denoted the concomitant cleavage of caspase-9 in support of these observations [
10]. These findings are concordant with those obtained earlier for vascular smooth muscle cells [
39] but contradict the previous assumptions that the dissolution of CPPs provokes lysosomal rupture by creating an osmotic pressure gradient between the lysosomes and cytosol [
40] (since the dissolution of MPPs would also simulate this scenario, yet these particles lack cytotoxicity). Another mechanism of CPP-related cytotoxicity may be the generation of reactive oxygen species [
24,
41]; however, we did not document conclusive signs of this phenomenon.
The augmented release of proinflammatory cytokines (i.e., IL-6, IL-8, MIF, and CXCL1) enhanced leukocyte adhesion and increased the expression of transcription factors Snail and Slug, suggesting endothelial activation and endothelial-to-mesenchymal transition as prominent molecular consequences upon CPP internalisation by ECs. This corresponds to earlier data obtained on ECs [
10], vascular smooth muscle cells [
42], and macrophages [
24], collectively suggesting chronic vascular inflammation as a major mechanism mediating the pathogenic effects of CPPs on blood vessels. Yet, vascular cell populations showed a differential response to CPPs, as ECs demonstrated an augmented production of IL-6 and IL-8, while perivascular adipocytes ceased IL-8 secretion upon CPP treatment. The orchestration of vascular inflammation by CPPs (e.g., the paracrine effects of CPP-exposed ECs, vascular smooth muscle cells, adventitial macrophages, and perivascular adipocytes) and its molecular mechanisms in vivo are yet to be elucidated. Although CPPs mount a pro-inflammatory response in ECs [
10], vascular smooth muscle cells [
42], and macrophages [
19,
24,
41], these effects may be cell- and context-specific and require further investigation in co-culture or conditioned medium experiments.
In contrast to magnesium phosphate hydrate particles containing a negligible amount of calcium (MPPs), both amorphous and spherical aggregates of calcium, phosphate, and serum proteins (CPP-P) and crystalline and spindle-shaped calcium phosphate protein-coated particles (CPP-S) caused pathogenic effects on arteries upon intravenous administration to normolipidemic/normotensive animals and after the addition to EC cultures. The pathogenicity of CPPs for ECs regardless of their ripening suggests that these particles have detrimental effects immediately upon their formation, thereby emphasising the clinical relevance of excessive serum’s propensity to form CPPs.
The limitations of our study include:
We did not apply the T50 test, which reflects the rate of transformation of CPP-P to CPP-S in the human serum, as we think the total propensity of serum to form CPPs (measured by OD650 increment) is a suitable and pathophysiologically relevant alternative that might be less sophisticated for routine clinical use. The T50 test has its advantages (e.g., validation of multiple patient cohorts), yet the mineral stress test (OD650 increase) applied in this study is not contradictory and represents another variation in serum calcification propensity measurement.
The concentration of +2 mmol/L CaCl2 and Na2HPO4 added is supraphysiological and such excessive Ca/P concentrations are rarely encountered, except in patients with severe osteopenia/osteoporosis and hyperparathyroidism who frequently show hypercalcemia and those with ESRD who generally suffer from hyperphosphatemia. Yet, this Ca/P increase represents a mineral stress test for the measurement of serum’s propensity to form CPPs; lower amounts of added CaCl2 and Na2HPO4 concentrations inconsistently triggered CPP generation. Increased CPP generation propensity (higher OD650 increase values) was associated with higher ionised calcium (Ca2+) and lower albumin, thereby reflecting disturbed mineral homeostasis in cohorts of patients with coronary artery disease and cerebrovascular disease compared with healthy blood donors.
In our study, we did not perform animal section immunostainings as these sections were formalin-fixed for significantly longer than 24 h and therefore were unsuitable for IHC-P or IHC-Fr applications, although electron microscopy analysis well-distinguishes blood microvessels from the background by the combination of clearly visible vessel lumen, endothelial layer, and (optionally) red blood cells within the lumen. In addition, macrophages also have morphological patterns distinct from other adventitial cell populations (e.g., fibroblasts and lymphocytes), in particular when assembled into clusters, which is not characteristic for fibroblasts. IHC stainings of the aortic sections from another experiment showed positive staining of MCs for F4/80 and myeloperoxidase but not for T lymphocyte CD3 marker (
Figure 5E, insets).
We did not study CPP’s effects on vascular smooth muscle cells. However, we stained aortas from both animal models with alizarin red S and found no signs of vascular calcification, suggesting that the osteogenic differentiation of vascular smooth muscle cells is not among the major consequences of CPP administration in vivo. In a balloon injury model, endothelial denudation and mechanical vascular injury collectively led to intimal hyperplasia because of damaged internal elastic lamina and the possible proliferative activation of vascular smooth muscle cells. We suggest that exposure of balloon-injured blood vessel to CPPs might also contribute to the contractile-to-synthetic phenotypic switch of vascular smooth muscle cells, although we could not confirm this because the sections could not be used for immunohistochemistry. Whether this scenario occurs in intact blood vessels is currently unclear, whereas endothelial dysfunction might result in pathological paracrine signalling to adjacent vascular smooth muscle cells.
To summarise (
Figure 9), we here found that patients with CVD are prone to the formation of CPPs as a result of disturbed mineral homeostasis (in particular to reduced Ca
2+-binding capacity). In other words, decreased albumin (the most abundant Ca
2+-binding protein) leads to the reciprocal increase in ionised calcium (Ca
2+), which provides a chemical substrate for the elevated CPP generation. We for the first time showed that CPPs are able to cause intimal hyperplasia due to endothelial dysfunction and adventitial/perivascular inflammation, potentially through the increased leukocyte adhesion and excessive release of pro-inflammatory cytokines by ECs. Hence, we propose CPPs as a mechanistic link explaining the increased incidence of CVD in individuals with altered mineral homeostasis. We suggest disturbed mineral homeostasis (in particular reduced albumin, increased ionised calcium, and elevated serum CPP formation propensity) as an additional risk factor of cardiovascular disease, although its contribution might be less significant in comparison with other major established cardiovascular risk factors (i.e., dyslipidaemia, arterial hypertension, overweight/obesity, and carbohydrate metabolism disorders), in concert with the epidemiological studies that showed an association between increased calcium, decreased albumin, and major adverse cardiovascular events [
1,
2,
3,
4]. The pathophysiological importance of excessive CPP formation in experimental cardiovascular disease settings and relevant clinical scenarios is underscored by the association of elevated CPP formation propensity with coronary artery disease and cerebrovascular disease (especially myocardial infarction and ischaemic stroke), the development of intimal hyperplasia in normolipidemic/normotensive rats upon regular intravenous administration of CPPs, the aggravation of intimal hyperplasia in balloon-injured aortas after CPP injections, and the pronounced endothelial dysfunction in primary arterial EC cultures treated with CPPs.
From a translational perspective, CPPs may be targeted with Ca
2+ chelators, which demonstrated certain efficiency in the prevention of cardiovascular events after index myocardial infarction [
43], especially in patients with a concomitant diabetes mellitus [
44] and peripheral artery disease [
45]. Another promising therapeutic option is Mg
2+ supplementation, which delays the maturation of primary-to-secondary CPPs in a dose-dependent manner [
46], reduces CPP load and abates inflammation in haemodialysed patients [
47], and inhibits vascular calcification in uremic rats [
48]. Alternatively, 4,6-di-O-(methoxy-diethyleneglycol)-myo-inositol-1,2,3,5-tetrakis(phosphate), an inositol phosphate analogue, displayed similar effects [
49]. A recent study showed that replacement of calcium carbonate with lanthanum carbonate as a phosphate binder lowers serum CPP levels in patients with ESRD [
50], in addition to attenuating aortic calcification [
51]. Collectively, our results provide a rationale for the development and testing of the aforementioned and novel modalities of anti-CPP therapy potentially beneficial for patients with disturbed mineral homeostasis.
4. Materials and Methods
4.1. Evaluation of Serum Propensity for Calciprotein Particle (CPP) Formation
This study was approved by the local ethical committee of the Research Institute for Complex Issues of Cardiovascular Diseases (Kemerovo, Russia, protocol numbers 20160404 and 20180803, dates of approval: 4 April 2016 and 3 August 2018, respectively), and a written informed consent was provided by all study participants after receiving a full explanation of the study. The investigation was carried out in accordance with the Good Clinical Practices and the Declaration of Helsinki. The criteria for inclusion were: (1) performance of carotid endarterectomy due to brain ischemia or ischemic stroke or coronary artery bypass graft surgery because of stable angina or hospitalisation due to myocardial infarction or participance in the Prospective Urban Rural Epidemiology Study in combination with the absence of symptomatic carotid or coronary atherosclerosis; (2) a signed written informed consent to be enrolled. One criterion of exclusion was incomplete investigation regardless of the reason; in this case, we enrolled another subject with similar age, sex, and clinicopathological features who met the inclusion criteria. In total, we consecutively enrolled 88 healthy volunteers, 44 patients with brain ischemia who required carotid endarterectomy, 44 patients with ischemic stroke, 44 patients with stable angina who required coronary artery bypass graft surgery, and 44 patients with myocardial infarction.
Cerebrovascular disease (brain ischemia and ischemic stroke), stable angina, and myocardial infarction, as well as comorbid conditions (arterial hypertension, chronic heart failure, chronic obstructive pulmonary disease, asthma, chronic kidney disease, diabetes mellitus, overweight, and obesity) were diagnosed and treated according to the respective guidelines of European Society of Cardiology, Global Initiative for Chronic Obstructive Lung Disease, Global Initiative for Asthma, Kidney Disease: Improving Global Outcomes, American Diabetes Association, and European Association for the Study of Obesity. eGFR was calculated according to the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation. The left ventricular ejection fraction was evaluated by means of echocardiography (Sonos 2500 Diagnostic Ultrasound System, Hewlett Packard, Palo Alto, CA, USA). The number of affected coronary arteries in patients with myocardial infarction was defined during coronary angiography (Innova 3100 Cardiac Angiography System, General Electric Healthcare, Chicago, IL, USA), whereas extracranial artery stenosis in those with cerebrovascular disease was assessed using colour duplex screening (Vivid 7 Dimension Ultrasound System, General Electric Healthcare, Chicago, IL, USA). Data on age, sex, smoking status, and pharmacological anamnesis were collected at the time of admission. The detailed characteristics of the study sample are presented in
Table S2.
To determine the optimal amount of added CaCl2 and Na2HPO4 for the assay, the serum of healthy (i.e., asymptomatic) volunteers (n = 49) was supersaturated with calcium and phosphate (0.5, 1, 2, 3, 4, or 5 mmol/L) by adding equal concentrations of CaCl2 (21115, Sigma-Aldrich, St. Louis, MO, USA) and Na2HPO4 (94046, Sigma-Aldrich, St. Louis, MO, USA). Upon 24 h incubation in cell culture conditions (37 °C, 5% CO2, and high humidity), we monitored the change in optical density at a wavelength of 650 nm (OD650, Multiskan Sky, Thermo Fisher Scientific, Waltham, MA, USA) s compared to the same serum without calcium/phosphate supplementation. To evaluate CPP formation over time, equimolar (2 mmol/L) concentrations of CaCl2 and Na2HPO4 were added to the serum of healthy individuals (n = 42) with the subsequent time-lapse (1 h time frame) measurement of OD650 during 24 h and calculation of the: (1) average one-half maximal transition time (T50) reflecting the average time required for reaching the one-half of the maximal OD650 increase across the samples; (2) one-half maximal OD650 increase reflecting the average OD650 value equal to the half of the maximal OD650 increase across the samples. To characterise serum-generated CPPs, we diluted the supersaturated and 24 h incubated serum 4-fold to reduce its viscosity, centrifuged it at 200,000× g for 2 h (Optima MAX-XP, Beckman Coulter, Brea, CA, USA), and lysed the sediment in RIPA buffer (89901, Thermo Fisher Scientific, Waltham, MA, USA) for 30 min to lyse the extracellular vesicles. Then, the lysate was centrifuged again at 200,000× g for 1 h, and the pellet was washed in sterile-filtered double-distilled water with the following centrifugation at 200,000× g for 1 h. The serum-generated CPPs were characterised by means of scanning electron microscopy (S-3400N, Hitachi, Tokyo, Japan) and elemental analysis (energy-dispersive X-ray spectroscopy, XFlash 4010, Bruker, Billerica, MA, USA) upon the resuspension of washed CPP pellet in sterile-filtered double distilled water and pipetting the resuspended CPPs on a double-sided adhesive conductive carbon tape (16084-7, Ted Pella, Redding, CA, USA).
Equimolar (2 mmol/L) concentrations of CaCl2 and Na2HPO4 were added to the serum of healthy volunteers (n = 88) or patients with brain ischemia (n = 44), ischemic stroke (n = 44), stable angina (n = 44), or myocardial infarction (n = 44) in 96-well plates (100 μL per well). Upon 24 h incubation in cell culture conditions, we monitored the change in OD650 compared to the same serum without calcium/phosphate supplementation. Serum concentrations of ionised calcium, phosphate, total protein, and albumin were measured using an automated biochemical analyser (Konelab 60i, Thermo Fisher Scientific, Waltham, MA, USA), whereas fetuin-A level was determined by enzyme-linked immunosorbent assay (RD191037100, BioVendor, Heidelberg, Germany) according to the manufacturer’s protocol.
4.2. Artificial Synthesis and Quantification of CPPs
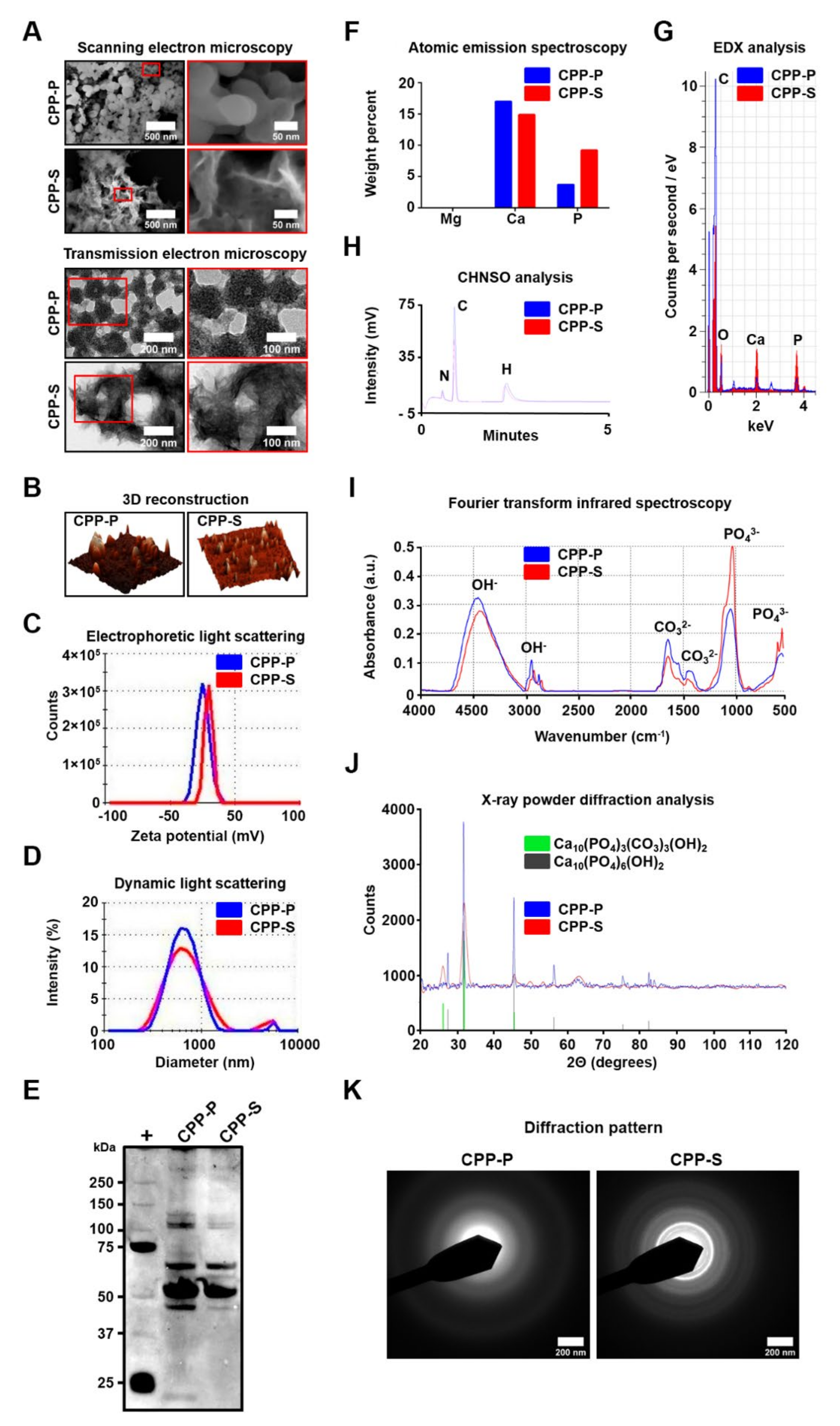
To synthesise primary (CPP-P) and secondary (CPP-S) CPPs, stock solutions of CaCl2 and Na2HPO4 were diluted to equal concentrations of 3 (CPP-P) or 7.5 (CPP-S) mmol/L in Dulbecco’s modified Eagle’s medium (DMEM, 31330038, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (CPP-P) or 1% foetal bovine serum (CPP-S). For the synthesis of magnesiprotein particles (MPPs), stock solutions of MgCl2 (97062-848, VWR, Radnor, PA, USA) and Na2HPO4 were diluted to equal concentrations of 20 mmol/L in DMEM supplemented with 10% foetal bovine serum (FBS, 10270106, Thermo Fisher Scientific, Waltham, MA, USA). The reagents were added into DMEM in the following order: (1) FBS; (2) CaCl2 or MgCl2; (3) Na2HPO4, with a vortexing between the added reagents. Following 24 h incubation in cell culture conditions, the medium was centrifuged at 200,000 g for 1 h (Optima MAX-XP, Beckman Coulter, Brea, CA, USA), and the particle sediment was resuspended in double-distilled water for the microscopy and chemical analysis, phosphate-buffered saline (PBS) for cell culture experiments, or 0.9% NaCl for animal studies. Quantification of CPPs and MPPs was performed using three approaches: (1) OD650 measurement of MPP/CPP suspension; (2) determination of Ca2+ amount per microlitre of MPP/CPP suspension by means of the respective colorimetric kit (ab102505, Abcam, Cambridge, U.K.) at an optical density of 575 nm; (3) quantitation of OsteoSense 680EX-positive PKH67-negative events per microlitre of MPP/CPP suspension in strict accordance with a recent fluorescent probe-based flow cytometry assay [ Briefly, 15 μL of the CPP suspension was added to 75 μL sterile-filtered Tris-buffered saline (pH 7.4); then, 67 μL of this mix was blended with 83 μL fluorescent-labelled bisphosphonate OsteoSense 680EX (1:75 dilution, NEV10020EX, PerkinElmer, Waltham, MA, USA) and incubated in the dark for 50 min at 4 °C, with the subsequent addition of 8.3 μL lipophilic dye PKH67 (1:100 dilution, MIDI67-1KT, Sigma-Aldrich, St. Louis, MO, USA) and further incubation in the dark for another 10 min at 4 °C before sample acquisition (CytoFLEX, Beckman Coulter, Brea, CA, USA). In this experimental setup, OsteoSense 680EX bound to CPPs while PKH67 discriminated CPPs from similar-sized extracellular vesicles; therefore, CPPs were defined as OsteoSense 680EX-positive PKH67-negative events. Generally, OD650 values of 0.08–0.10 (≈0.5 μg/μL calcium and ≈1.2 × 103 OsteoSense 680EX-positive PKH67-negative events/μL for both CPP-P and CPP-S) were considered as proper for the experimentation as this is the minimum reliable threshold of the particle density in the solution. MPPs were almost devoid of calcium (≈0.03 μg/μL calcium and <10 OsteoSense 680EX-positive PKH67-negative events/μL). The concept was to apply as low a dose of CPPs as possible.
4.3. Electron and Atomic Force Microscopy
MPPs, CPP-P, and CPP-S were visualised using scanning electron microscopy (SEM), transmission electron microscopy (TEM), and atomic force microscopy (AFM). For SEM, we pipetted a few drops of the particle suspension on a glass microscope slide, dried the slides at room temperature overnight, mounted the slides on a double-sided adhesive conductive carbon tape (16084-7, Ted Pella, Redding, CA, USA), sputter-coated with gold and palladium (EM ACE200, Leica Microsystems, Wetzlar, Germany), and finally performed the SEM (SU8220, Hitachi, Tokyo, Japan). For TEM, we pipetted a few drops of the particle suspension on a carbon-coated copper grid (3520C-FA, Structure Probe, Inc., West Chester, PA, USA), stained the sample with 2% uranyl acetate (22400-2, Electron Microscopy Sciences, Hatfield, PA, USA) for 20 min, and carried out the TEM (JEM-2100, Jeol, Tokyo, Japan). For AFM, we pipetted a few drops of the particle suspension on a mica disc (50-12, Ted Pella, Redding, CA, USA), and conducted the AFM (Cypher, Asylum Research, Santa Barbara, CA, USA).
4.4. Measurement of Particle-Size Distribution and Surface Charge
The particle-size distribution curve and surface charge (zeta potential) of MPPs, CPP-P, and CPP-S were assessed by dynamic and electrophoretic light scattering, respectively (Zetasizer Nano ZS, Malvern Instruments, Malvern, U.K.). Before the measurement, samples were incubated at 25 °C for 10 min. All measurements were performed thrice (30 runs per measurement) with the further calculation of the average distribution.
4.5. Chemical Profiling
The chemical elements composing MPPs, CPP-P, and CPP-S were determined by energy-dispersive X-ray spectroscopy (EDX), atomic emission spectroscopy (AES), and CHNSO analysis. For EDX, we pipetted a few drops of the particle suspension on a double-sided adhesive conductive carbon tape, dried it for 2 h at 37 °C, and performed elemental analysis (XFlash 4010, Bruker, Billerica, MA, USA). For each sample, we defined three quadrants where particles were clearly observed, and then calculated the average atomic percent for each element. AES (iCAP 6500, Thermo Fisher Scientific, Waltham, MA, USA) was carried out upon dissolution of the particles in HNO3 (438073, Sigma-Aldrich, St. Louis, MO, USA) for 1 h at 80 °C. CHNSO analysis was performed by catalytic oxidation of the particles at 1060 °C (Flash 2000, Thermo Fisher Scientific, Waltham, MA, USA).
To determine which functional groups were formed by the particle-associated elements, we applied Fourier transform infrared spectroscopy (FTIR, Vertex 80v, Bruker, Billerica, MA, USA) and Raman spectroscopy (LabRam HR800, Horiba Scientific, Piscataway, NJ, USA). FTIR and Raman spectra were obtained at a resolution of 4 cm−1 (FTIR) or 0.222 cm−1 (Raman spectroscopy) and at wavelengths ranging from 4000 to 500 cm−1 (FTIR) or 100 cm−1 (Raman spectroscopy).
The chemical formulas of the compounds that constitute MPPs, CPP-P, and CPP-S were deciphered by X-ray powder diffractometry (XRD, D8 ADVANCE, Bruker, Billerica, MA, USA) with an X-ray copper tube operating at 40 kV. Data were collected over a 2θ angle ranging from 20 to 120° at a speed of 0.02°/s. Diffraction spectra were compared with the database of the Joint Committee on Powder Diffraction and Standards. Particle crystallinity was defined during TEM by an analysis of the selected area diffraction patterns that resulted from the electron beam scattered by the sample lattice.
The protein content of CPPs and MPPs was defined by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) with subsequent silver staining. Equal aliquots (20 μL, OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 10 μg calcium or 2.4 × 104 OsteoSense 680EX-positive PKH67-negative events) of MPPs, CPP-P, or CPP-S suspensions were mixed with NuPAGE LDS sample buffer (NP0007, Thermo Fisher Scientific, Waltham, MA, USA) in a 4:1 ratio and a NuPAGE sample reducing agent (NP0009, Thermo Fisher Scientific, Waltham, MA, USA) at a 10:1 ratio, and then loaded on a 1.5 mm NuPAGE 4–12% Bis-Tris protein gel (NP0335BOX, Thermo Fisher Scientific, Waltham, MA, USA). Precision Plus protein standard (1610374, Bio-Rad, Hercules, CA, USA) was loaded as a molecular weight marker. Proteins were separated by SDS-PAGE at 100 V for 1 h. The gel was stained using a Silver Stain Plus staining kit (1610449, Bio-Rad, Hercules, CA, USA) according to the manufacturer’s protocols. Ethylenediaminetetraacetic acid disodium salt (E4884, Sigma-Aldrich, St. Louis, MO, USA) at a 39 mmol/L concentration was added to stop the reaction. Gels were photographed using an HP Scanjet Enterprise Flow 7500 Flatbed Scanner (Hewlett Packard, Palo Alto, CA, USA).
The extraction of lipids was performed by the Folch method using a conventional protocol. Gas chromatography-mass spectrometry (GC-MS) was performed using an MDN-1 column (nonpolar methylsilicone, 30 m × 0.25 mm, 24259, Sigma-Aldrich, St. Louis, MO, USA) and a GCMS-QP2010 Ultra (Shimadzu, Kyoto, Japan) according to the following parameters: injection volume, 1 μL; injector temperature, 200 °C; split ratio, 1:10; interface temperature, 210 °C; detector temperature, 200 °C; carrier (He) flow rate, 0.8 mL/min; and temperature program, 100 °C for 2 min, 5°/min up to 120 °C, 20°/min up to 260 °C, then 260 °C for 2 min. Mass range was from 1.5 to 1900 m/z.
4.6. Animal Models
Male Wistar rats weighing 250–300 g, 12–14 weeks of age, provided by the Research Institute for Complex Issue of Cardiovascular Diseases Core Facility, were used for all animal experiments (n = 130). Animals were allocated to polypropylene cages (5 rats per cage) lined with wood chips and had access to the water and food (rat chow) ad libitum. Throughout the duration of the experiment, the standard conditions of temperature (24 ± 1 °C), relative humidity (55 ± 10%), and 12 h light/dark cycles were carefully maintained, and the health status of all rats was monitored daily. No randomisation was performed to allocate animals to experimental groups or cages. There were no specific inclusion or exclusion criteria. Experiments were performed in a blinded fashion. All procedures were approved by the Local Ethical Committee of the Research Institute for Complex Issues of Cardiovascular Diseases (Kemerovo, Russia, protocol number 20160404, date of approval: 4 April 2016). All procedures conformed to the guidelines of Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes and to the NIH Guide for the Care and Use of Laboratory Animals.
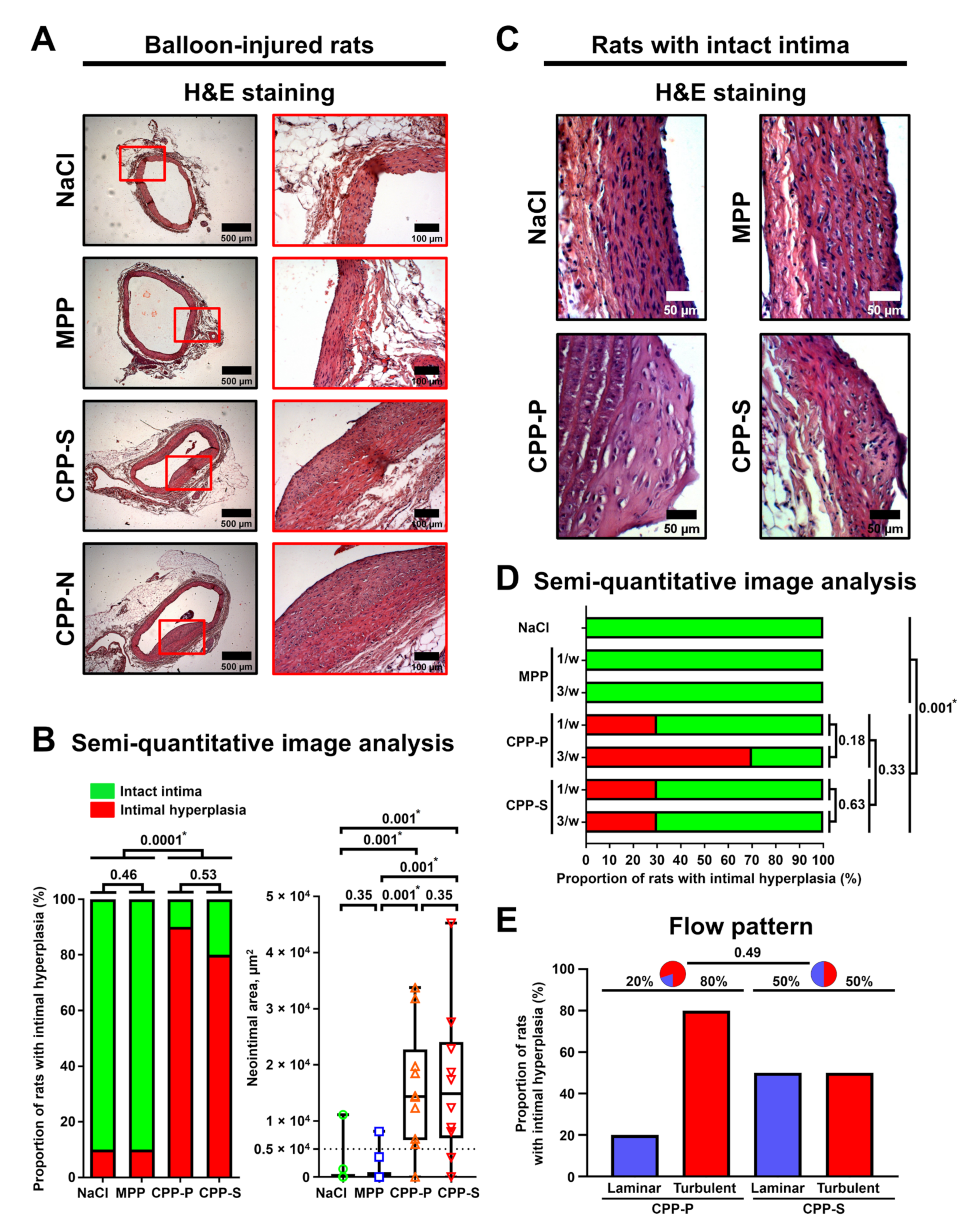
To assess whether CPPs are able to aggravate pre-existing endothelial injury, we applied an experimental model of rat aorta angioplasty with a coronary angioplasty balloon catheter. After the induction of anaesthesia with 3% isoflurane (Aerrane), all animals received inhalation anaesthesia with 1.5% isoflurane throughout surgery. Briefly, the aorta was punctured in the proximal direction with a 21-gauge needle; a DIOR 2.0 × 15 mm balloon catheter with a 0.014 inch guidewire was then inserted into the aortic lumen, and an angioplasty was finally carried out with inflation pressure of 5 atm for 30 sec. Immediately after the surgery, a suspension of MPPs, CPP-P, or CPP-S (900 μL, OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 450 μg calcium or 1.08 × 106 OsteoSense 680EX-positive PKH67-negative events) or equal volume of 0.9% NaCl (n = 10 rats per group, 40 rats in total) was injected into the tail vein.
Five weeks postoperation, all rats were euthanised by an intraperitoneal injection of a sodium pentobarbital (100 mg/kg body weight). At the site of injury, the aorta was excised and fixed in two changes of 10% neutral phosphate-buffered formalin for 24 h at 4 °C, dehydrated in ascending ethanol series (70%, 80%, and 95%; 1 h each) and isopropanol (1 h), impregnated and embedded into paraffin (3 changes, 1 h each, Paraplast REGULAR, 39601006, Leica Biosystems, Wetzlar, Germany), cooled at 4 °C overnight, frozen at −20 °C, and cut (5 μm sections) on a microtome (Microm HM 325, Thermo Fisher Scientific, Waltham, MA, USA). To ensure proper histological examination, we prepared 12 sections, evenly distributed across the entire aortic segment, per slide. Sections were then H&E- and alizarin red S stained (ab245880 and ab146374, Abcam, Cambridge, U.K.) according to the manufacturer’s protocols for general examination. Sections were evaluated by light microscopy (AxioImager.A1, Carl Zeiss, Stuttgart, Germany) in a blinded fashion for the extent of intimal hyperplasia or adventitial/perivascular inflammation.
Neointimal area as well as total number and area of macrophage clusters were evaluated using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Intimal hyperplasia was defined as neointimal area ≥5000 μm2, as these values indicate a clearly visible neointimal lesion. Macrophage phenotype in such clusters was verified by immunohistochemistry (Novolink Max Polymer Detection System, RE7280-K, Leica, Wetzlar, Germany) after antigen retrieval (ab93678, Abcam, Cambridge, UK) according to the manufacturers’ protocols using the antibodies to a macrophage marker F4/80 (ab100790, Abcam, Cambridge, U.K.; 1:200 dilution), macrophage activity-reflecting enzyme myeloperoxidase (ab208670, Abcam, Cambridge, U.K.; 1:200), pan-leukocyte marker CD45 (ab10558, Abcam, Cambridge, U.K.; 1:200), and T cell marker CD3 (ab16669, Abcam, Cambridge, U.K.; 1:200). Haematoxylin was used as the counterstain.
Alternatively, to test the ability of CPPs to cause endothelial injury per se, we performed consecutive tail vein injections of MPPs, CPP-P, or CPP-S (900 μL of particles per injection, OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 450 μg calcium or 1.08 × 106 OsteoSense 680EX-positive PKH67-negative events) or an equal volume of 0.9% NaCl (once or thrice per week during 5 weeks, n = 10 rats per group, 70 rats in total) without any surgical intervention. Five weeks following the start of the injections, all rats were euthanised, and aortic arches and descending aortas (i.e., aortic segments with a turbulent and laminar flow, respectively) were excised and treated as described above. Sections were then examined for the presence of intimal hyperplasia.
As the proper examination of the vasa vasorum requires electron microscopy, which is incompatible with routine histological examination, for the comprehensive investigation of adventitial/perivascular inflammation, we applied another animal model in which rats underwent balloon angioplasty followed by daily tail vein injections of MPPs, CPP-P, or CPP-S (900 μL of particles per injection, OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 450 μg calcium or 1.08 × 106 OsteoSense 680EX-positive PKH67-negative events) or an equal volume of 0.9% NaCl for 5 days (n = 5 rats per group, 20 rats in total). Rats were euthanised 5 weeks postoperation as described above, with subsequent excision of the injured and intact aortic segments. Vessels were then fixed in two changes of 10% neutral phosphate-buffered formalin for 24 h at 4 °C, postfixed in 1% osmium tetroxide (OsO4, 19110, Electron Microscopy Sciences, Hatfield, PA, USA) for 24 h, stained in 2% osmium tetroxide for 48 h, dehydrated in ascending ethanol series (50%, 60%, 70%, 80% and 95%, 15 min each), stained in 2% alcoholic uranyl acetate for 5 h, dehydrated in isopropanol (5 h) and acetone (1 h), impregnated with an acetone:epoxy resin (Epon, 14120, Electron Microscopy Sciences, Hatfield, PA, USA) mixture (1:1) for 6 h and with epoxy resin for 24 h, and were finally embedded into fresh epoxy resin at 60 °C. Samples were then ground, polished (TegraPol-11, Struers, Copenhagen, Denmark), and counterstained with Reynolds’s lead citrate (17810, Electron Microscopy Sciences, Hatfield, PA, USA) for 7 min. After a washing in double-distilled water, samples were sputter-coated (10 nm thickness) with carbon (EM ACE200, Leica Biosystems, Wetzlar, Germany) and visualised by means of backscattered scanning electron microscopy at a 10 kV voltage (S-3400N, Hitachi, Tokyo, Japan). The total number and area of VV and MCs in the examined vessels, further normalised by the area of adventitia and perivascular adipose tissue to calculate VV and MC density, were evaluated using ImageJ software (National Institutes of Health, Bethesda, MD, USA). VV was defined by the combination of clearly visible vessel lumen, endothelial layer, and (optionally) red blood cells within the lumen.
4.7. Cell Culture
Primary cultures of human coronary artery endothelial cells (HCAECs, 300K-05a, Cell Applications, San Diego, CA, USA) and human internal thoracic artery endothelial cells (HITAECs, 308K-05a, Cell Applications, San Diego, CA, USA) were cultured according to the manufacturer’s protocols. For the extraction of primary perivascular adipocytes, adipose tissue samples (2 mm3) obtained from the coronary artery and internal mammary artery anastomosis during the coronary artery bypass graft surgery (the patient provided a written informed consent for the procedure after receiving a full explanation of the study, Local Ethical Committee of the Research Institute for Complex Issues of Cardiovascular Diseases (Kemerovo, Russia) protocol number 20190704, date of approval: 4 July 2019) were incubated in a collagenase solution (0.5 mg/mL, 17100017, Thermo Fisher Scientific, Waltham, MA, USA) containing 200 nmol/L adenosine (1160CBC, Merck Millipore, Burlington, MA, USA) in a water bath at 37 °C for 30 min. The floated fraction of the digested perivascular adipose tissue was then transferred into M199 culture medium (12340030, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with glucose (5 mmol/L) and 10% FBS and filtered through a 100 μm cell strainer (21008-950, VWR, Radnor, PA, USA). The flow-through containing perivascular adipocytes was further centrifuged for 2 min at 200× g, resuspended in 1 mL culture medium, and finally seeded into a 24-well plate.
4.8. Cytotoxicity Assays
Following the addition of ab112118 (Abcam, Cambridge, U.K.) reagent, HCAECs and HITAECs cultured in 96-well plates (85–90% confluence) were exposed to 10 μL of MPPs, CPP-P, CPP-S (OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 5 μg calcium or 1.2 × 104 OsteoSense 680EX-positive PKH67-negative events), or PBS (n = 24 wells per group) for 4 h with the subsequent calculation of the OD530/OD650 ratio to evaluate cell proliferation and viability (percent in relation to the vehicle control (PBS) group). The cells were visualised by phase contrast microscopy (AxioObserver.Z1, Carl Zeiss, Stuttgart, Germany) before adding ab112118. Alternatively, HCAECs and HITAECs cultured in 6-well plates (85–90% confluence) were exposed to 100 μL of MPPs, CPP-P, CPP-S (OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 50 μg calcium or 1.2 × 105 OsteoSense 680EX-positive PKH67-negative events), or PBS (n = 11 wells per group) for 4 h with subsequent Hoechst 33342 (2 μg/mL, H3570, Thermo Fisher Scientific, Waltham, MA, USA) and ethidium bromide (10 μg/mL, IB40075, VWR, Radnor, PA, USA) staining for 5 min, washing in a dye-free medium, and fluorescence microscopy (3 fields of view per well, AxioObserver.Z1, Carl Zeiss, Stuttgart, Germany). Quantitative image analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
4.9. Internalisation Assays
HCAECs cultured in T-75 flasks (85–90% confluence) were exposed to 1000 μL of CPP-P (OD650 = 0.08–0.10, equal to 500 μg calcium or 1.2 × 106 OsteoSense 680EX-positive PKH67-negative events) or PBS for 4 h. The cell pellet was fixed in 2.5% glutaraldehyde (16320, Electron Microscopy Sciences, Hatfield, PA, USA) for 1 h, postfixed in 1% osmium tetroxide for 1 h, embedded into 2% agarose (97062-244, VWR, Radnor, PA, USA), and dehydrated in ascending ethanol series (50%, 60%, 70%, 80%, and 95%, 15 min each) and acetone (1 h). The sections of the agarose gel were then impregnated with an acetone:epoxy resin mixture (1:1) mixture for 2 h and with epoxy resin for 24 h, and were finally embedded into fresh epoxy resin at 60 °C. Ultrafine sections were prepared using an ultramicrotome (LKB Bromma Nova Ultra, LKB, Stockholm, Sweden) placed on a carbon-coated copper grid, then stained with 2% uranyl acetate for 20 min, counterstained with a Reynolds’s lead citrate for 1 h, and examined with a transmission electron microscope.
Alternatively, HCAECs (85–90% confluence) cultured in T-75 flasks (85–90% confluence) were exposed to 1000 μL of MPPs, CPP-P, CPP-S (OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 500 μg calcium or 1.2 × 106 OsteoSense 680EX-positive PKH67-negative events) or PBS for 1 h, and the cell pellet was then treated as described above but without performing ultrafine sectioning. Epoxy resin blocks were then ground, polished, stained with 2% uranyl acetate at 60 °C for 1 h, and counterstained with Reynolds’s lead citrate for 7 min. After a washing in double distilled water, samples were sputter-coated (10 nm thickness) with carbon and visualised by means of backscattered scanning electron microscopy at a 10 kV voltage. The calcium presence in electron-dense dots indicative of internalised CPPs was confirmed by EDX analysis as described above.
To examine whether CPPs enter the lysosomes upon the internalisation, we cultured HCAECs on chambered coverslips (80826, Ibidi, Grafelfing, Germany) to 85–90% confluence, exposed them to 25 μL of fluorescein isothiocyanate (FITC)-labelled CPP-P (OD650 = 0.08–0.10, equal to 12.5 μg calcium or 3 × 104 OsteoSense 680EX-positive PKH67-negative events) or PBS for 1 or 4 h, and concurrently stained with LysoTracker Red (500 nmol/L, L7528, Thermo Fisher Scientific, Waltham, MA, USA) for 1 h according to the manufacturer’s protocol. Labeling of CPP-P was performed by their incubation with FITC-labelled albumin (A23015, Thermo Fisher Scientific, Waltham, MA, USA) for 1 h. Nuclear counterstaining was performed by incubation with Hoechst 33342 (2 μg/mL) for 5 min. Visualisation was performed using confocal microscopy (LSM 700, Carl Zeiss, Stuttgart, Germany) after a washing with a dye-free medium.
4.10. Lysosome Permeabilisation Assay
HCAECs cultured in 96-well culture plates (85–90% confluence) were exposed to 100 μL of MPPs, CPP-P, CPP-S (OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 50 μg calcium or 1.2 × 105 OsteoSense 680EX-positive PKH67-negative events) or PBS (n = 12 wells per group) for 4 or 24 h with or without specific inhibitor of vacuolar-type H+-ATPase bafilomycin A1 (ab120497 (Abcam, Cambridge, UK), 0.1 or 1 μmol/L). Cell proliferation and viability were monitored by a concurrent addition of ab112118 reagent (Abcam, Cambridge, UK) for 4 h and further calculations as described above.
4.11. Ca2+ Translocation Assay
HCAECs cultured on chambered coverslips (80826, Ibidi, Grafelfing, Germany) to 85–90% confluence were exposed to 25 μL of CPP-P (OD650 = 0.08–0.10, equal to 12.5 μg calcium or 3 × 104 OsteoSense 680EX-positive PKH67-negative events) or PBS for 30 min and concurrently stained with a specific Ca2+ indicator fluo-3 AM (5 μmol/L, F14218, Thermo Fisher Scientific, Waltham, MA, USA) for 30 min with the subsequent washing with an indicator-free medium and incubation for 30 min to allow complete de-esterification of intracellular AM esters according to the manufacturer’s protocol. Nuclear counterstaining was performed by incubation with Hoechst 33342 (2 μg/mL) for 5 min. Visualisation was performed using confocal microscopy after washing with a dye-free medium.
4.12. Assessment of Oxidative Stress
HCAECs cultured on chambered coverslips (80826, Ibidi, Grafelfing, Germany) to 85–90% confluence were exposed to 25 μL of MPPs, CPP-P, CPP-S (OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 12.5 μg calcium or 3 × 104 OsteoSense 680EX-positive PKH67-negative events) or PBS for 1 or 4 h and concurrently stained with MitoSOX Red (5 μmol/L, M36008, Thermo Fisher Scientific, Waltham, MA, USA), a fluorogenic probe for superoxide, for 10 min; or with CellROX Green (5 μmol/L, C10444, Thermo Fisher Scientific, Waltham, MA, USA), a fluorogenic probe for total reactive oxygen species, for 30 min. Nuclear counterstaining was performed by incubation with Hoechst 33342 (2 μg/mL) for 5 min. Visualisation was performed using confocal microscopy after washing with PBS.
Alternatively, HCAECs cultured in 96-well culture plates (85–90% confluence) were exposed to 100 μL of MPPs, CPP-P, CPP-S (OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 50 μg calcium or 1.2 × 105 OsteoSense 680EX-positive PKH67-negative events) or PBS (n = 12 wells per group) for 4 or 24 h with or without antioxidant enzymes superoxide dismutase (SOD, 250 U/mL) and catalase (CAT, 500 U/mL). Both SOD and CAT were added 3 h before exposure to the particles to ensure their internalisation through the plasma membrane. Cell proliferation and viability were monitored by the concurrent addition of ab112118 reagent (Abcam, Cambridge, U.K.) for 4 h and further calculations as described above.
4.13. Thiobarbituric Acid Reactive Substances (TBARS) Assay
HCAECs and HITAECs cultured in 6-well plates (85–90% confluence) were exposed to 100 μL of MPPs, CPP-P, CPP-S (OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 50 μg calcium or 1.2 × 105 OsteoSense 680EX-positive PKH67-negative events) or PBS (n = 11 wells per group) for 24 h. Conditioned medium (2 mL per well) was then collected and measured for thiobarbituric acid reactive substances (TBARS), by-products of lipid peroxidation, by the addition of 1 mL of thiobarbituric acid (T5500, Sigma-Aldrich, St. Louis, MO, USA) and 3 mL of H3PO4 (695017, Sigma-Aldrich, St. Louis, MO, USA) to 250 μL of the sample. After 60 min incubation at 100 °C and cooling at 8 °C for 5 min, 1 mL of 1-butanol (360465, Sigma-Aldrich, St. Louis, MO, USA) was added for the enrichment of the TBARS fraction. Upon 10 min centrifugation at 3000 rpm, the supernatant was aliquoted in 96-well plates (100 μL per well, each sample was aliquoted in triplicate), followed by the measurement of OD450. The level of TBARS in each sample was then calculated through the division of OD450 by the molar extinction coefficient of malondialdehyde (0.156) and then multiplication by the sample dilution factor (16).
4.14. Gene Expression Profiling
After exposure of HCAECs and HITAECs cultured in 6-well plates (85–90% confluence) to 100 μL of MPPs, CPP-P, CPP-S (OD
650 = 0.08–0.10, for CPP-P and CPP-S: equal to 50 μg calcium or 1.2 × 10
5 OsteoSense 680EX-positive PKH67-negative events), or PBS (3 wells per group) for 4 h, total RNA was extracted using TRIzol (15596018, Thermo Fisher Scientific, Waltham, MA, USA) and then reverse-transcribed using a High Capacity cDNA Reverse Transcription Kit (4368814, Thermo Fisher Scientific, Waltham, MA, USA). Gene expression was measured by quantitative polymerase chain reaction (qPCR) using customised primers (500 nmol/L each, Evrogen, Moscow, Russia,
Table S3), cDNA (20 ng) and PowerUp SYBR Green Master Mix (A25778, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol for T
m ≥ 60 °C (fast cycling mode). Technical replicates (n = 3 per sample collected from one well) were performed in all qPCR experiments. Quantification of the mRNA levels was performed using the 2
−ΔΔCt method. Relative transcript levels are expressed as a value relative to the average of three housekeeping genes (
GAPDH,
ACTB, and
B2M) and to the PBS group (2
−ΔΔCt).
4.15. Cytokine Secretion Profiling
HCAECs and HITAECs cultured in 6-well plates (85–90% confluence) were exposed to 100 μL of MPPs, CPP-P, CPP-S (OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 50 μg calcium or 1.2 × 105 OsteoSense 680EX-positive PKH67-negative events), or PBS for 4 h. Upon the collection of the conditioned medium (300 μL per well), the levels of interleukin-6 and -8 in cell culture supernatant were measured by enzyme-linked immunosorbent assay using the respective kits (ab178013 and ab46032, Abcam, Cambridge, UK) according to the manufacturer’s protocol.
Alternatively, HCAECs and HITAECs cultured in T-75 flasks (85–90% confluence) were exposed to 1000 μL of CPP-P (OD650 = 0.08–0.10, equal to 500 μg calcium or 1.2 × 106 OsteoSense 680EX-positive PKH67-negative events) or PBS for 4 h. Upon the collection of the conditioned medium (2 mL per flask) and cell lysis by RIPA buffer (89901, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with protease and phosphatase inhibitors (78444, Thermo Fisher Scientific, Waltham, MA, USA), we profiled the conditioned medium and cell lysate (1 mg protein) for a wide spectrum of cytokines using the respective dot blotting kit (ARY005B, R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s protocol. Additionally, primary human perivascular adipocytes were treated with CPP-P (50 μL of particles per well of a 24-well plate, OD650 = 0.08–0.10, equal to 25 μg calcium or 0.6 × 105 OsteoSense 680EX-positive PKH67-negative events) or PBS for 24 h. Conditioned medium (1 mL per well) and cell lysate (200 μg protein) were then collected and profiled for adipokines and cytokines using the respective dot blotting kit (ARY024, R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s protocol. Chemiluminescent detection was performed using a C-DiGit blot scanner (LI-COR Biosciences, Lincoln, NE, USA). Densitometry was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
4.16. Leukocyte Adhesion Assay
HCAECs and HITAECs were cultured in flow system chambers (80126, Ibidi, Grafelfing, Germany) to 85–90% confluence, preconditioned by a 15 dyn/cm2 laminar flow (10964, Ibidi, Grafelfing, Germany) for 24 h, and then exposed to 500 μL of MPPs, CPP-P, CPP-S (OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 250 μg calcium or 0.6 × 106 OsteoSense 680EX-positive PKH67-negative events), or PBS for 4 h. Primary human peripheral blood mononuclear cells (PBMCs) were isolated from a healthy volunteer using Histopaque-1077 (10771, Sigma-Aldrich, St. Louis, MO, USA) and labelled with a CellTracker Green CMFDA Dye (5 μmol/L, C7025, Thermo Fisher Scientific, Waltham, MA, USA) for 30 min according to the respective manufacturer’s protocols. PBMCs were then added to the flow system (125,000 cells per mL; 1,500,000 cells per flow system unit) for 1 h (1 h before the end of the incubation with the particles). Upon the end of the incubation, nuclear counterstaining was performed (Hoechst 33342, 2 μg/mL, 5 min). Fluorescence was visualised after thorough washing in a dye-free medium (40 fields of view per flow system chamber, AxioObserver.Z1, Carl Zeiss, Stuttgart, Germany). Quantitative image analysis was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
4.17. Western Blotting
After exposure of HCAECs and HITAECs (85–90% confluence) to MPPs, CPP-P, CPP-S (1000 μL of particles per T-75 flask, OD650 = 0.08–0.10, for CPP-P and CPP-S: equal to 500 μg calcium or 1.2 × 106 OsteoSense 680EX-positive PKH67-negative events), or PBS for 4 h, the total protein was extracted as described above. Equal amounts of protein (16 μg) were mixed with NuPAGE LDS sample buffer in a 4:1 ratio and NuPAGE sample reducing agent at a 10:1 ratio, and then loaded on a 1.5 mm NuPAGE 4–12% Bis-Tris protein gel. The 1:1 mixture of Novex Sharp prestained protein standard (LC5800, Thermo Fisher Scientific, Waltham, MA, USA) and MagicMark XP Western protein standard (LC5602, Thermo Fisher Scientific, Waltham, MA, USA) was loaded as a molecular weight marker. Proteins were separated by SDS-PAGE at 150 V for 90 min. Protein transfer was performed using polyvinylidene difluoride transfer stacks (IB24001, Thermo Fisher Scientific, Waltham, MA, USA) and iBlot 2 Gel Transfer Device (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocols.
Blots were probed with rabbit antibodies to cleaved caspase-3 (9661, Cell Signaling Technology, Danvers, MA, USA; 1:1000 dilution), poly [ADP-ribose] polymerase 1 (PARP-1, 9542, Cell Signaling Technology, Danvers, MA, USA 1:1000), X-linked inhibitor of apoptosis (XIAP, 2042, Cell Signaling Technology, Danvers, MA, USA; 1:1000), plasminogen activator inhibitor 1 (PAI-1, 11907, Cell Signaling Technology, Danvers, MA, USA; 1:1000), vascular cell adhesion molecule 1 (VCAM1, ab134047, Abcam, Cambridge, U.K.; 1:2000), intercellular cell adhesion molecule 1 (ICAM1, ab109361, Abcam, Cambridge, U.K.; 1:1000), Snail and Slug transcription factors (ab180714, Abcam, Cambridge, U.K.; 1:500), kinase insert domain receptor (KDR, ab39256, Abcam, Cambridge, U.K.; 1:1000), vascular endothelial cadherin (VE-cadherin, 361900, Thermo Fisher Scientific, Waltham, MA, USA 1:100), mouse antibodies to CD31 (ab9498, Abcam, Cambridge, U.K.; 1:1000), N-cadherin (MA515633, Thermo Fisher Scientific, Waltham, MA, USA; 1:500), and histone H3 (3638, Cell Signaling Technology, Danvers, MA, USA; 1:1000), or goat antibody to β-tubulin (ab21057, Abcam, Cambridge, U.K.; 1:1000). Horseradish-peroxidase-conjugated goat anti-rabbit (7074, Cell Signaling Technology, Danvers, MA, USA) or goat anti-mouse secondary antibodies (AP130P, Sigma-Aldrich, St. Louis, MO, USA) were used at 1:200 and 1:1000 dilution, respectively. Incubation with the antibodies was performed using iBind Flex Solution Kit (SLF2020, Thermo Fisher Scientific, Waltham, MA, USA), iBind Flex Cards (SLF2010, Thermo Fisher Scientific, Waltham, MA, USA) and Bind Flex Western Device (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocols. Chemiluminescent detection was performed using SuperSignal West Pico PLUS chemiluminescent substrate (34580, Thermo Fisher Scientific, Waltham, MA, USA) and a C-DiGit blot scanner (LI-COR Biosciences, Lincoln, NE, USA). Densitometry was performed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
4.18. Statistical Analysis
Statistical analysis was performed using GraphPad Prism 7 (GraphPad Software, San Diego, CA, USA). For descriptive statistics, data are presented as proportion or media, 25th and 75th percentiles, and range. Two independent groups were compared by Pearson’s chi-squared test with Yates’s correction for continuity or by the Mann–Whitney U-test, whereas three or more groups were compared by the Kruskal–Wallis test with post hoc calculation of false discovery rate (FDR) by the two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli. Calculation of the odds ratio and the respective 95% confidence intervals was conducted employing a MedCalc online calculator (MedCalc Software, Ostend, Belgium). Correlation analysis was performed using Spearman’s rank correlation coefficient. P-values, or q-values if FDR was applied (q-values are the name given to the adjusted p-values found using an optimised FDR approach), ≤0.05 were regarded as statistically significant.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}