
A Comprehensive Analysis of Human Endogenous Retroviruses HERV-K (HML.2) from Teratocarcinoma Cell Lines and Detection of Viral Cargo in Microvesicles


Abstract
1. Introduction
2. Results
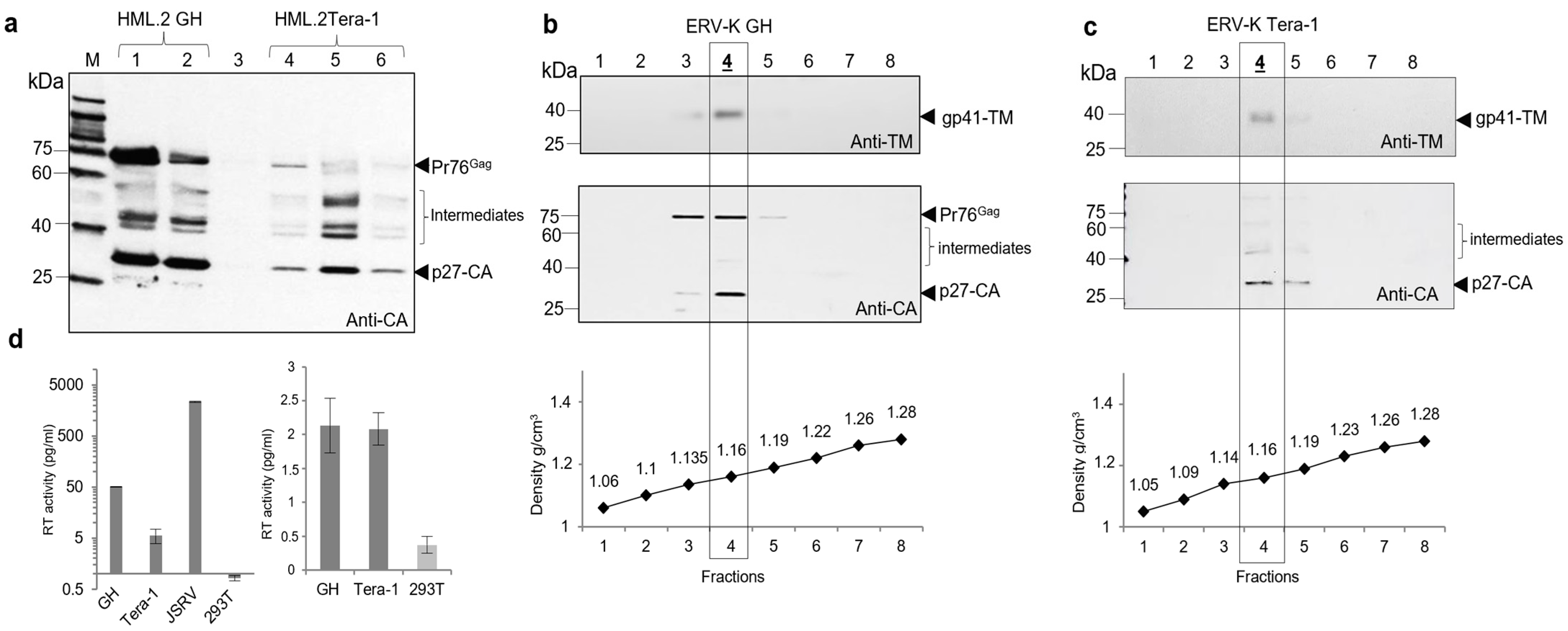
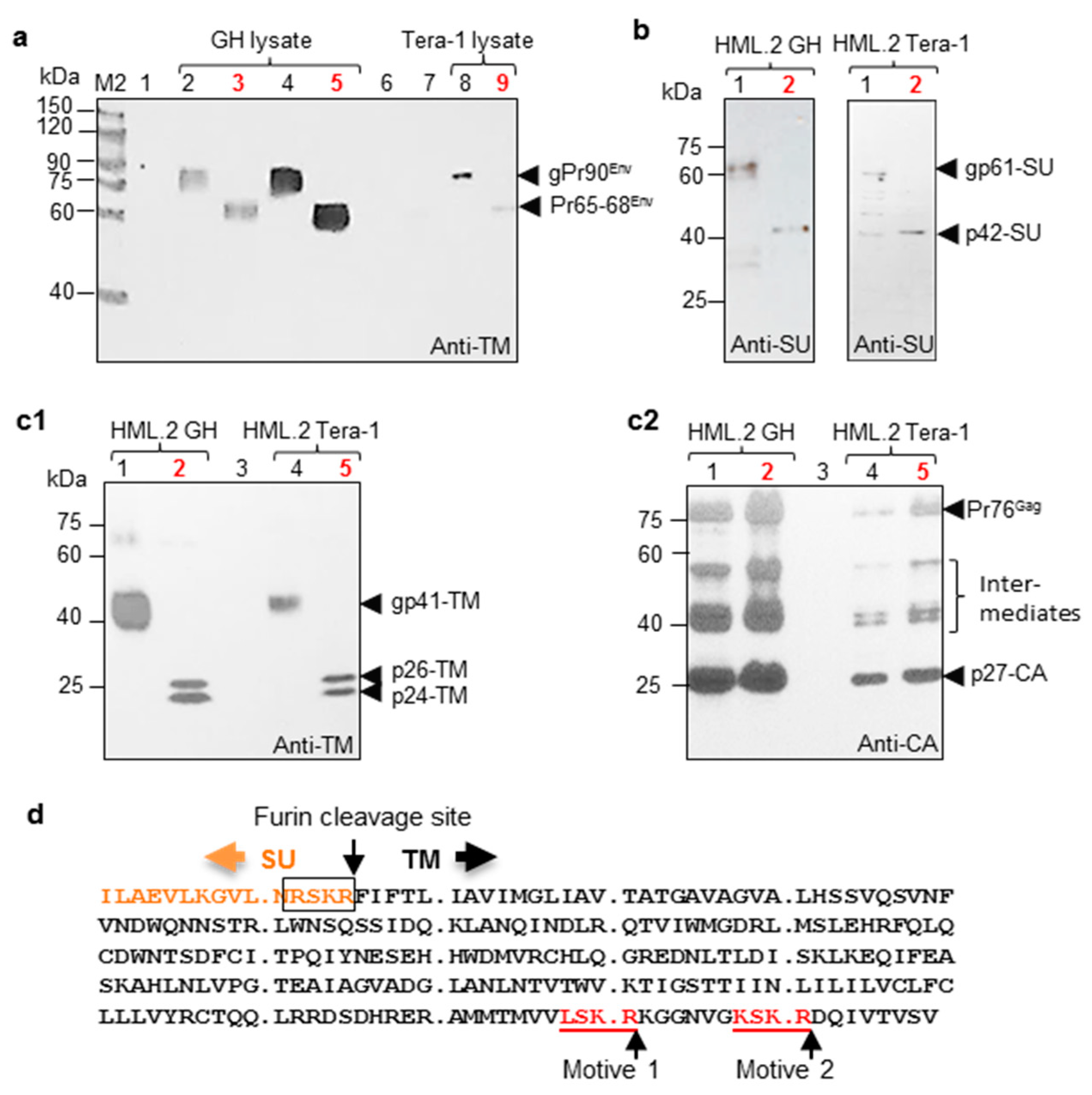
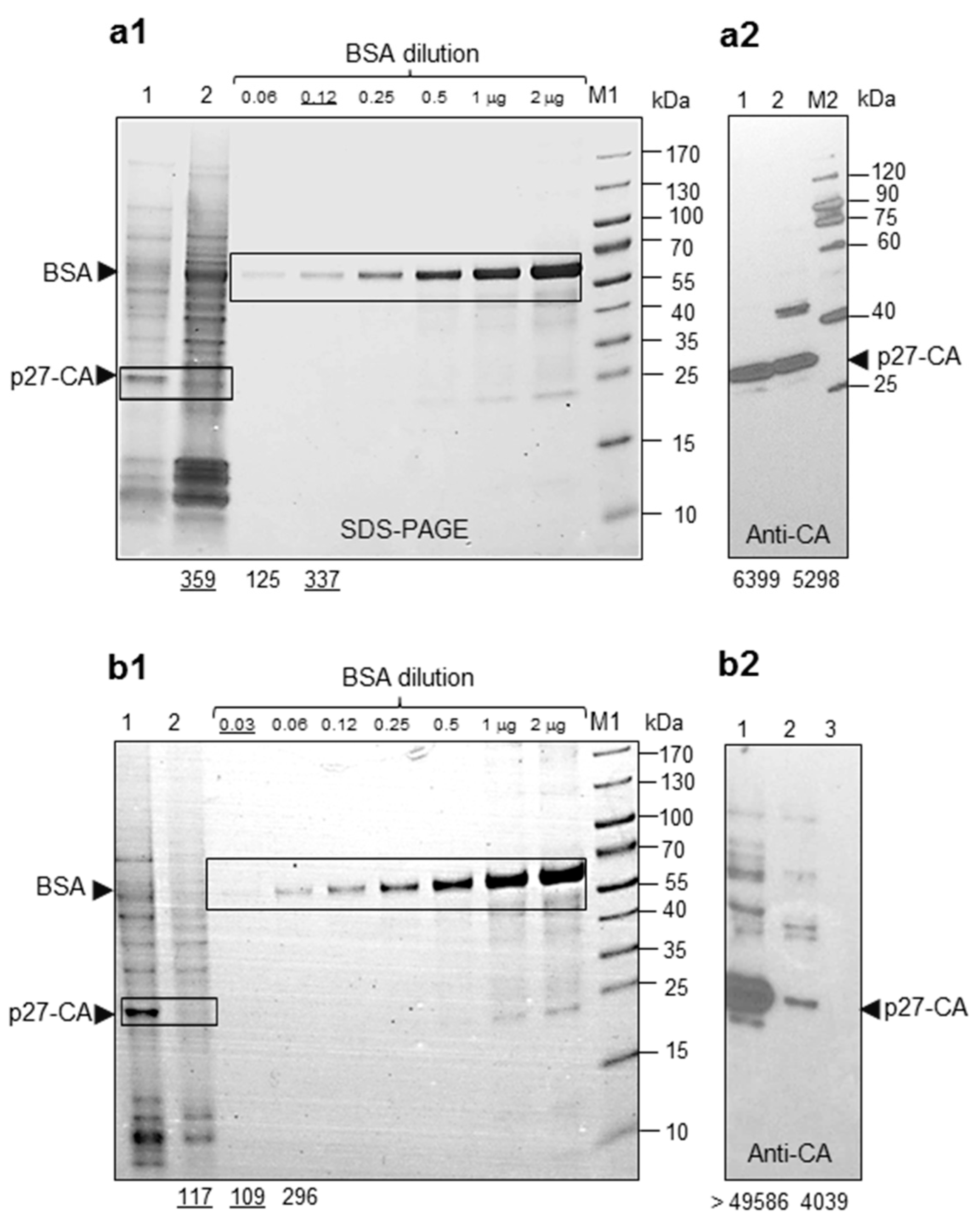
2.1. Proteins and Parameters of HML.2 from Teratocarcinoma Cell Lines GH and Tera-1
2.2. Reverse Transcriptase (RT) Activity in HML.2 GH and Tera-1
2.3. Deglycosylation of HML.2 GH and HML.2 Tera-1
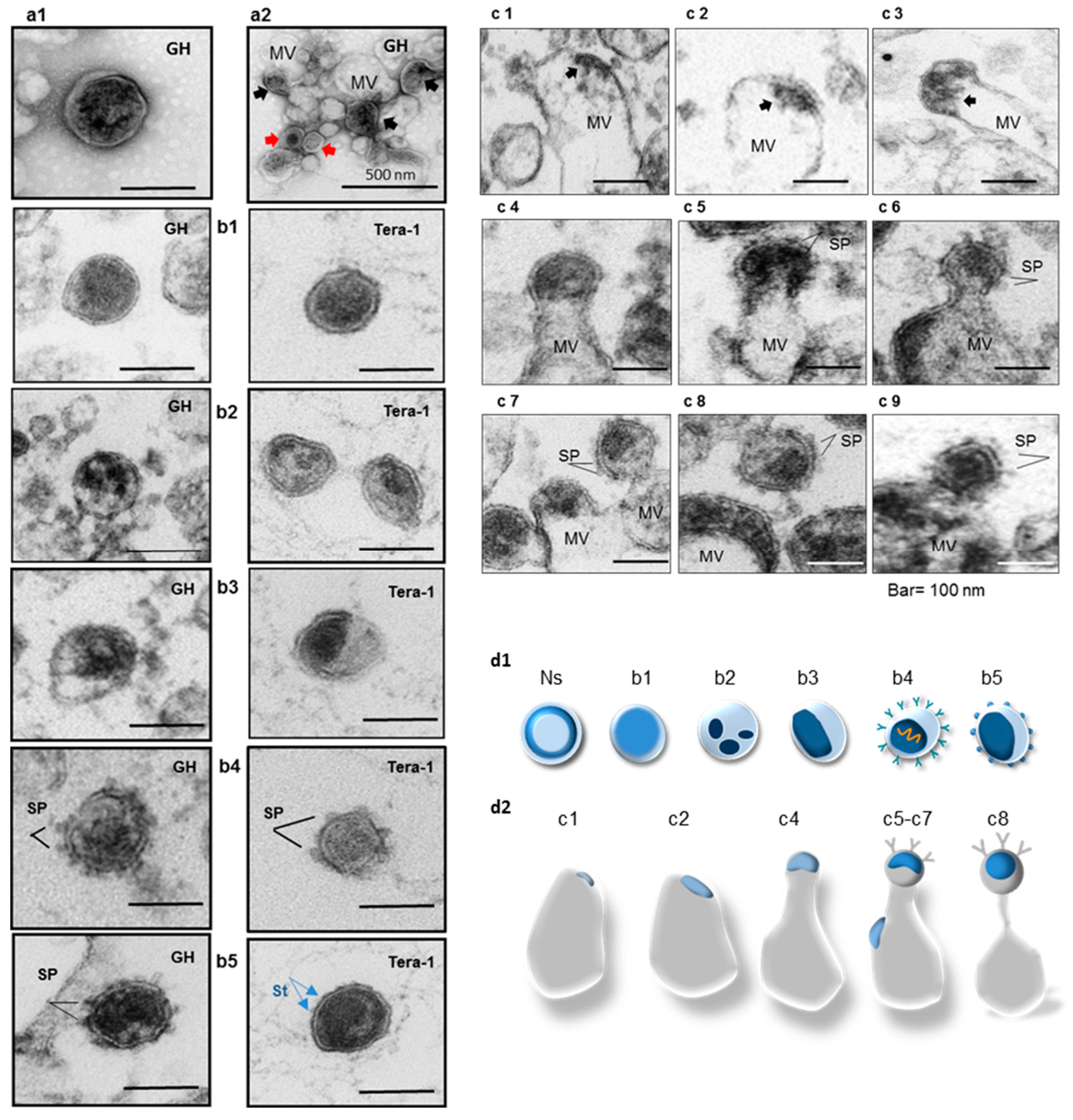
2.4. Analyses of Viral Particles and Extracellular Vesicles by Negative Staining and Ultrathin Section TEM
- Group 1—Bald particles with amorphous capsids (Figure 3(b1)). About 60–70%.
- Group 2—Bald particles with fragmented capsids (Figure 3(b2)). Not frequent.
- Group 4—Particles with 6–8 nm long SP and condensed or partially condensed capsids about 65–80 nm in diameter (Figure 3(b4,b5) left). Such particles were not frequent (~1%). Several virions with partially condensed capsids and small surface protrusions (~2–3 nm) located at nearly equal distance between each other were detected. These protrusions are likely representing the TM stumps (Figure 3(b5) right).
- Group 5—Extracellular vesicles with captured “viral elements” displayed a variety of vesicular assemblies containing HML.2 GH budding structures with and without SP (Figure 3(c1–c9)). Association of HML.2 Tera-1 with MVs was exceptionally rare.
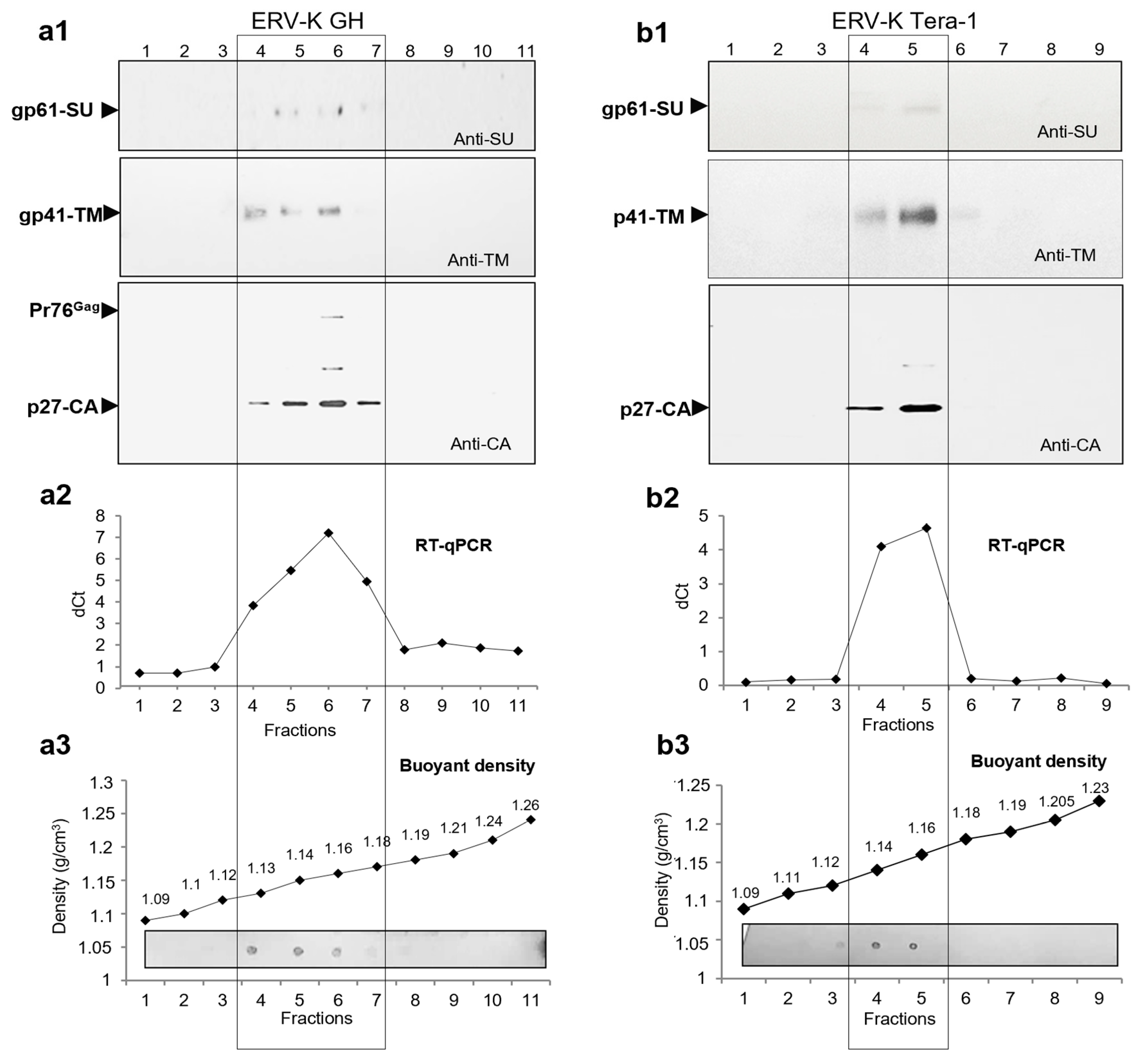
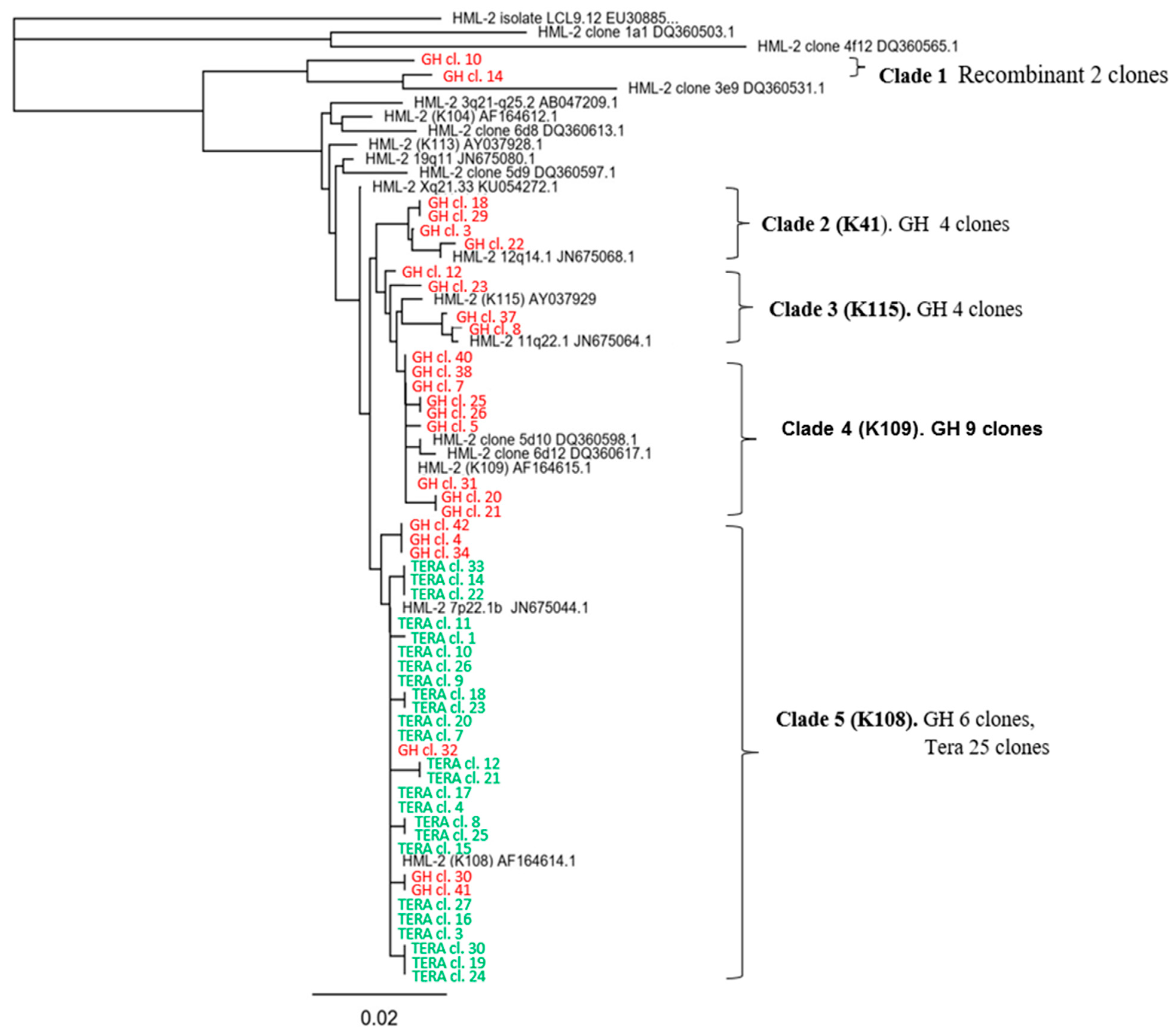
2.5. Search for Virus Diversification in HML.2 Pool and Analyses of SU and vRNA in Fractions of the Sucrose Density Gradient
2.6. Viral RNA Packaged in HML.2 GH and HML.2 Tera-1, Analyses of env Type 2 Gene
2.7. Quantification of HML.2 Particles Released from GH and Tera-1 Cells
3. Discussion
4. Materials and Methods
4.1. Cells and Cellular Lysates
4.2. Antibodies
4.3. Virus Isolation, Sucrose Density Gradient Centrifugation (Dgc)
4.4. Transfection of Cells and Purification of Recombinant HML.2 Particles
4.5. Western Blotting and Dot Blot
4.6. PNGase F Treatment
4.7. RNA Extraction, RT-PCR and RT-qPCR
4.8. Reverse Transcriptase Assay
4.9. Cloning and Sequence Analysis
4.10. Transmission Electron Microscopy (TEM)
4.11. Software
4.12. Sequences Deposited to the GenBank
5. Summary and Conclusions
- -
- As a consequence of active expression of several proviruses, GH cells produced 4–5 times more viruses, compared to Tera-1 cells. It is likely that SU and TM of HML.2 are completely glycosylated. Truncated TM variant (p24-TM), a low SU content and reduced RT activity were disclosed in examined HML.2.
- -
- Bald virions with amorphous capsids were the most common morphological variants in HML.2 pools. Virus particles with SP and condensed (collapsed) capsids were rarely detected.
- -
- GH cells actively release MVs. During budding, MVs can capture fragments of plasma membrane with HML.2 elements at different stages of assembly. Judging from the TEM images, maturation of virus particles inside MVs may take place. The release of MVs from Tera-1 cells was insignificant.
- -
- We assume that MVs with HML.2 elements upon delivery to target cells can trigger cell signaling systems, promote inflammation and autoimmune diseases.
- -
- Six different HML.2 transcripts including two recombinant variants were packaged into virus particles from GH cells. However, HML.2 Tera-1 contained only HERV-K108 env transcripts and similar sequences.
- -
- It was assessed that, in 24 h, one GH cell can produce ~27 virions, while Tera-1 cell ~6 virions, whereas human lymphocytes chronically infected with HIV-1 release about 1 × 104 virions.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bannert, N.; Kurth, R. Retroelements and the human genome: New perspectives on an old relation. Proc. Natl. Acad. Sci. USA 2004, 101, 14572–14579. [Google Scholar] [CrossRef]
- Weiss, R.A.; Stoye, J.P. Virology. Our viral inheritance. Sci. N. Y. 2013, 340, 820–821. [Google Scholar] [CrossRef]
- Harrow, J.; Nagy, A.; Reymond, A.; Alioto, T.; Patthy, L.; Antonarakis, S.E.; Guigó, R. Identifying protein-coding genes in genomic sequences. Genome Biol. 2009, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, R.P.; Wildschutte, J.H.; Russo, C.; Coffin, J.M. Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 2011, 8, 90. [Google Scholar] [CrossRef]
- Demirov, D.G.; Freed, E.O. Retrovirus budding. Virus Res. 2004, 106, 87–102. [Google Scholar] [CrossRef]
- Dewannieux, M.; Blaise, S.; Heidmann, T. Identification of a functional envelope protein from the HERV-K family of human endogenous retroviruses. J. Virol. 2005, 79, 15573–15577. [Google Scholar] [CrossRef]
- Wildschutte, J.H.; Williams, Z.H.; Montesion, M.; Subramanian, R.P.; Kidd, J.M.; Coffin, J.M. Discovery of unfixed endogenous retrovirus insertions in diverse human populations. Proc. Natl. Acad. Sci. USA 2016, 113, E2326–E2334. [Google Scholar] [CrossRef]
- Turner, G.; Barbulescu, M.; Su, M.; Jensen-Seaman, M.I.; Kidd, K.K.; Lenz, J. Insertional polymorphisms of full-length endogenous retroviruses in humans. Curr. Biol. CB 2001, 11, 1531–1535. [Google Scholar] [CrossRef]
- Lower, R.; Boller, K.; Hasenmaier, B.; Korbmacher, C.; Muller-Lantzsch, N.; Lower, J.; Kurth, R. Identification of human endogenous retroviruses with complex mRNA expression and particle formation. Proc. Natl. Acad. Sci. USA 1993, 90, 4480–4484. [Google Scholar] [CrossRef] [PubMed]
- Gross, H.; Barth, S.; Pfuhl, T.; Willnecker, V.; Spurk, A.; Gurtsevitch, V.; Sauter, M.; Hu, B.; Noessner, E.; Mueller-Lantzsch, N.; et al. The NP9 protein encoded by the human endogenous retrovirus HERV-K(HML-2) negatively regulates gene activation of the Epstein-Barr virus nuclear antigen 2 (EBNA2). J. Int. Cancer 2011, 129, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Heyne, K.; Kolsch, K.; Bruand, M.; Kremmer, E.; Grasser, F.A.; Mayer, J.; Roemer, K. Np9, a cellular protein of retroviral ancestry restricted to human, chimpanzee and gorilla, binds and regulates ubiquitin ligase MDM2. Cell Cycle 2015, 14, 2619–2633. [Google Scholar] [CrossRef]
- Bronson, D.L.; Fraley, E.E.; Fogh, J.; Kalter, S.S. Induction of retrovirus particles in human testicular tumor (Tera-1) cell cultures: An electron microscopic study. J. Natl. Cancer Inst. 1979, 63, 337–339. [Google Scholar]
- Goering, W.; Schmitt, K.; Dostert, M.; Schaal, H.; Deenen, R.; Mayer, J.; Schulz, W.A. Human endogenous retrovirus HERV-K(HML-2) activity in prostate cancer is dominated by a few loci. Prostate 2015, 75, 1958–1971. [Google Scholar] [CrossRef]
- Lower, R.; Lower, J.; Frank, H.; Harzmann, R.; Kurth, R. Human teratocarcinomas cultured in vitro produce unique retrovirus-like viruses. J. Gen. Virol. 1984, 65, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Muster, T.; Waltenberger, A.; Grassauer, A.; Hirschl, S.; Caucig, P.; Romirer, I.; Fodinger, D.; Seppele, H.; Schanab, O.; Magin-Lachmann, C.; et al. An endogenous retrovirus derived from human melanoma cells. Cancer Res. 2003, 63, 8735–8741. [Google Scholar] [PubMed]
- Ono, M.; Kawakami, M.; Ushikubo, H. Stimulation of expression of the human endogenous retrovirus genome by female steroid hormones in human breast cancer cell line T47D. J. Virol. 1987, 61, 2059–2062. [Google Scholar] [CrossRef]
- Wang-Johanning, F.; Radvanyi, L.; Rycaj, K.; Plummer, J.B.; Yan, P.; Sastry, K.J.; Piyathilake, C.J.; Hunt, K.K.; Johanning, G.L. Human endogenous retrovirus K triggers an antigen-specific immune response in breast cancer patients. Cancer Res. 2008, 68, 5869–5877. [Google Scholar] [CrossRef]
- Christensen, T.; Dissing Sorensen, P.; Riemann, H.; Hansen, H.J.; Moller-Larsen, A. Expression of sequence variants of endogenous retrovirus RGH in particle form in multiple sclerosis. Lancet 1998, 352, 1033. [Google Scholar] [CrossRef]
- Kury, P.; Nath, A.; Creange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef]
- Arru, G.; Sechi, E.; Mariotto, S.; Zarbo, I.R.; Ferrari, S.; Gajofatto, A.; Monaco, S.; Deiana, G.A.; Bo, M.; Sechi, L.A.; et al. Antibody response against HERV-W in patients with MOG-IgG associated disorders, multiple sclerosis and NMOSD. J. Neuroimmunol. 2020, 338, 577110. [Google Scholar] [CrossRef]
- Arru, G.; Mameli, G.; Deiana, G.A.; Rassu, A.L.; Piredda, R.; Sechi, E.; Caggiu, E.; Bo, M.; Nako, E.; Urso, D.; et al. Humoral immunity response to human endogenous retroviruses K/W differentiates between amyotrophic lateral sclerosis and other neurological diseases. Eur. J. Neurol. 2018, 25, 1076-e84. [Google Scholar] [CrossRef] [PubMed]
- Mameli, G.; Erre, G.L.; Caggiu, E.; Mura, S.; Cossu, D.; Bo, M.; Cadoni, M.L.; Piras, A.; Mundula, N.; Colombo, E.; et al. Identification of a HERV-K env surface peptide highly recognized in Rheumatoid Arthritis (RA) patients: A cross-sectional case-control study. Clin. Exp. Immunol. 2017, 189, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Bannert, N.; Hofmann, H.; Block, A.; Hohn, O. HERVs New Role in Cancer: From Accused Perpetrators to Cheerful Protectors. Front. Microbiol. 2018, 9, 178. [Google Scholar] [CrossRef]
- Kassiotis, G. Endogenous retroviruses and the development of cancer. J. Immunol. Baltim. Md. 1950 2014, 192, 1343–1349. [Google Scholar] [CrossRef]
- Contreras-Galindo, R.; Lopez, P.; Velez, R.; Yamamura, Y. HIV-1 infection increases the expression of human endogenous retroviruses type K (HERV-K) in vitro. AIDS Res. Hum. Retrovir. 2007, 23, 116–122. [Google Scholar] [CrossRef]
- Bhardwaj, N.; Maldarelli, F.; Mellors, J.; Coffin, J.M. HIV-1 infection leads to increased transcription of human endogenous retrovirus HERV-K (HML-2) proviruses in vivo but not to increased virion production. J. Virol. 2014, 88, 11108–11120. [Google Scholar] [CrossRef]
- Toufaily, C.; Landry, S.; Leib-Mosch, C.; Rassart, E.; Barbeau, B. Activation of LTRs from different human endogenous retrovirus (HERV) families by the HTLV-1 tax protein and T-cell activators. Viruses 2011, 3, 2146. [Google Scholar] [CrossRef]
- Fogh, J.T.G. (Ed.) New Human Tumor Cell Lines; Springer: Boston, MA, USA, 1975; pp. 115–159. [Google Scholar] [CrossRef]
- Boller, K.; Frank, H.; Lower, J.; Lower, R.; Kurth, R. Structural organization of unique retrovirus-like particles budding from human teratocarcinoma cell lines. J. Gen. Virol. 1983, 64, 2549–2559. [Google Scholar] [CrossRef] [PubMed]
- Boller, K.; Schonfeld, K.; Lischer, S.; Fischer, N.; Hoffmann, A.; Kurth, R.; Tonjes, R.R. Human endogenous retrovirus HERV-K113 is capable of producing intact viral particles. J. Gen. Virol. 2008, 89, 567–572. [Google Scholar] [CrossRef]
- Bieda, K.; Hoffmann, A.; Boller, K. Phenotypic heterogeneity of human endogenous retrovirus particles produced by teratocarcinoma cell lines. J. Gen. Virol. 2001, 82, 591–596. [Google Scholar] [CrossRef]
- Kramer, P.; Lausch, V.; Volkwein, A.; Hanke, K.; Hohn, O.; Bannert, N. The human endogenous retrovirus K(HML-2) has a broad envelope-mediated cellular tropism and is prone to inhibition at a post-entry, pre-integration step. Virology 2016, 487, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Tonjes, R.R.; Boller, K.; Limbach, C.; Lugert, R.; Kurth, R. Characterization of human endogenous retrovirus type K virus-like particles generated from recombinant baculoviruses. Virology 1997, 233, 280–291. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kraus, B.; Boller, K.; Reuter, A.; Schnierle, B.S. Characterization of the human endogenous retrovirus K Gag protein: Identification of protease cleavage sites. Retrovirology 2011, 8, 21. [Google Scholar] [CrossRef]
- Hanke, K.; Kramer, P.; Seeher, S.; Beimforde, N.; Kurth, R.; Bannert, N. Reconstitution of the ancestral glycoprotein of human endogenous retrovirus k and modulation of its functional activity by truncation of the cytoplasmic domain. J. Virol. 2009, 83, 12790–12800. [Google Scholar] [CrossRef]
- Walsh, D.; Mathews, M.B.; Mohr, I. Tinkering with translation: Protein synthesis in virus-infected cells. Cold Spring Harb. Perspect. Biol. 2013, 5, a012351. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.N.; Bieniasz, P.D. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 2007, 3, e10. [Google Scholar] [CrossRef] [PubMed]
- Dewannieux, M.; Harper, F.; Richaud, A.; Letzelter, C.; Ribet, D.; Pierron, G.; Heidmann, T. Identification of an infectious progenitor for the multiple-copy HERV-K human endogenous retroelements. Genome Res. 2006, 16, 1548–1556. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef]
- Morozov, V.A.; Weiss, R.A. Two types of HTLV-1 particles are released from MT-2 cells. Virology 1999, 255, 279–284. [Google Scholar] [CrossRef]
- Parker, S.D.; Wall, J.S.; Hunter, E. Analysis of Mason-Pfizer monkey virus Gag particles by scanning transmission electron microscopy. J. Virol. 2001, 75, 9543–9548. [Google Scholar] [CrossRef]
- Briggs, J.A.; Wilk, T.; Welker, R.; Krausslich, H.G.; Fuller, S.D. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 2003, 22, 1707–1715. [Google Scholar] [CrossRef]
- Julias, J.G.; Ferris, A.L.; Boyer, P.L.; Hughes, S.H. Replication of phenotypically mixed human immunodeficiency virus type 1 virions containing catalytically active and catalytically inactive reverse transcriptase. J. Virol. 2001, 75, 6537–6546. [Google Scholar] [CrossRef][Green Version]
- Taube, R.; Loya, S.; Avidan, O.; Perach, M.; Hizi, A. Reverse transcriptase of mouse mammary tumour virus: Expression in bacteria, purification and biochemical characterization. Biochem. J. 1998, 329, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Golan, M.; Hizi, A.; Resau, J.H.; Yaal-Hahoshen, N.; Reichman, H.; Keydar, I.; Tsarfaty, I. Human endogenous retrovirus (HERV-K) reverse transcriptase as a breast cancer prognostic marker. Neoplasia N. Y. 2008, 10, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Yuste, E.; Bixby, J.; Lifson, J.; Sato, S.; Johnson, W.; Desrosiers, R. Glycosylation of gp41 of simian immunodeficiency virus shields epitopes that can be targets for neutralizing antibodies. J. Virol. 2008, 82, 12472–12486. [Google Scholar] [CrossRef]
- Montero, M.; van Houten, N.E.; Wang, X.; Scott, J.K. The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: Dominant site of antibody neutralization and target for vaccine design. Microbiol. Mol. Biol. Rev. MMBR 2008, 72, 54–84. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr.; Raab-Traub, N. Microvesicles and viral infection. J. Virol. 2011, 85, 12844–12854. [Google Scholar] [CrossRef]
- Brody, B.A.; Kimball, M.G.; Hunter, E. Mutations within the transmembrane glycoprotein of Mason-Pfizer monkey virus: Loss of SU-TM association and effects on infectivity. Virology 1994, 202, 673–683. [Google Scholar] [CrossRef]
- Wang, W.; Nie, J.; Prochnow, C.; Truong, C.; Jia, Z.; Wang, S.; Chen, X.S.; Wang, Y. A systematic study of the N-glycosylation sites of HIV-1 envelope protein on infectivity and antibody-mediated neutralization. Retrovirology 2013, 10, 14. [Google Scholar] [CrossRef]
- Muriaux, D.; Mirro, J.; Harvin, D.; Rein, A. RNA is a structural element in retrovirus particles. Proc. Natl. Acad. Sci. USA 2001, 98, 5246–5251. [Google Scholar] [CrossRef] [PubMed]
- Acton, O.; Grant, T.; Nicastro, G.; Ball, N.J.; Goldstone, D.C.; Robertson, L.E.; Sader, K.; Nans, A.; Ramos, A.; Stoye, J.P.; et al. Structural basis for Fullerene geometry in a human endogenous retrovirus capsid. Nat. Commun. 2019, 10, 5822. [Google Scholar] [CrossRef] [PubMed]
- Record, M.; Silvente-Poirot, S.; Poirot, M.; Wakelam, M.J.O. Extracellular vesicles: Lipids as key components of their biogenesis and functions. J. Lipid Res. 2018, 59, 1316–1324. [Google Scholar] [CrossRef]
- Willms, E.; Johansson, H.J.; Mäger, I.; Lee, Y.; Blomberg, K.E.; Sadik, M.; Alaarg, A.; Smith, C.I.; Lehtiö, J.; El Andaloussi, S.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 2016, 6, 22519. [Google Scholar] [CrossRef]
- Sedgwick, A.E.; D’Souza-Schorey, C. The biology of extracellular microvesicles. Traffic Cph. Den. 2018, 19, 319–327. [Google Scholar] [CrossRef]
- Boller, K.; Janssen, O.; Schuldes, H.; Tönjes, R.R.; Kurth, R. Characterization of the antibody response specific for the human endogenous retrovirus HTDV/HERV-K. J. Virol. 1997, 71, 4581–4588. [Google Scholar] [CrossRef] [PubMed]
- Morozov, V.A.; Dao Thi, V.L.; Denner, J. The transmembrane protein of the human endogenous retrovirus--K (HERV-K) modulates cytokine release and gene expression. PLoS ONE 2013, 8, e70399. [Google Scholar] [CrossRef]
- Kassiotis, G.; Stoye, J.P. Immune responses to endogenous retroelements: Taking the bad with the good. Nature reviews. Immunology 2016, 16, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Hsu, A.C.; Pang, Z.; Pan, H.; Zuo, X.; Wang, G.; Zheng, J.; Wang, F. Role of the Innate Cytokine Storm Induced by the Influenza A Virus. Viral Immunol. 2019, 32, 244–251. [Google Scholar] [CrossRef]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Ren, S.; Chen, X.; Huang, R.; Zhou, G.G.; Yuan, Z. SOCS4 expressed by recombinant HSV protects against cytokine storm in a mouse model. Oncol. Rep. 2019, 41, 1509–1520. [Google Scholar] [CrossRef]
- Nolte, E.; Cremer, T.; Gallo, R.C.; Margolis, L.B. Extracellular vesicles and viruses: Are they close relatives? Proc. Natl. Acad. Sci. USA 2016, 113, 9155–9161. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, K.; Ferreira, H.; Flockerzi, A.; Wahl, S.; Sauter, M.; Mayer, J.; Mueller-Lantzsch, N. Human endogenous retrovirus family HERV-K(HML-2) RNA transcripts are selectively packaged into retroviral particles produced by the human germ cell tumor line Tera-1 and originate mainly from a provirus on chromosome 22q11.21. J. Virol. 2008, 82, 10008–10016. [Google Scholar] [CrossRef]
- Bhardwaj, N.; Montesion, M.; Roy, F.; Coffin, J.M. Differential expression of HERV-K (HML-2) proviruses in cells and virions of the teratocarcinoma cell line Tera-1. Viruses 2015, 7, 939. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.J.; Ribeiro, R.M.; Perelson, A.S. Current estimates for HIV-1 production imply rapid viral clearance in lymphoid tissues. PLoS Comput. Biol. 2010, 6, e1000906. [Google Scholar] [CrossRef]
- Hohn, O.; Hanke, K.; Lausch, V.; Zimmermann, A.; Mostafa, S.; Bannert, N. CMV-promoter driven codon-optimized expression alters the assembly type and morphology of a reconstituted HERV-K(HML-2). Viruses 2014, 6, 4332. [Google Scholar] [CrossRef]
- Vermeire, J.; Naessens, E.; Vanderstraeten, H.; Landi, A.; Iannucci, V.; Van Nuffel, A.; Taghon, T.; Pizzato, M.; Verhasselt, B. Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLoS ONE 2012, 7, e50859. [Google Scholar] [CrossRef]
- Moller, L.; Schunadel, L.; Nitsche, A.; Schwebke, I.; Hanisch, M.; Laue, M. Evaluation of virus inactivation by formaldehyde to enhance biosafety of diagnostic electron microscopy. Viruses 2015, 7, 666. [Google Scholar] [CrossRef] [PubMed]
- Giannessi, F.; Aiello, A.; Franchi, F.; Percario, Z.A.; Affabris, E. The role of extracellular vesicles as allies of HIV, HCV and SARS viruses. Viruses 2020, 12, 571. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Viruses | Total gls | SU gls | TM gls | TM Ectodomain (a.a./gls) | TM Ectodomain IG (a.a./gls) |
|---|---|---|---|---|---|
| HERV-K108 (HML.2) | 11 | 7 * | 4 | 168/4 | 42 |
| HERV-K109 (HML.2) | 11 | 7 * | 4 | 168/4 | 42 |
| HERV-K113 (HML.2) | 11 | 7 * | 4 | 170/4 | ~42.5 |
| HERV-K115 (HML.2) | 11 | 7 | 4 | 170/4 | ~42.5 |
| HERV-K108 (HML.2) | 11 | 7 | 4 | 170/4 | ~42.5 |
| JSRV | 9 | 6 | 3 | 171/3 | 57 |
| ENTV | 8 | 5 | 3 | 170/3 | ~56.6 |
| MMTV | 5 | 3 | 2 | 168/2 | 84 |
| MPMV | 11 | 10 | 1 | 167/1 | 167 |
| HIV-1 | 31 | 24 | 7 (-3) ** | 170/4 | ~42.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morozov, V.A.; Morozov, A.V. A Comprehensive Analysis of Human Endogenous Retroviruses HERV-K (HML.2) from Teratocarcinoma Cell Lines and Detection of Viral Cargo in Microvesicles. Int. J. Mol. Sci. 2021, 22, 12398. https://doi.org/10.3390/ijms222212398
Morozov VA, Morozov AV. A Comprehensive Analysis of Human Endogenous Retroviruses HERV-K (HML.2) from Teratocarcinoma Cell Lines and Detection of Viral Cargo in Microvesicles. International Journal of Molecular Sciences. 2021; 22(22):12398. https://doi.org/10.3390/ijms222212398
Chicago/Turabian StyleMorozov, Vladimir A., and Alexey V. Morozov. 2021. "A Comprehensive Analysis of Human Endogenous Retroviruses HERV-K (HML.2) from Teratocarcinoma Cell Lines and Detection of Viral Cargo in Microvesicles" International Journal of Molecular Sciences 22, no. 22: 12398. https://doi.org/10.3390/ijms222212398
APA StyleMorozov, V. A., & Morozov, A. V. (2021). A Comprehensive Analysis of Human Endogenous Retroviruses HERV-K (HML.2) from Teratocarcinoma Cell Lines and Detection of Viral Cargo in Microvesicles. International Journal of Molecular Sciences, 22(22), 12398. https://doi.org/10.3390/ijms222212398