
Inhibitory Potential of New Phenolic Hydrazide-Hydrazones with a Decoy Substrate Fragment towards Laccase from a Phytopathogenic Fungus: SAR and Molecular Docking Studies


Abstract
:1. Introduction
2. Results and Discussion
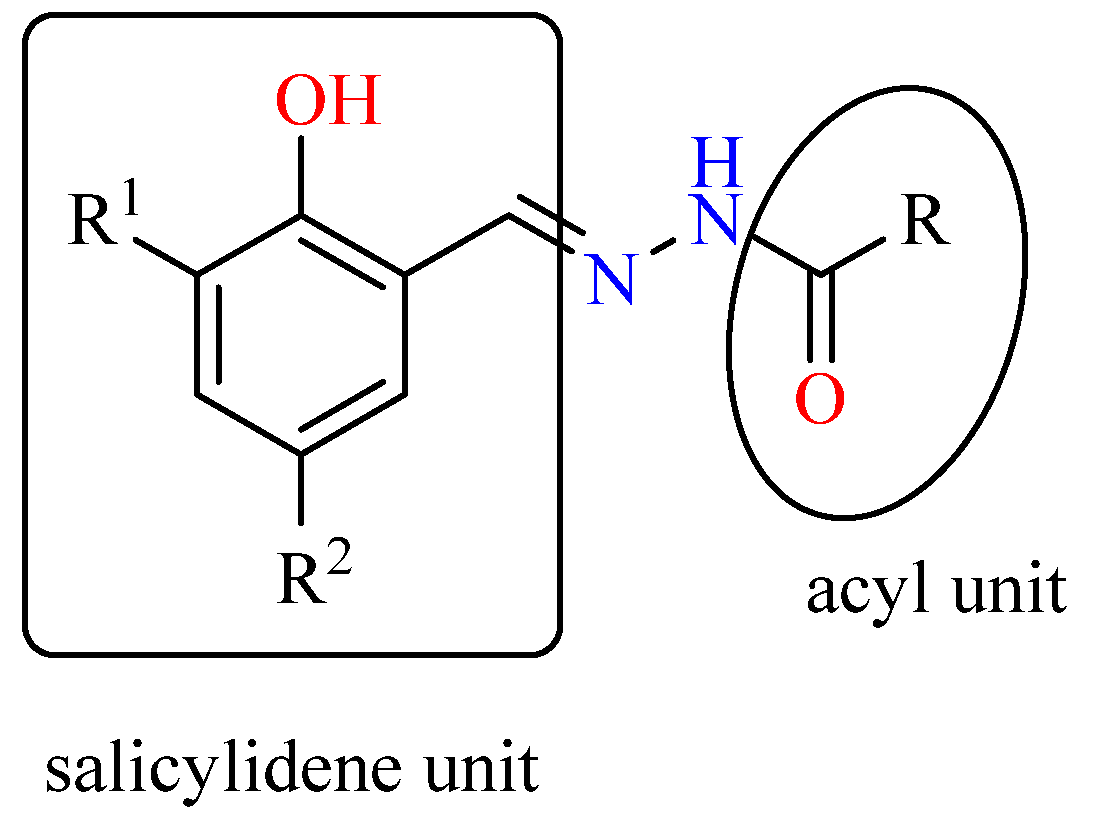
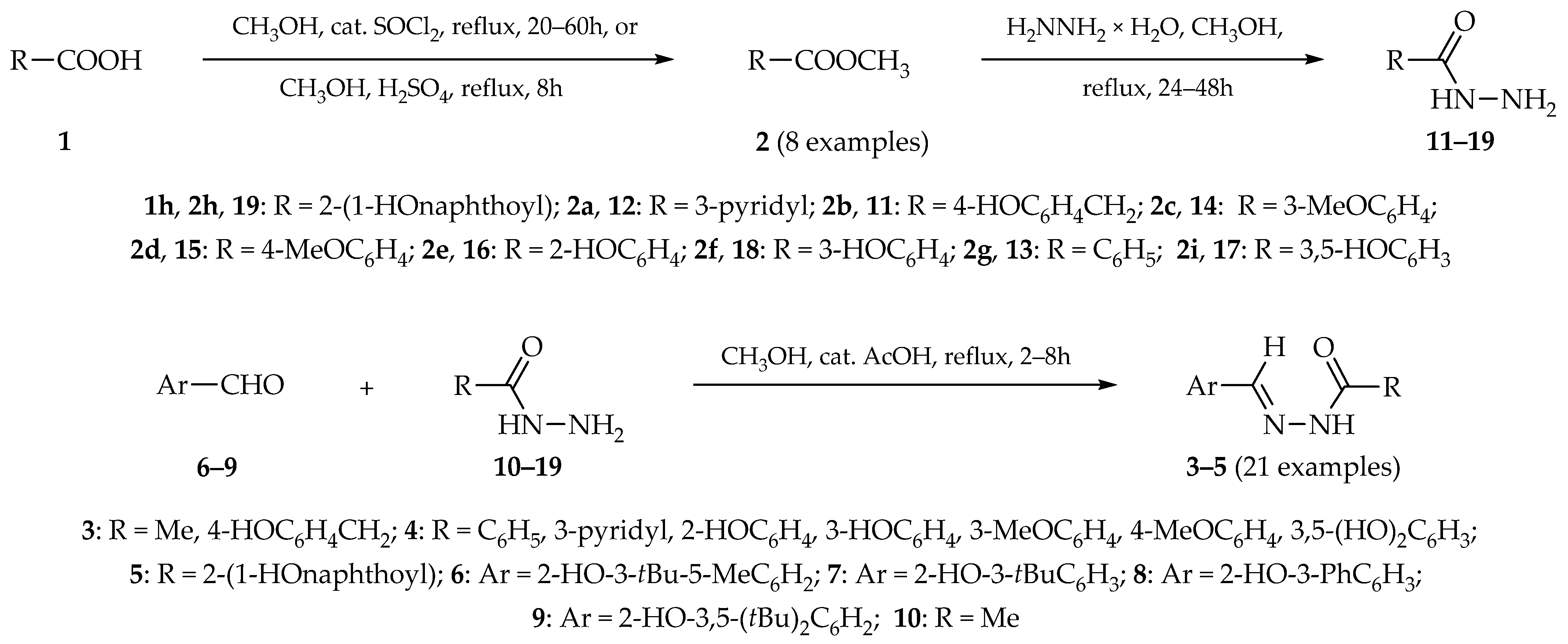
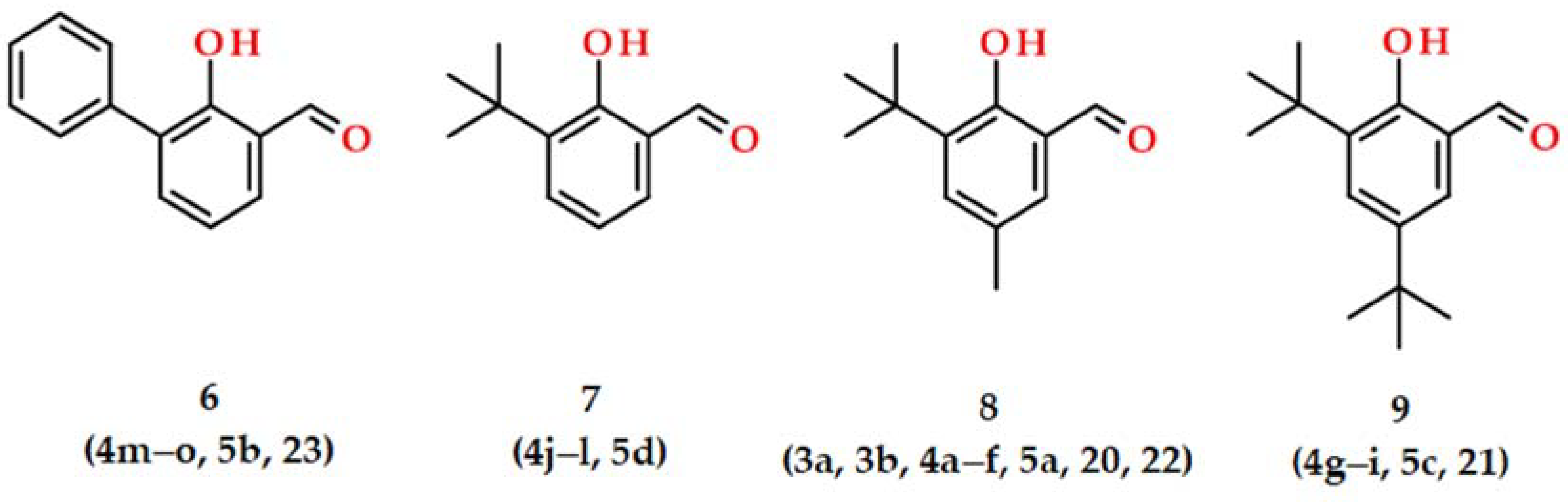
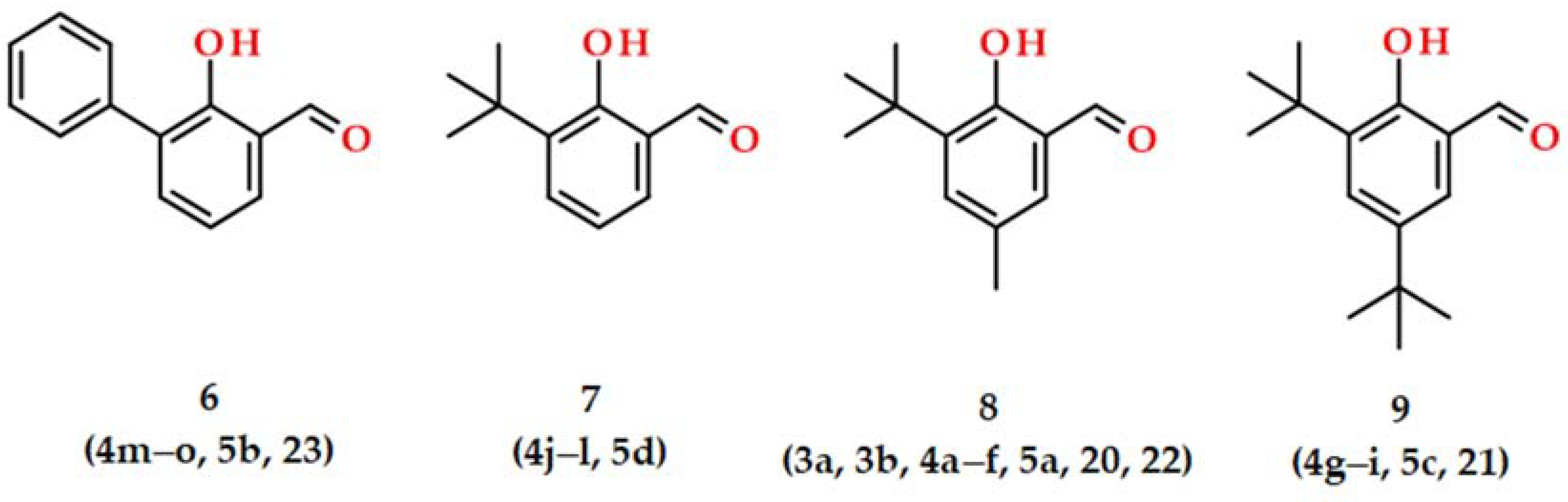
2.1. Syntheses and Characterizations
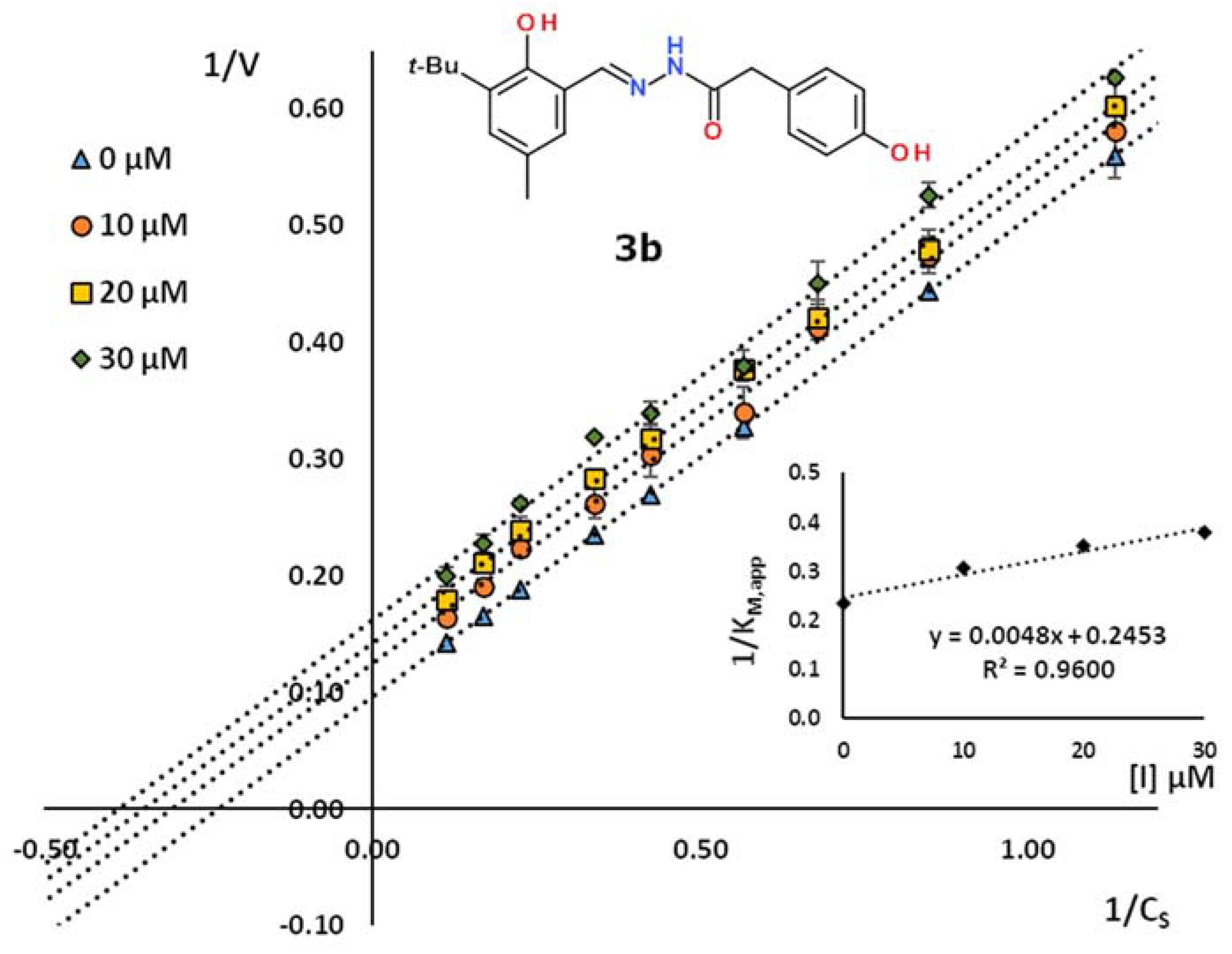
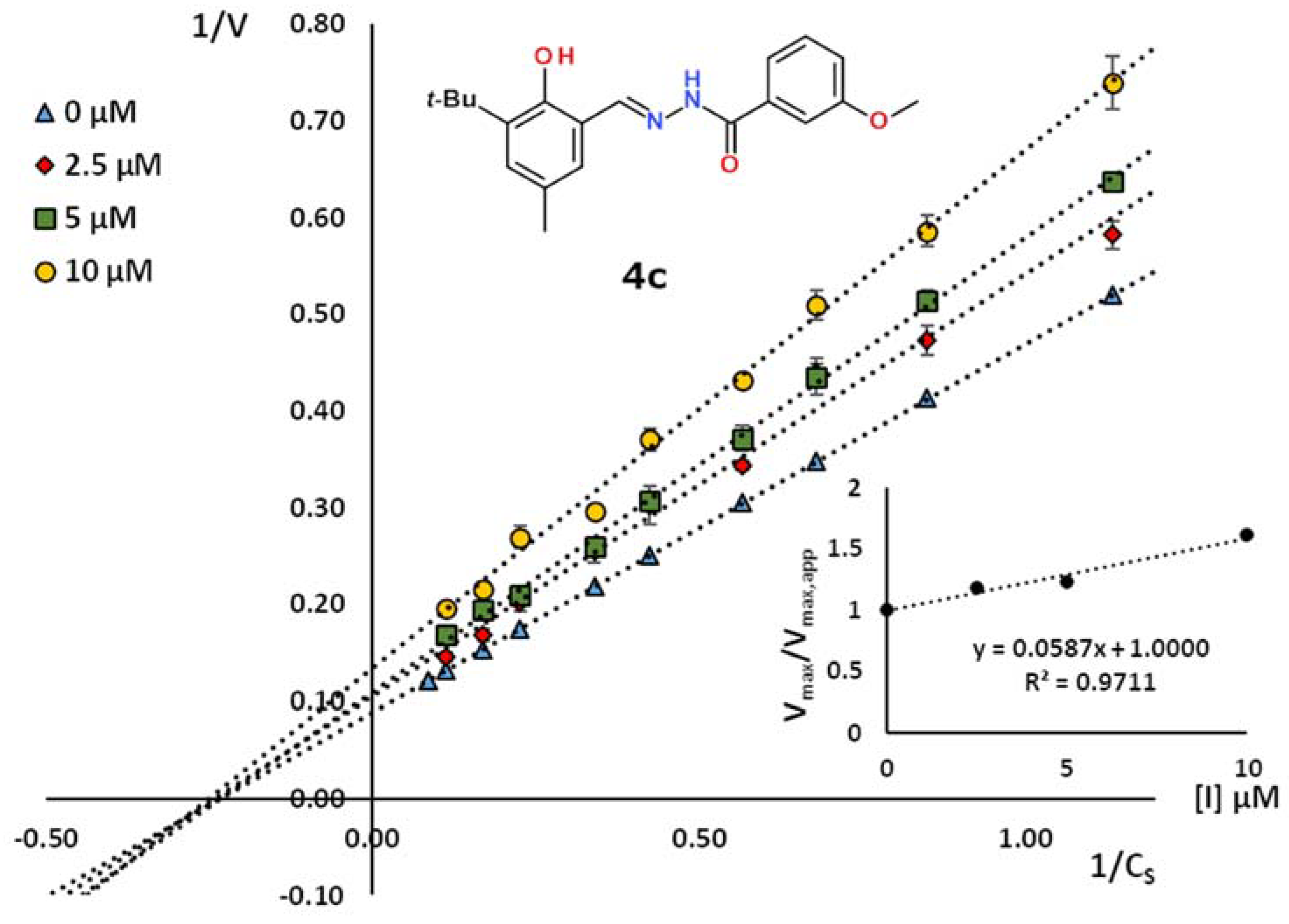
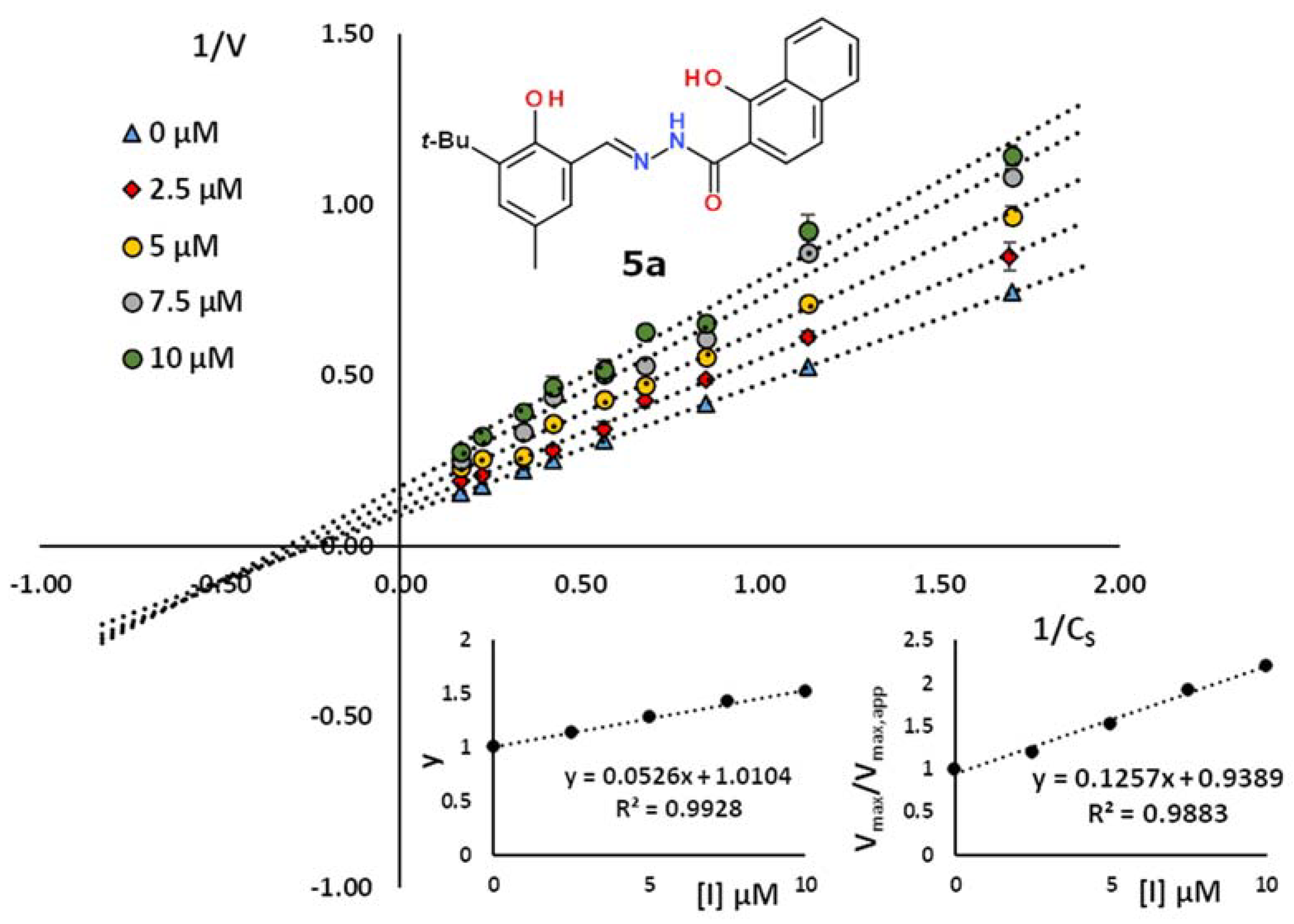
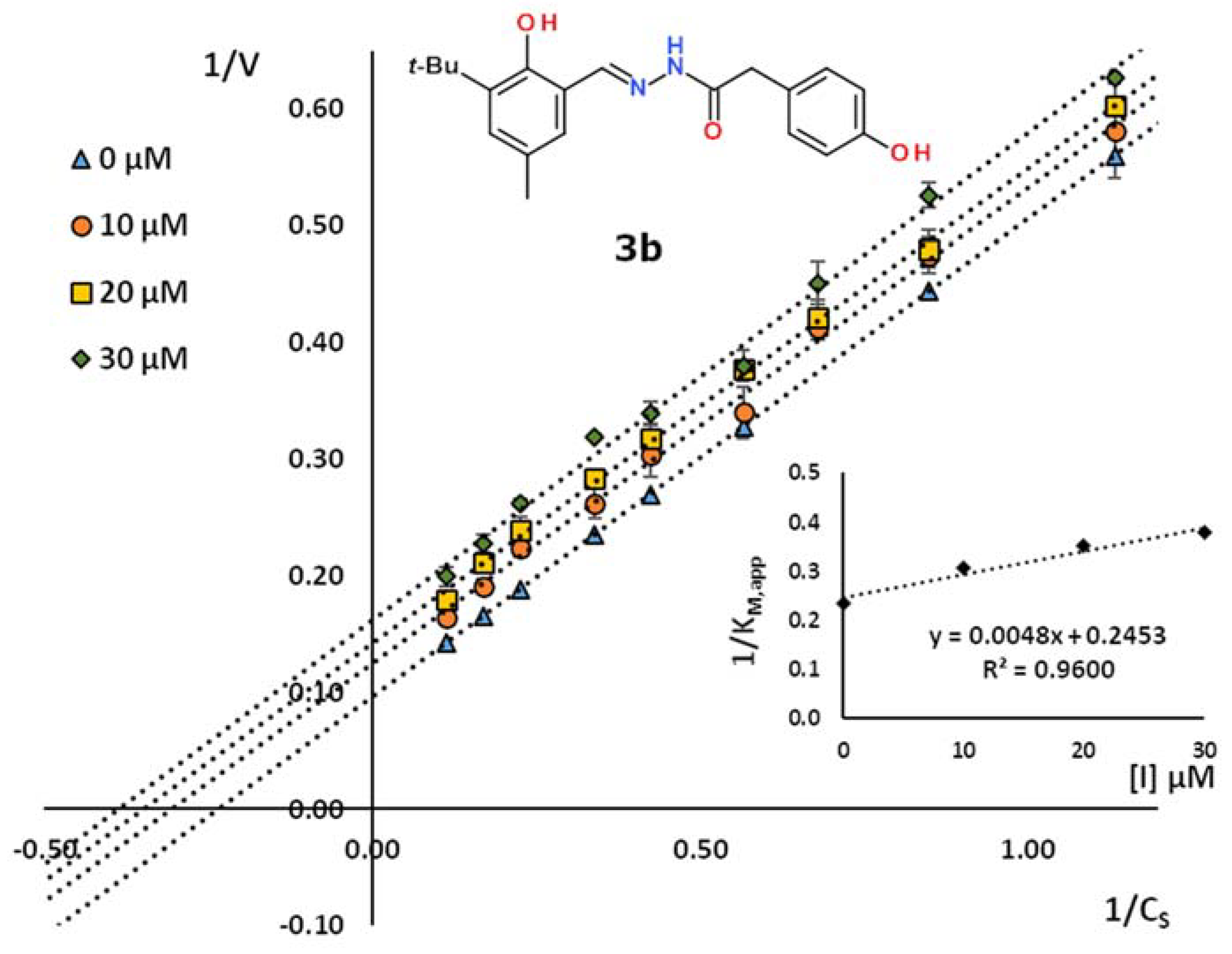
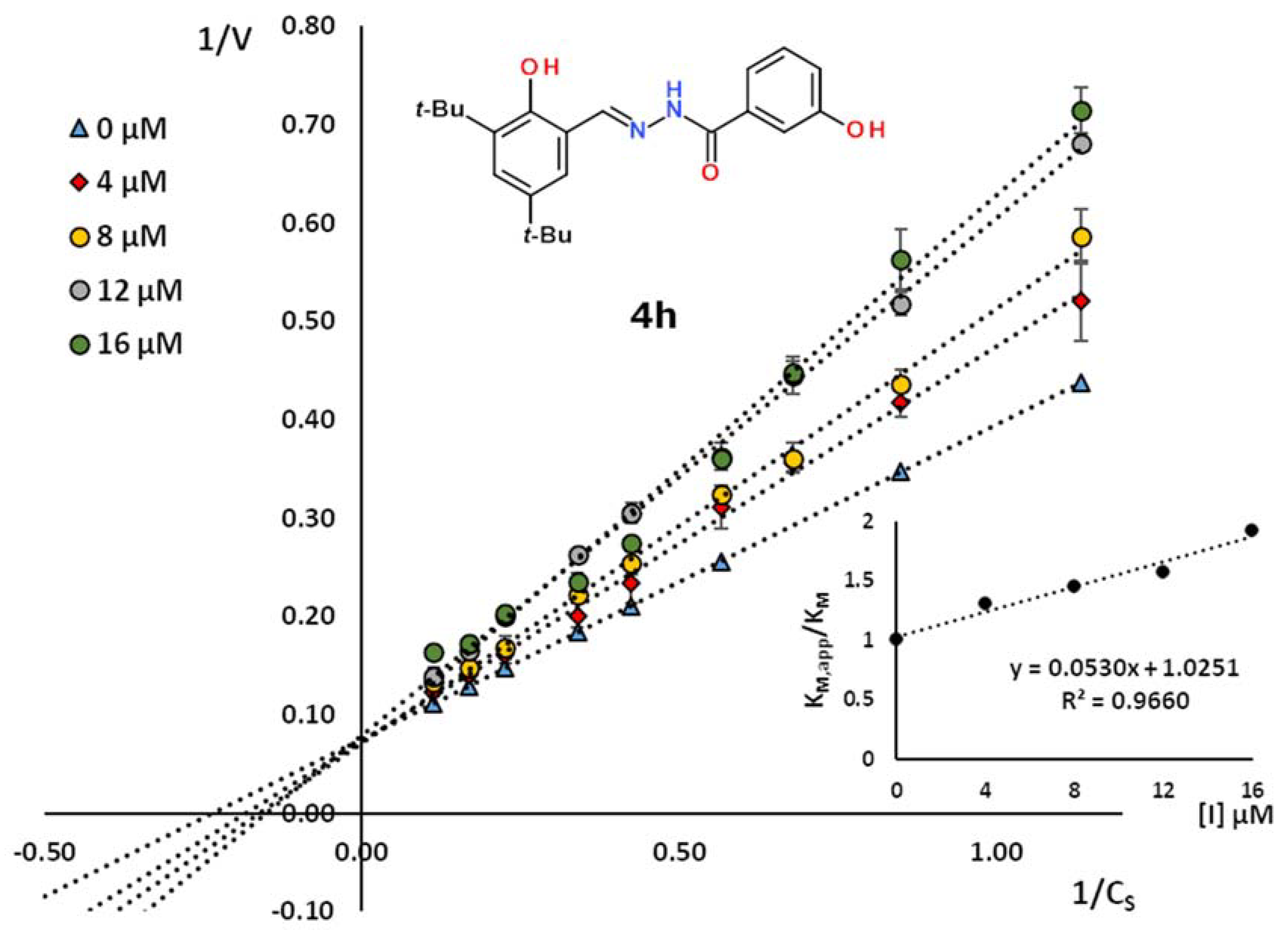
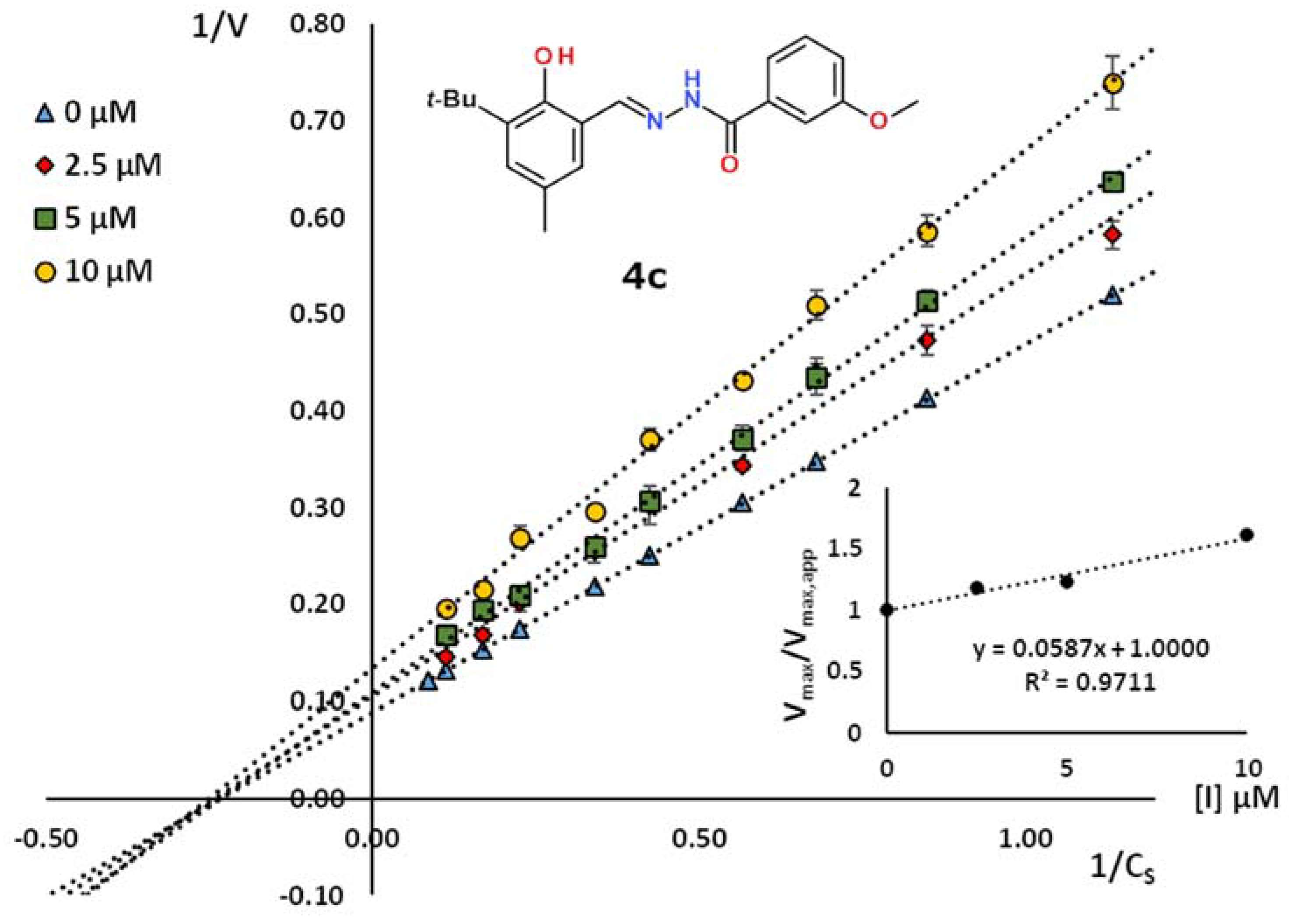
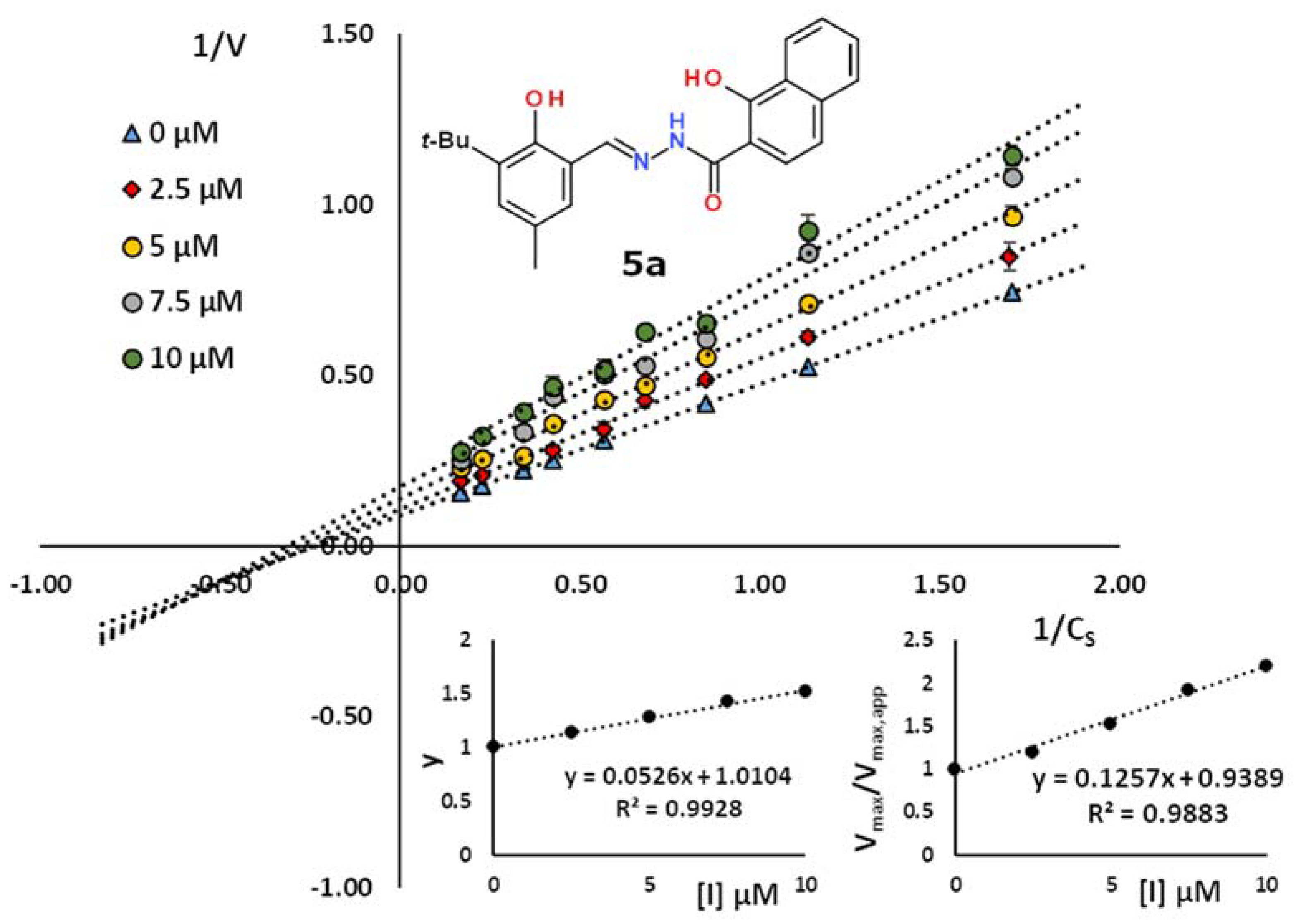
2.2. Kinetic Studies
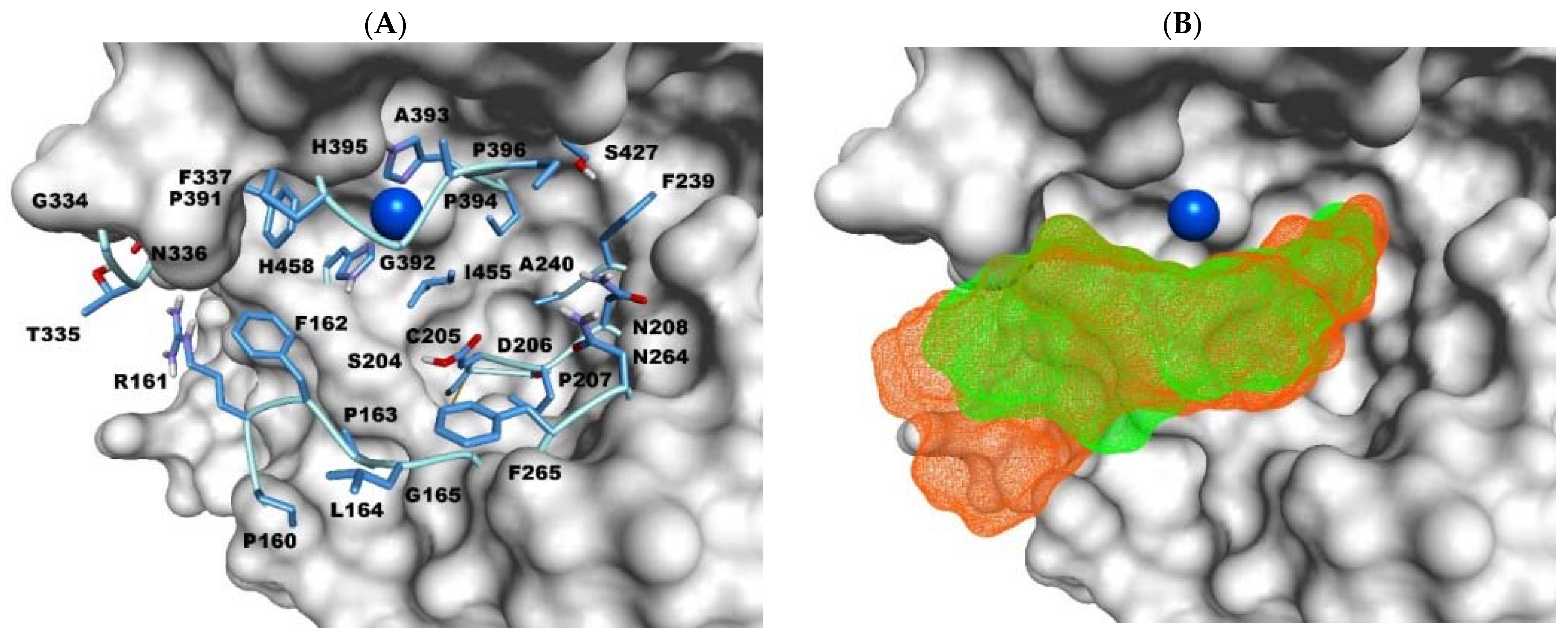
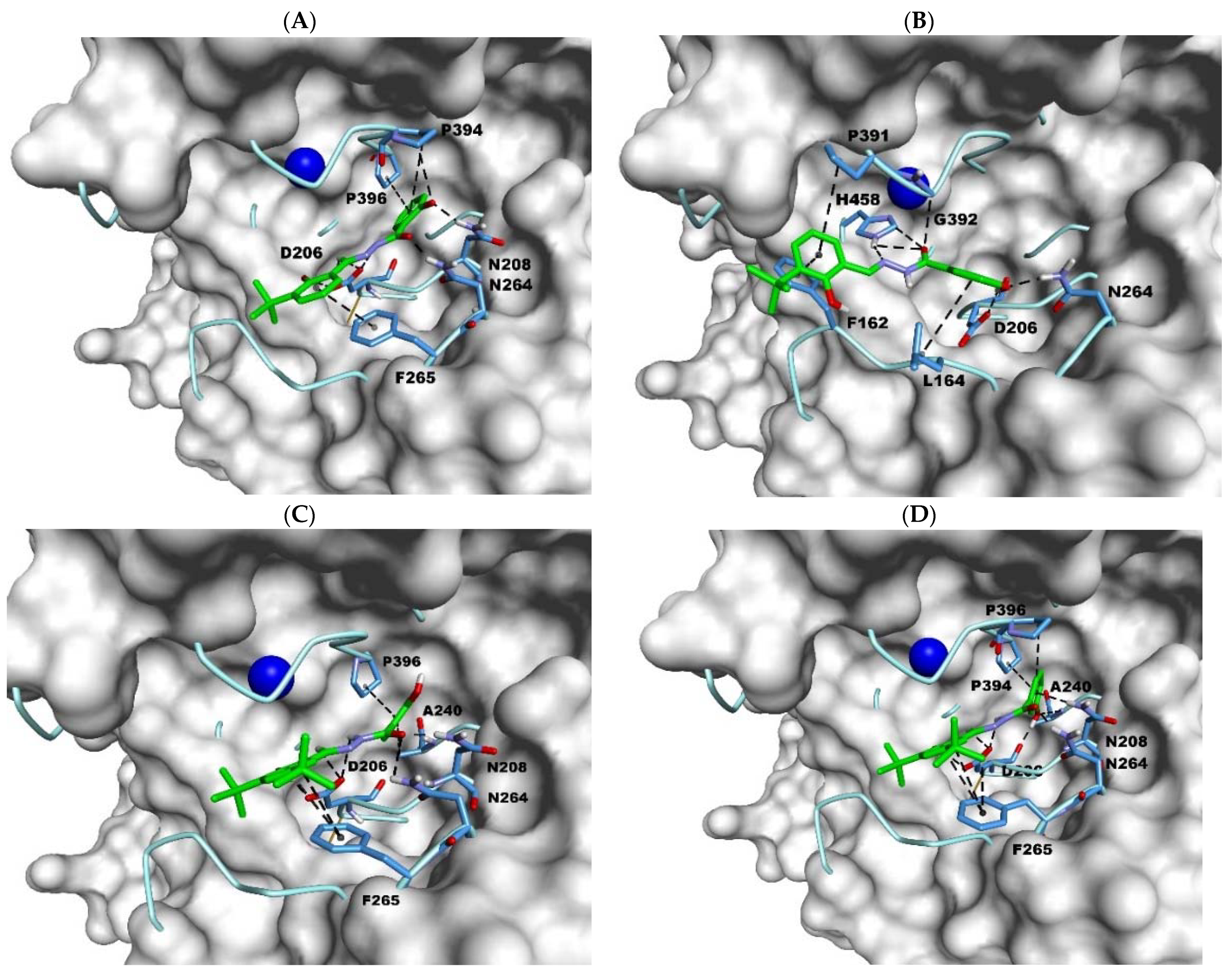
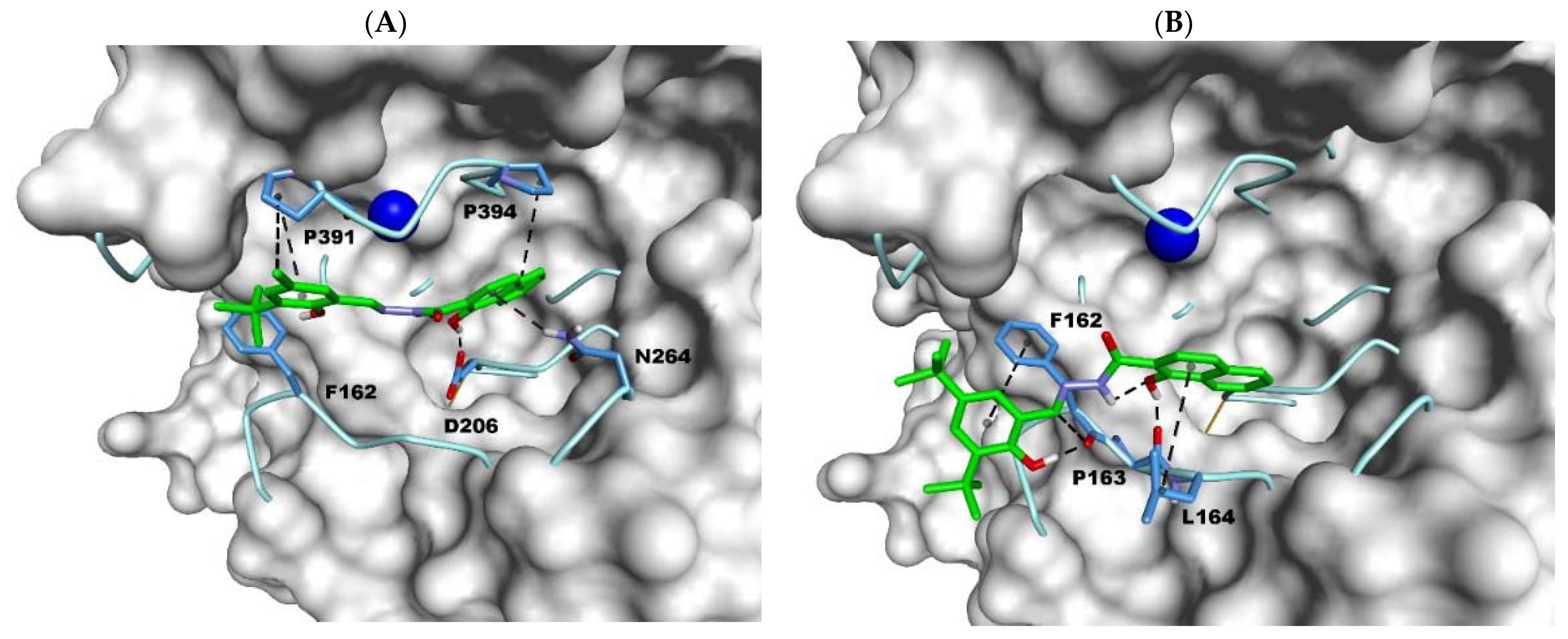
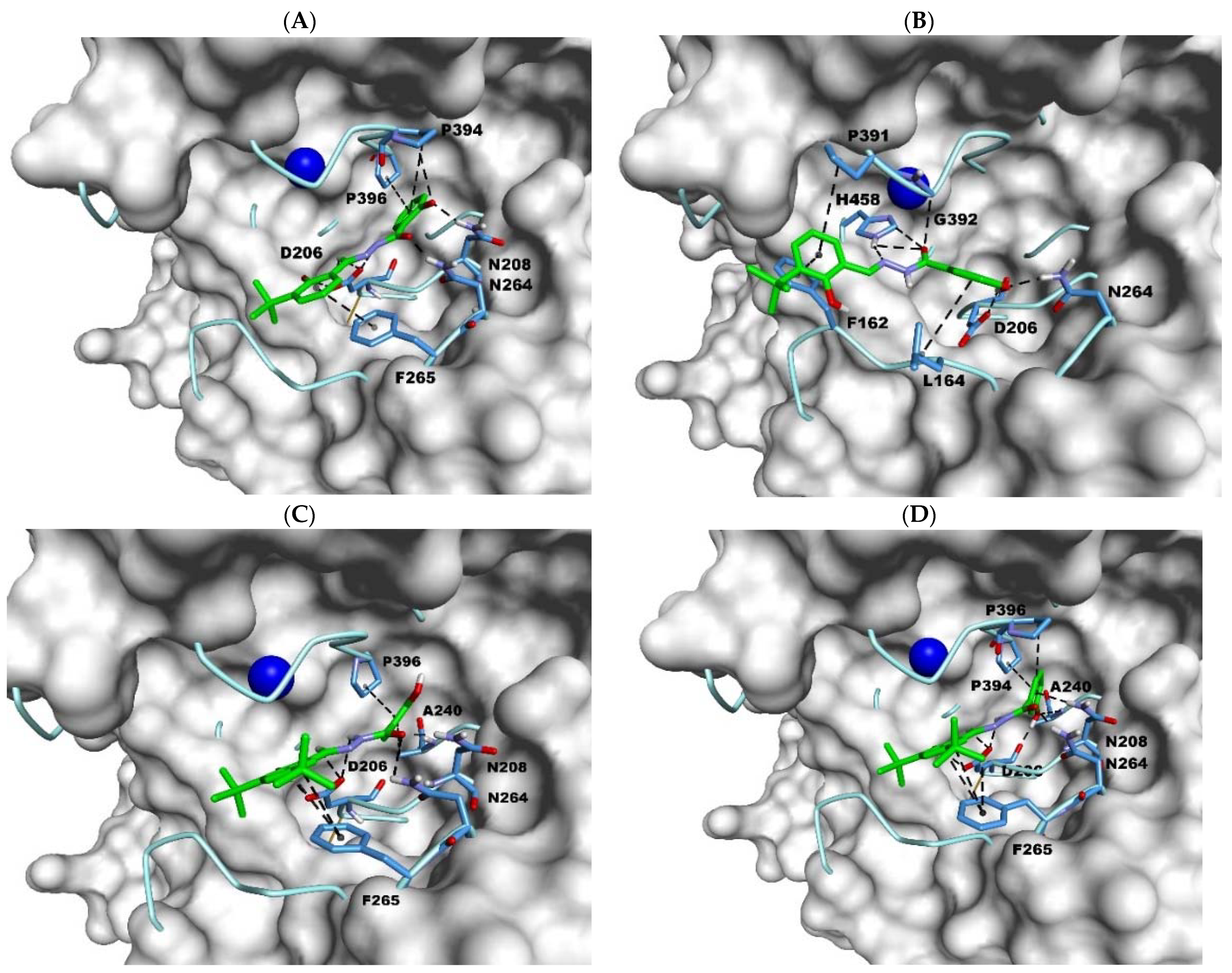
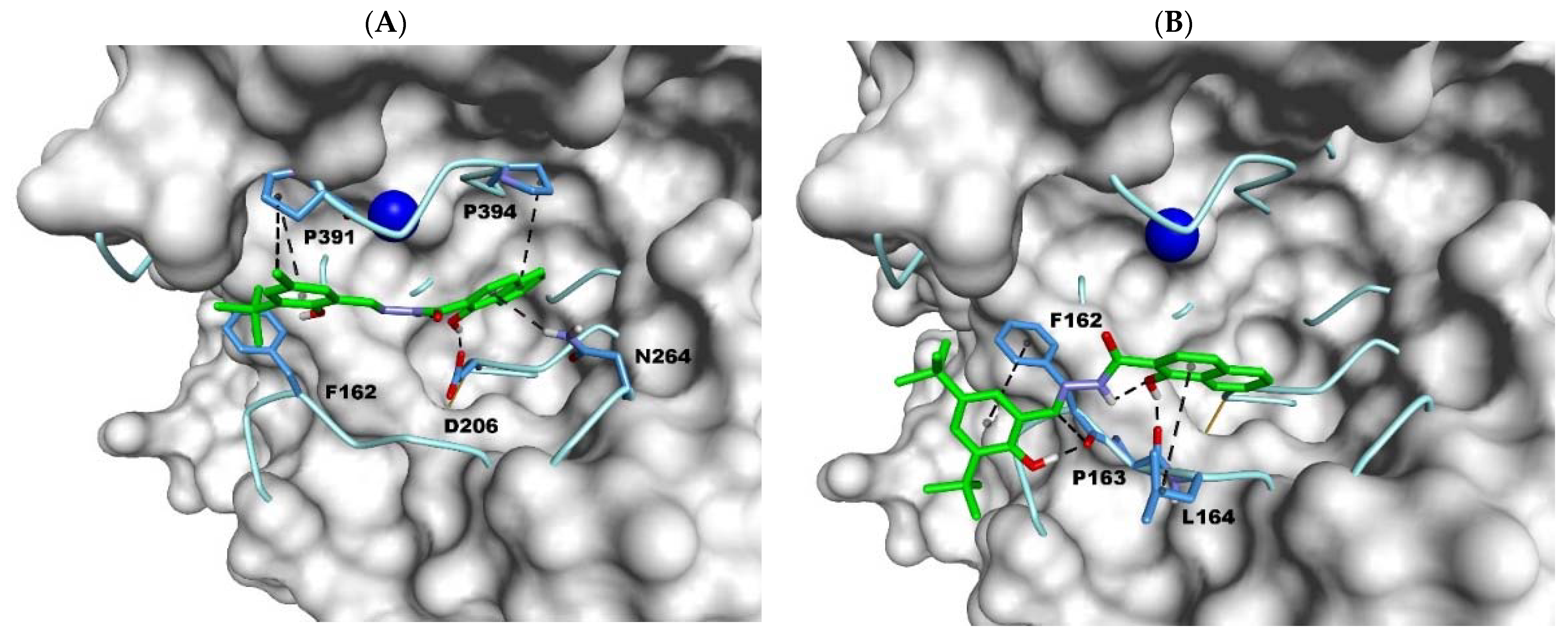
2.3. Docking Studies
3. Materials and Methods
3.1. Synthesis of Aroylhydrazones 3–5
3.2. Reaction Condition and Kinetic Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Witayakran, S.; Ragauskas, A.J. Synthetic Applications of Laccase in Green Chemistry. Adv. Synth. Catal. 2009, 351, 1187–1209. [Google Scholar] [CrossRef]
- Sousa, A.; Martins, L.; Robalo, M. Laccases: Versatile Biocatalysts for the Synthesis of Heterocyclic Cores. Molecules 2021, 26, 3719. [Google Scholar] [CrossRef]
- Kim, S. Mushroom Ligninolytic Enzymes―Features and Application of Potential Enzymes for Conversion of Lignin into Bio-Based Chemicals and Materials. Appl. Sci. 2021, 11, 6161. [Google Scholar] [CrossRef]
- Ardila-Leal, L.; Poutou-Piñales, R.; Pedroza-Rodríguez, A.; Quevedo-Hidalgo, B. A Brief History of Colour, the Environmental Impact of Synthetic Dyes and Removal by Using Laccases. Molecules 2021, 26, 3813. [Google Scholar] [CrossRef]
- Bassanini, I.; Ferrandi, E.; Riva, S.; Monti, D. Biocatalysis with Laccases: An Updated Overview. Catalysts 2020, 11, 26. [Google Scholar] [CrossRef]
- Singh, G.; Bhalla, A.; Kaur, P.; Capalash, N.; Sharma, P. Laccase from prokaryotes: A new source for an old enzyme. Rev. Environ. Sci. Bio. Technol. 2011, 10, 309–326. [Google Scholar] [CrossRef]
- Asano, T.; Seto, Y.; Hashimoto, K.; Kurushima, H. Mini-review an insect-specific system for terrestrialization: Laccase-mediated cuticle formation. Insect Biochem. Mol. Biol. 2019, 108, 61–70. [Google Scholar] [CrossRef]
- Dwivedi, U.N.; Singh, P.; Pandey, V.; Kumar, A. Structure–function relationship among bacterial, fungal and plant laccases. J. Mol. Catal. B Enzym. 2011, 68, 117–128. [Google Scholar] [CrossRef]
- Leonowicz, A.; Cho, N.; Luterek, J.; Jarosz-Wilkolazka, A.; Wojtas-Wasilewska, M.; Matuszewska, A.; Hofrichter, M.; Wesenberg, D.; Rogalski, J. Fungal laccase: Properties and activity on lignin. J. Basic Microbiol. 2001, 41, 185–227. [Google Scholar] [CrossRef]
- Williamson, P.R. Laccase and melanin in the pathogenesis of Cryptococcus neoformans. Front. Biosci. 1997, 2, e99–e107. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Williamson, P.R. Role of laccase in the biology and virulence of Cryptococcus neoformans. FEMS Yeast Res. 2004, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Emidio, E.C.P.; Urán, M.E.J.; Silva, L.B.R.; Dias, L.S.; Doprado, M.; Nosanchuk, J.D.; Taborda, C.P. Melanin as a Virulence Factor in Different Species of Genus Paracoccidioides. J. Fungi 2020, 6, 291. [Google Scholar] [CrossRef]
- Labois, C.; Stempien, E.; Schneider, J.; Schaeffer-Reiss, C.; Bertsch, C.; Goddard, M.-L.; Chong, J. Comparative Study of Secreted Proteins, Enzymatic Activities of Wood Degradation and Stilbene Metabolization in Grapevine Botryosphaeria Dieback Fungi. J. Fungi 2021, 7, 568. [Google Scholar] [CrossRef]
- Degani, O.; Goldblat, Y. Potential Role of Laccases in the Relationship of the Maize Late Wilt Causal Agent, Magnaporthiopsis maydis, and Its Host. J. Fungi 2020, 6, 63. [Google Scholar] [CrossRef]
- Favaron, F.; Lucchetta, M.; Odorizzi, S.; Pais da Cunha, A.; Sella, L. The role of grape polyphenols on trans-rescerastrol activity against Botrytis cinerea and of fungal laccase on the solubility of putiative grape PR proteins. J. Plant Pathol. 2009, 91, 579–588. [Google Scholar]
- Rigling, D.; Van Alfen, N.K. Regulation of laccase biosynthesis in the plant-pathogenic fungus Cryphonectria parasitica by double-stranded RNA. J. Bacteriol. 1991, 173, 8000–8003. [Google Scholar] [CrossRef] [Green Version]
- Quijada-Morin, N.; Garcia, F.; Lambert, K.; Walker, A.-S.; Tiers, L.; Viaud, M.; Sauvage, F.-X.; Hirtz, C.; Saucier, C. Strain effect on extracellular laccase activities fromBotrytis cinerea. Aust. J. Grape Wine Res. 2018, 24, 241–251. [Google Scholar] [CrossRef]
- Claus, H. Laccases of Botrytis cinerea. In Biology of Microorganisms on Grapes, in Must and in Wine; Konig, H., Unden, G., Frohlich, Ü., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 339–356. ISBN 9783319600208. [Google Scholar]
- Coman, C.; Moţ, A.C.; Gal, E.; Pârvu, M.; Silaghi-Dumitrescu, R. Laccase is upregulated via stress pathways in the phytopathogenic fungus Sclerotinia sclerotiorum. Fungal Biol. 2013, 117, 528–539. [Google Scholar] [CrossRef]
- Wahleithner, J.A.; Xu, F.; Brown, K.M.; Brown, S.H.; Golightly, E.J.; Halkier, T.; Kauppinen, S.; Pederson, A.; Schneider, P. The identification and characterization of four laccases from the plant pathogenic fungusRhizoctonia solani. Curr. Genet. 1996, 29, 395–403. [Google Scholar] [CrossRef]
- Rogalski, J.; Janusz, G.; Legiec, D.; Shin, S.-J.; Ohga, S. Purification of Extracellular Laccase from Rhizoctonia praticola. J. Fac. Agric. Kyushu Univ. 2011, 56, 1–7. [Google Scholar] [CrossRef]
- Peng, Y.; Li, S.J.; Yan, J.; Tang, Y.; Cheng, J.P.; Gao, A.J.; Yao, X.; Ruan, J.J.; Xu, B.L. Research Progress on Phytopathogenic Fungi and Their Role as Biocontrol Agents. Front. Microbiol. 2021, 12, 670135. [Google Scholar] [CrossRef]
- Agrios, G. Plant Pathogens and Disease: General Introduction. In Encyclopedia of Microbiology; Elsevier BV: Amsterdam, The Netherlands, 2009; pp. 613–646. [Google Scholar]
- Couto, S.; Herrera, L. Inhibitors of Laccases: A Review. Curr. Enzym. Inhib. 2006, 2, 343–352. [Google Scholar] [CrossRef]
- Martínez-Sotres, C.; Rutiaga-Quiñones, J.G.; Herrera-Bucio, R.; Gallo, M.; López-Albarrán, P. Molecular docking insights into the inhibition of laccase activity by medicarpin. Wood Sci. Technol. 2015, 49, 857–868. [Google Scholar] [CrossRef]
- Dalisay, D.S.; Saludes, J.P.; Molinski, T.F. Ptilomycalin A inhibits laccase and melanization in Cryptococcus neoformans. Bioorganic Med. Chem. 2011, 19, 6654–6657. [Google Scholar] [CrossRef] [Green Version]
- Zavarzina, A.; Leontievsky, A.; Golovleva, L.; Trofimov, S. Biotransformation of soil humic acids by blue laccase of Panus tigrinus 8/18: An in vitro study. Soil Biol. Biochem. 2004, 36, 359–369. [Google Scholar] [CrossRef]
- Tišma, M.; Molnar, M.; Skarica, M.; Čačić, M.; Zelić, B. Laccase Inhibiting Activity of Some Coumarin Derivatives. Lett. Org. Chem. 2014, 11, 583–589. [Google Scholar] [CrossRef]
- Ladd, J.; Butler, J. Inhibition by soil humic acids of native and acetylated proteolytic enzymes. Soil Biol. Biochem. 1971, 3, 157–160. [Google Scholar] [CrossRef]
- Krátký, M.; Svrčková, K.; Vu, Q.; Štěpánková, Š.; Vinšová, J. Hydrazones of 4-(Trifluoromethyl)benzohydrazide as New Inhibitors of Acetyl- and Butyrylcholinesterase. Molecules 2021, 26, 989. [Google Scholar] [CrossRef]
- Maniak, H.; Talma, M.; Matyja, K.; Trusek, A.; Giurg, M. Synthesis and Structure-Activity Relationship Studies of Hydrazide-Hydrazones as Inhibitors of Laccase from Trametes versicolor. Molecules 2020, 25, 1255. [Google Scholar] [CrossRef] [Green Version]
- Popiołek, Ł. The bioactivity of benzenesulfonyl hydrazones: A short review. Biomed. Pharmacother. 2021, 141, 111851. [Google Scholar] [CrossRef]
- Sava, A.; Buron, F.; Routier, S.; Panainte, A.; Bibire, N.; Constantin, S.; Lupașcu, F.; Focșa, A.; Profire, L. Design, Synthesis, In Silico and In Vitro Studies for New Nitric Oxide-Releasing Indomethacin Derivatives with 1,3,4-Oxadiazole-2-thiol Scaffold. Int. J. Mol. Sci. 2021, 22, 7079. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Hu, D.; Chen, Z.; Wang, Y.; Song, B. Synthesis and antiviral evaluation of novel 1,3,4-oxadiazole/thiadiazole-chalcone conjugates. Bioorganic Med. Chem. Lett. 2017, 27, 4298–4301. [Google Scholar] [CrossRef]
- Roztocki, K.; Formalik, F.; Krawczuk, A.; Senkovska, I.; Kuchta, B.; Kaskel, S.; Matoga, D. Collective Breathing in an Eightfold Interpenetrated Metal–Organic Framework: From Mechanistic Understanding towards Threshold Sensing Architectures. Angew. Chem. 2020, 132, 4521–4527. [Google Scholar] [CrossRef]
- Hardy, J.G. Metallosupramolecular grid complexes: Towards nanostructured materials with high-tech applications. Chem. Soc. Rev. 2013, 42, 7881–7899. [Google Scholar] [CrossRef] [Green Version]
- Gale, P.A.; Busschaert, N.; Haynes, C.J.E.; Karagiannidis, L.E.; Kirby, I.L. Anion receptor chemistry: Highlights from 2011 and 2012. Chem. Soc. Rev. 2014, 43, 205–241. [Google Scholar] [CrossRef] [Green Version]
- Gan, C.; Cui, J.; Su, S.; Lin, Q.; Jia, L.; Fan, L.; Huang, Y. Synthesis and antiproliferative activity of some steroidal thiosemicarbazones, semicarbazones and hydrozones. Steroids 2014, 87, 99–107. [Google Scholar] [CrossRef]
- Cho, H.J.; Kim, T.; Kim, H.; Song, C. Kim Solid-State Emissive Metallo-Supramolecular Assemblies of Quinoline-Based Acyl Hydrazone. Sensors 2020, 20, 600. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Mori, Y.; Fossey, J.; Salter, M.M. Catalytic Enantioselective Formation of C−C Bonds by Addition to Imines and Hydrazones: A Ten-Year Update. Chem. Rev. 2011, 111, 2626–2704. [Google Scholar] [CrossRef]
- Yang, J.; Huang, L.; Guo, Z.; Ren, W.; Wang, Q. A Cu(II)-benzoyl hydrazone based fluorescent probe for lipopolysaccharides. J. Lumin. 2016, 172, 290–296. [Google Scholar] [CrossRef]
- Patil, S.; Pandey, S.; Singh, A.; Radhakrishna, M.; Basu, S. Hydrazide–Hydrazone Small Molecules as AIEgens: Illuminating Mitochondria in Cancer Cells. Chem.-A Eur. J. 2019, 25, 8229–8235. [Google Scholar] [CrossRef]
- Nasr, T.; Bondock, S.; Rashed, H.M.; Fayad, W.; Youns, M.; Sakr, T.M. Novel hydrazide-hydrazone and amide substituted coumarin derivatives: Synthesis, cytotoxicity screening, microarray, radiolabeling and in vivo pharmacokinetic studies. Eur. J. Med. Chem. 2018, 151, 723–739. [Google Scholar] [CrossRef]
- Wu, B.-X.; Chang, H.-Y.; Liao, Y.-S.; Yeh, M.-Y. Synthesis, photochemical isomerization and photophysical properties of hydrazide–hydrazone derivatives. New J. Chem. 2021, 45, 1651–1657. [Google Scholar] [CrossRef]
- Tai, X.-S.; Meng, Q.-G.; Liu, L.-L. Synthesis, Crystal Structure, and Cytotoxic Activity of a Novel Eight-Coordinated Dinuclear Ca(II)-Schiff Base Complex. Crystals 2016, 6, 109. [Google Scholar] [CrossRef] [Green Version]
- Pilichos, E.; Spanakis, E.; Maniaki, E.-K.; Raptopoulou, C.P.; Psycharis, V.; Turnbull, M.M.; Perlepes, S.P. Diversity of Coordination Modes in a Flexible Ditopic Ligand Containing 2-Pyridyl, Carbonyl and Hydrazone Functionalities: Mononuclear and Dinuclear Cobalt(III) Complexes, and Tetranuclear Copper(II) and Nickel(II) Clusters. Magnetochemistry 2019, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Prakash, G.; Nirmala, M.; Ramachandran, R.; Viswanathamurthi, P.; Malecki, J.G.; Sanmartin, J. Heteroleptic binuclear copper(I) complexes bearing bis(salicylidene)hydrazone ligands: Synthesis, crystal structure and application in catalytic N-alkylation of amines. Polyhedron 2015, 89, 62–69. [Google Scholar] [CrossRef]
- Qin, J.; Zhao, S.-S.; Liu, Y.-P.; Man, Z.-W.; Wang, P.; Wang, L.-N.; Xu, Y.; Zhu, H.-L. Preparations, characterization, and biological features of mononuclear Cu(II) complexes based on hydrazone ligands. Bioorganic Med. Chem. Lett. 2016, 26, 4925–4929. [Google Scholar] [CrossRef] [PubMed]
- Affan, A.; Foo, S.W.; Jusoh, I.; Hanapi, S.; Tiekink, E.R. Synthesis, characterization and biological studies of organotin(IV) complexes with hydrazone ligand. Inorg. Chim. Acta 2009, 362, 5031–5037. [Google Scholar] [CrossRef]
- Mishra, M.; Tiwari, K.; Singh, A.K.; Singh, V.P. Synthesis, structural and corrosion inhibition studies on Mn(II), Cu(II) and Zn(II) complexes with a Schiff base derived from 2-hydroxypropiophenone. Polyhedron 2014, 77, 57–65. [Google Scholar] [CrossRef]
- Chaouiki, A.; Chafiq, M.; Lgaz, H.; Al-Hadeethi, M.R.; Ali, I.H.; Masroor, S.; Chung, I.-M. Green Corrosion Inhibition of Mild Steel by Hydrazone Derivatives in 1.0 M HCl. Coatings 2020, 10, 640. [Google Scholar] [CrossRef]
- El-Wahab, H.A. The synthesis and characterization of the hydrazone ligand and its metal complexes and their performance in epoxy formulation surface coatings. Prog. Org. Coat. 2015, 89, 106–113. [Google Scholar] [CrossRef]
- Khamaysa, O.M.A.; Selatnia, I.; Lgaz, H.; Sid, A.; Lee, H.-S.; Zeghache, H.; Benahmed, M.; Ali, I.H.; Mosset, P. Hydrazone-based green corrosion inhibitors for API grade mild steel in HCl: Insights from Electrochemical, XPS, and Computational studies. Colloids Surf. A Physicochem. Eng. Asp. 2021, 626, 127047. [Google Scholar] [CrossRef]
- Narang, R.; Narasimhan, B.; Sharma, S. A Review on Biological Activities and Chemical Synthesis of Hydrazide Derivatives. Curr. Med. Chem. 2012, 19, 569–612. [Google Scholar] [CrossRef] [PubMed]
- Angelova, V.T.; Vassilev, N.G.; Nikolova-Mladenova, B.; Vitas, J.; Malbaša, R.; Momekov, G.; Djukic, M.; Saso, L. Antiproliferative and antioxidative effects of novel hydrazone derivatives bearing coumarin and chromene moiety. Med. Chem. Res. 2016, 25, 2082–2092. [Google Scholar] [CrossRef]
- Onnis, V.; Cocco, M.T.; Fadda, R.; Congiu, C. Synthesis and evaluation of anticancer activity of 2-arylamino-6-trifluoromethyl-3-(hydrazonocarbonyl)pyridines. Bioorganic Med. Chem. 2009, 17, 6158–6165. [Google Scholar] [CrossRef] [PubMed]
- Vicini, P.; Incerti, M.; Doytchinova, I.; La Colla, P.; Busonera, B.; Loddo, R. Synthesis and antiproliferative activity of benzo[d]isothiazole hydrazones. Eur. J. Med. Chem. 2006, 41, 624–632. [Google Scholar] [CrossRef]
- Iliev, I.; Kontrec, D.; Detcheva, R.; Georgieva, M.; Balacheva, A.; Galić, N.; Pajpanova, T. Cancer cell growth inhibition by aroylhydrazone derivatives. Biotechnol. Biotechnol. Equip. 2019, 33, 756–763. [Google Scholar] [CrossRef] [Green Version]
- Pham, V.H.; Phan, T.P.D.; Phan, D.C.; Vu, B.D. Synthesis and Bioactivity of Hydrazide-Hydrazones with the 1-Adamantyl-Carbonyl Moiety. Molecules 2019, 24, 4000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sayed, H.A.; Moustafa, A.H.; El-moneim, M.A.; Awad, H.M.; Esmat, A. A new series of hydrazide-hydrazones based 2-oxonicotinonitriles as antimicrobial agents: Design, synthesis and antimicrobial evaluation. Afinidad 2018, 75, 305–310. [Google Scholar]
- Reis, D.C.; Despaigne, A.A.R.; Da Silva, J.G.; Silva, N.F.; Vilela, C.F.; Mendes, I.C.; Takahashi, J.; Beraldo, H. Structural Studies and Investigation on the Activity of Imidazole-Derived Thiosemicarbazones and Hydrazones against Crop-Related Fungi. Molecules 2013, 18, 12645–12662. [Google Scholar] [CrossRef] [Green Version]
- Popiołek, Ł.; Biernasiuk, A.; Berecka, A.; Gumieniczek, A.; Malm, A.; Wujec, M. New hydrazide-hydrazones of isonicotinic acid: Synthesis, lipophilicity and in vitro antimicrobial screening. Chem. Biol. Drug Des. 2018, 91, 915–923. [Google Scholar] [CrossRef]
- Şenkardeş, S.; Kaushik-Basu, N.; Durmaz, I.; Manvar, D.; Basu, A.; Cetin-Atalay, R.; Küçükgüzel, Ş.G. Synthesis of novel diflunisal hydrazide–hydrazones as anti-hepatitis C virus agents and hepatocellular carcinoma inhibitors. Eur. J. Med. Chem. 2016, 108, 301–308. [Google Scholar] [CrossRef] [PubMed]
- El-Sabbagh, O.I.; Rady, H. Synthesis of new acridines and hydrazones derived from cyclic β-diketone for cytotoxic and antiviral evaluation. Eur. J. Med. Chem. 2009, 44, 3680–3686. [Google Scholar] [CrossRef] [PubMed]
- Siemann, S.; Evanoff, D.P.; Marrone, L.; Clarke, A.J.; Viswanatha, T.; Dmitrienko, G.I. N-Arylsulfonyl hydrazones as inhibitors of N-arylsulfonyl hydrazones as inhibitors of β-lactamase. Antimicrob. Agents Chemother. 2002, 46, 2450–2457. [Google Scholar] [CrossRef] [Green Version]
- Ravish, I. Synthesis, Pharmacological Evaluation and Molecular Docking of Some Pyrimidinyl Hydrazones. Biochem. Anal. Biochem. 2016, 5, S3. [Google Scholar] [CrossRef]
- Can, N.Ö.; Osmaniye, D.; Levent, S.; Sağlık, B.N.; Inci, B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of New Hydrazone Derivatives for MAO Enzymes Inhibitory Activity. Molecules 2017, 22, 1381. [Google Scholar] [CrossRef]
- Wilde, F.; Lemmerhirt, H.; Emmrich, T.; Bednarski, P.J.; Link, A. Microwave-assisted synthesis and evaluation of acylhydrazones as potential inhibitors of bovine glutathione peroxidase. Mol. Divers. 2014, 18, 307–322. [Google Scholar] [CrossRef]
- Khan, M.; Alam, A.; Khan, K.M.; Salar, U.; Chigurupati, S.; Wadood, A.; Ali, F.; Mohammad, J.I.; Riaz, M.; Perveen, S. Flurbiprofen derivatives as novel α-amylase inhibitors: Biology-oriented drug synthesis (BIODS), in vitro, and in silico evaluation. Bioorganic Chem. 2018, 81, 157–167. [Google Scholar] [CrossRef]
- Taha, M.; Irshad, M.; Imran, S.; Rahim, F.; Selvaraj, M.; Almandil, N.; Mosaddik, A.; Chigurupati, S.; Nawaz, F.; Ismail, N.H.; et al. Thiazole Based Carbohydrazide Derivatives as α-Amylase Inhibitor and Their Molecular Docking Study. Heteroat. Chem. 2019, 2019, 7502347. [Google Scholar] [CrossRef] [Green Version]
- Atli, Ö.; Özkay, Y. Synthesis and MAO inhibitory activity of novel thiazole-hydrazones. Turk. J. Chem. 2017, 41, 685–699. [Google Scholar] [CrossRef]
- Islam, R.; Koizumi, F.; Kodera, Y.; Inoue, K.; Okawara, T.; Masutani, M. Design and synthesis of phenolic hydrazide hydrazones as potent poly(ADP-ribose) glycohydrolase (PARG) inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 3802–3806. [Google Scholar] [CrossRef]
- Wang, G.; Chen, M.; Wang, J.; Peng, Y.; Li, L.; Xie, Z.; Deng, B.; Chen, S.; Li, W. Synthesis, biological evaluation and molecular docking studies of chromone hydrazone derivatives as α -glucosidase inhibitors. Bioorganic Med. Chem. Lett. 2017, 27, 2957–2961. [Google Scholar] [CrossRef]
- Mayer, N.; Schweiger, M.; Melcher, M.-C.; Fledelius, C.; Zechner, R.; Zimmermann, R.; Breinbauer, R. Structure–activity studies in the development of a hydrazone based inhibitor of adipose-triglyceride lipase (ATGL). Bioorganic Med. Chem. 2015, 23, 2904–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasool, S.; Rehman, A.U.; Abbasi, M.A.; Siddiqui, S.Z.; Gondal, A.S.; Noor, H.A.; Sheral, S.; Ahmad, I. Antibacterial and Enzyme Inhibition Study of Hydrazone Derivatives Bearing 1, 3, 4-Oxadiazole. Pak. J. Chem. 2015, 5, 14–22. [Google Scholar] [CrossRef]
- Ahmad, F.; Alam, I.; Huff, S.E.; Pink, J.; Flanagan, S.A.; Shewach, D.; Misko, T.A.; Oleinick, N.L.; Harte, W.E.; Viswanathan, R.; et al. Potent competitive inhibition of human ribonucleotide reductase by a nonnucleoside small molecule. Proc. Natl. Acad. Sci. USA 2017, 114, 8241–8246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.A.; Antholine, W.E.; Wong, S.J.; Richardson, D.; Chitambar, C.R. Inhibition of malignant cell growth by 311, a novel iron chelator of the pyridoxal isonicotinoyl hydrazone class: Effect on the R2 subunit of ribonucleotide reductase. Clin. Cancer Res. 2001, 7, 3574–3579. [Google Scholar] [PubMed]
- Hanna, M.L.; Tarasow, T.M.; Perkins, J. Mechanistic differences between in vitro assays for hydrazone-based small molecule inhibitors of anthrax lethal factor. Bioorganic Chem. 2007, 35, 50–58. [Google Scholar] [CrossRef]
- Evranos-Aksöz, B.; Yabanoğlu-Çiftçi, S.; Uçar, G.; Yelekçi, K.; Ertan, R. Synthesis of some novel hydrazone and 2-pyrazoline derivatives: Monoamine oxidase inhibitory activities and docking studies. Bioorganic Med. Chem. Lett. 2014, 24, 3278–3284. [Google Scholar] [CrossRef] [PubMed]
- Min, D.-H.; Tang, W.-J.; Mrksich, M. Chemical screening by mass spectrometry to identify inhibitors of anthrax lethal factor. Nat. Biotechnol. 2004, 22, 717–723. [Google Scholar] [CrossRef]
- Yaropolov, A.I.; Skorobogatko, O.V.; Vartanov, S.S.; Varflomeyev, S.D. Laccase. Appl. Biochem. Biotechnol. 1994, 49, 257–280. [Google Scholar] [CrossRef]
- Polak, J.; Jarosz-Wilkolazka, A. Structure/Redox potential relationship of simple organic compounds as potential precursors of dyes for laccase-mediated transformation. Biotechnol. Prog. 2012, 28, 93–102. [Google Scholar] [CrossRef]
- Baldrian, P. Fungal laccases—Occurrence and properties. FEMS Microbiol. Rev. 2006, 30, 215–242. [Google Scholar] [CrossRef] [Green Version]
- Johannes, C.; Majcherczyk, A. Natural Mediators in the Oxidation of Polycyclic Aromatic Hydrocarbons by Laccase Mediator Systems. Appl. Environ. Microbiol. 2000, 66, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Węglarz-Tomczak, E.; Burda-Grabowska, M.; Giurg, M.; Mucha, A. Identification of methionine aminopeptidase 2 as a molecular target of the organoselenium drug ebselen and its derivatives/analogues: Synthesis, inhibitory activity and molecular modeling study. Bioorganic Med. Chem. Lett. 2016, 26, 5254–5259. [Google Scholar] [CrossRef] [PubMed]
- Weglarz-Tomczak, E.; Tomczak, J.M.; Talma, M.; Burda-Grabowska, M.; Giurg, M.; Brul, S. Identification of ebselen and its analogues as potent covalent inhibitors of papain-like protease from SARS-CoV-2. Sci. Rep. 2021, 11, 3640. [Google Scholar] [CrossRef] [PubMed]
- Giurg, M.; Gołąb, A.; Suchodolski, J.; Kaleta, R.; Krasowska, A.; Piasecki, E.; Piętka-Ottlik, M. Reaction of bis[(2-chlorocarbonyl)phenyl] Diselenide with Phenols, Aminophenols, and Other Amines towards Diphenyl Diselenides with Antimicrobial and Antiviral Properties. Molecules 2017, 22, 974. [Google Scholar] [CrossRef] [Green Version]
- Wanat, W.; Talma, M.; Dziuk, B.; Pirat, J.-L.; Kafarski, P. Phosphonic Acid Analogs of Fluorophenylalanines as Inhibitors of Human and Porcine Aminopeptidases N: Validation of the Importance of the Substitution of the Aromatic Ring. Biomolecules 2020, 10, 579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Węglarz-Tomczak, E.; Talma, M.; Giurg, M.; Westerhoff, H.V.; Janowski, R.; Mucha, A. Neutral metalloaminopeptidases APN and MetAP2 as newly discovered anticancer molecular targets of actinomycin D and its simple analogs. Oncotarget 2018, 9, 29365–29378. [Google Scholar] [CrossRef] [Green Version]
- Helios, K.; Maniak, H.; Sowa, M.; Zierkiewicz, W.; Wąsińska-Kałwa, M.; Giurg, M.; Drożdżewski, P.; Trusek-Hołownia, A.; Malik-Gajewska, M.; Krauze, K. Silver(I) complex with 2-amino-4,4α-dihydro-4α,7-dimethyl-3H-phenoxazin-3-one (Phx-1) ligand: Crystal structure, vibrational spectra and biological studies. J. Coord. Chem. 2017, 70, 3471–3487. [Google Scholar] [CrossRef]
- Ndikuryayo, F.; Kang, W.-M.; Wu, F.-X.; Yang, W.-C.; Yang, G.-F. Hydrophobicity-oriented drug design (HODD) of new human 4-hydroxyphenylpyruvate dioxygenase inhibitors. Eur. J. Med. Chem. 2019, 166, 22–31. [Google Scholar] [CrossRef]
- Nomura, N.; Ishii, R.; Yamamoto, Y.; Kondo, T. Stereoselective Ring-Opening Polymerization of a Racemic Lactide by Using Achiral Salen– and Homosalen–Aluminum Complexes. Chem.-A Eur. J. 2007, 13, 4433–4451. [Google Scholar] [CrossRef]
- Casiraghi, G.; Casnati, G.; Puglia, G.; Sartori, G.; Terenghi, G. Selective reactions between phenols and formaldehyde. A novel route to salicylaldehydes. J. Chem. Soc. Perkin Trans. 1 1980, 1, 1862–1865. [Google Scholar] [CrossRef]
- Grimster, N.P.; Connelly, S.; Baranczak, A.; Dong, J.; Krasnova, L.B.; Sharpless, K.B.; Powers, E.T.; Wilson, I.A.; Kelly, J.W. Aromatic Sulfonyl Fluorides Covalently Kinetically Stabilize Transthyretin to Prevent Amyloidogenesis while Affording a Fluorescent Conjugate. J. Am. Chem. Soc. 2013, 135, 5656–5668. [Google Scholar] [CrossRef] [Green Version]
- Ghatak, S.; Vyas, A.; Misra, S.; O’Brien, P.; Zambre, A.; Fresco, V.M.; Markwald, R.R.; Swamy, K.V.; Afrasiabi, Z.; Choudhury, A.; et al. Novel di-tertiary-butyl phenylhydrazones as dual cyclooxygenase-2/5-lipoxygenase inhibitors: Synthesis, COX/LOX inhibition, molecular modeling, and insights into their cytotoxicities. Bioorganic Med. Chem. Lett. 2014, 24, 317–324. [Google Scholar] [CrossRef]
- Peng, X.; Tang, X.; Qin, W.; Dou, W.; Guo, Y.; Zheng, J.; Liu, W.; Wang, D. Aroylhydrazone derivative as fluorescent sensor for highly selective recognition of Zn2+ ions: Syntheses, characterization, crystal structures and spectroscopic properties. Dalton Trans. 2011, 40, 5271–5277. [Google Scholar] [CrossRef] [PubMed]
- Drożdżewski, P.; Zasłona, H.; Kubiak, M. Vibrational spectroscopy of 4-hydroxybenzhydrazide and its O,N-deuterated isotopologue accompanied by X-ray structure refinement. Vib. Spectrosc. 2009, 50, 185–192. [Google Scholar] [CrossRef]
- Durig, J.R.; Harris, W.C. Infrared and Raman Spectra of Substituted Hydrazines. II Unsymmetrical Dimethyl Hydrazine. J. Chem. Phys. 1969, 51, 4457–4468. [Google Scholar] [CrossRef]
- Segel, I.H. Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems; A Wiley-Interscience publication; Wiley: Hoboken, NJ, USA, 1993; ISBN 978-0-471-30309-1. [Google Scholar]
- Pierpoint, W.S. The natural history of salicylic acid Plant product and mammalian medicine. Interdiscip. Sci. Rev. 1997, 22, 45–52. [Google Scholar] [CrossRef]
- Cui, Z.; Ito, J.; Dohi, H.; Amemiya, Y.; Nishida, Y. Molecular Design and Synthesis of Novel Salicyl Glycoconjugates as Elicitors against Plant Diseases. PLoS ONE 2014, 9, e108338. [Google Scholar] [CrossRef] [Green Version]
- Faize, L.; Faize, M. Functional Analogues of Salicylic Acid and Their Use in Crop Protection. Agronomy 2018, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Sýs, M.; Metelka, R.; Frangu, A.; Vytřas, K.; Arbneshi, T. Electrochemical Study of Trametes Versicolor Laccase Compatibility to Different Polyphenolic Substrates. Chemosensors 2017, 5, 9. [Google Scholar] [CrossRef]
- Polak, J.; Jarosz-Wilkołazka, A. Whole-cell fungal transformation of precursors into dyes. Microb. Cell Factories 2010, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Arora, D.; Sandhu, D. Laccase production and wood degradation by a white-rot fungus Daedalea flavida. Enzym. Microb. Technol. 1985, 7, 405–408. [Google Scholar] [CrossRef]
- Cañas, A.; Camarero, S. Laccases and their natural mediators: Biotechnological tools for sustainable eco-friendly processes. Biotechnol. Adv. 2010, 28, 694–705. [Google Scholar] [CrossRef]
- Maniak, H.; Witkowska, D.; Giurg, M. Zastosowanie Hydrazydu Kwasu 4-Hydroksybenzoesowego Oraz Zastosowanie Hydrazydo-Hydrazonów Pochodnych Kwasu 4-Hydroksybenzoesowego Aldehydów Zawierających Fragment Aromatyczny. Polska Patent A1 238660, 20 September 2021. [Google Scholar]
- Skarżewski, J.; Ostrycharz, E.; Siedlecka, R.; Zielińska-Błajet, M.; Pisarski, B. Substituted N-Salicylidene β-Aminoalcohols: Preparation and use as Chiral Ligands in Enantioselective Sulfoxidation and Conjugate Addition. J. Chem. Res. 2001, 2001, 263–264. [Google Scholar] [CrossRef]
- White, E.H.; Bursey, M.M.; Roswell, D.F.; Hill, J.H.M. Chemiluminescence of some monoacylhydrazides. J. Org. Chem. 1967, 32, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Leonowicz, A.; Grzywnowicz, K. Quantitative estimation of laccase forms in some white-rot fungi using syringaldazine as a substrate. Enzym. Microb. Technol. 1981, 3, 55–58. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Ar | R | Yield, (%) | Mp, (°C) | C=O a,b | N–H b,c | C=N b | CAr-O b |
|---|---|---|---|---|---|---|---|---|
| 3a | 3-tBu-2-HO-5-MeC6H2 | Me | 84 | 231–233 | 1654 | 1614 | 1565 | 1239 |
| 3b | 3-tBu-2-HO-5-MeC6H2 | 4-HOC6H4CH2 | 85 | 242–244 | 1654 | 1612 | 1552 | 1261 |
| 4a | 3-tBu-2-HO-5-MeC6H2 | 3-pyridyl | 93 | 230–232 | 1638 | 1590 | 1552 | 1261 |
| 4b | 3-tBu-2-HO-5-MeC6H2 | C6H5 | 96 | 246–248 | 1635 | 1612 | 1561 | 1264 |
| 4c | 3-tBu-2-HO-5-MeC6H2 | 3-MeOC6H4 | 93 | 238–240 | 1633 | 1584 | 1560 | 1241 |
| 4d | 3-tBu-2-HO-5-MeC6H2 | 4-MeOC6H4 | 81 | 243–244 | 1636 | 1605 | 1537 | 1235 |
| 4e | 3-tBu-2-HO-5-MeC6H2 | 2-HOC6H4 | 95 | 252–254 | 1637 | 1615 | 1560 | 1235 |
| 4f | 3-tBu-2-HO-5-MeC6H2 | 3,5-(HO)2C6H3 | 83 | 246 d | 1643 | 1583 | 1556 | 1265 |
| 4g | 3,5-(tBu)2-2-HOC6H2 | 2-HOC6H4 | 97 | 233–235 e | 1636 | 1584 | 1554 | 1247 |
| 4h | 3,5-(tBu)2-2-HOC6H2 | 3-HOC6H4 | 98 | 273–274 | 1638 | 1611 | 1567 | 1251 |
| 4i | 3,5-(tBu)2-2-HOC6H2 | 3,5-(HO)2C6H3 | 99 | 266 d | 1599 | 1583 | 1549 | 1234 |
| 4j | 3-tBu-2-HOC6H3 | 2-HOC6H4 | 98 | 218–220 | 1630 | 1610 | 1552 | 1231 |
| 4k | 3-tBu-2-HOC6H3 | 3-HOC6H4 | 90 | 225–228 | 1648 | 1590 | 1567 | 1240 |
| 4l | 3-tBu-2-HOC6H3 | 3,5-(HO)2C6H3 | 84 | 217–220 | 1591 | 1591 | 1557 | 1262 |
| 4m | 2-HO-3-PhC6H3 | 2-HOC6H4 | 83 | 224–227 | 1601 | 1580 | 1559 | 1231 |
| 4n | 2-HO-3-PhC6H3 | 3-HOC6H4 | 94 | 207–209 | 1651 | 1592 | 1527 | 1227 |
| 4o | 2-HO-3-PhC6H3 | 3,5-(HO)2C6H3 | 91 | 241–243 | 1655 | 1592 | 1544 | 1254 |
| 5a | 3-tBu-2-HO-5-MeC6H2 | 2-(1-hydroxynaphthyl) | 92 | 215–217 | 1616 | 1581 | 1532 | 1250 |
| 5b | 2-HO-3-PhC6H3 | 2-(1-hydroxynaphthyl) | 82 | 226–228 | 1628 | 1600 | 1532 | 1249 |
| 5c | 3,5-(tBu)2-2-HOC6H2 | 2-(1-hydroxynaphthyl) | 95 f | 121–123 | 1611 | 1584 | 1563 | 1250 |
| 5d | 3-tBu-2-HOC6H3 | 2-(1-hydroxynaphthyl) | 99 | 220–222 | 1623 | 1602 | 1565 | 1252 |
| No. | Ar | R | KI, µM | R2 | Inhibition Type |
|---|---|---|---|---|---|
| 3a | 3-tBu-2-HO-5-MeC6H2 | Me | – a | – | – b |
| 3b | 3-tBu-2-HO-5-MeC6H2 | 4-HOC6H4CH2 | 49.2 | 0.960 | uncompetitive |
| 4a | 3-tBu-2-HO-5-MeC6H2 | 3-pyridyl | – c | – | – b |
| 4b | 3-tBu-2-HO-5-MeC6H2 | C6H5 | 82.0 | 0.996 | competitive |
| 4c | 3-tBu-2-HO-5-MeC6H2 | 3-MeOC6H4 | 17.4 | 0.974 | non-competitive |
| 4d | 3-tBu-2-HO-5-MeC6H2 | 4-MeOC6H4 | 25.8 | 0.984 | competitive |
| 4e | 3-tBu-2-HO-5-MeC6H2 | 2-HOC6H4 | 150.0 | 0.958 | uncompetitive |
| 4f | 3-tBu-2-HO-5-MeC6H2 | 3,5-(HO)2C6H3 | 32.3 | 0.982 | competitive |
| 4g | 3,5-(tBu)2-2-HOC6H2 | 2-HOC6H4 | 52.6 | 0.998 | non-competitive |
| 4h | 3,5-(tBu)2-2-HOC6H2 | 3-HOC6H4 | 18.9 | 0.996 | competitive |
| 4i | 3,5-(tBu)2-2-HOC6H2 | 3,5-(HO)2C6H3 | 55.6 | 0.961 | competitive |
| 4j | 3-tBu-2-HOC6H3 | 2-HOC6H4 | 65.6 | 0.965 | uncompetitive |
| 4k | 3-tBu-2-HOC6H3 | 3-HOC6H4 | 35.8 | 0.994 | competitive |
| 4l | 3-tBu-2-HOC6H3 | 3,5-(HO)2C6H3 | 38.0 | 0.923 | competitive |
| 4m | 2-HO-3-PhC6H3 | 2-HOC6H4 | 26.4 | 0.982 | competitive |
| 57.1 | 0.959 | uncompetitive | |||
| 4n | 2-HO-3-PhC6H3 | 3-HOC6H4 | 233 | 0.976 | competitive |
| 139 | 0.964 | non-competitive | |||
| 4o | 2-HO-3-PhC6H3 | 3,5-(HO)2C6H3 | 69.9 | 0.977 | non-competitive |
| 5a | 3-tBu-2-HO-5-MeC6H2 | 2-(1-hydroxynaphthyl) | 19.0 | 0.993 | competitive |
| 8.0 | 0.982 | non-competitive | |||
| 5b | 2-HO-3-PhC6H3 | 2-(1-hydroxynaphthyl) | 25.8 | 0.950 | competitive |
| 24.3 | 0.999 | non-competitive | |||
| 5c | 3,5-(tBu)2-2-HOC6H2 | 2-(1-hydroxynaphthyl) | 16.2 | 0.998 | non-competitive |
| 5d | 3-tBu-2-HOC6H3 | 2-(1-hydroxynaphthyl) | 75.2 | 0.971 | competitive |
| 55.6 | 0.999 | non-competitive | |||
| 20 d | 3-tBu-2-HO-5-MeC6H2 | 4-HOC6H4 | 26.4 [31] | – | competitive |
| 21 d | 3,5-(tBu)2-2-HOC6H2 | 4-HOC6H4 | 17.9 [31] | – | uncompetitive |
| 22 d | 3-tBu-2-HO-5-MeC6H2 | 3-HOC6H4 | 32.0 [31] | – | non-competitive |
| 23 d | 2-HO-3-PhC6H3 | 4-MeOC6H4 | ≥1000 [31] | – | – b |
| – | NaN3 d | 2.7 [31] | non-competitive | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maniak, H.; Talma, M.; Giurg, M. Inhibitory Potential of New Phenolic Hydrazide-Hydrazones with a Decoy Substrate Fragment towards Laccase from a Phytopathogenic Fungus: SAR and Molecular Docking Studies. Int. J. Mol. Sci. 2021, 22, 12307. https://doi.org/10.3390/ijms222212307
Maniak H, Talma M, Giurg M. Inhibitory Potential of New Phenolic Hydrazide-Hydrazones with a Decoy Substrate Fragment towards Laccase from a Phytopathogenic Fungus: SAR and Molecular Docking Studies. International Journal of Molecular Sciences. 2021; 22(22):12307. https://doi.org/10.3390/ijms222212307
Chicago/Turabian StyleManiak, Halina, Michał Talma, and Mirosław Giurg. 2021. "Inhibitory Potential of New Phenolic Hydrazide-Hydrazones with a Decoy Substrate Fragment towards Laccase from a Phytopathogenic Fungus: SAR and Molecular Docking Studies" International Journal of Molecular Sciences 22, no. 22: 12307. https://doi.org/10.3390/ijms222212307
APA StyleManiak, H., Talma, M., & Giurg, M. (2021). Inhibitory Potential of New Phenolic Hydrazide-Hydrazones with a Decoy Substrate Fragment towards Laccase from a Phytopathogenic Fungus: SAR and Molecular Docking Studies. International Journal of Molecular Sciences, 22(22), 12307. https://doi.org/10.3390/ijms222212307