
Acetylenic Synthetic Betulin Derivatives Inhibit Akt and Erk Kinases Activity, Trigger Apoptosis and Suppress Proliferation of Neuroblastoma and Rhabdomyosarcoma Cell Lines

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
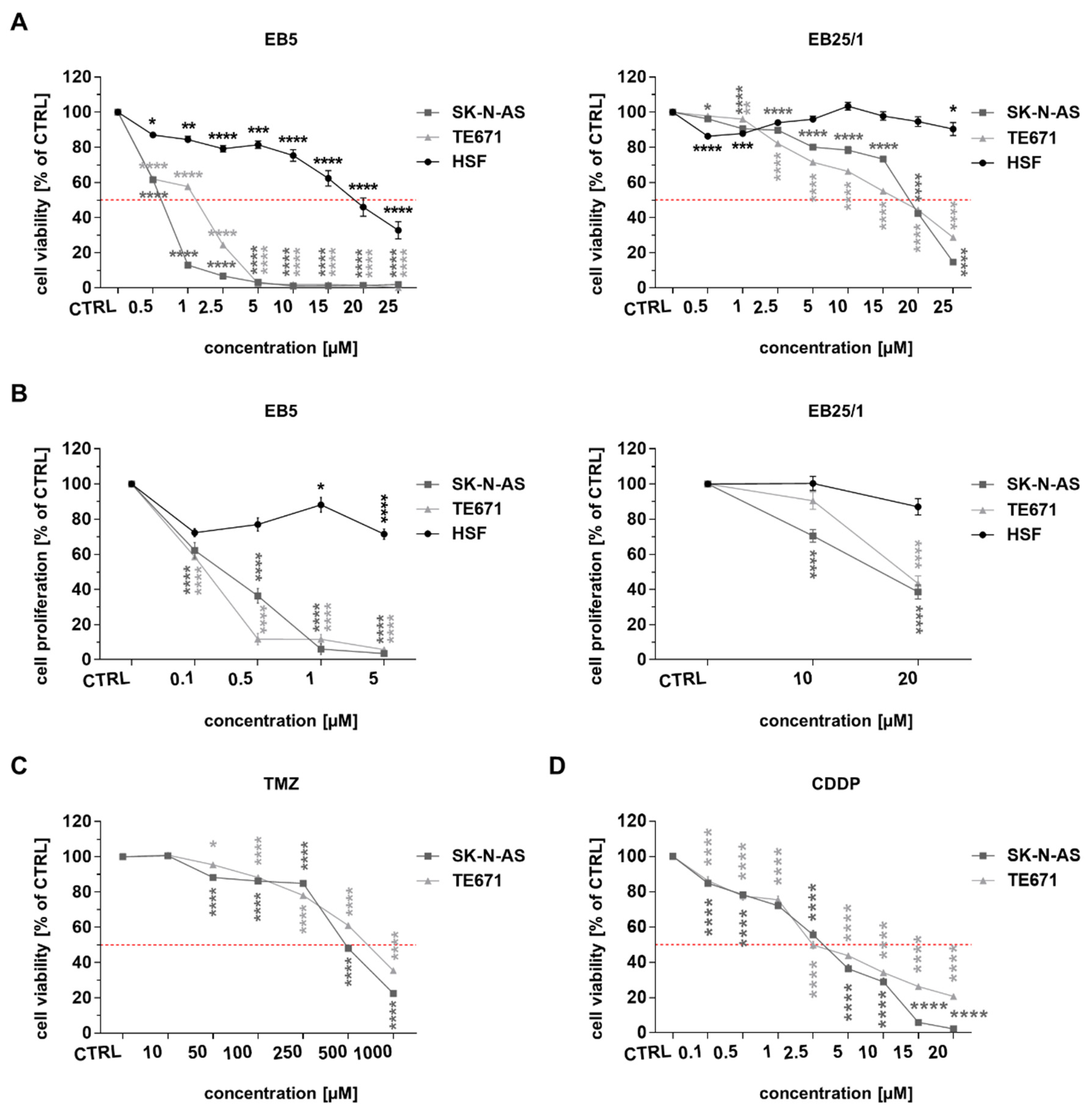
2.1. ASBDs Reduce Viability and Proliferation of Pediatric Cancer Cells Stronger than TMZ and CDDP In Vitro, Whereas They Show Moderate Activity Against Normal Cells
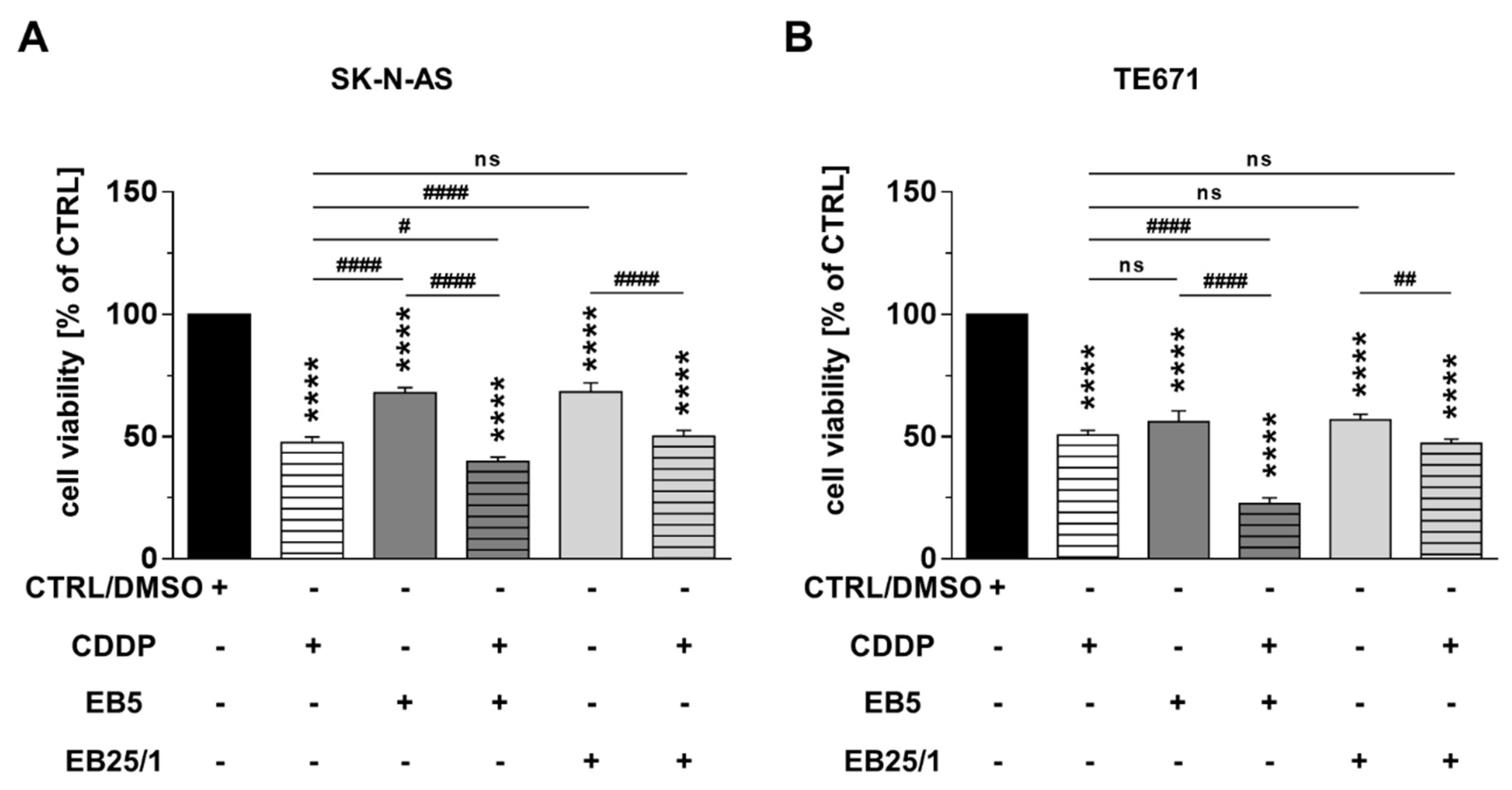
2.2. Combination of ASBDs with CDDP Enhances Cytotoxicity of Both CDDP and EB5 Administered Singly
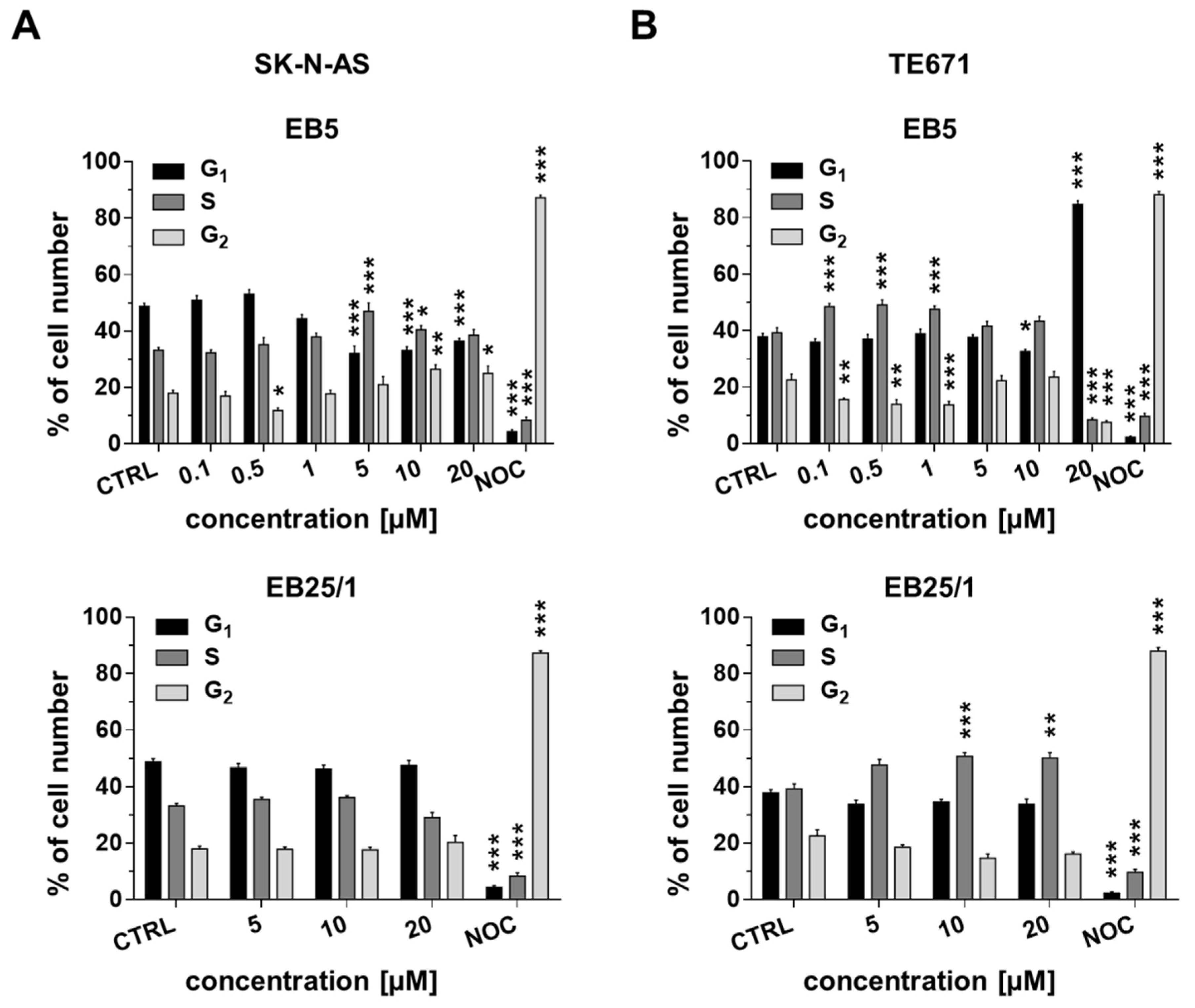
2.3. ASBDs Inhibit Cell Cycle Progression of Pediatric Cancer Cells by Affecting S Phase
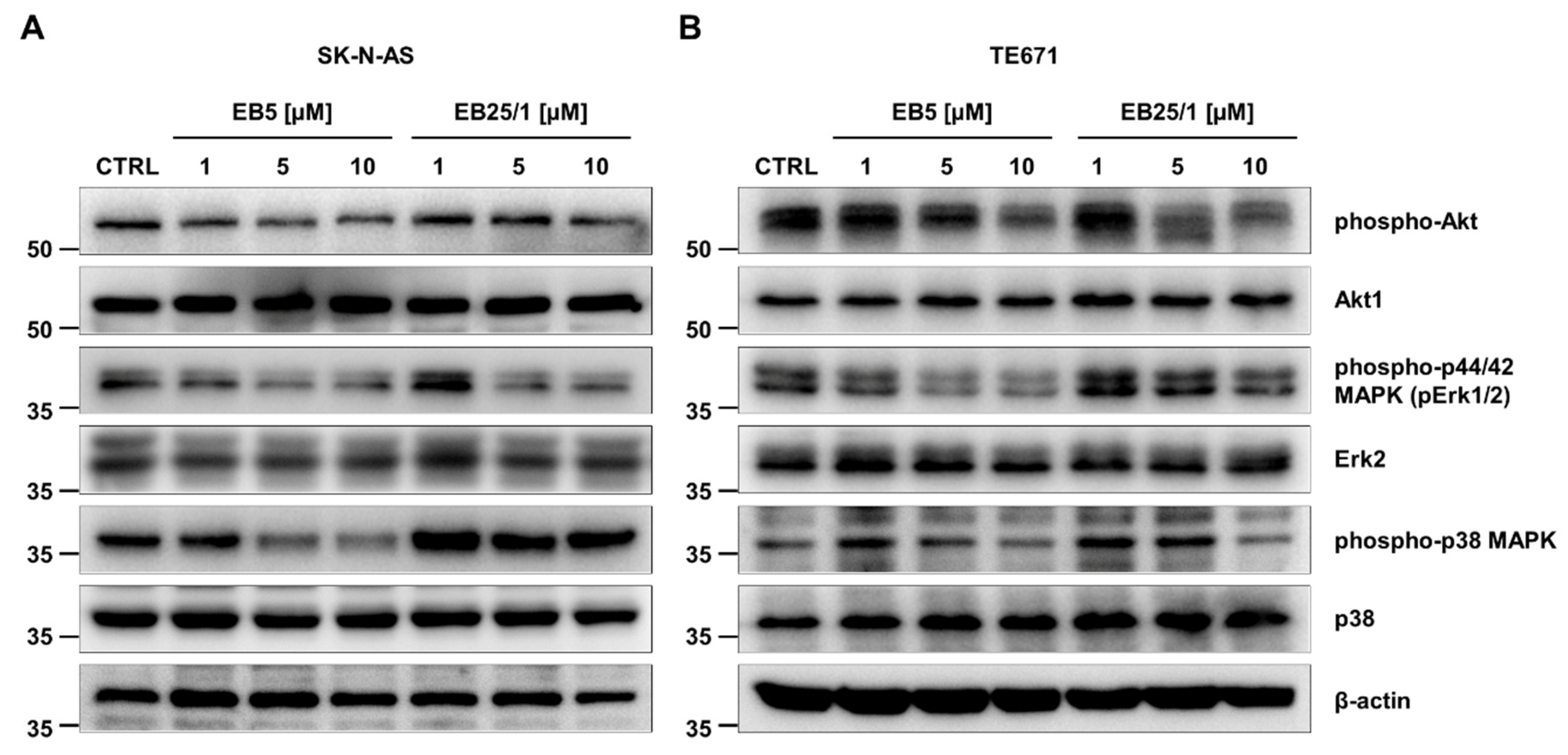
2.4. ASBDs Inhibit Phosphorylation of Kinases Crucial for Growth and Proliferation of Cancer Cells
2.5. EB5 Induces Apoptosis of Pediatric Cancer Cells In Vitro in a Concentration- and Time-Dependent Manner
2.6. In Silico Study of Physicochemical Parameters, Pharmacokinetic Profile and Druglikeness of ASBDs
3. Discussion
4. Material and Methods
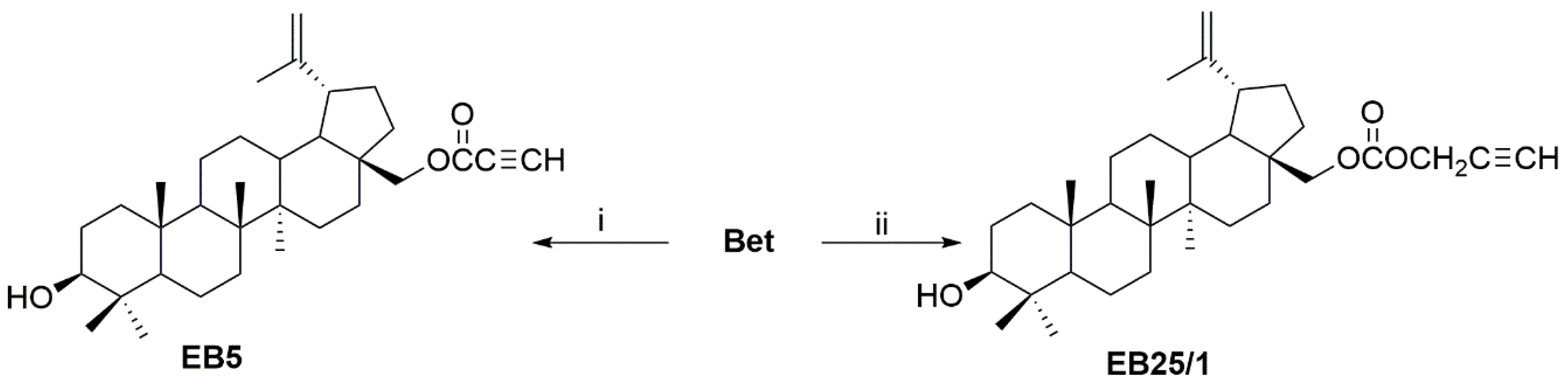
4.1. Synthesis of ASBDs
4.2. Cell Culture and Treatment
4.3. MTT Metabolism Assay
4.4. Calculation of Half Maximal Inhibitory Concentration and Selectivity Index
4.5. BrdU Incorporation Test
4.6. Cell Cycle Analysis by FACS Flow Cytometry
4.7. Analysis of Apoptosis by FACS Flow Cytometry
4.8. Protein Extraction and Western Blotting
4.9. In Silico Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The Ever-Increasing Importance of Cancer as a Leading Cause of Premature Death Worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef]
- Newman, E.A.; Abdessalam, S.; Aldrink, J.H.; Austin, M.; Heaton, T.E.; Bruny, J.; Ehrlich, P.; Dasgupta, R.; Baertschiger, R.M.; Lautz, T.B.; et al. Update on Neuroblastoma. J. Pediatr. Surg. 2019, 54, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Pastor, E.R.; Mousa, S.A. Current Management of Neuroblastoma and Future Direction. Crit. Rev. Oncol. Hematol. 2019, 138, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Matthay, K.K.; Maris, J.M.; Schleiermacher, G.; Nakagawara, A.; Mackall, C.L.; Diller, L.; Weiss, W.A. Neuroblastoma. Nat. Rev. Dis. Primer 2016, 2, 16078. [Google Scholar] [CrossRef]
- Allen-Rhoades, W.; Whittle, S.B.; Rainusso, N. Pediatric Solid Tumors of Infancy: An Overview. Pediatr. Rev. 2018, 39, 57–67. [Google Scholar] [CrossRef]
- Zhu, J.; Zhen, Z.; Wang, J.; Chen, T.; Lu, S.; Sun, F.; Huang, J.; Que, Y.; Zhang, L.; Zhang, Y.; et al. Vincristine, Irinotecan, and Temozolomide in Patients with Relapsed and Refractory Neuroblastoma. J. Clin. Oncol. 2021, 39, e22009. [Google Scholar] [CrossRef]
- Piskareva, O.; Harvey, H.; Nolan, J.; Conlon, R.; Alcock, L.; Buckley, P.; Dowling, P.; Henry, M.; O’Sullivan, F.; Bray, I.; et al. The Development of Cisplatin Resistance in Neuroblastoma Is Accompanied by Epithelial to Mesenchymal Transition In Vitro. Cancer Lett. 2015, 364, 142–155. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, R.; Kubota, N.; Hidaka, E.; Sakashita, K.; Tanaka, M.; Nakazawa, Y.; Nakamura, T. Cisplatin-Induced Nephrotoxicity in Patients with Advanced Neuroblastoma. Pediatr. Blood Cancer 2018, 65, e27253. [Google Scholar] [CrossRef]
- Rodrigo, M.A.M.; Buchtelova, H.; Jimenez, A.M.J.; Adam, P.; Babula, P.; Heger, Z.; Adam, V. Transcriptomic Landscape of Cisplatin-Resistant Neuroblastoma Cells. Cells 2019, 8, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, R.; Fuchs, J.; Rodeberg, D. Rhabdomyosarcoma. Semin. Pediatr. Surg. 2016, 25, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Ingley, K.M.; Cohen-Gogo, S.; Gupta, A.A. Systemic Therapy in Pediatric-Type Soft-Tissue Sarcoma. Curr. Oncol. 2020, 27, 6–16. [Google Scholar] [CrossRef]
- Chen, C.; Dorado Garcia, H.; Scheer, M.; Henssen, A.G. Current and Future Treatment Strategies for Rhabdomyosarcoma. Front. Oncol. 2019, 9, 1458. [Google Scholar] [CrossRef] [PubMed]
- Patočka, J. Biologically Active Pentacyclic Triterpenes and Their Current Medicine Signification. J. Appl. Biomed. 2003, 1, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Król, S.K.; Kiełbus, M.; Rivero-Müller, A.; Stepulak, A. Comprehensive Review on Betulin as a Potent Anticancer Agent. BioMed Res. Int. 2015, 2015, e584189. [Google Scholar] [CrossRef] [Green Version]
- Drąg-Zalesińska, M.; Borska, S. Betulin and Its Derivatives—Precursors of New Drugs. World Sci. News 2019, 127, 123–138. [Google Scholar]
- Rzeski, W.; Stepulak, A.; Szymański, M.; Juszczak, M.; Grabarska, A.; Sifringer, M.; Kaczor, J.; Kandefer-Szerszeń, M. Betulin Elicits Anti-Cancer Effects in Tumour Primary Cultures and Cell Lines In Vitro. Basic Clin. Pharmacol. Toxicol. 2009, 105, 425–432. [Google Scholar] [CrossRef]
- Hata, K.; Hori, K.; Ogasawara, H.; Takahashi, S. Anti-Leukemia Activities of Lup-28-al-20(29)-En-3-One, a Lupane Triterpene. Toxicol. Lett. 2003, 143, 1–7. [Google Scholar] [CrossRef]
- Li, Y.; He, K.; Huang, Y.; Zheng, D.; Gao, C.; Cui, L.; Jin, Y.-H. Betulin Induces Mitochondrial Cytochrome c Release Associated Apoptosis in Human Cancer Cells. Mol. Carcinog. 2010, 49, 630–640. [Google Scholar] [CrossRef]
- Gauthier, C.; Legault, J.; Lebrun, M.; Dufour, P.; Pichette, A. Glycosidation of Lupane-Type Triterpenoids as Potent In Vitro Cytotoxic Agents. Bioorg. Med. Chem. 2006, 14, 6713–6725. [Google Scholar] [CrossRef]
- Gauthier, C.; Legault, J.; Lavoie, S.; Rondeau, S.; Tremblay, S.; Pichette, A. Synthesis and Cytotoxicity of Bidesmosidic Betulin and Betulinic Acid Saponins. J. Nat. Prod. 2009, 72, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.S.; Roh, S.H.; Kim, D.K.; Lee, J.G.; Lee, Y.Y.; Hong, S.S.; Kwon, S.W.; Park, J.H. Anti-Cancer Effect of Betulin on a Human Lung Cancer Cell Line: A Pharmacoproteomic Approach Using 2 D SDS PAGE Coupled with Nano-HPLC Tandem Mass Spectrometry. Planta Med. 2009, 75, 127–131. [Google Scholar] [CrossRef]
- Dehelean, C.A.; Feflea, S.; Molnár, J.; Zupko, I.; Soica, C. Betulin as an Antitumor Agent Tested In Vitro on A431, HeLa and MCF7, and as an Angiogenic Inhibitor In Vivo in the CAM Assay. Nat. Prod. Commun. 2012, 7. [Google Scholar] [CrossRef] [Green Version]
- Boryczka, S.; Bębenek, E.; Wietrzyk, J.; Kempińska, K.; Jastrzębska, M.; Kusz, J.; Nowak, M. Synthesis, Structure and Cytotoxic Activity of New Acetylenic Derivatives of Betulin. Molecules 2013, 18, 4526–4543. [Google Scholar] [CrossRef] [Green Version]
- Amico, V.; Barresi, V.; Condorelli, D.; Spatafora, C.; Tringali, C. Antiproliferative Terpenoids from Almond Hulls (Prunus Dulcis): Identification and Structure−Activity Relationships. J. Agric. Food Chem. 2006, 54, 810–814. [Google Scholar] [CrossRef]
- Sarek, J.; Kvasnica, M.; Urban, M.; Klinot, J.; Hajduch, M. Correlation of Cytotoxic Activity of Betulinines and Their Hydroxy Analogues. Bioorg. Med. Chem. Lett. 2005, 15, 4196–4200. [Google Scholar] [CrossRef] [PubMed]
- Drag, M.; Surowiak, P.; Drag-Zalesinska, M.; Dietel, M.; Lage, H.; Oleksyszyn, J. Comparision of the Cytotoxic Effects of Birch Bark Extract, Betulin and Betulinic Acid Towards Human Gastric Carcinoma and Pancreatic Carcinoma Drug-Sensitive and Drug-Resistant Cell Lines. Molecules 2009, 14, 1639–1651. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Hori, K.; Takahashi, S. Differentiation- and Apoptosis-Inducing Activities by Pentacyclic Triterpenes on a Mouse Melanoma Cell Line. J. Nat. Prod. 2002, 65, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.H.L.; Pezzuto, J.M.; Pisha, E. Synthesis of Betulinic Acid Derivatives with Activity against Human Melanoma. Bioorg. Med. Chem. Lett. 1998, 8, 1707–1712. [Google Scholar] [CrossRef]
- Şoica, C.; Dehelean, C.; Danciu, C.; Wang, H.M.; Wenz, G.; Ambrus, R.; Bojin, F.; Anghel, M. Betulin Complex in γ-Cyclodextrin Derivatives: Properties and Antineoplasic Activities in In Vitro and In Vivo Tumor Models. Int. J. Mol. Sci. 2012, 13, 14992–15011. [Google Scholar] [CrossRef] [PubMed]
- Şoica, C.M.; Dehelean, C.A.; Peev, C.; Aluas, M.; Zupkó, I.; Kása, P.; Alexa, E. Physico-Chemical Comparison of Betulinic Acid, Betulin and Birch Bark Extract and in Vitro Investigation of Their Cytotoxic Effects towards Skin Epidermoid Carcinoma (A431), Breast Carcinoma (MCF7) and Cervix Adenocarcinoma (HeLa) Cell Lines. Nat. Prod. Res. 2012, 26, 968–974. [Google Scholar] [CrossRef]
- Dehelean, C.A.; Şoica, C.; Ledeţi, I.; Aluaş, M.; Zupko, I.; Gǎluşcan, A.; Cinta-Pinzaru, S.; Munteanu, M. Study of the Betulin Enriched Birch Bark Extracts Effects on Human Carcinoma Cells and Ear Inflammation. Chem. Cent. J. 2012, 6, 137. [Google Scholar] [CrossRef] [Green Version]
- Dehelean, C.A.; Feflea, S.; Gheorgheosu, D.; Ganta, S.; Cimpean, A.M.; Muntean, D.; Amiji, M.M. Anti-Angiogenic and Anti-Cancer Evaluation of Betulin Nanoemulsion in Chicken Chorioallantoic Membrane and Skin Carcinoma in Balb/c Mice. J. Biomed. Nanotechnol. 2013, 9, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.-H.; Choi, J.-E.; Lim, S.-C. Protection of Betulin against Cadmium-Induced Apoptosis in Hepatoma Cells. Toxicology 2006, 220, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, J.; Yin, M.; Li, X.; Lou, G.; Liu, Y.; Chen, X. Betulin Induces Apoptosis of HeLa Cell Lines In Vitro and Its Possible Mechanism. Tumori 2012, 32, 234–238. [Google Scholar]
- Mullauer, F.B.; Kessler, J.H.; Medema, J.P. Betulin Is a Potent Anti-Tumor Agent That Is Enhanced by Cholesterol. PLoS ONE 2009, 4, e1. [Google Scholar] [CrossRef] [Green Version]
- Zhanataev, A.K.; Presnova, G.A.; Chistyakov, A.N.; Durnev, A.D. Effect of Betula Bark Extract on Spontaneous and Induced Mutagenesis in Mice. Bull. Exp. Biol. Med. 2004, 138, 475–478. [Google Scholar] [CrossRef]
- Ciurlea, S.; Tiulea, C.; Csányi, E.; Berkó, S.; Toma, C.; Dehelean, C.; Loghin, F. A Pharmacotoxicological Evaluation of a Betulin Topical Formulation Tested on C57BL/6J Mouse Experimental Nevi and Skin Lesions. Stud. Univ. Vasile Goldis Arad Ser. Stiintele Vietii 2010, 20, 5–9. [Google Scholar]
- Krasutsky, P.A. Birch Bark Research and Development. Nat. Prod. Rep. 2006, 23, 919–942. [Google Scholar] [CrossRef]
- Hwang, B.Y.; Chai, H.-B.; Kardono, L.B.S.; Riswan, S.; Farnsworth, N.R.; Cordell, G.A.; Pezzuto, J.M.; Douglas Kinghorn, A. Cytotoxic Triterpenes from the Twigs of Celtis Philippinensis. Phytochemistry 2003, 62, 197–201. [Google Scholar] [CrossRef]
- Liu, M.; Yang, S.; Jin, L.; Hu, D.; Wu, Z.; Yang, S. Chemical Constituents of the Ethyl Acetate Extract of Belamcanda chinensis (L.) DC Roots and Their Antitumor Activities. Molecules 2012, 17, 6156–6169. [Google Scholar] [CrossRef]
- Yang, S.; Liu, M.; Liang, N.; Zhao, Q.; Zhang, Y.; Xue, W.; Yang, S. Discovery and Antitumor Activities of Constituents from Cyrtomium fortumei (J.) Smith Rhizomes. Chem. Cent. J. 2013, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- Alakurtti, S.; Mäkelä, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological Properties of the Ubiquitous Natural Product Betulin. Eur. J. Pharm. Sci. 2006, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Amiri, S.; Dastghaib, S.; Ahmadi, M.; Mehrbod, P.; Khadem, F.; Behrouj, H.; Aghanoori, M.-R.; Machaj, F.; Ghamsari, M.; Rosik, J.; et al. Betulin and Its Derivatives as Novel Compounds with Different Pharmacological Effects. Biotechnol. Adv. 2020, 38, 107409. [Google Scholar] [CrossRef]
- Giannatale, A.D.; Dias-Gastellier, N.; Devos, A.; Hugh, K.M.; Boubaker, A.; Courbon, F.; Verschuur, A.; Ducassoul, S.; Malekzadeh, K.; Casanova, M.; et al. Phase II Study of Temozolomide in Combination with Topotecan (TOTEM) in Relapsed or Refractory Neuroblastoma: A European Innovative Therapies for Children with Cancer-SIOP-European Neuroblastoma Study. Eur. J. Cancer 2014, 50, 170–177. [Google Scholar] [CrossRef]
- Mody, R.; Yu, A.L.; Naranjo, A.; Zhang, F.F.; London, W.B.; Shulkin, B.L.; Parisi, M.T.; Servaes, S.-E.-N.; Diccianni, M.B.; Hank, J.A.; et al. Irinotecan, Temozolomide, and Dinutuximab with GM-CSF in Children with Refractory or Relapsed Neuroblastoma: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2020, 38, 2160–2169. [Google Scholar] [CrossRef] [PubMed]
- Setty, B.A.; Stanek, J.R.; Mascarenhas, L.; Miller, A.; Bagatell, R.; Okcu, F.; Nicholls, L.; Lysecki, D.; Gupta, A.A. VIncristine, Irinotecan, and Temozolomide in Children and Adolescents with Relapsed Rhabdomyosarcoma. Pediatr. Blood Cancer 2018, 65, e26728. [Google Scholar] [CrossRef] [PubMed]
- Defachelles, A.S.; Bogart, E.; Casanova, M.; Merks, H.; Bisogno, G.; Calareso, G.; Gallego Melcon, S.; Gatz, S.; Le Deley, M.-C.; McHugh, K.; et al. Randomized Phase 2 Trial of the Combination of Vincristine and Irinotecan with or without Temozolomide, in Children and Adults with Refractory or Relapsed Rhabdomyosarcoma (RMS). J. Clin. Oncol. 2019, 37, 10000. [Google Scholar] [CrossRef]
- Ju, H.Y.; Park, M.; Lee, J.A.; Park, H.J.; Park, S.Y.; Kim, J.H.; Kang, H.G.; Yang, H.C.; Park, B.-K. Vincristine, Irinotecan, and Temozolomide as a Salvage Regimen for Relapsed or Refractory Sarcoma in Children and Young Adults. Cancer Res. Treat. 2021, 37, 10040. [Google Scholar] [CrossRef]
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Huang, D.; Zhao, W.; Sun, L.; Xiong, H.; Zhang, Y.; Jin, M.; Zhang, D.; Huang, C.; Wang, H.; et al. Clinical Characteristics and Prognosis of Childhood Rhabdomyosarcoma: A Ten-Year Retrospective Multicenter Study. Int. J. Clin. Exp. Med. 2015, 8, 17196–17205. [Google Scholar]
- Hosoi, H. Current Status of Treatment for Pediatric Rhabdomyosarcoma in the USA and Japan: Rhabdomyosarcoma. Pediatr. Int. 2016, 58, 81–87. [Google Scholar] [CrossRef]
- Paulino, A.C.; Okcu, M.F. Rhabdomyosarcoma. Curr. Probl. Cancer 2008, 32, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.-H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Revathidevi, S.; Munirajan, A.K. Akt in Cancer: Mediator and More. Semin. Cancer Biol. 2019, 59, 80–91. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Klekota, J.; Roth, F.P. Chemical Substructures That Enrich for Biological Activity. Bioinformatics 2008, 24, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Bębenek, E.; Kadela-Tomanek, M.; Chrobak, E.; Wietrzyk, J.; Sadowska, J.; Boryczka, S. New Acetylenic Derivatives of Betulin and Betulone, Synthesis and Cytotoxic Activity. Med. Chem. Res. 2017, 26, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Bębenek, E.; Bober-Majnusz, K.; Siudak, S.; Chrobak, E.; Kadela-Tomanek, M.; Wietrzyk, J.; Boryczka, S. Application of TLC to Evaluate the Lipophilicity of Newly Synthesized Betulin Derivatives. J. Chromatogr. Sci. 2020, 58, 323–333. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. ILOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Zhao, Y.; Li, X.; Lin, F.; Xu, Y.; Zhang, X.; Li, Y.; Wang, R.; Lai, L. Computation of Octanol−Water Partition Coefficients by Guiding an Additive Model with Knowledge. J. Chem. Inf. Model. 2007, 47, 2140–2148. [Google Scholar] [CrossRef]
- Wildman, S.A.; Crippen, G.M. Prediction of Physicochemical Parameters by Atomic Contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Moriguchi, I.; Hirono, S.; Liu, Q.; Nakagome, I.; Matsushita, Y. Simple Method of Calculating Octanol/Water Partition Coefficient. Chem. Pharm. Bull. 1992, 40, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, I.; Hirono, S.; Nakagome, I.; Hirano, H. Comparison of Reliability of Log P Values for Drugs Calculated by Several Methods. Chem. Pharm. Bull. 1994, 42, 976–978. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Silicos-It|Welcome. Available online: http://silicos-it.be.s3-website-eu-west-1.amazonaws.com/ (accessed on 22 August 2021).
- Delaney, J.S. ESOL: Estimating Aqueous Solubility Directly from Molecular Structure. J. Chem. Inf. Comput. Sci. 2004, 44, 1000–1005. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Camilleri, P.; Brown, M.B.; Hutt, A.J.; Kirton, S.B. In Silico Prediction of Aqueous Solubility Using Simple QSPR Models: The Importance of Phenol and Phenol-like Moieties. J. Chem. Inf. Model. 2012, 52, 2950–2957. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Martin, Y.C. A Bioavailability Score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. AdmetSAR 2.0: Web-Service for Prediction and Optimization of Chemical ADMET Properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, L.; Cheng, F.; Wu, Z.; Bian, H.; Xu, C.; Li, W.; Liu, G.; Shen, X.; Tang, Y. In Silico Prediction of Chemical Acute Oral Toxicity Using Multi-Classification Methods. J. Chem. Inf. Model. 2014, 54, 1061–1069. [Google Scholar] [CrossRef]
- Wu, Z.; Cheng, F.; Li, J.; Li, W.; Liu, G.; Tang, Y. SDTNBI: An Integrated Network and Chemoinformatics Tool for Systematic Prediction of Drug–Target Interactions and Drug Repositioning. Brief. Bioinform. 2017, 18, 333–347. [Google Scholar] [CrossRef]
- Wu, Z.; Peng, Y.; Yu, Z.; Li, W.; Liu, G.; Tang, Y. NetInfer: A Web Server for Prediction of Targets and Therapeutic and Adverse Effects via Network-Based Inference Methods. J. Chem. Inf. Model. 2020, 60, 3687–3691. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Z.; Peng, Y.; Li, W.; Liu, G.; Tang, Y. Pathway-Based Drug Repurposing with DPNetinfer: A Method to Predict Drug–Pathway Associations via Network-Based Approaches. J. Chem. Inf. Model. 2021, 61, 2475–2485. [Google Scholar] [CrossRef]
- Wu, Z.; Lu, W.; Wu, D.; Luo, A.; Bian, H.; Li, J.; Li, W.; Liu, G.; Huang, J.; Cheng, F.; et al. In Silico Prediction of Chemical Mechanism of Action via an Improved Network-Based Inference Method. Br. J. Pharmacol. 2016, 173, 3372–3385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Nakaya, A. The KEGG Databases at GenomeNet. Nucleic Acids Res. 2002, 30, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Moutselos, K.; Kanaris, I.; Chatziioannou, A.; Maglogiannis, I.; Kolisis, F.N. KEGGconverter: A Tool for the in-Silico Modelling of Metabolic Networks of the KEGG Pathways Database. BMC Bioinformatics 2009, 10, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahal, Z.; Abdulhai, F.; Kadara, H.; Saab, R. Genomics of Adult and Pediatric Solid Tumors. Am. J. Cancer Res. 2018, 8, 1356–1386. [Google Scholar]
- Sweet-Cordero, E.A.; Biegel, J.A. The Genomic Landscape of Pediatric Cancers: Implications for Diagnosis and Treatment. Science 2019, 363, 1170–1175. [Google Scholar] [CrossRef]
- Jones, D.T.W.; Banito, A.; Grünewald, T.G.P.; Haber, M.; Jäger, N.; Kool, M.; Milde, T.; Molenaar, J.J.; Nabbi, A.; Pugh, T.J.; et al. Molecular Characteristics and Therapeutic Vulnerabilities across Paediatric Solid Tumours. Nat. Rev. Cancer 2019, 19, 420–438. [Google Scholar] [CrossRef] [PubMed]
- Oberlin, O.; Rey, A.; Lyden, E.; Bisogno, G.; Stevens, M.C.G.; Meyer, W.H.; Carli, M.; Anderson, J.R. Prognostic Factors in Metastatic Rhabdomyosarcomas: Results of a Pooled Analysis from United States and European Cooperative Groups. J. Clin. Oncol. 2008, 26, 2384–2389. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.A.; Seibel, N.L.; Altekruse, S.F.; Ries, L.A.G.; Melbert, D.L.; O’Leary, M.; Smith, F.O.; Reaman, G.H. Outcomes for Children and Adolescents with Cancer: Challenges for the Twenty-First Century. J. Clin. Oncol. 2010, 28, 2625–2634. [Google Scholar] [CrossRef]
- Saletta, F.; Seng, M.S.; Lau, L.M.S. Advances in Paediatric Cancer Treatment. Transl. Pediatr. 2014, 3, 156–182. [Google Scholar]
- Król, S.K.; Kapka-Skrzypczak, L. Aktywność farmakologiczna olejków eterycznych i ich składników w leczeniu schorzeń układu pokarmowego. Med. Ogólna Nauki Zdrowiu 2011, 17, 202–205. [Google Scholar]
- Król, S.K.; Skalicka-Woźniak, K.; Kandefer-Szerszeń, M.; Stepulak, A. Aktywność Biologiczna i Farmakologiczna Olejków Eterycznych w Leczeniu i Profilaktyce Chorób Infekcyjnych. Postępy Hig. Med. Dośw 2013, 67, 1000–1007. [Google Scholar] [CrossRef]
- Król, S.K.; Kapka-Skrzypczak, L. Nowotwory jelita grubego jako poważny problem w Polsce i na świecie—Kwestie medyczne i środowiskowe. Medycyna Środowiskowa-Environmental Med. 2011, 14, 75–80. [Google Scholar]
- Paduch, R.; Trytek, M.; Król, S.K.; Kud, J.; Frant, M.; Kandefer-Szerszeń, M.; Fiedurek, J. Biological Activity of Terpene Compounds Produced by Biotechnological Methods. Pharm. Biol. 2016, 54, 1096–1107. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Paterson, I.; Anderson, E.A. The Renaissance of Natural Products as Drug Candidates. Science 2005, 310, 451–453. [Google Scholar] [CrossRef]
- Nirmala, M.J.; Samundeeswari, A.; Sankar, P.D. Natural Plant Resources in Anti-Cancer Therapy-A Review. Res. Plant Biol. 2011, 1, 1–14. [Google Scholar]
- Fridlender, M.; Kapulnik, Y.; Koltai, H. Plant Derived Substances with Anti-Cancer Activity: From Folklore to Practice. Front. Plant Sci. 2015, 6, 799. [Google Scholar] [CrossRef]
- Choudhari, A.S.; Mandave, P.C.; Deshpande, M.; Ranjekar, P.; Prakash, O. Phytochemicals in Cancer Treatment: From Preclinical Studies to Clinical Practice. Front. Pharmacol. 2020, 10, 1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, L.; de Blanco, E.J.C.; Kinghorn, A.D. Plant-derived natural products as leads for drug discovery. In Plant-Derived Natural Products: Synthesis, Function, and Application; Osbourn, A.E., Lanzotti, V., Eds.; Springer: New York, NY, USA, 2009; pp. 547–567. ISBN 978-0-387-85498-4. [Google Scholar]
- Seidel, V. Plant-Derived Chemicals: A Source of Inspiration for New Drugs. Plants 2020, 9, 1562. [Google Scholar] [CrossRef] [PubMed]
- Csuk, R.; Barthel, A.; Kluge, R.; Ströhl, D. Synthesis, Cytotoxicity and Liposome Preparation of 28-Acetylenic Betulin Derivatives. Bioorg. Med. Chem. 2010, 18, 7252–7259. [Google Scholar] [CrossRef]
- Orchel, A.; Kulczycka, A.; Chodurek, E.; Bębenek, E.; Borkowska, P.; Boryczka, S.; Kowalski, J.; Dzierżewicz, Z. Influence of Betulin and 28-O-Propynoylbetulin on Proliferation and Apoptosis of Human Melanoma Cells (G-361). Postepy Hig. Med. Doswiadczalnej Online 2014, 68, 191–197. [Google Scholar] [CrossRef]
- Ben-Zvi, Z.; Danon, A. Pharmacology of Acetylenic Derivatives. In Triple Bonded Functional Groups (1994); John Wiley & Sons, Ltd.: New Jersey, USA, 1994; pp. 739–788. ISBN 978-0-470-02477-5. [Google Scholar]
- Boryczka, S.; Mól, W.; Milczarek, M.; Wietrzyk, J.; Bębenek, E. Synthesis and in Vitro Antiproliferative Activity of Novel (4-Chloro- and 4-Acyloxy-2-Butynyl)Thioquinolines. Med. Chem. Res. 2011, 20, 1402–1410. [Google Scholar] [CrossRef] [Green Version]
- Król, S.K.; Bębenek, E.; Sławińska-Brych, A.; Dmoszyńska-Graniczka, M.; Boryczka, S.; Stepulak, A. Synthetic Betulin Derivatives Inhibit Growth of Glioma Cells In Vitro. Anticancer Res. 2020, 40, 6151–6158. [Google Scholar] [CrossRef]
- Moke, D.J.; Luo, C.; Millstein, J.; Knight, K.R.; Rassekh, S.R.; Brooks, B.; Ross, C.J.D.; Wright, M.; Mena, V.; Rushing, T.; et al. Prevalence and Risk Factors for Cisplatin-Induced Hearing Loss in Children, Adolescents, and Young Adults: A Multi-Institutional North American Cohort Study. Lancet Child Adolesc. Health 2021, 5, 274–283. [Google Scholar] [CrossRef]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical Development Success Rates for Investigational Drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, B.; Abed, S.N.; Al-Attraqchi, O.; Kuche, K.; Tekade, R.K. Chapter 21—Computer-Aided Prediction of Pharmacokinetic (ADMET) Properties. In Dosage Form Design Parameters; Tekade, R.K., Ed.; Advances in Pharmaceutical Product Development and Research; Academic Press: Cambridge, MA, USA, 2018; pp. 731–755. ISBN 978-0-12-814421-3. [Google Scholar]
- Achrem-Achremowicz, J.; Kępczyńska, E.; Żylewski, M.; Janeczko, Z. Synthesis of Betulin Derivatives and the Determination of Their Relative Lipophilicities Using Reversed-Phase Thin-Layer Chromatography. Biomed. Chromatogr. 2010, 24, 261–267. [Google Scholar] [CrossRef]
- Şoica, C.M.; Peev, C.; Sorina, C.; Ambrus, R.; Dehelean, C. Physico-Chemical and Toxicological Evaluations of Betulin and Betulinic Acid Interactions with Hydrophilic Cyclodextrins. Farmacia 2010, 58, 611–619. [Google Scholar]
- Stocks, M. Chapter 3—The small molecule drug discovery process—From target selection. In Introduction to Biological and Small Molecule Drug Research and Development; Ganellin, R., Roberts, S., Jefferis, R., Eds.; Elsevier: Oxford, UK, 2013; pp. 81–126. ISBN 978-0-12-397176-0. [Google Scholar]
- Di, L.; Kerns, E.H. Chapter 5—Lipophilicity. In Drug-Like Properties, 2nd ed.; Di, L., Kerns, E.H., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 39–50. ISBN 978-0-12-801076-1. [Google Scholar]
- Pardridge, W.M. Transport of Small Molecules through the Blood-Brain Barrier: Biology and Methodology. Adv. Drug Deliv. Rev. 1995, 15, 5–36. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H. Chapter 10—Blood-Brain Barrier. In Drug-Like Properties, 2nd ed.; Di, L., Kerns, E.H., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 141–159. ISBN 978-0-12-801076-1. [Google Scholar]
- Kelder, J.; Grootenhuis, P.D.J.; Bayada, D.M.; Delbressine, L.P.C.; Ploemen, J.-P. Polar Molecular Surface as a Dominating Determinant for Oral Absorption and Brain Penetration of Drugs. Pharm. Res. 1999, 16, 1514–1519. [Google Scholar] [CrossRef]
- Abd El-Karim, S.S.; Anwar, M.M.; Mohamed, N.A.; Nasr, T.; Elseginy, S.A. Design, Synthesis, Biological Evaluation and Molecular Docking Studies of Novel Benzofuran–Pyrazole Derivatives as Anticancer Agents. Bioorganic Chem. 2015, 63, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral Druggable Space beyond the Rule of 5: Insights from Drugs and Clinical Candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsson, P.; Doak, B.C.; Over, B.; Kihlberg, J. Cell Permeability beyond the Rule of 5. Adv. Drug Deliv. Rev. 2016, 101, 42–61. [Google Scholar] [CrossRef] [PubMed]
- Doak, B.C.; Kihlberg, J. Drug Discovery beyond the Rule of 5—Opportunities and Challenges. Expert Opin. Drug Discov. 2017, 12, 115–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Li, Y.; Yu, H.; Zhang, L.; Hou, T. Computational Models for Predicting Substrates or Inhibitors of P-Glycoprotein. Drug Discov. Today 2012, 17, 343–351. [Google Scholar] [CrossRef]
- Desai, P.V.; Sawada, G.A.; Watson, I.A.; Raub, T.J. Integration of in Silico and In Vitro Tools for Scaffold Optimization during Drug Discovery: Predicting P-Glycoprotein Efflux. Mol. Pharm. 2013, 10, 1249–1261. [Google Scholar] [CrossRef] [PubMed]
- Alam, K.; Crowe, A.; Wang, X.; Zhang, P.; Ding, K.; Li, L.; Yue, W. Regulation of Organic Anion Transporting Polypeptides (OATP) 1B1- and OATP1B3-Mediated Transport: An Updated Review in the Context of OATP-Mediated Drug-Drug Interactions. Int. J. Mol. Sci. 2018, 19, 855. [Google Scholar] [CrossRef] [Green Version]
- Schulte, R.R.; Ho, R.H. Organic Anion Transporting Polypeptides: Emerging Roles in Cancer Pharmacology. Mol. Pharmacol. 2019, 95, 490–506. [Google Scholar] [CrossRef] [Green Version]
- Nies, A.T.; Koepsell, H.; Damme, K.; Schwab, M. Organic Cation Transporters (OCTs, MATEs), In Vitro and In Vivo Evidence for the Importance in Drug Therapy. In Drug Transporters; Fromm, M.F., Kim, R.B., Eds.; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2011; pp. 105–167. ISBN 978-3-642-14541-4. [Google Scholar]
- Koepsell, H. Role of Organic Cation Transporters in Drug–Drug Interaction. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1619–1633. [Google Scholar] [CrossRef]
- Koepsell, H. Organic Cation Transporters in Health and Disease. Pharmacol. Rev. 2020, 72, 253–319. [Google Scholar] [CrossRef] [PubMed]
- Kubitz, R.; Dröge, C.; Stindt, J.; Weissenberger, K.; Häussinger, D. The Bile Salt Export Pump (BSEP) in Health and Disease. Clin. Res. Hepatol. Gastroenterol. 2012, 36, 536–553. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y. 5—The bile salt export pump (BSEP/ABCB11). In Transporters in Drug Discovery and Development; Lai, Y., Ed.; Woodhead Publishing Series in Biomedicine; Woodhead Publishing: Sawston, UK, 2013; pp. 327–352. ISBN 978-1-907568-21-3. [Google Scholar]
- Saha, N. Chapter 6—Clinical Pharmacokinetics and Drug Interactions. In Pharmaceutical Medicine and Translational Clinical Research; Vohora, D., Singh, G., Eds.; Academic Press: Boston, MA, USA, 2018; pp. 81–106. ISBN 978-0-12-802103-3. [Google Scholar]
- Zanger, U.M.; Schwab, M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, A.M.; Dang, C.H. Basic Review of the Cytochrome P450 System. J. Adv. Pract. Oncol. 2013, 4, 263. [Google Scholar] [CrossRef]
- Lappano, R.; Maggiolini, M. G Protein-Coupled Receptors: Novel Targets for Drug Discovery in Cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shavit, R.; Maoz, M.; Kancharla, A.; Nag, J.K.; Agranovich, D.; Grisaru-Granovsky, S.; Uziely, B. G Protein-Coupled Receptors in Cancer. Int. J. Mol. Sci. 2016, 17, 1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, M.A. Maturing of the Nuclear Receptor Family. J. Clin. Investig. 2017, 127, 1123–1125. [Google Scholar] [CrossRef] [Green Version]
- Mazaira, G.I.; Zgajnar, N.R.; Lotufo, C.M.; Daneri-Becerra, C.; Sivils, J.C.; Soto, O.B.; Cox, M.B.; Galigniana, M.D. The Nuclear Receptor Field: A Historical Overview and Future Challenges. Nucl. Recept. Res. 2018, 5, 101320. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Cidlowski, J.A.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Nuclear Hormone Receptors. Br. J. Pharmacol. 2019, 176, S229–S246. [Google Scholar] [CrossRef] [Green Version]
- Alexander, S.P.H.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G Protein-Coupled Receptors. Br. J. Pharmacol. 2019, 176, S21–S141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilghman, R.W.; Parsons, J.T. Focal Adhesion Kinase as a Regulator of Cell Tension in the Progression of Cancer. Semin. Cancer Biol. 2008, 18, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Eke, I.; Cordes, N. Focal Adhesion Signaling and Therapy Resistance in Cancer. Semin. Cancer Biol. 2015, 31, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Noorolyai, S.; Shajari, N.; Baghbani, E.; Sadreddini, S.; Baradaran, B. The Relation between PI3K/AKT Signalling Pathway and Cancer. Gene 2019, 698, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/MTOR Inhibitors in Cancer: At the Bench and Bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ Signaling and Metabolism: The Good, the Bad and the Future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Vitale, S.G.; Laganà, A.S.; Nigro, A.; La Rosa, V.L.; Rossetti, P.; Rapisarda, A.M.C.; La Vignera, S.; Condorelli, R.A.; Corrado, F.; Buscema, M.; et al. Peroxisome Proliferator-Activated Receptor Modulation during Metabolic Diseases and Cancers: Master and Minions. PPAR Res. 2016, 2016, e6517313. [Google Scholar] [CrossRef] [Green Version]
- Glazer, R.I. PPARδ as a Metabolic Initiator of Mammary Neoplasia and Immune Tolerance. PPAR Res. 2016, 2016, e3082340. [Google Scholar] [CrossRef] [Green Version]
- Fanale, D.; Amodeo, V.; Caruso, S. The Interplay between Metabolism, PPAR Signaling Pathway, and Cancer. PPAR Res. 2017, 2017, e1830626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a New Form of Cell Death: Opportunities and Challenges in Cancer. J. Hematol. Oncol.J Hematol Oncol 2019, 12, 34. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Molecular Mechanisms of Ferroptosis and Its Role in Cancer Therapy. J. Cell. Mol. Med. 2019, 23, 4900–4912. [Google Scholar] [CrossRef] [Green Version]
- Grabarska, A.; Skalicka-Woźniak, K.; Kiełbus, M.; Dmoszyńska-Graniczka, M.; Miziak, P.; Szumiło, J.; Nowosadzka, E.; Kowalczuk, K.; Khalifa, S.; Smok-Kalwat, J.; et al. Imperatorin as a Promising Chemotherapeutic Agent against Human Larynx Cancer and Rhabdomyosarcoma Cells. Molecules 2020, 25, 2046. [Google Scholar] [CrossRef] [PubMed]
- Was, H.; Krol, S.K.; Rotili, D.; Mai, A.; Wojtas, B.; Kaminska, B.; Maleszewska, M. Histone Deacetylase Inhibitors Exert Anti-Tumor Effects on Human Adherent and Stem-like Glioma Cells. Clin. Epigenetics 2019, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, T.M.; Kiełbus, M.; Kaczor, A.A.; Kryštof, V.; Karczmarzyk, Z.; Wysocki, W.; Fruziński, A.; Król, S.K.; Grabarska, A.; Stepulak, A.; et al. Discovery of Nitroaryl Urea Derivatives with Antiproliferative Properties. J. Enzyme Inhib. Med. Chem. 2016, 31, 608–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Król, S.K.; Kaczmarczyk, A.; Wojnicki, K.; Wojtas, B.; Gielniewski, B.; Grajkowska, W.; Kotulska, K.; Szczylik, C.; Czepko, R.; Banach, M.; et al. Aberrantly Expressed RECQL4 Helicase Supports Proliferation and Drug Resistance of Human Glioma Cells and Glioma Stem Cells. Cancers 2020, 12, 2919. [Google Scholar] [CrossRef]
- Sławińska-Brych, A.; Mizerska-Kowalska, M.; Król, S.K.; Stepulak, A.; Zdzisińska, B. Xanthohumol Impairs the PMA-Driven Invasive Behaviour of Lung Cancer Cell Line A549 and Exerts Anti-EMT Action. Cells 2021, 10, 1484. [Google Scholar] [CrossRef]
- Sławińska-Brych, A.; Król, S.K.; Dmoszyńska-Graniczka, M.; Zdzisińska, B.; Stepulak, A.; Gagoś, M. Xanthohumol Inhibits Cell Cycle Progression and Proliferation of Larynx Cancer Cells In Vitro. Chem. Biol. Interact. 2015, 240, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 | ||||
|---|---|---|---|---|---|
| CDDP | TMZ | EB5 | EB25/1 | Bet | |
| SK-N-AS | 3.22 µM | 486.59 µM | 0.62 µM | 18.77 µM | 2.5 µM [18] |
| TE671 | 2.48 µM | 714.48 µM | 1.34 µM | 17.35 µM | 10.3 µM [18] |
| HSF | – 1 | – | 17.15 µM | N/A 2 | – |
| Compound | Bet | EB5 | EB25/1 | |
|---|---|---|---|---|
| Parameter | ||||
| Formula | C30H50O2 | C33H50O3 | C34H52O4 | |
| Molecular Weight [g/mol] | 442.72 | 494.75 | 524.77 | |
| Lipophilicity | ||||
| LogPTLC | 5.41 [61] | 7.08 [61] | 7.76 [61] | |
| LogP (iLOGP 1) | 4.47 | 5.11 | 5.44 | |
| LogP (XLOGP3 2) | 8.28 | 9.34 | 9.38 | |
| LogP (WLOGP 3) | 7.00 | 7.26 | 7.87 | |
| LogP (MLOGP 4) | 6.00 | 6.29 | 6.03 | |
| LogP (SILICOS-IT 5) | 6.21 | 6.96 | 6.92 | |
| Consensus LogP 6 | 6.39 | 6.99 | 7.13 | |
| Water Solubility | ||||
| LogS (ESOL 7) | −7.67 | −8.53 | −8.61 | |
| Solubility | 9.48 × 10−6 mg/mL; 2.14 × 10−8 mol/l | 1.47 × 10−6 mg/mL; 2.97 × 10−9 mol/l | 1.30 × 10−6 mg/mL; 2.47 × 10−9 mol/l | |
| Class 8 | Poorly soluble | Poorly soluble | Poorly soluble | |
| LogS (Ali 9) | −8.99 | −10.22 | −10.46 | |
| Solubility | 4.50 × 10−7 mg/mL; 1.02 × 10−9 mol/l | 2.98 × 10−8 mg/mL; 6.02 × 10−11 mol/l | 1.84 × 10−8 mg/mL; 3.50 × 10−11 mol/l | |
| Class | Poorly soluble | Insoluble | Insoluble | |
| LogS (SILICOS-IT 10) | −6.17 | −6.46 | −6.57 | |
| Solubility | 2.99 × 10−4 mg/mL; 6.75 × 10−7 mol/l | 1.70 × 10−4 mg/mL; 3.44 × 10−7 mol/l | 1.40 × 10−4 mg/mL; 2.67 × 10−7 mol/l | |
| Class | Poorly soluble | Poorly soluble | Poorly soluble | |
| Compound | Bet | EB5 | EB25/1 | |
|---|---|---|---|---|
| Violation | ||||
| Lipinski 1 | Yes; 1 violation: MLOGP > 4.15 | Yes; 1 violation: MLOGP > 4.15 | No; 2 violations: MW > 500, MLOGP > 4.15 | |
| Ghose 2 | No; 3 violations: WLOGP > 5.6, MR > 130, #atoms > 70 | No; 4 violations: MW > 480, WLOGP > 5.6, MR > 130, #atoms > 70 | No; 4 violations: MW > 480, WLOGP > 5.6, MR > 130, #atoms > 70 | |
| Veber 3 | Yes | Yes | Yes | |
| Egan 4 | No; 1 violation: WLOGP > 5.88 | No; 1 violation: WLOGP > 5.88 | No; 1 violation: WLOGP > 5.88 | |
| Muegge 5 | No; 1 violation: XLOGP3 > 5 | No; 1 violation: XLOGP3 > 5 | No; 1 violation: XLOGP3 > 5 | |
| Bioavailability Score 6 | 0.55 | 0.55 | 0.17 | |
| Compound | Bet | EB5 | EB25/1 | ||||
|---|---|---|---|---|---|---|---|
| Parameter | |||||||
| ADMET Profile Classifications | Value | Probability | Value | Probability | Value | Probability | |
| Absorption | |||||||
| HIA 1 | + | 0.9884 | + | 0.9892 | + | 0.9818 | |
| Caco-2 permeability | − | 0.5542 | − | 0.6911 | − | 0.7424 | |
| Human oral bioavailability | − | 0.5857 | − | 0.6571 | − | 0.6714 | |
| Distribution | |||||||
| Subcellular localization | Lys 2 | 0.4831 | Mito 3 | 0.8480 | Mito | 0.8300 | |
| BBB 4 permeant | − | 0.4533 | + | 0.8120 | + | 0.9081 | |
| P-glycoprotein inhibitor | − | 0.8836 | − | 0.7952 | − | 0.4746 | |
| P-glycoprotein substrate | − | 0.7347 | − | 0.8347 | − | 0.6175 | |
| BSEP 5 inhibitor | + | 0.6370 | + | 0.8859 | + | 0.7854 | |
| OATP 6 1B1 inhibitor | + | 0.9413 | + | 0.9013 | + | 0.9004 | |
| OATP 1B3 inhibitor | + | 0.9480 | + | 0.8936 | + | 0.8682 | |
| OATP 2B1 inhibitor | − | 0.7184 | − | 0.7112 | − | 0.5653 | |
| OCT2 7 inhibitor | − | 0.6385 | − | 0.6000 | − | 0.6526 | |
| MATE1 8 inhibitor | − | 1.0000 | − | 0.8200 | − | 0.9600 | |
| Metabolism | |||||||
| CYP450 9 3A4 substrate | + | 0.6751 | − | 0.6453 | + | 0.7183 | |
| CYP450 2C9 substrate | − | 0.6284 | − | 0.5974 | + | 0.5886 | |
| CYP450 2D6 substrate | − | 0.7222 | − | 0.5760 | − | 0.7448 | |
| CYP450 1A2 inhibition | − | 0.9045 | − | 0.9286 | − | 0.8561 | |
| CYP450 2C9 inhibition | − | 0.9071 | − | 0.8779 | − | 0.7006 | |
| CYP450 2C19 inhibition | − | 0.9026 | − | 0.6899 | − | 0.6710 | |
| CYP450 2D6 inhibition | − | 0.9297 | − | 0.6000 | − | 0.9188 | |
| CYP450 3A4 inhibition | − | 0.8309 | − | 0.9281 | − | 0.6587 | |
| CYP inhibitory promiscuity | − | 0.6441 | − | 0.6416 | − | 0.7258 | |
| Toxicity | |||||||
| Carcinogenicity | − | 0.9857 | − | 0.8347 | − | 0.9073 | |
| Ames mutagenesis | − | 0.7500 | − | 0.7000 | − | 0.6250 | |
| Eye corrosion | − | 0.9892 | − | 0.9923 | + | 0.7267 | |
| Eye irritation | − | 0.9008 | − | 0.9001 | − | 0.7900 | |
| Hepatotoxicity | − | 0.6250 | − | 0.5500 | + | 0.7678 | |
| Acute Oral Toxicity | III 10 | 0.7441 | III | 0.6655 | − | 0.5236 | |
| Compound | Target ID 1 | Protein Name 2 | Protein Family | Gene Symbol 3 | Gene ID 4 | Organism | Score |
|---|---|---|---|---|---|---|---|
| Bet | P04278 | Sex hormone-binding globulin | - | SHBG | 6462 | Homo sapiens (Human) | 0.000334142 |
| P34972 | Cannabinoid receptor 2 (CB-2) | G protein coupled receptor 1 family | CNR2 | 1269 | 0.000277126 | ||
| Q8TDU6 | G protein coupled bile acid receptor 1 | GPBAR1 | 151306 | 0.000266207 | |||
| P04150 | Glucocorticoid receptor (GR) | Nuclear hormone receptor family | NR3C1 | 2908 | 0.000243245 | ||
| P41145 | Kappa-type opioid receptor (K-OR-1) | G protein coupled receptor 1 family | OPRK1 | 4986 | 0.000236339 | ||
| P41143 | Delta-type opioid receptor (D-OR-1) | OPRD1 | 4985 | 0.000213809 | |||
| P35372 | Mu-type opioid receptor (M-OR-1) | OPRM1 | 4988 | 0.000201019 | |||
| P32249 | Epstein–Barr virus-induced molecule 2 (G protein coupled receptor 183) | GPR183 | 1880 | 0.000196558 | |||
| Q9NUW8 | Tyrosyl-DNA phosphodiesterase 1 | Tyrosyl-DNA phosphodiesterase family | TDP1 | 55775 | 0.000188861 | ||
| P03372 | Estrogen receptor (ER-α) | Nuclear hormone receptor family | ESR1 | 2099 | 0.000188836 | ||
| EB5 | P04278 | Sex hormone-binding globulin | - | SHBG | 6462 | Homo sapiens (Human) | 0.000271256 |
| P41594 | Metabotropic glutamate receptor 5 (mGluR5) | G protein coupled receptor 3 family | GRM5 | 2915 | 0.000224581 | ||
| P04150 | Glucocorticoid receptor (GR) | Nuclear hormone receptor family | NR3C1 | 2908 | 0.000220946 | ||
| P04035 | 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA reductase) | HMG-CoA reductase family | HMGCR | 3156 | 0.000208677 | ||
| P41145 | Kappa-type opioid receptor (K-OR-1) | G protein coupled receptor 1 family | OPRK1 | 4986 | 0.00020737 | ||
| P34972 | Cannabinoid receptor 2 (CB-2) | CNR2 | 1269 | 0.000204833 | |||
| Q9NUW8 | Tyrosyl-DNA phosphodiesterase 1 | Tyrosyl-DNA phosphodiesterase family | TDP1 | 55775 | 0.00019616 | ||
| Q8TDU6 | G protein coupled bile acid receptor 1 | G protein coupled receptor 1 family | GPBAR1 | 151306 | 0.00018902 | ||
| P41143 | Delta-type opioid receptor (D-OR-1) | OPRD1 | 4985 | 0.000166818 | |||
| Q16236 | Nuclear factor erythroid 2-related factor 2 (NRF2) | BZIP family | NFE2L2 | 4780 | 0.000160777 | ||
| EB25/1 | P04278 | Sex hormone-binding globulin | - | SHBG | 6462 | Homo sapiens (Human) | 0.000268805 |
| P04150 | Glucocorticoid receptor (GR) | Nuclear hormone receptor family | NR3C1 | 2908 | 0.000226021 | ||
| P41594 | Metabotropic glutamate receptor 5 (mGluR5) | G protein coupled receptor 3 family | GRM5 | 2915 | 0.000225983 | ||
| Q9NUW8 | Tyrosyl-DNA phosphodiesterase 1 | Tyrosyl-DNA phosphodiesterase family | TDP1 | 55775 | 0.000223637 | ||
| P34972 | Cannabinoid receptor 2 (CB-2) | G protein coupled receptor 1 family | CNR2 | 1269 | 0.000205072 | ||
| P41145 | Kappa-type opioid receptor (K-OR-1) | OPRK1 | 4986 | 0.000200803 | |||
| P04035 | 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA reductase) | HMG-CoA reductase family | HMGCR | 3156 | 0.000190366 | ||
| Q8TDU6 | G protein coupled bile acid receptor 1 | G protein coupled receptor 1 family | GPBAR1 | 151306 | 0.000186738 | ||
| P20309 | Muscarinic acetylcholine receptor M3 (mAChR M3) | CHRM3 | 1131 | 0.000169216 | |||
| P41143 | Delta-type opioid receptor (D-OR-1) | OPRD1 | 4985 | 0.000162948 |
| Compound | Pathway ID 1 | Description | Score |
|---|---|---|---|
| Bet | hsa04610 | Complement and coagulation cascades | 0.000706300 |
| hsa04510 | Focal adhesion | 0.000645716 | |
| hsa04151 | PI3K-Akt signaling pathway | 0.000573224 | |
| hsa04915 | Estrogen signaling pathway | 0.000562535 | |
| hsa03320 | PPAR signaling pathway | 0.000518514 | |
| hsa04974 | Protein digestion and absorption | 0.000484971 | |
| hsa04978 | Mineral absorption | 0.000438870 | |
| hsa04216 | Ferroptosis | 0.000430336 | |
| hsa04640 | Hematopoietic cell lineage | 0.000409757 | |
| hsa04115 | p53 signaling pathway | 0.000404527 | |
| EB5 | hsa04610 | Complement and coagulation cascades | 0.000683879 |
| hsa04510 | Focal adhesion | 0.000611875 | |
| hsa04151 | PI3K-Akt signaling pathway | 0.000543412 | |
| hsa04915 | Estrogen signaling pathway | 0.000542487 | |
| hsa03320 | PPAR signaling pathway | 0.000497120 | |
| hsa04974 | Protein digestion and absorption | 0.000470290 | |
| hsa04216 | Ferroptosis | 0.000432794 | |
| hsa04978 | Mineral absorption | 0.000420103 | |
| hsa04640 | Hematopoietic cell lineage | 0.000407986 | |
| hsa04115 | p53 signaling pathway | 0.000386938 | |
| EB25/1 | hsa04610 | Complement and coagulation cascades | 0.000659049 |
| hsa04510 | Focal adhesion | 0.000598057 | |
| hsa04915 | Estrogen signaling pathway | 0.000535527 | |
| hsa04151 | PI3K-Akt signaling pathway | 0.000531424 | |
| hsa03320 | PPAR signaling pathway | 0.000482445 | |
| hsa04974 | Protein digestion and absorption | 0.000455536 | |
| hsa04216 | Ferroptosis | 0.000423241 | |
| hsa04978 | Mineral absorption | 0.000419440 | |
| hsa04640 | Hematopoietic cell lineage | 0.000393377 | |
| hsa04115 | p53 signaling pathway | 0.000373228 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Król, S.K.; Bębenek, E.; Dmoszyńska-Graniczka, M.; Sławińska-Brych, A.; Boryczka, S.; Stepulak, A. Acetylenic Synthetic Betulin Derivatives Inhibit Akt and Erk Kinases Activity, Trigger Apoptosis and Suppress Proliferation of Neuroblastoma and Rhabdomyosarcoma Cell Lines. Int. J. Mol. Sci. 2021, 22, 12299. https://doi.org/10.3390/ijms222212299
Król SK, Bębenek E, Dmoszyńska-Graniczka M, Sławińska-Brych A, Boryczka S, Stepulak A. Acetylenic Synthetic Betulin Derivatives Inhibit Akt and Erk Kinases Activity, Trigger Apoptosis and Suppress Proliferation of Neuroblastoma and Rhabdomyosarcoma Cell Lines. International Journal of Molecular Sciences. 2021; 22(22):12299. https://doi.org/10.3390/ijms222212299
Chicago/Turabian StyleKról, Sylwia K., Ewa Bębenek, Magdalena Dmoszyńska-Graniczka, Adrianna Sławińska-Brych, Stanisław Boryczka, and Andrzej Stepulak. 2021. "Acetylenic Synthetic Betulin Derivatives Inhibit Akt and Erk Kinases Activity, Trigger Apoptosis and Suppress Proliferation of Neuroblastoma and Rhabdomyosarcoma Cell Lines" International Journal of Molecular Sciences 22, no. 22: 12299. https://doi.org/10.3390/ijms222212299
APA StyleKról, S. K., Bębenek, E., Dmoszyńska-Graniczka, M., Sławińska-Brych, A., Boryczka, S., & Stepulak, A. (2021). Acetylenic Synthetic Betulin Derivatives Inhibit Akt and Erk Kinases Activity, Trigger Apoptosis and Suppress Proliferation of Neuroblastoma and Rhabdomyosarcoma Cell Lines. International Journal of Molecular Sciences, 22(22), 12299. https://doi.org/10.3390/ijms222212299