
Genomic Instability and Cancer Risk Associated with Erroneous DNA Repair


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
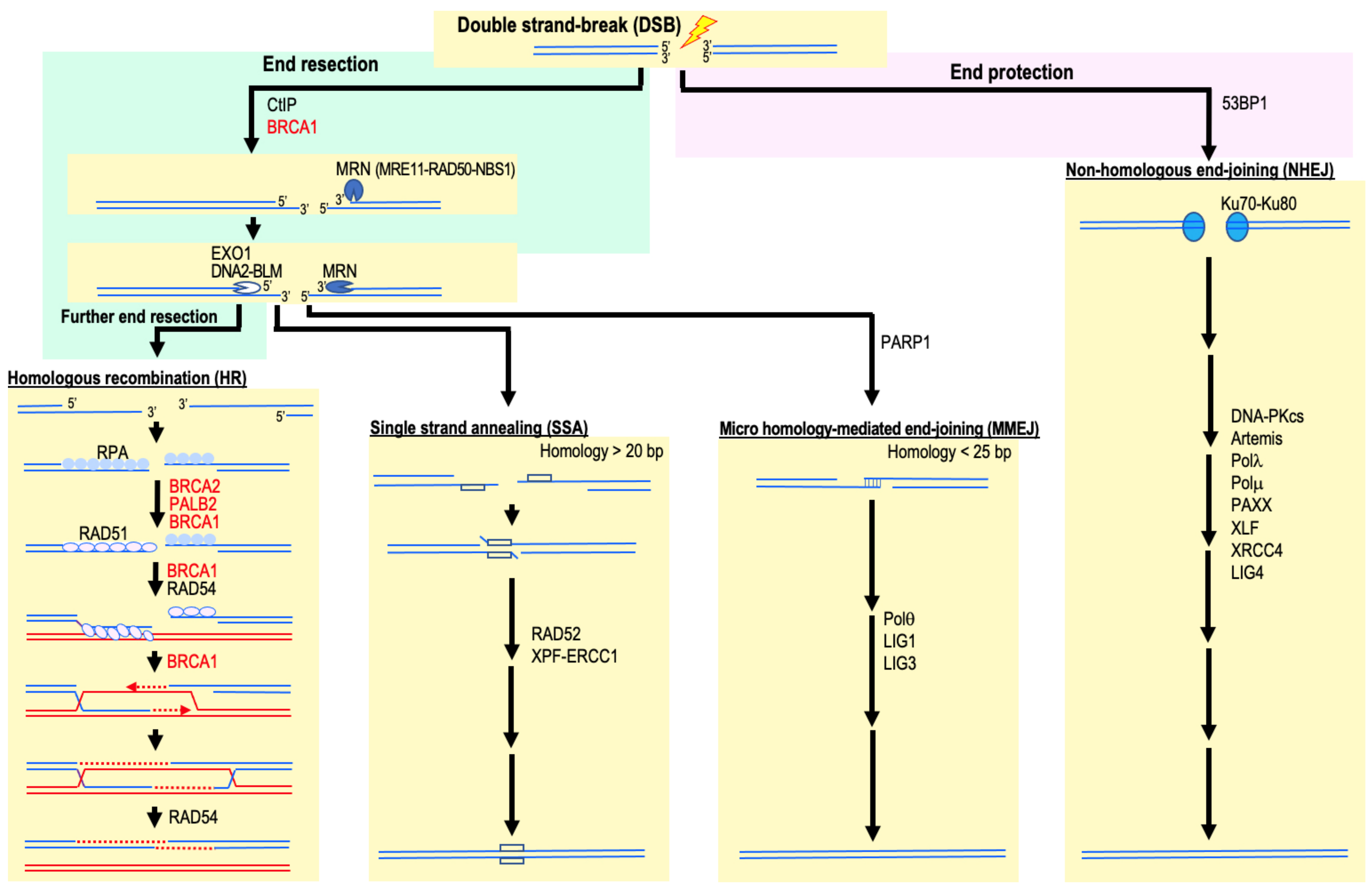
2. HR Deficiency and Increased Cancer Risk
3. FA Factor Mutation and Increased Cancer Risk
4. Involvement of NHEJ and MMEJ in Cancer Development
5. NER Deficiency and Increased Cancer Risk
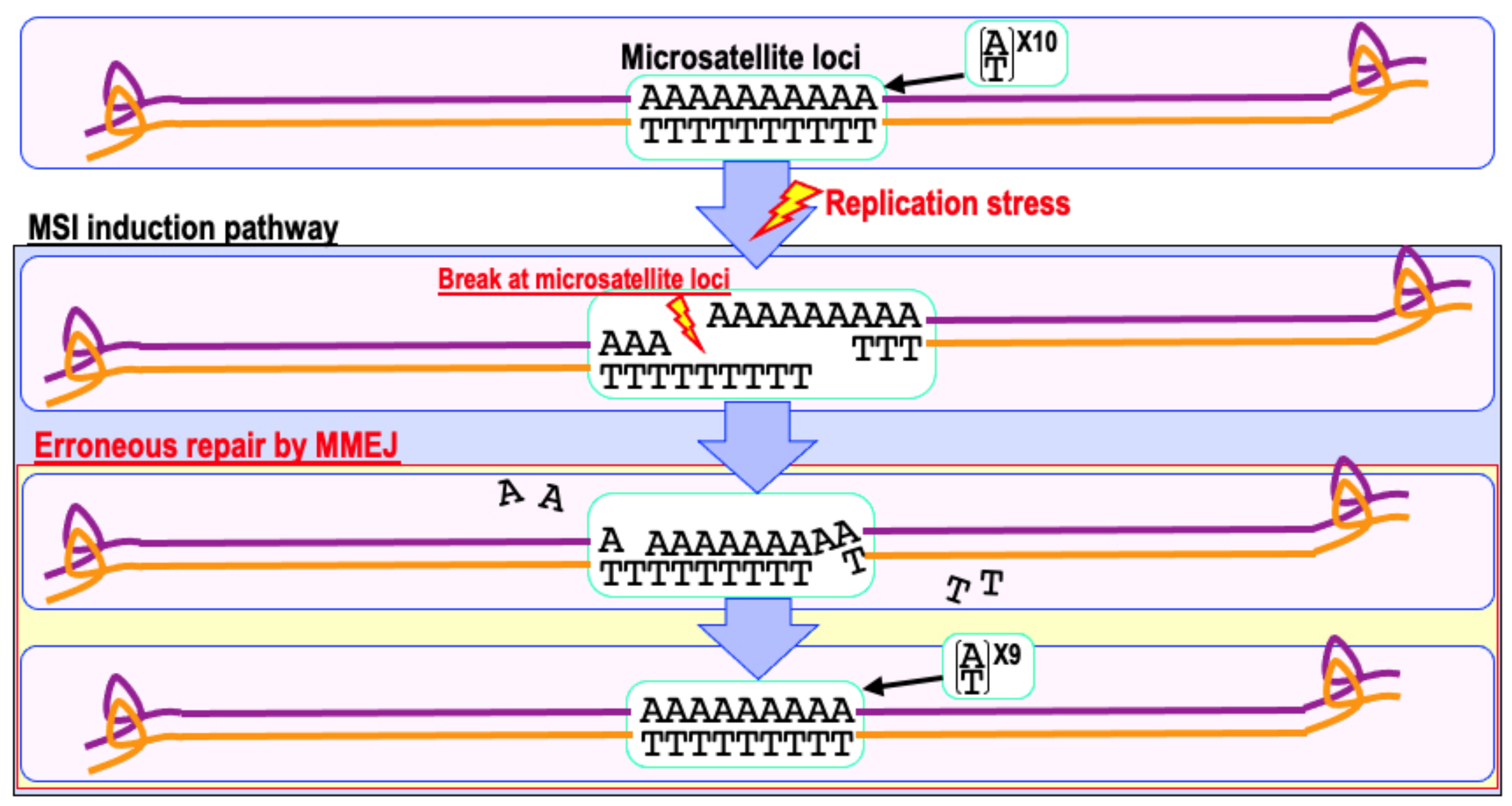
6. Mismatch Repair Deficiency and Increased Cancer Risk
7. Base Excision Repair and Cancer Risk
8. Telomere Maintenance and Cancer
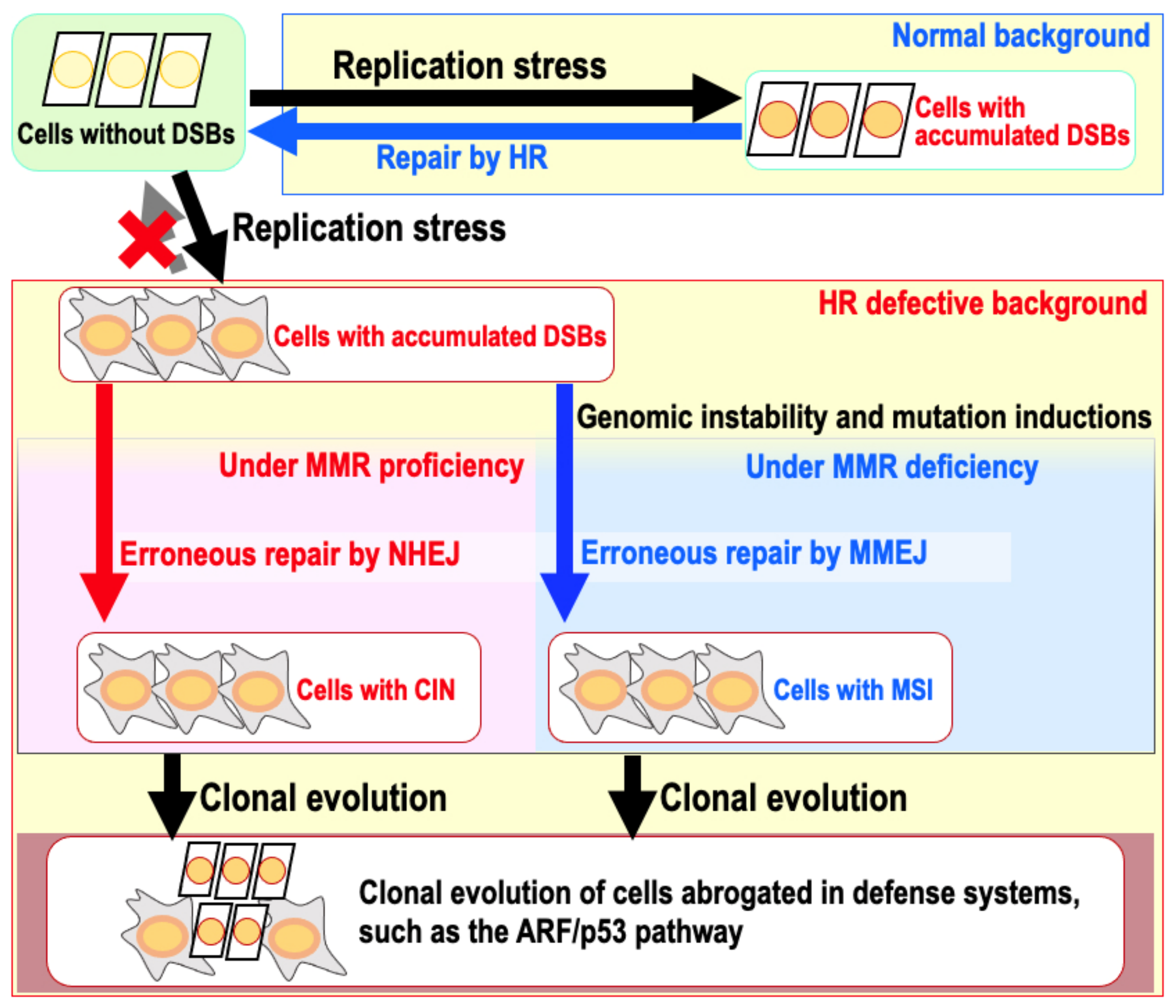
9. The Risk of Genomic Destabilization in a Repair-Proficient Background
10. Repair Pathways Activated by Chemo- and Radiotherapy-Induced DNA Damage
11. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic Instability in Colorectal Cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic Instabilities in Human Cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic Instability—An Evolving Hallmark of Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, Y.; Atsumi, Y.; Shimizu, A.; Katayama, K.; Fujimori, H.; Hyodo, M.; Minakawa, Y.; Nakatsu, Y.; Kaneko, S.; Hamamoto, R.; et al. Replication Stress Triggers Microsatellite Destabilization and Hypermutation Leading to Clonal Expansion in Vitro. Nat. Commun. 2019, 10, 3925. [Google Scholar] [CrossRef] [Green Version]
- Matsuno, Y.; Hyodo, M.; Suzuki, M.; Tanaka, Y.; Horikoshi, Y.; Murakami, Y.; Torigoe, H.; Mano, H.; Tashiro, S.; Yoshioka, K. Replication-Stress-Associated DSBs Induced by Ionizing Radiation Risk Genomic Destabilization and Associated Clonal Evolution. iScience 2021, 24, 102313. [Google Scholar] [CrossRef]
- Rajagopalan, H.; Lengauer, C. Aneuploidy and Cancer. Nature 2004, 432, 338–341. [Google Scholar] [CrossRef] [PubMed]
- Korbel, J.O.; Campbell, P.J. Criteria for Inference of Chromothripsis in Cancer Genomes. Cell 2013, 152, 1226–1236. [Google Scholar] [CrossRef] [Green Version]
- Woerner, S.M.; Kloor, M.; von Knebel Doeberitz, M.; Gebert, J.F. Microsatellite Instability in the Development of DNA Mismatch Repair Deficient Tumors. Cancer Biomark. 2006, 2, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Bishop, A.J.R.; Schiestl, R.H. Homologous Recombination and Its Role in Carcinogenesis. J. Biomed. Biotechnol. 2002, 2002, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Hoeijmakers, J.H.J. DNA Damage, Aging, and Cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Sebastian, R.; Raghavan, S.C. Induction of DNA Damage and Erroneous Repair Can Explain Genomic Instability Caused by Endosulfan. Carcinogenesis 2016, 37, 929–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, R.T. Cell Cycle Checkpoint Signaling through the ATM and ATR Kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H.J. Understanding Nucleotide Excision Repair and Its Roles in Cancer and Ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Modrich, P.; Lahue, R. Mismatch Repair in Replication Fidelity, Genetic Recombination, and Cancer Biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef]
- Sedelnikova, O.A.; Horikawa, I.; Zimonjic, D.B.; Popescu, N.C.; Bonner, W.M.; Barrett, J.C. Senescing Human Cells and Ageing Mice Accumulate DNA Lesions with Unrepairable Double-Strand Breaks. Nat. Cell Biol. 2004, 6, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA Damage Signaling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat. Cell Biol. 2009, 11, 973. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA Damage Is Irreparable and Causes Persistent DNA-Damage-Response Activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Fujimori, H.; Hyodo, M.; Matsuno, Y.; Shimizu, A.; Minakawa, Y.; Atsumi, Y.; Nakatsu, Y.; Tsuzuki, T.; Murakami, Y.; Yoshioka, K. Mismatch Repair Dependence of Replication Stress-Associated DSB Recognition and Repair. Heliyon 2019, 5, e03057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-Stalled Replication Forks Become Progressively Inactivated and Require Two Different RAD51-Mediated Pathways for Restart and Repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkova, A.; Ozerov, I.V.; Pustovalova, M.; Grekhova, A.; Eremin, P.; Vorobyeva, N.; Eremin, I.; Pulin, A.; Zorin, V.; Kopnin, P.; et al. ΓH2AX, 53BP1 and Rad51 Protein Foci Changes in Mesenchymal Stem Cells during Prolonged X-Ray Irradiation. Oncotarget 2017, 8, 64317–64329. [Google Scholar] [CrossRef]
- Tarsounas, M.; Sung, P. The Antitumorigenic Roles of BRCA1–BARD1 in DNA Repair and Replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA Double-Strand Break Repair-Pathway Choice in Somatic Mammalian Cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Bayraktar, S.; Arun, B. BRCA Mutation Genetic Testing Implications in the United States. Breast 2017, 31, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; Disaia, P.; Gabra, H.; Glenn, P.; et al. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar]
- Riaz, N.; Blecua, P.; Lim, R.S.; Shen, R.; Higginson, D.S.; Weinhold, N.; Norton, L.; Weigelt, B.; Powell, S.N.; Reis-Filho, J.S. Pan-Cancer Analysis of Bi-Allelic Alterations in Homologous Recombination DNA Repair Genes. Nat. Commun. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Niraj, J.; Färkkilä, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Nalepa, G.; Clapp, D.W. Fanconi Anaemia and Cancer: An Intricate Relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef]
- Ishiai, M.; Sato, K.; Tomida, J.; Kitao, H.; Kurumizaka, H.; Takata, M. Activation of the FA Pathway Mediated by Phosphorylation and Ubiquitination. Mutat. Res.-Fundam. Mol. Mech. Mutagenesis 2017, 803–805, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Madireddy, A.; Kosiyatrakul, S.T.; Boisvert, R.A.; Herrera-Moyano, E.; García-Rubio, M.L.; Gerhardt, J.; Vuono, E.A.; Owen, N.; Yan, Z.; Olson, S.; et al. FANCD2 Facilitates Replication through Common Fragile Sites. Mol. Cell 2016, 64, 388–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, Y.; Iwasaki, W.M.; Kugou, K.; Takahashi, K.K.; Oda, A.; Sato, K.; Kobayashi, W.; Kawai, H.; Sakasai, R.; Takaori-Kondo, A.; et al. Replication Stress Induces Accumulation of FANCD2 at Central Region of Large Fragile Genes. Nucleic Acids Res. 2018, 46, 2932–2944. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, Y.; Hejna, J.; Takata, M. Regulation of R-Loops and Genome Instability in Fanconi Anemia. J. Biochem. 2019, 165, 465–470. [Google Scholar] [CrossRef]
- Fang, C.B.; Wu, H.T.; Zhang, M.L.; Liu, J.; Zhang, G.J. Fanconi Anemia Pathway: Mechanisms of Breast Cancer Predisposition Development and Potential Therapeutic Targets. Front. Cell Dev. Biol. 2020, 8, 160. [Google Scholar] [CrossRef]
- Liu, W.; Palovcak, A.; Li, F.; Zafar, A.; Yuan, F.; Zhang, Y. Fanconi Anemia Pathway as a Prospective Target for Cancer Intervention. Cell Biosci. 2020, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, K.; Matsuno, Y. Genomic Destabilization and Its Associated Mutagenesis Increase with Senescence-associated Phenotype Expression. Cancer Sci. 2020, 112, cas.14746. [Google Scholar] [CrossRef]
- Difilippantonio, M.J.; Zhu, J.; Chen, H.T.; Meffre, E.; Nussenzweig, M.C.; Max, E.E.; Ried, T.; Nussenzweig, A. DNA Repair Protein Ku80 Suppresses Chromosomal Aberrations and Malignant Transformation. Nature 2000, 404, 510–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espejel, S.; Martín, M.; Klatt, P.; Martín-Caballero, J.; Flores, J.M.; Blasco, M.A. Shorter Telomeres, Accelerated Ageing and Increased Lymphoma in DNA-PKcs-deficient Mice. EMBO Rep. 2004, 5, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, D.W.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.D.; Gupta, G.P.; Ramsden, D.A. Essential Roles for Polymerase θ-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Patterson-Fortin, J.; D’Andrea, A.D. Exploiting the Microhomology-Mediated End-Joining Pathway in Cancer Therapy. Cancer Res. 2020, 80, 4593–4600. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Atsumi, Y.; Inase, A.; Osawa, T.; Sugihara, E.; Sakasai, R.; Fujimori, H.; Teraoka, H.; Saya, H.; Kanno, M.; Tashiro, F.; et al. The Arf/P53 Protein Module, Which Induces Apoptosis, Down-Regulates Histone H2AX to Allow Normal Cells to Survive in the Presence of Anti-Cancer Drugs. J. Biol. Chem. 2013, 288, 13269–13277. [Google Scholar] [CrossRef] [Green Version]
- Matsuno, Y.; Hyodo, M.; Fujimori, H.; Shimizu, A.; Yoshioka, K. Sensitization of Cancer Cells to Radiation and Topoisomerase I Inhibitor Camptothecin Using Inhibitors of PARP and Other Signaling Molecules. Cancers 2018, 10, 364. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, T.; Fujihara, H.; Watanabe, M.; Tsutsumi, M.; Nakamoto, K.; Kusuoka, O.; Kamada, N.; Suzuki, H.; Nakagama, H.; Sugimura, T.; et al. Parp-1 Deficiency Implicated in Colon and Liver Tumorigenesis Induced by Azoxymethane. Cancer Sci. 2003, 94, 497–500. [Google Scholar] [CrossRef] [Green Version]
- Wilson, T.E.; Sunder, S. Double-Strand Breaks in Motion: Implications for Chromosomal Rearrangement. Curr. Genet. 2020, 66, 1. [Google Scholar] [CrossRef] [PubMed]
- Van Bussel, M.T.J.; Awada, A.; de Jonge, M.J.A.; Mau-Sørensen, M.; Nielsen, D.; Schöffski, P.; Verheul, H.M.W.; Sarholz, B.; Berghoff, K.; el Bawab, S.; et al. A First-in-Man Phase 1 Study of the DNA-Dependent Protein Kinase Inhibitor Peposertib (Formerly M3814) in Patients with Advanced Solid Tumours. Br. J. Cancer 2021, 124, 728–735. [Google Scholar] [CrossRef]
- Lee, M.N.; Tseng, R.C.; Hsu, H.S.; Chen, J.Y.; Tzao, C.; Ho, W.L.; Wang, Y.C. Epigenetic Inactivation of the Chromosomal Stability Control Genes BRCA1 BRCA2, and XRCC5 in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2007, 13, 832–838. [Google Scholar] [CrossRef] [Green Version]
- Tryndyak, V.P.; Kovalchuk, O.; Muskhelishvili, L.; Montgomery, B.; Rodrigues-Juarez, R.; Melnyk, S.; Ross, S.A.; Beland, F.A.; Pogribny, I.P. Epigenetic Reprogramming of Liver Cells in Tamoxifen-Induced Rat Hepatocarcinogenesis. Mol. Carcinog. 2007, 46, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Tang, H.; Yu, J.; Zhuang, D.; Zhang, H. Blood-Based DNA Methylation of DNA Repair Genes in the Non-Homologous End-Joining (NEHJ) Pathway in Patient with Glioma. Int. J. Clin. Exp. Pathol. 2015, 8, 9463–9467. [Google Scholar] [PubMed]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; ASM Press: Washington DC, USA, 2005. [Google Scholar]
- Hu, J.; Selby, C.P.; Adar, S.; Adebali, O.; Sancar, A. Molecular Mechanisms and Genomic Maps of DNA Excision Repair in Escherichia Coli and Humans. J. Biol. Chem. 2017, 292, 15588–15597. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, J.; Seebode, C.; Martens, M.C.; Emmert, S. Xeroderma Pigmentosum—Facts and Perspectives. Anticancer Res. 2018, 38, 1159–1164. [Google Scholar] [PubMed]
- Reid-Bayliss, K.S.; Arron, S.T.; Loeb, L.A.; Bezrookove, V.; Cleaver, J.E. Why Cockayne Syndrome Patients Do Not Get Cancer despite Their DNA Repair Deficiency. Proc. Natl. Acad. Sci. USA 2016, 113, 10151–10156. [Google Scholar] [CrossRef] [Green Version]
- Sugasawa, K. Mechanism and regulation of DNA damage recognition in mammalian nucleotide excision repair. In Enzymes; Academic Press: Cambridge, UK, 2019; Volume 45, pp. 99–138. ISBN 9780128173961. [Google Scholar]
- Van der Weegen, Y.; Golan-Berman, H.; Mevissen, T.E.T.; Apelt, K.; González-Prieto, R.; Goedhart, J.; Heilbrun, E.E.; Vertegaal, A.C.O.; van den Heuvel, D.; Walter, J.C.; et al. The Cooperative Action of CSB, CSA, and UVSSA Target TFIIH to DNA Damage-Stalled RNA Polymerase II. Nat. Commun. 2020, 11, 1–16. [Google Scholar]
- Oksenych, V.; de Jesus, B.B.; Zhovmer, A.; Egly, J.M.; Coin, F. Molecular Insights into the Recruitment of TFIIH to Sites of DNA Damage. EMBO J. 2009, 28, 2971–2980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogi, T.; Limsirichaikul, S.; Overmeer, R.M.; Volker, M.; Takenaka, K.; Cloney, R.; Nakazawa, Y.; Niimi, A.; Miki, Y.; Jaspers, N.G.; et al. Three DNA Polymerases, Recruited by Different Mechanisms, Carry Out NER Repair Synthesis in Human Cells. Mol. Cell 2010, 37, 714–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, J.; Kool, H.; Giakzidis, I.; Caldecott, K.; Mullenders, L.H.F.; Fousteri, M.I. Sealing of Chromosomal DNA Nicks during Nucleotide Excision Repair Requires XRCC1 and DNA Ligase IIIα in a Cell-Cycle-Specific Manner. Mol. Cell 2007, 27, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Geijer, M.E.; Zhou, D.; Selvam, K.; Steurer, B.; Mukherjee, C.; Evers, B.; Cugusi, S.; van Toorn, M.; van der Woude, M.; Janssens, R.C.; et al. Elongation Factor ELOF1 Drives Transcription-Coupled Repair and Prevents Genome Instability. Nat. Cell Biol. 2021, 23, 608–619. [Google Scholar] [CrossRef]
- Van der Weegen, Y.; de Lint, K.; van den Heuvel, D.; Nakazawa, Y.; Mevissen, T.E.T.; van Schie, J.J.M.; San Martin Alonso, M.; Boer, D.E.C.; González-Prieto, R.; Narayanan, I.V.; et al. ELOF1 Is a Transcription-Coupled DNA Repair Factor That Directs RNA Polymerase II Ubiquitylation. Nat. Cell Biol. 2021, 23, 595–607. [Google Scholar] [CrossRef]
- Burgers, P.M.J.; Kunkel, T.A. Eukaryotic DNA Replication Fork. Annu. Rev. Biochem. 2017, 86, 417–438. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D. Replication-Coupled DNA Repair. Mol. Cell 2019, 74, 866–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baretti, M.; Le, D.T. DNA Mismatch Repair in Cancer. Pharmacol. Ther. 2018, 189, 45–62. [Google Scholar] [CrossRef]
- Biller, L.H.; Syngal, S.; Yurgelun, M.B. Recent Advances in Lynch Syndrome. Fam. Cancer 2019, 18, 211–219. [Google Scholar] [CrossRef]
- Gao, A.; Guo, M. Epigenetic Based Synthetic Lethal Strategies in Human Cancers. Biomark. Res. 2020, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Heinen, C.D. The Mismatch Repair-Dependent DNA Damage Response: Mechanisms and Implications. DNA Repair 2019, 78, 60–69. [Google Scholar] [CrossRef]
- Anand, R.; Beach, A.; Li, K.; Haber, J. Rad51-Mediated Double-Strand Break Repair and Mismatch Correction of Divergent Substrates. Nature 2017, 544, 377–380. [Google Scholar] [CrossRef]
- Thomas, A.; Tanaka, M.; Trepel, J.; Reinhold, W.C.; Rajapakse, V.N.; Pommier, Y. Temozolomide in the Era of Precision Medicine. Cancer Res. 2017, 77, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Ciriano, I.; Lee, S.; Park, W.-Y.; Kim, T.-M.; Park, P.J. A Molecular Portrait of Microsatellite Instability across Multiple Cancers. Nat. Commun. 2017, 8, 15180. [Google Scholar] [CrossRef] [Green Version]
- Cortés-Ciriano, I.; Lee, J.J.K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S.; et al. Comprehensive Analysis of Chromothripsis in 2,658 Human Cancers Using Whole-Genome Sequencing. Nat. Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Palombo, F.; Iaccarino, I.; Nakajima, E.; Ikejima, M.; Shimada, T.; Jiricny, J. HMutSβ, a Heterodimer of HMSH2 and HMSH3, Binds to Insertion/Deletion Loops in DNA. Curr. Biol. 1996, 6, 1181–1184. [Google Scholar] [CrossRef] [Green Version]
- Li, G.-M. Mechanisms and Functions of DNA Mismatch Repair. Cell Res. 2007, 18, 85–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gannon, A.-M.M.; Frizzell, A.; Healy, E.; Lahue, R.S. MutSβ and Histone Deacetylase Complexes Promote Expansions of Trinucleotide Repeats in Human Cells. Nucleic Acids Res. 2012, 40, 10324–10333. [Google Scholar] [CrossRef]
- Williams, G.M.; Surtees, J.A. MSH3 Promotes Dynamic Behavior of Trinucleotide Repeat Tracts In Vivo. Genetics 2015, 200, 737–754. [Google Scholar] [CrossRef] [Green Version]
- Allen, D.J.; Makhov, A.; Grilley, M.; Taylor, J.; Thresher, R.; Modrich, P.; Griffith, J. MutS Mediates Heteroduplex Loop Formation by a Translocation Mechanism. EMBO J. 1997, 16, 4467–4476. [Google Scholar] [CrossRef] [Green Version]
- Habraken, Y.; Sung, P.; Prakash, L.; Prakash, S. Binding of Insertion/Deletion DNA Mismatches by the Heterodimer of Yeast Mismatch Repair Proteins MSH2 and MSH3. Curr. Biol. 1996, 6, 1185–1187. [Google Scholar] [CrossRef] [Green Version]
- Matsuno, Y.; Atsumi, Y.; Alauddin, M.D.; Rana, M.D.M.; Fujimori, H.; Hyodo, M.; Shimizu, A.; Ikuta, T.; Tani, H.; Torigoe, H.; et al. Resveratrol and Its Related Polyphenols Contribute to the Maintenance of Genome Stability. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- McVey, M.; Khodaverdian, V.Y.; Meyer, D.; Cerqueira, P.G.; Heyer, W.-D. Eukaryotic DNA Polymerases in Homologous Recombination. Annu. Rev. Genet. 2016, 50, 393–421. [Google Scholar] [CrossRef] [Green Version]
- Conde, C.D.; Petronczki, Ö.Y.; Baris, S.; Willmann, K.L.; Girardi, E.; Salzer, E.; Weitzer, S.; Ardy, R.C.; Krolo, A.; Ijspeert, H.; et al. Polymerase δ Deficiency Causes Syndromic Immunodeficiency with Replicative Stress. J. Clin. Investig. 2019, 129, 4194–4206. [Google Scholar] [CrossRef]
- Wong, R.P.; García-Rodríguez, N.; Zilio, N.; Hanulová, M.; Ulrich, H.D. Processing of DNA Polymerase-Blocking Lesions during Genome Replication Is Spatially and Temporally Segregated from Replication Forks. Mol. Cell 2020, 77, 3–16.e4. [Google Scholar] [CrossRef]
- Ma, X.; Tang, T.-S.; Guo, C. Regulation of Translesion DNA Synthesis in Mammalian Cells. Environ. Mol. Mutagenesis 2020, 61, 680–692. [Google Scholar] [CrossRef]
- Nayak, S.; Calvo, J.A.; Cong, K.; Peng, M.; Berthiaume, E.; Jackson, J.; Zaino, A.M.; Vindigni, A.; Hadden, M.K.; Cantor, S.B. Inhibition of the Translesion Synthesis Polymerase REV1 Exploits Replication Gaps as a Cancer Vulnerability. Sci. Adv. 2020, 6, 7808–7818. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela, A.; Leuzzi, G.; Sannino, V.; Cuella-Martin, R.; Huang, J.-W.; Wu-Baer, F.; Baer, R.; Costanzo, V.; Ciccia, A. REV1-Polζ Maintains the Viability of Homologous Recombination-Deficient Cancer Cells through Mutagenic Repair of PRIMPOL-Dependent SsDNA Gaps. Mol. Cell 2021, 81, 4008–4025.e7. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J. Oxyradicals and DNA Damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Sedgwick, B.; Bates, P.A.; Paik, J.; Jacobs, S.C.; Lindahl, T. Repair of Alkylated DNA: Recent Advances. DNA Repair 2007, 6, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA Damage: Mechanisms, Mutation, and Disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [Green Version]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing Repair and Tolerance of DNA Damage Caused by Alkylating Agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.H.; Kang, T.H. DNA Oxidation and Excision Repair Pathways. Int. J. Mol. Sci. 2019, 20, 6092. [Google Scholar] [CrossRef] [Green Version]
- Chow, E.; Thirlwell, C.; Macrae, F.; Lipton, L. Colorectal Cancer and Inherited Mutations in Base-Excision Repair. Lancet Oncol. 2004, 5, 600–606. [Google Scholar] [CrossRef]
- Das, L.; Quintana, V.G.; Sweasy, J.B. NTHL1 in Genomic Integrity, Aging and Cancer. DNA Repair 2020, 93, 102920. [Google Scholar] [CrossRef]
- Weren, R.D.A.; Ligtenberg, M.J.L.; Geurts van Kessel, A.; de Voer, R.M.; Hoogerbrugge, N.; Kuiper, R.P. NTHL1 and MUTYH Polyposis Syndromes: Two Sides of the Same Coin? J. Pathol. 2018, 244, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 2019, 75, 117–130.e6. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; de Lange, T. Telomeres in Cancer: Tumour Suppression and Genome Instability. Nat. Rev. Mol. Cell Biol. 2017, 18, 175–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal, A.; Tusell, L. Telomeres: Implications for Cancer Development. Int. J. Mol. Sci. 2018, 19, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannen, R.; Bartsch, J.W. Essential Roles of Telomerase Reverse Transcriptase HTERT in Cancer Stemness and Metastasis. FEBS Lett. 2018, 592, 2023–2031. [Google Scholar] [CrossRef]
- Campbell, P.J.; Getz, G.; Korbel, J.O.; Stuart, J.M.; Jennings, J.L.; Stein, L.D.; Perry, M.D.; Nahal-Bose, H.K.; Ouellette, B.F.F.; Li, C.H.C.; et al. Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93. [Google Scholar]
- Diplas, B.H.; He, X.; Brosnan-Cashman, J.A.; Liu, H.; Chen, L.H.; Wang, Z.; Moure, C.J.; Killela, P.J.; Loriaux, D.B.; Lipp, E.S.; et al. The Genomic Landscape of TERT Promoter Wildtype-IDH Wildtype Glioblastoma. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Williams, E.A.; Miller, J.J.; Tummala, S.S.; Penson, T.; Iafrate, A.J.; Juratli, T.A.; Cahill, D.P. TERT Promoter Wild-Type Glioblastomas Show Distinct Clinical Features and Frequent PI3K Pathway Mutations. Acta Neuropathol. Commun. 2018, 6, 106. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Liu, J.; Zhang, Z.; Zhang, H.; Liu, H.; Gao, S.; Rong, Y.S.; Zhao, Y. Homologous Recombination-Dependent Repair of Telomeric DSBs in Proliferating Human Cells. Nat. Commun. 2016, 7, 12154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keefe, D.L. Telomeres and Genomic Instability during Early Development. Eur. J. Med. Genet. 2020, 63, 103638. [Google Scholar] [CrossRef] [PubMed]
- Noureen, N.; Wu, S.; Lv, Y.; Yang, J.; Alfred Yung, W.K.; Gelfond, J.; Wang, X.; Koul, D.; Ludlow, A.; Zheng, S. Integrated Analysis of Telomerase Enzymatic Activity Unravels an Association with Cancer Stemness and Proliferation. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Stephens, P.J.; McBride, D.J.; Lin, M.-L.; Varela, I.; Pleasance, E.D.; Simpson, J.T.; Stebbings, L.A.; Leroy, C.; Edkins, S.; Mudie, L.J.; et al. Complex Landscapes of Somatic Rearrangement in Human Breast Cancer Genomes. Nature 2009, 462, 1005–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.-L.; Ordóñez, G.R.; Bignell, G.R.; et al. A Comprehensive Catalogue of Somatic Mutations from a Human Cancer Genome. Nature 2010, 463, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem Cell Divisions, Somatic Mutations, Cancer Etiology, and Cancer Prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The Repertoire of Mutational Signatures in Human Cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Gorgoulis, V.G.; Vassiliou, L.-V.V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.T.; Venere, M.; DiTullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA Damage Checkpoint and Genomic Instability in Human Precancerous Lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA Damage Response as a Candidate Anti-Cancer Barrier in Early Human Tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate Immune Sensing of Cytosolic Chromatin Fragments through CGAS Promotes Senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Peto, J. Cancer Epidemiology in the Last Century and the next Decade. Nature 2001, 411, 390–395. [Google Scholar] [CrossRef]
- Ahmad, A.S.; Ormiston-Smith, N.; Sasieni, P.D. Trends in the Lifetime Risk of Developing Cancer in Great Britain: Comparison of Risk for Those Born from 1930 to 1960. Br. J. Cancer 2015, 112, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Amor-Guéret, M. Bloom Syndrome, Genomic Instability and Cancer: The SOS-like Hypothesis. Cancer Lett. 2006, 236, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kaur, E.; Agrawal, R.; Sengupta, S. Functions of BLM Helicase in Cells: Is It Acting Like a Double-Edged Sword? Front. Genet. 2021, 12, 277. [Google Scholar] [CrossRef]
- Atsumi, Y.; Fujimori, H.; Fukuda, H.; Inase, A.; Shinohe, K.; Yoshioka, Y.; Shikanai, M.; Ichijima, Y.; Unno, J.; Mizutani, S.; et al. Onset of Quiescence Following P53 Mediated Down-Regulation of H2AX in Normal Cells. PLoS ONE 2011, 6, e23432. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, Y.; Minakawa, Y.; Ono, M.; Dobashi, S.; Shinohe, K.; Shinohara, A.; Takeda, S.; Takagi, M.; Takamatsu, N.; Nakagama, H.; et al. ATM and SIRT6/SNF2H Mediate Transient H2AX Stabilization When DSBs Form by Blocking HUWE1 to Allow Efficient ΓH2AX Foci Formation. Cell Rep. 2015, 13, 2728–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minakawa, Y.; Atsumi, Y.; Shinohara, A.; Murakami, Y.; Yoshioka, K. Gamma-Irradiated Quiescent Cells Repair Directly Induced Double-Strand Breaks but Accumulate Persistent Double-Strand Breaks during Subsequent DNA Replication. Genes Cells 2016, 21, 789–797. [Google Scholar] [CrossRef] [Green Version]
- Lukačišinová, M.; Novak, S.; Paixão, T. Stress-Induced Mutagenesis: Stress Diversity Facilitates the Persistence of Mutator Genes. PLoS Comput. Biol. 2017, 13, e1005609. [Google Scholar] [CrossRef]
- Fitzgerald, D.M.; Hastings, P.J.; Rosenberg, S.M. Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance. Annu. Rev. Cancer Biol. 2017, 1, 119–140. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Stevens, M.F.G.; Bradshaw, T.D. Temozolomide: Mechanisms of Action, Repair and Resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Yoshioka, K.; Yoshioka, Y.; Hsieh, P. ATR Kinase Activation Mediated by MutSα and MutLα in Response to Cytotoxic O6-Methylguanine Adducts. Mol. Cell 2006, 22, 501–510. [Google Scholar] [CrossRef]
- Van Nifterik, K.A.; van den Berg, J.; Stalpers, L.J.A.; Lafleur, M.V.M.; Leenstra, S.; Slotman, B.J.; Hulsebos, T.J.M.; Sminia, P. Differential Radiosensitizing Potential of Temozolomide in MGMT Promoter Methylated Glioblastoma Multiforme Cell Lines. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Ichijima, Y.; Yoshioka, K.; Yoshioka, Y.; Shinohe, K.; Fujimori, H.; Unno, J.; Takagi, M.; Goto, H.; Inagaki, M.; Mizutani, S.; et al. DNA Lesions Induced by Replication Stress Trigger Mitotic Aberration and Tetraploidy Development. PLoS ONE 2010, 5, e8821. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoshioka, K.-i.; Kusumoto-Matsuo, R.; Matsuno, Y.; Ishiai, M. Genomic Instability and Cancer Risk Associated with Erroneous DNA Repair. Int. J. Mol. Sci. 2021, 22, 12254. https://doi.org/10.3390/ijms222212254
Yoshioka K-i, Kusumoto-Matsuo R, Matsuno Y, Ishiai M. Genomic Instability and Cancer Risk Associated with Erroneous DNA Repair. International Journal of Molecular Sciences. 2021; 22(22):12254. https://doi.org/10.3390/ijms222212254
Chicago/Turabian StyleYoshioka, Ken-ichi, Rika Kusumoto-Matsuo, Yusuke Matsuno, and Masamichi Ishiai. 2021. "Genomic Instability and Cancer Risk Associated with Erroneous DNA Repair" International Journal of Molecular Sciences 22, no. 22: 12254. https://doi.org/10.3390/ijms222212254
APA StyleYoshioka, K.-i., Kusumoto-Matsuo, R., Matsuno, Y., & Ishiai, M. (2021). Genomic Instability and Cancer Risk Associated with Erroneous DNA Repair. International Journal of Molecular Sciences, 22(22), 12254. https://doi.org/10.3390/ijms222212254