
Nearly 30 Years of Animal Models to Study Amyotrophic Lateral Sclerosis: A Historical Overview and Future Perspectives

 ,
,  ,
,  ,
,  and
and 
{kind=link}
Abstract
1. Introduction
2. Genes Involved in ALS
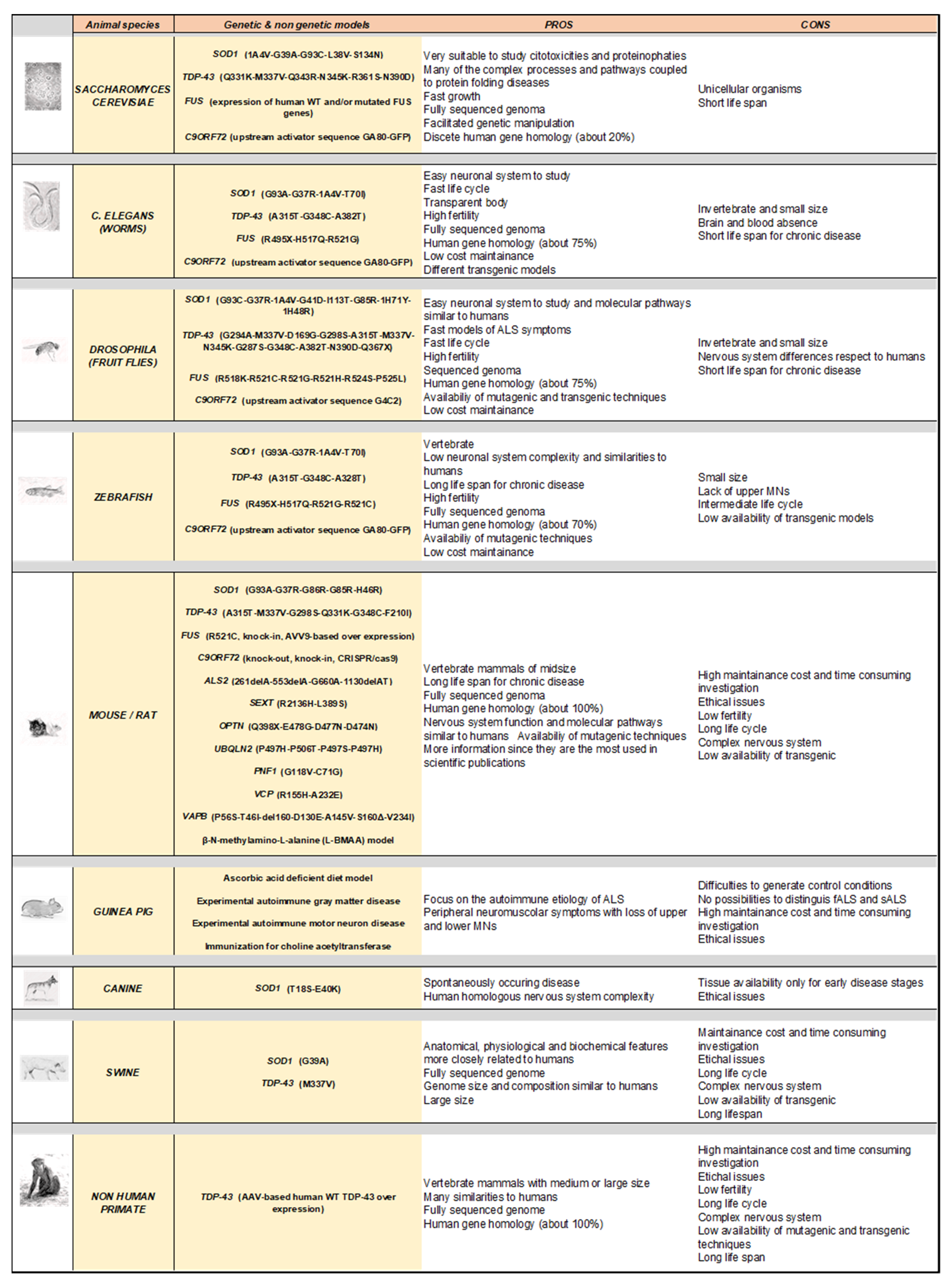
3. Modelling Human ALS in Living Organisms
4. Rodent Models
4.1. Rodents Carrying Cu/Zn Superoxide Dismutase 1 (SOD1) Mutations
4.2. Rodents Carrying Tar DNA-Binding Protein 43 (TDP-43) Mutation
4.3. Rodents Carrying the RNA-Binding Protein Fused in Sarcoma (FUS) Mutations
4.4. Rodents Carrying Chromosome 9 Open Reading Frame 72 (C9orf72) Mutations
5. Rodent Models Carrying Rare Mutations Linked to ALS
5.1. Rodents Carrying Alsin Mutations
5.2. Rodents Carrying Senataxin Mutations
5.3. Rodents Carrying Optineurin Mutations
5.4. Rodents Carrying Ubiquilin-2 Mutations
5.5. Rodents Carrying Profilin 1 Mutations
5.6. Rodents Carrying Valosin Containing Protein Mutations
5.7. Rodents Carrying Vesicle-Associated Membrane Protein (VAMP)-Associated Protein B Mutations
6. Non-Genetic Rodent Models
6.1. Guinea Pig ALS Models
6.2. L-BMAA-Induced ALS Rodent Models
7. Drosophila Melanogaster (Fruit Fly) Models
7.1. Drosophila Melanogaster Carrying SOD1 Mutations
7.2. Drosophila Melanogaster Carrying TDP-43 Mutations
7.3. Drosophila Melanogaster Carrying FUS Mutations
7.4. Drosophila Melanogaster Carrying C9orf72 Mutations
8. Danio Rerio (Zebrafish) Models
8.1. Zebrafish Carrying Cu/Zn SOD1 Mutations
8.2. Zebrafish Carrying TDP-43 Mutations
8.3. Zebrafish Carrying FUS Mutations
8.4. Zebrafish Carrying C9orf72 Mutations
9. Caenorhabditis Elegans Models
9.1. C. elegans Carrying SOD1 Mutations
9.2. C. elegans Carrying TDP-43 Mutations
9.3. C. elegans Carrying FUS Mutations
9.4. C. elegans Carrying Deletion or Overexpression of C9orf72
10. Saccharomyces Cerevisiae Models
10.1. Saccharomyces Cerevisiae Carrying SOD1 Mutations
10.2. Saccharomyces Cerevisiae Carrying TDP-43 Mutations
10.3. Saccharomyces Cerevisiae Carrying FUS Mutations
11. Other Animal Models of ALS
11.1. Canine Models
11.2. Swine Models
11.3. Non-Human Primate Models
12. ALS-Related Protein Mutations for Novel or Potential Animal Models
12.1. MATR3 Mutations
12.2. CHCHD10 Mutations
12.3. TBK1 Mutations
12.4. CCNF Mutations
12.5. Other ALS-Related Mutations
13. Translational Burdens and Usefulness of In Vivo ALS Models
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, R.H.J.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 1602. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17085. [Google Scholar] [CrossRef] [PubMed]
- Štětkářová, I.; Ehler, E. Diagnostics of Amyotrophic Lateral Sclerosis: Up to Date. Diagnostics 2021, 11, 231. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef]
- Taylor, J.P.; Brown, R.H.J.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef]
- Dukkipati, S.S.; Garrett, T.L.; Elbasiouny, S.M. The vulnerability of spinal motoneurons and soma size plasticity in a mouse model of amyotrophic lateral sclerosis. J. Physiol. 2018, 596, 1723–1745. [Google Scholar] [CrossRef]
- Milanese, M.; Zappettini, S.; Onofri, F.; Musazzi, L.; Tardito, D.; Bonifacino, T.; Messa, M.; Racagni, G.; Usai, C.; Benfenati, F.; et al. Abnormal exocytotic release of glutamate in a mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2011, 116, 1028–1042. [Google Scholar] [CrossRef]
- Pieri, M.; Caioli, S.; Canu, N.; Mercuri, N.B.; Guatteo, E.; Zona, C. Over-expression of N-type calcium channels in cortical neurons from a mouse model of Amyotrophic Lateral Sclerosis. Exp. Neurol. 2013, 247, 349–358. [Google Scholar] [CrossRef]
- Bonifacino, T.; Musazzi, L.; Milanese, M.; Seguini, M.; Marte, A.; Gallia, E.; Cattaneo, L.; Onofri, F.; Popoli, M.; Bonanno, G. Altered mechanisms underlying the abnormal glutamate release in amyotrophic lateral sclerosis at a pre-symptomatic stage of the disease. Neurobiol. Dis. 2016, 95, 122–133. [Google Scholar] [CrossRef]
- Sugiyama, K.; Tanaka, K. Spinal cord-specific deletion of the glutamate transporter GLT1 causes motor neuron death in mice. Biochem. Biophys. Res. Commun. 2018, 497, 689–693. [Google Scholar] [CrossRef]
- Tedeschi, V.; Petrozziello, T.; Secondo, A. Calcium Dyshomeostasis and Lysosomal Ca(2+) Dysfunction in Amyotrophic Lateral Sclerosis. Cells 2019, 8, 1216. [Google Scholar] [CrossRef]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Milanese, M.; Bonifacino, T.; Fedele, E.; Rebosio, C.; Cattaneo, L.; Benfenati, F.; Usai, C.; Bonanno, G. Exocytosis regulates trafficking of GABA and glycine heterotransporters in spinal cord glutamatergic synapses: A mechanism for the excessive heterotransporter-induced release of glutamate in experimental amyotrophic lateral sclerosis. Neurobiol. Dis. 2015, 74, 314–324. [Google Scholar] [CrossRef]
- Stifanese, R.; Averna, M.; De Tullio, R.; Pedrazzi, M.; Milanese, M.; Bonifacino, T.; Bonanno, G.; Salamino, F.; Pontremoli, S.; Melloni, E. Role of calpain-1 in the early phase of experimental ALS. Arch. Biochem. Biophys. 2014, 562, 1–8. [Google Scholar] [CrossRef]
- Hadzhieva, M.; Kirches, E.; Wilisch-Neumann, A.; Pachow, D.; Wallesch, M.; Schoenfeld, P.; Paege, I.; Vielhaber, S.; Petri, S.; Keilhoff, G.; et al. Dysregulation of iron protein expression in the G93A model of amyotrophic lateral sclerosis. Neuroscience 2013, 230, 94–101. [Google Scholar] [CrossRef]
- Burlando, B.; Milanese, M.; Giordano, G.; Bonifacino, T.; Ravera, S.; Blanchini, F.; Bonanno, G. A multistationary loop model of ALS unveils critical molecular interactions involving mitochondria and glucose metabolism. PLoS ONE 2020, 15, e0244234. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef]
- Morfini, G.A.; Bosco, D.A.; Brown, H.; Gatto, R.; Kaminska, A.; Song, Y.; Molla, L.; Baker, L.; Marangoni, M.N.; Berth, S.; et al. Inhibition of fast axonal transport by pathogenic SOD1 involves activation of p38 MAP kinase. PLoS ONE 2013, 8, e65235. [Google Scholar] [CrossRef]
- Gibbs, K.L.; Kalmar, B.; Rhymes, E.R.; Fellows, A.D.; Ahmed, M.; Whiting, P.; Davies, C.H.; Greensmith, L.; Schiavo, G. Inhibiting p38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS. Cell Death Dis. 2018, 9, 596. [Google Scholar] [CrossRef]
- Burk, K.; Pasterkamp, R.J. Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol. 2019, 137, 859–877. [Google Scholar] [CrossRef]
- Suzuki, N.; Akiyama, T.; Warita, H.; Aoki, M. Omics Approach to Axonal Dysfunction of Motor Neurons in Amyotrophic Lateral Sclerosis (ALS). Front. Neurosci. 2020, 14, 194. [Google Scholar] [CrossRef]
- Brites, D.; Vaz, A.R. Microglia centered pathogenesis in ALS: Insights in cell interconnectivity. Front. Cell. Neurosci. 2014, 8, 117. [Google Scholar] [CrossRef]
- Chiarotto, G.B.; Cartarozzi, L.P.; Perez, M.; Biscola, N.P.; Spejo, A.B.; Gubert, F.; França Junior, M.; Mendez-Otero, R.; de Oliveira, A.L.R. Tempol improves neuroinflammation and delays motor dysfunction in a mouse model (SOD1(G93A)) of ALS. J. Neuroinflamm. 2019, 16, 218. [Google Scholar] [CrossRef]
- Zetterström, P.; Stewart, H.G.; Bergemalm, D.; Jonsson, P.A.; Graffmo, K.S.; Andersen, P.M.; Brännström, T.; Oliveberg, M.; Marklund, S.L. Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proc. Natl. Acad. Sci. USA 2007, 104, 14157–14162. [Google Scholar] [CrossRef]
- Ramesh, N.; Pandey, U.B. Autophagy Dysregulation in ALS: When Protein Aggregates Get Out of Hand. Front. Mol. Neurosci. 2017, 10, 263. [Google Scholar] [CrossRef]
- Bendotti, C.; Marino, M.; Cheroni, C.; Fontana, E.; Crippa, V.; Poletti, A.; De Biasi, S. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: Implication for protein aggregation and immune response. Prog. Neurobiol. 2012, 97, 101–126. [Google Scholar] [CrossRef]
- Yerbury, J.J.; Farrawell, N.E.; McAlary, L. Proteome Homeostasis Dysfunction: A Unifying Principle in ALS Pathogenesis. Trends Neurosci. 2020, 43, 274–284. [Google Scholar] [CrossRef]
- Atkin, J.D.; Farg, M.A.; Walker, A.K.; McLean, C.; Tomas, D.; Horne, M.K. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol. Dis. 2008, 30, 400–407. [Google Scholar] [CrossRef]
- Jaronen, M.; Goldsteins, G.; Koistinaho, J. ER stress and unfolded protein response in amyotrophic lateral sclerosis-a controversial role of protein disulphide isomerase. Front. Cell. Neurosci. 2014, 8, 402. [Google Scholar] [CrossRef]
- Vandoorne, T.; De Bock, K.; Van Den Bosch, L. Energy metabolism in ALS: An underappreciated opportunity? Acta Neuropathol. 2018, 135, 489–509. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador-Palmer, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. The Link between Oxidative Stress, Redox Status, Bioenergetics and Mitochondria in the Pathophysiology of ALS. Int. J. Mol. Sci. 2021, 22, 6352. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Torazza, C.; Bonifacino, T.; Provenzano, F.; Rebosio, C.; Milanese, M.; Usai, C.; Panfoli, I.; Bonanno, G. Altered glucose catabolism in the presynaptic and perisynaptic compartments of SOD1(G93A) mouse spinal cord and motor cortex indicates that mitochondria are the site of bioenergetic imbalance in ALS. J. Neurochem. 2019, 151, 336–350. [Google Scholar] [CrossRef] [PubMed]
- Volkening, K.; Leystra-Lantz, C.; Yang, W.; Jaffee, H.; Strong, M.J. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res. 2009, 1305, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Droppelmann, C.A.; Campos-Melo, D.; Ishtiaq, M.; Volkening, K.; Strong, M.J. RNA metabolism in ALS: When normal processes become pathological. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 321–336. [Google Scholar] [CrossRef]
- Gentile, F.; Scarlino, S.; Falzone, Y.M.; Lunetta, C.; Tremolizzo, L.; Quattrini, A.; Riva, N. The Peripheral Nervous System in Amyotrophic Lateral Sclerosis: Opportunities for Translational Research. Front. Neurosci. 2019, 13, 601. [Google Scholar] [CrossRef]
- Filipi, T.; Hermanova, Z.; Tureckova, J.; Vanatko, O.; Anderova, A.M. Glial Cells-The Strategic Targets in Amyotrophic Lateral Sclerosis Treatment. J. Clin. Med. 2020, 9, 261. [Google Scholar] [CrossRef]
- Crabé, R.; Aimond, F.; Gosset, P.; Scamps, F.; Raoul, C. How Degeneration of Cells Surrounding Motoneurons Contributes to Amyotrophic Lateral Sclerosis. Cells 2020, 9, 2550. [Google Scholar] [CrossRef]
- Abramzon, Y.A.; Fratta, P.; Traynor, B.J.; Chia, R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front. Neurosci. 2020, 14, 42. [Google Scholar] [CrossRef]
- Boylan, K. Familial Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 807–830. [Google Scholar] [CrossRef]
- Mejzini, R.; Flynn, L.L.; Pitout, I.L.; Fletcher, S.; Wilton, S.D.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef]
- Shatunov, A.; Al-Chalabi, A. The genetic architecture of ALS. Neurobiol. Dis. 2021, 147, 105156. [Google Scholar] [CrossRef]
- Zou, Z.-Y.; Zhou, Z.-R.; Che, C.-H.; Liu, C.-Y.; He, R.-L.; Huang, H.-P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef]
- Shepheard, S.R.; Parker, M.D.; Cooper-Knock, J.; Verber, N.S.; Tuddenham, L.; Heath, P.; Beauchamp, N.; Place, E.; Sollars, E.S.A.; Turner, M.R.; et al. Value of systematic genetic screening of patients with amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2021, 92, 510–518. [Google Scholar] [CrossRef]
- Schram, S.; Loeb, J.A.; Song, F. Disease propagation in amyotrophic lateral sclerosis (ALS): An interplay between genetics and environment. J. Neuroinflamm. 2020, 17, 175. [Google Scholar] [CrossRef]
- Grad, L.I.; Cashman, N.R. Prion-like activity of Cu/Zn superoxide dismutase: Implications for amyotrophic lateral sclerosis. Prion 2014, 8, 33–41. [Google Scholar] [CrossRef]
- Chen, Q.; Zhou, J.; Huang, C.; Huang, B.; Bi, F.; Zhou, H.; Xiao, B. Temporal Expression of Mutant TDP-43 Correlates with Early Amyotrophic Lateral Sclerosis Phenotype and Motor Weakness. Curr. Neurovasc. Res. 2018, 15, 3–9. [Google Scholar] [CrossRef]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef]
- Ederle, H.; Dormann, D. TDP-43 and FUS en route from the nucleus to the cytoplasm. FEBS Lett. 2017, 591, 1489–1507. [Google Scholar] [CrossRef]
- Ciryam, P.; Lambert-Smith, I.A.; Bean, D.M.; Freer, R.; Cid, F.; Tartaglia, G.G.; Saunders, D.N.; Wilson, M.R.; Oliver, S.G.; Morimoto, R.I.; et al. Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E3935–E3943. [Google Scholar] [CrossRef]
- Wood, A.; Gurfinkel, Y.; Polain, N.; Lamont, W.; Lyn Rea, S. Molecular Mechanisms Underlying TDP-43 Pathology in Cellular and Animal Models of ALS and FTLD. Int. J. Mol. Sci. 2021, 22, 4705. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, C.; Kirby, J.; Highley, J.R.; Hartley, J.A.; Hibberd, R.; Hollinger, H.C.; Williams, T.L.; Ince, P.G.; McDermott, C.J.; Shaw, P.J. Novel FUS/TLS mutations and pathology in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2010, 67, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef]
- McGoldrick, P.; Zhang, M.; van Blitterswijk, M.; Sato, C.; Moreno, D.; Xiao, S.; Zhang, A.B.; McKeever, P.M.; Weichert, A.; Schneider, R.; et al. Unaffected mosaic C9orf72 case: RNA foci, dipeptide proteins, but upregulated C9orf72 expression. Neurology 2018, 90, e323–e331. [Google Scholar] [CrossRef]
- Pang, W.; Hu, F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J. Neurochem. 2021, 157, 334–350. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, T.; Lu, K.; Qi, S. The progress in C9orf72 research: ALS/FTD pathogenesis, functions and structure. Small GTPases 2021, 1–21. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; van Es, M.A.; Koppers, M.; van Rheenen, W.; Medic, J.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; Veldink, J.H.; van den Berg, L.H. VAPB and C9orf72 mutations in 1 familial amyotrophic lateral sclerosis patient. Neurobiol. Aging 2012, 33, e1–e4. [Google Scholar] [CrossRef]
- Gitler, A.D.; Tsuiji, H. There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain Res. 2016, 1647, 19–29. [Google Scholar] [CrossRef]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Beal, M.F.; Becher, M.W.; Schulz, J.B.; Wong, P.C.; Price, D.L.; Cleveland, D.W. Elevated free nitrotyrosine levels, but not protein-bound nitrotyrosine or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide d. Proc. Natl. Acad. Sci. USA 1997, 94, 7606–7611. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Ezzi, S.A.; Urushitani, M.; Julien, J.-P. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 2007, 102, 170–178. [Google Scholar] [CrossRef]
- Chen, K.; Bennett, S.A.; Rana, N.; Yousuf, H.; Said, M.; Taaseen, S.; Mendo, N.; Meltser, S.M.; Torrente, M.P. Neurodegenerative Disease Proteinopathies Are Connected to Distinct Histone Post-translational Modification Landscapes. ACS Chem. Neurosci. 2018, 9, 938–948. [Google Scholar] [CrossRef]
- Jhanji, R.; Behl, T.; Sehgal, A.; Bungau, S. Mitochondrial dysfunction and traffic jams in amyotrophic lateral sclerosis. Mitochondrion 2021, 58, 102–110. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Tsai, P.-Y.; Chern, Y. Energy Homeostasis and Abnormal RNA Metabolism in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci. 2017, 11, 126. [Google Scholar] [CrossRef]
- Piancone, F.; La Rosa, F.; Marventano, I.; Saresella, M.; Clerici, M. The Role of the Inflammasome in Neurodegenerative Diseases. Molecules 2021, 26, 953. [Google Scholar] [CrossRef]
- Leblond, C.S.; Kaneb, H.M.; Dion, P.A.; Rouleau, G.A. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp. Neurol. 2014, 262, 91–101. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet. Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Spencer, P.S.; Lagrange, E.; Camu, W. ALS and environment: Clues from spatial clustering? Rev. Neurol. 2019, 175, 652–663. [Google Scholar] [CrossRef]
- Babin, P.J.; Goizet, C.; Raldúa, D. Zebrafish models of human motor neuron diseases: Advantages and limitations. Prog. Neurobiol. 2014, 118, 36–58. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Rodent Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 5.67.1–5.67.21. [Google Scholar] [CrossRef]
- Lutz, C. Mouse models of ALS: Past, present and future. Brain Res. 2018, 1693, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liguori, F.; Amadio, S.; Volonté, C. Where and Why Modeling Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2021, 22, 3977. [Google Scholar] [CrossRef] [PubMed]
- Al-Chalabi, A.; van den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2017, 13, 96–104. [Google Scholar] [CrossRef]
- Venkova-Hristova, K.; Christov, A.; Kamaluddin, Z.; Kobalka, P.; Hensley, K. Progress in therapy development for amyotrophic lateral sclerosis. Neurol. Res. Int. 2012, 2012, 187234. [Google Scholar] [PubMed]
- Zou, Y.-H.; Guan, P.-P.; Zhang, S.-Q.; Guo, Y.-S.; Wang, P. Rofecoxib Attenuates the Pathogenesis of Amyotrophic Lateral Sclerosis by Alleviating Cyclooxygenase-2-Mediated Mechanisms. Front. Neurosci. 2020, 14, 817. [Google Scholar] [CrossRef]
- Medina, D.X.; Chung, E.P.; Teague, C.D.; Bowser, R.; Sirianni, R.W. Intravenously Administered, Retinoid Activating Nanoparticles Increase Lifespan and Reduce Neurodegeneration in the SOD1(G93A) Mouse Model of ALS. Front. Bioeng. Biotechnol. 2020, 8, 224. [Google Scholar] [CrossRef]
- Traynor, B.J.; Bruijn, L.; Conwit, R.; Beal, F.; O’Neill, G.; Fagan, S.C.; Cudkowicz, M.E. Neuroprotective agents for clinical trials in ALS: A systematic assessment. Neurology 2006, 67, 20–27. [Google Scholar] [CrossRef]
- Ito, H.; Wate, R.; Zhang, J.; Ohnishi, S.; Kaneko, S.; Ito, H.; Nakano, S.; Kusaka, H. Treatment with edaravone, initiated at symptom onset, slows motor decline and decreases SOD1 deposition in ALS mice. Exp. Neurol. 2008, 213, 448–455. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- McCombe, P.A.; Henderson, R.D. Effects of gender in amyotrophic lateral sclerosis. Gend. Med. 2010, 7, 557–570. [Google Scholar] [CrossRef]
- Bame, M.; Pentiak, P.A.; Needleman, R.; Brusilow, W.S.A. Effect of sex on lifespan, disease progression, and the response to methionine sulfoximine in the SOD1 G93A mouse model for ALS. Gend. Med. 2012, 9, 524–535. [Google Scholar] [CrossRef]
- Heiman-Patterson, T.D.; Sher, R.B.; Blankenhorn, E.A.; Alexander, G.; Deitch, J.S.; Kunst, C.B.; Maragakis, N.; Cox, G. Effect of genetic background on phenotype variability in transgenic mouse models of amyotrophic lateral sclerosis: A window of opportunity in the search for genetic modifiers. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Gr. Mot. Neuron Dis. 2011, 12, 79–86. [Google Scholar] [CrossRef]
- Marino, M.; Papa, S.; Crippa, V.; Nardo, G.; Peviani, M.; Cheroni, C.; Trolese, M.C.; Lauranzano, E.; Bonetto, V.; Poletti, A.; et al. Differences in protein quality control correlate with phenotype variability in 2 mouse models of familial amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 36, 492–504. [Google Scholar] [CrossRef]
- Joyce, P.I.; Fratta, P.; Fisher, E.M.C.; Acevedo-Arozena, A. SOD1 and TDP-43 animal models of amyotrophic lateral sclerosis: Recent advances in understanding disease toward the development of clinical treatments. Mamm. Genome 2011, 22, 420–448. [Google Scholar] [CrossRef]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar] [CrossRef]
- Ripps, M.E.; Huntley, G.W.; Hof, P.R.; Morrison, J.H.; Gordon, J.W. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 1995, 92, 689–693. [Google Scholar] [CrossRef]
- Howland, D.S.; Liu, J.; She, Y.; Goad, B.; Maragakis, N.J.; Kim, B.; Erickson, J.; Kulik, J.; DeVito, L.; Psaltis, G.; et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc. Natl. Acad. Sci. USA 2002, 99, 1604–1609. [Google Scholar] [CrossRef]
- Sanagi, T.; Yuasa, S.; Nakamura, Y.; Suzuki, E.; Aoki, M.; Warita, H.; Itoyama, Y.; Uchino, S.; Kohsaka, S.; Ohsawa, K. Appearance of phagocytic microglia adjacent to motoneurons in spinal cord tissue from a presymptomatic transgenic rat model of amyotrophic lateral sclerosis. J. Neurosci. Res. 2010, 88, 2736–2746. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Aoki, M.; Miyoshi, I.; Kato, M.; Pasinelli, P.; Kasai, N.; Brown, R.H.J.; Itoyama, Y. Rats expressing human cytosolic copper-zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: Associated mutations develop motor neuron disease. J. Neurosci. 2001, 21, 9246–9254. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- De Giorgio, F.; Maduro, C.; Fisher, E.M.C.; Acevedo-Arozena, A. Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Dis. Model. Mech. 2019, 12, dmm037424. [Google Scholar] [CrossRef]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef]
- Xu, Y.-F.; Gendron, T.F.; Zhang, Y.-J.; Lin, W.-L.; D’Alton, S.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef]
- Xu, Y.-F.; Zhang, Y.-J.; Lin, W.-L.; Cao, X.; Stetler, C.; Dickson, D.W.; Lewis, J.; Petrucelli, L. Expression of mutant TDP-43 induces neuronal dysfunction in transgenic mice. Mol. Neurodegener. 2011, 6, 73. [Google Scholar] [CrossRef]
- Stallings, N.R.; Puttaparthi, K.; Luther, C.M.; Burns, D.K.; Elliott, J.L. Progressive motor weakness in transgenic mice expressing human TDP-43. Neurobiol. Dis. 2010, 40, 404–414. [Google Scholar] [CrossRef]
- Wils, H.; Kleinberger, G.; Janssens, J.; Pereson, S.; Joris, G.; Cuijt, I.; Smits, V.; Ceuterick-de Groote, C.; Van Broeckhoven, C.; Kumar-Singh, S. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 3858–3863. [Google Scholar] [CrossRef]
- Shan, X.; Chiang, P.-M.; Price, D.L.; Wong, P.C. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci. USA 2010, 107, 16325–16330. [Google Scholar] [CrossRef]
- Arnold, E.S.; Ling, S.-C.; Huelga, S.C.; Lagier-Tourenne, C.; Polymenidou, M.; Ditsworth, D.; Kordasiewicz, H.B.; McAlonis-Downes, M.; Platoshyn, O.; Parone, P.A.; et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. USA 2013, 110, E736–E745. [Google Scholar] [CrossRef]
- Stribl, C.; Samara, A.; Trümbach, D.; Peis, R.; Neumann, M.; Fuchs, H.; Gailus-Durner, V.; Hrabě de Angelis, M.; Rathkolb, B.; Wolf, E.; et al. Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J. Biol. Chem. 2014, 289, 10769–10784. [Google Scholar] [CrossRef]
- Mitchell, J.C.; Constable, R.; So, E.; Vance, C.; Scotter, E.; Glover, L.; Hortobagyi, T.; Arnold, E.S.; Ling, S.-C.; McAlonis, M.; et al. Wild type human TDP-43 potentiates ALS-linked mutant TDP-43 driven progressive motor and cortical neuron degeneration with pathological features of ALS. Acta Neuropathol. Commun. 2015, 3, 36. [Google Scholar] [CrossRef]
- Morrice, J.R.; Gregory-Evans, C.Y.; Shaw, C.A. Animal models of amyotrophic lateral sclerosis: A comparison of model validity. Neural Regen. Res. 2018, 13, 2050–2054. [Google Scholar]
- Swarup, V.; Phaneuf, D.; Bareil, C.; Robertson, J.; Rouleau, G.A.; Kriz, J.; Julien, J.-P. Pathological hallmarks of amyotrophic lateral sclerosis/frontotemporal lobar degeneration in transgenic mice produced with TDP-43 genomic fragments. Brain 2011, 134, 2610–2626. [Google Scholar] [CrossRef]
- Igaz, L.M.; Kwong, L.K.; Lee, E.B.; Chen-Plotkin, A.; Swanson, E.; Unger, T.; Malunda, J.; Xu, Y.; Winton, M.J.; Trojanowski, J.Q.; et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J. Clin. Investig. 2011, 121, 726–738. [Google Scholar] [CrossRef]
- Walker, A.K.; Spiller, K.J.; Ge, G.; Zheng, A.; Xu, Y.; Zhou, M.; Tripathy, K.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M.-Y. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 2015, 130, 643–660. [Google Scholar] [CrossRef]
- Fratta, P.; Sivakumar, P.; Humphrey, J.; Lo, K.; Ricketts, T.; Oliveira, H.; Brito-Armas, J.M.; Kalmar, B.; Ule, A.; Yu, Y.; et al. Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. EMBO J. 2018, 37, 37. [Google Scholar] [CrossRef]
- Bendotti, C.; Bonetto, V.; Pupillo, E.; Logroscino, G.; Al-Chalabi, A.; Lunetta, C.; Riva, N.; Mora, G.; Lauria, G.; Weishaupt, J.H.; et al. Focus on the heterogeneity of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 485–495. [Google Scholar] [CrossRef]
- Zhou, H.; Huang, C.; Chen, H.; Wang, D.; Landel, C.P.; Xia, P.Y.; Bowser, R.; Liu, Y.-J.; Xia, X.G. Transgenic rat model of neurodegeneration caused by mutation in the TDP gene. PLoS Genet. 2010, 6, e1000887. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Cleveland, D.W. Rethinking ALS: The FUS about TDP-43. Cell 2009, 136, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Nolan, M.; Talbot, K.; Ansorge, O. Pathogenesis of FUS-associated ALS and FTD: Insights from rodent models. Acta Neuropathol. Commun. 2016, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Kino, Y.; Washizu, C.; Kurosawa, M.; Yamada, M.; Miyazaki, H.; Akagi, T.; Hashikawa, T.; Doi, H.; Takumi, T.; Hicks, G.G.; et al. FUS/TLS deficiency causes behavioral and pathological abnormalities distinct from amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2015, 3, 24. [Google Scholar] [CrossRef]
- Sharma, A.; Lyashchenko, A.K.; Lu, L.; Nasrabady, S.E.; Elmaleh, M.; Mendelsohn, M.; Nemes, A.; Tapia, J.C.; Mentis, G.Z.; Shneider, N.A. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat. Commun. 2016, 7, 10465. [Google Scholar] [CrossRef] [PubMed]
- Scekic-Zahirovic, J.; Oussini, H.E.l.; Mersmann, S.; Drenner, K.; Wagner, M.; Sun, Y.; Allmeroth, K.; Dieterlé, S.; Sinniger, J.; Dirrig-Grosch, S.; et al. Motor neuron intrinsic and extrinsic mechanisms contribute to the pathogenesis of FUS-associated amyotrophic lateral sclerosis. Acta Neuropathol. 2017, 133, 887–906. [Google Scholar] [CrossRef] [PubMed]
- Soo, K.Y.; Halloran, M.; Sundaramoorthy, V.; Parakh, S.; Toth, R.P.; Southam, K.A.; McLean, C.A.; Lock, P.; King, A.; Farg, M.A.; et al. Rab1-dependent ER-Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP-43 and FUS-associated ALS. Acta Neuropathol. 2015, 130, 679–697. [Google Scholar] [CrossRef]
- Deĭkin, A.V.; Kovrazhkina, E.A.; Ovchinnikov, R.K.; Bronovitskiĭ, E.V.; Razinskaia, O.D.; Smirnov, A.P.; Ermolkevich, T.G.; Eliakov, A.B.; Popov, A.N.; Fedorov, E.N.; et al. A mice model of amyotrophic lateral sclerosis expressing mutant human FUS protein. Zhurnal Nevrol. 2014, 114, 62–69. [Google Scholar]
- Devoy, A.; Kalmar, B.; Stewart, M.; Park, H.; Burke, B.; Noy, S.J.; Redhead, Y.; Humphrey, J.; Lo, K.; Jaeger, J.; et al. Humanized mutant FUS drives progressive motor neuron degeneration without aggregation in “FUSDelta14” knockin mice. Brain 2017, 140, 2797–2805. [Google Scholar] [CrossRef]
- Shelkovnikova, T.A.; Peters, O.M.; Deykin, A.V.; Connor-Robson, N.; Robinson, H.; Ustyugov, A.A.; Bachurin, S.O.; Ermolkevich, T.G.; Goldman, I.L.; Sadchikova, E.R.; et al. Fused in sarcoma (FUS) protein lacking nuclear localization signal (NLS) and major RNA binding motifs triggers proteinopathy and severe motor phenotype in transgenic mice. J. Biol. Chem. 2013, 288, 25266–25274. [Google Scholar] [CrossRef]
- Mitchell, J.C.; McGoldrick, P.; Vance, C.; Hortobagyi, T.; Sreedharan, J.; Rogelj, B.; Tudor, E.L.; Smith, B.N.; Klasen, C.; Miller, C.C.J.; et al. Overexpression of human wild-type FUS causes progressive motor neuron degeneration in an age- and dose-dependent fashion. Acta Neuropathol. 2013, 125, 273–288. [Google Scholar] [CrossRef]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.-Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999. [Google Scholar] [CrossRef]
- Jackson, K.L.; Dhaibar, H.A.; Dayton, R.D.; Cananzi, S.G.; Mayhan, W.G.; Glasscock, E.; Klein, R.L. Severe respiratory changes at end stage in a FUS-induced disease state in adult rats. BMC Neurosci. 2016, 17, 69. [Google Scholar] [CrossRef]
- Huang, C.; Zhou, H.; Tong, J.; Chen, H.; Liu, Y.-J.; Wang, D.; Wei, X.; Xia, X.-G. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011, 7, e1002011. [Google Scholar] [CrossRef]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef]
- Tabet, R.; Schaeffer, L.; Freyermuth, F.; Jambeau, M.; Workman, M.; Lee, C.-Z.; Lin, C.-C.; Jiang, J.; Jansen-West, K.; Abou-Hamdan, H.; et al. CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts. Nat. Commun. 2018, 9, 152. [Google Scholar] [CrossRef]
- Smeyers, J.; Banchi, E.-G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell. Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef]
- Xu, Z.; Poidevin, M.; Li, X.; Li, Y.; Shu, L.; Nelson, D.L.; Li, H.; Hales, C.M.; Gearing, M.; Wingo, T.S.; et al. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 7778–7783. [Google Scholar] [CrossRef]
- Herranz-Martin, S.; Chandran, J.; Lewis, K.; Mulcahy, P.; Higginbottom, A.; Walker, C.; Valenzuela, I.M.-P.Y.; Jones, R.A.; Coldicott, I.; Iannitti, T.; et al. Viral delivery of C9orf72 hexanucleotide repeat expansions in mice leads to repeat-length-dependent neuropathology and behavioural deficits. Dis. Model. Mech. 2017, 10, 859–868. [Google Scholar]
- Chew, J.; Gendron, T.F.; Prudencio, M.; Sasaguri, H.; Zhang, Y.-J.; Castanedes-Casey, M.; Lee, C.W.; Jansen-West, K.; Kurti, A.; Murray, M.E.; et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 2015, 348, 1151–1154. [Google Scholar] [CrossRef]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P.W. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef]
- Schludi, M.H.; Becker, L.; Garrett, L.; Gendron, T.F.; Zhou, Q.; Schreiber, F.; Popper, B.; Dimou, L.; Strom, T.M.; Winkelmann, J.; et al. Spinal poly-GA inclusions in a C9orf72 mouse model trigger motor deficits and inflammation without neuron loss. Acta Neuropathol. 2017, 134, 241–254. [Google Scholar] [CrossRef]
- Koppers, M.; Blokhuis, A.M.; Westeneng, H.-J.; Terpstra, M.L.; Zundel, C.A.C.; Vieira de Sá, R.; Schellevis, R.D.; Waite, A.J.; Blake, D.J.; Veldink, J.H.; et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 2015, 78, 426–438. [Google Scholar] [CrossRef]
- O’Rourke, J.G.; Bogdanik, L.; Yáñez, A.; Lall, D.; Wolf, A.J.; Muhammad, A.K.M.G.; Ho, R.; Carmona, S.; Vit, J.P.; Zarrow, J.; et al. C9orf72 is required for proper macrophage and microglial function in mice. Science 2016, 351, 1324–1329. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Burberry, A.; Suzuki, N.; Wang, J.-Y.; Moccia, R.; Mordes, D.A.; Stewart, M.H.; Suzuki-Uematsu, S.; Ghosh, S.; Singh, A.; Merkle, F.T.; et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 2016, 8, 347ra93. [Google Scholar] [CrossRef] [PubMed]
- Atanasio, A.; Decman, V.; White, D.; Ramos, M.; Ikiz, B.; Lee, H.-C.; Siao, C.-J.; Brydges, S.; LaRosa, E.; Bai, Y.; et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci. Rep. 2016, 6, 23204. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, J.G.; Bogdanik, L.; Muhammad, A.K.M.G.; Gendron, T.F.; Kim, K.J.; Austin, A.; Cady, J.; Liu, E.Y.; Zarrow, J.; Grant, S.; et al. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron 2015, 88, 892–901. [Google Scholar] [CrossRef]
- Peters, O.M.; Cabrera, G.T.; Tran, H.; Gendron, T.F.; McKeon, J.E.; Metterville, J.; Weiss, A.; Wightman, N.; Salameh, J.; Kim, J.; et al. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron 2015, 88, 902–909. [Google Scholar] [CrossRef]
- Batra, R.; Lee, C.W. Mouse Models of C9orf72 Hexanucleotide Repeat Expansion in Amyotrophic Lateral Sclerosis/Frontotemporal Dementia. Front. Cell. Neurosci. 2017, 11, 196. [Google Scholar] [CrossRef]
- Schludi, M.H.; Edbauer, D. Targeting RNA G-quadruplexes as new treatment strategy for C9orf72 ALS/FTD. EMBO Mol. Med. 2018, 10, 4–6. [Google Scholar] [CrossRef]
- Dong, W.; Ma, Y.; Guan, F.; Zhang, X.; Chen, W.; Zhang, L.; Zhang, L. Ablation of C9orf72 together with excitotoxicity induces ALS in rats. FEBS J. 2021, 288, 1712–1723. [Google Scholar] [CrossRef]
- Dong, W.; Zhang, L.; Sun, C.; Gao, X.; Guan, F.; Li, J.; Chen, W.; Ma, Y.; Zhang, L. Knock in of a hexanucleotide repeat expansion in the C9orf72 gene induces ALS in rats. Anim. Model. Exp. Med. 2020, 3, 237–244. [Google Scholar] [CrossRef]
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 2001, 29, 166–173. [Google Scholar] [CrossRef]
- Yang, Y.; Hentati, A.; Deng, H.X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef]
- Kress, J.A.; Kühnlein, P.; Winter, P.; Ludolph, A.C.; Kassubek, J.; Müller, U.; Sperfeld, A.-D. Novel mutation in the ALS2 gene in juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2005, 58, 800–803. [Google Scholar] [CrossRef]
- Chandran, J.; Ding, J.; Cai, H. Alsin and the molecular pathways of amyotrophic lateral sclerosis. Mol. Neurobiol. 2007, 36, 224–231. [Google Scholar] [CrossRef]
- Cai, H.; Shim, H.; Lai, C.; Xie, C.; Lin, X.; Yang, W.J.; Chandran, J. ALS2/alsin knockout mice and motor neuron diseases. Neurodegener. Dis. 2008, 5, 359–366. [Google Scholar] [CrossRef]
- Cai, H.; Lin, X.; Xie, C.; Laird, F.M.; Lai, C.; Wen, H.; Chiang, H.-C.; Shim, H.; Farah, M.H.; Hoke, A.; et al. Loss of ALS2 function is insufficient to trigger motor neuron degeneration in knock-out mice but predisposes neurons to oxidative stress. J. Neurosci. 2005, 25, 7567–7574. [Google Scholar] [CrossRef]
- Hadano, S.; Benn, S.C.; Kakuta, S.; Otomo, A.; Sudo, K.; Kunita, R.; Suzuki-Utsunomiya, K.; Mizumura, H.; Shefner, J.M.; Cox, G.A.; et al. Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum. Mol. Genet. 2006, 15, 233–250. [Google Scholar] [CrossRef]
- Devon, R.S.; Orban, P.C.; Gerrow, K.; Barbieri, M.A.; Schwab, C.; Cao, L.P.; Helm, J.R.; Bissada, N.; Cruz-Aguado, R.; Davidson, T.-L.; et al. Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc. Natl. Acad. Sci. USA 2006, 103, 9595–9600. [Google Scholar] [CrossRef]
- Gros-Louis, F.; Kriz, J.; Kabashi, E.; McDearmid, J.; Millecamps, S.; Urushitani, M.; Lin, L.; Dion, P.; Zhu, Q.; Drapeau, P.; et al. Als2 mRNA splicing variants detected in KO mice rescue severe motor dysfunction phenotype in Als2 knock-down zebrafish. Hum. Mol. Genet. 2008, 17, 2691–2702. [Google Scholar] [CrossRef]
- Yamanaka, K.; Miller, T.M.; McAlonis-Downes, M.; Chun, S.J.; Cleveland, D.W. Progressive spinal axonal degeneration and slowness in ALS2-deficient mice. Ann. Neurol. 2006, 60, 95–104. [Google Scholar] [CrossRef]
- Bennett, C.L.; Dastidar, S.G.; Ling, S.-C.; Malik, B.; Ashe, T.; Wadhwa, M.; Miller, D.B.; Lee, C.; Mitchell, M.B.; van Es, M.A.; et al. Senataxin mutations elicit motor neuron degeneration phenotypes and yield TDP-43 mislocalization in ALS4 mice and human patients. Acta Neuropathol. 2018, 136, 425–443. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Hortobágyi, T.; Troakes, C.; Nishimura, A.L.; Vance, C.; van Swieten, J.C.; Seelaar, H.; King, A.; Al-Sarraj, S.; Rogelj, B.; Shaw, C.E. Optineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disorders. Acta Neuropathol. 2011, 121, 519–527. [Google Scholar] [CrossRef]
- Osawa, T.; Mizuno, Y.; Fujita, Y.; Takatama, M.; Nakazato, Y.; Okamoto, K. Optineurin in neurodegenerative diseases. Neuropathology 2011, 31, 569–574. [Google Scholar] [CrossRef]
- Ito, H.; Nakamura, M.; Komure, O.; Ayaki, T.; Wate, R.; Maruyama, H.; Nakamura, Y.; Fujita, K.; Kaneko, S.; Okamoto, Y.; et al. Clinicopathologic study on an ALS family with a heterozygous E478G optineurin mutation. Acta Neuropathol. 2011, 122, 223–229. [Google Scholar] [CrossRef]
- Kamada, M.; Izumi, Y.; Ayaki, T.; Nakamura, M.; Kagawa, S.; Kudo, E.; Sako, W.; Maruyama, H.; Nishida, Y.; Kawakami, H.; et al. Clinicopathologic features of autosomal recessive amyotrophic lateral sclerosis associated with optineurin mutation. Neuropathology 2014, 34, 64–70. [Google Scholar] [CrossRef]
- Gleason, C.E.; Ordureau, A.; Gourlay, R.; Arthur, J.S.C.; Cohen, P. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon β. J. Biol. Chem. 2011, 286, 35663–35674. [Google Scholar] [CrossRef]
- Zheng, T.; Yang, Y.; Castañeda, C.A. Structure, dynamics and functions of UBQLNs: At the crossroads of protein quality control machinery. Biochem. J. 2020, 477, 3471–3497. [Google Scholar] [CrossRef] [PubMed]
- Renaud, L.; Picher-Martel, V.; Codron, P.; Julien, J.-P. Key role of UBQLN2 in pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol. Commun. 2019, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Gorrie, G.H.; Fecto, F.; Radzicki, D.; Weiss, C.; Shi, Y.; Dong, H.; Zhai, H.; Fu, R.; Liu, E.; Li, S.; et al. Dendritic spinopathy in transgenic mice expressing ALS/dementia-linked mutant UBQLN2. Proc. Natl. Acad. Sci. USA 2014, 111, 14524–14529. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, M.; Huang, C.; Liu, X.; Huang, B.; Li, N.; Zhou, H.; Xia, X.-G. Pathogenic Ubqln2 gains toxic properties to induce neuron death. Acta Neuropathol. 2015, 129, 417–428. [Google Scholar] [CrossRef]
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; McWilliams, T.G.; Johnson, C.; Sahu, I.; Varghese, J.; Wood, N.; Wightman, M.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef]
- Le, N.T.T.; Chang, L.; Kovlyagina, I.; Georgiou, P.; Safren, N.; Braunstein, K.E.; Kvarta, M.D.; Van Dyke, A.M.; LeGates, T.A.; Philips, T.; et al. Motor neuron disease, TDP-43 pathology, and memory deficits in mice expressing ALS-FTD-linked UBQLN2 mutations. Proc. Natl. Acad. Sci. USA 2016, 113, E7580–E7589. [Google Scholar] [CrossRef]
- Picher-Martel, V.; Renaud, L.; Bareil, C.; Julien, J.-P. Neuronal Expression of UBQLN2(P497H) Exacerbates TDP-43 Pathology in TDP-43 (G348C) Mice through Interaction with Ubiquitin. Mol. Neurobiol. 2019, 56, 4680–4696. [Google Scholar] [CrossRef]
- Yang, C.; Huang, M.; DeBiasio, J.; Pring, M.; Joyce, M.; Miki, H.; Takenawa, T.; Zigmond, S.H. Profilin enhances Cdc42-induced nucleation of actin polymerization. J. Cell Biol. 2000, 150, 1001–1012. [Google Scholar] [CrossRef]
- Dominguez, R. Structural insights into de novo actin polymerization. Curr. Opin. Struct. Biol. 2010, 20, 217–225. [Google Scholar] [CrossRef]
- Wu, C.-H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012, 488, 499–503. [Google Scholar] [CrossRef]
- Alkam, D.; Feldman, E.Z.; Singh, A.; Kiaei, M. Profilin1 biology and its mutation, actin(g) in disease. Cell. Mol. Life Sci. 2017, 74, 967–981. [Google Scholar] [CrossRef]
- Boopathy, S.; Silvas, T.V.; Tischbein, M.; Jansen, S.; Shandilya, S.M.; Zitzewitz, J.A.; Landers, J.E.; Goode, B.L.; Schiffer, C.A.; Bosco, D.A. Structural basis for mutation-induced destabilization of profilin 1 in ALS. Proc. Natl. Acad. Sci. USA 2015, 112, 7984–7989. [Google Scholar] [CrossRef]
- Henty-Ridilla, J.L.; Juanes, M.A.; Goode, B.L. Profilin Directly Promotes Microtubule Growth through Residues Mutated in Amyotrophic Lateral Sclerosis. Curr. Biol. 2017, 27, 3535–3543.e4. [Google Scholar] [CrossRef]
- Brettle, M.; Suchowerska, A.K.; Chua, S.W.; Ittner, L.M.; Fath, T. Amyotrophic lateral sclerosis-associated mutant profilin 1 increases dendritic arborisation and spine formation in primary hippocampal neurons. Neurosci. Lett. 2015, 609, 223–228. [Google Scholar] [CrossRef]
- Fil, D.; DeLoach, A.; Yadav, S.; Alkam, D.; MacNicol, M.; Singh, A.; Compadre, C.M.; Goellner, J.J.; O’Brien, C.A.; Fahmi, T.; et al. Mutant Profilin1 transgenic mice recapitulate cardinal features of motor neuron disease. Hum. Mol. Genet. 2017, 26, 686–701. [Google Scholar] [CrossRef]
- Yang, C.; Danielson, E.W.; Qiao, T.; Metterville, J.; Brown, R.H.J.; Landers, J.E.; Xu, Z. Mutant PFN1 causes ALS phenotypes and progressive motor neuron degeneration in mice by a gain of toxicity. Proc. Natl. Acad. Sci. USA 2016, 113, E6209–E6218. [Google Scholar] [CrossRef]
- Brettle, M.; Stefen, H.; Djordjevic, A.; Fok, S.Y.Y.; Chan, J.W.; van Hummel, A.; van der Hoven, J.; Przybyla, M.; Volkerling, A.; Ke, Y.D.; et al. Developmental Expression of Mutant PFN1 in Motor Neurons Impacts Neuronal Growth and Motor Performance of Young and Adult Mice. Front. Mol. Neurosci. 2019, 12, 231. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef]
- González-Pérez, P.; Cirulli, E.T.; Drory, V.E.; Dabby, R.; Nisipeanu, P.; Carasso, R.L.; Sadeh, M.; Fox, A.; Festoff, B.W.; Sapp, P.C.; et al. Novel mutation in VCP gene causes atypical amyotrophic lateral sclerosis. Neurology 2012, 79, 2201–2208. [Google Scholar] [CrossRef]
- Sun, X.; Qiu, H. Valosin-Containing Protein, a Calcium-Associated ATPase Protein, in Endoplasmic Reticulum and Mitochondrial Function and Its Implications for Diseases. Int. J. Mol. Sci. 2020, 21, 1216. [Google Scholar] [CrossRef]
- Badadani, M.; Nalbandian, A.; Watts, G.D.; Vesa, J.; Kitazawa, M.; Su, H.; Tanaja, J.; Dec, E.; Wallace, D.C.; Mukherjee, J.; et al. VCP associated inclusion body myopathy and paget disease of bone knock-in mouse model exhibits tissue pathology typical of human disease. PLoS ONE 2010, 5, e13183. [Google Scholar] [CrossRef] [PubMed]
- Custer, S.K.; Neumann, M.; Lu, H.; Wright, A.C.; Taylor, J.P. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum. Mol. Genet. 2010, 19, 1741–1755. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.Z.; Nalbandian, A.; Hsu, C.-I.; Li, S.; Llewellyn, K.J.; Mozaffar, T.; Kimonis, V.E.; Weiss, J.H. Slow development of ALS-like spinal cord pathology in mutant valosin-containing protein gene knock-in mice. Cell Death Dis. 2012, 3, e374. [Google Scholar] [CrossRef] [PubMed]
- Clemen, C.S.; Winter, L.; Strucksberg, K.-H.; Berwanger, C.; Türk, M.; Kornblum, C.; Florin, A.; Aguilar-Pimentel, J.A.; Amarie, O.V.; Becker, L.; et al. The heterozygous R155C VCP mutation: Toxic in humans! Harmless in mice? Biochem. Biophys. Res. Commun. 2018, 503, 2770–2777. [Google Scholar] [CrossRef]
- Nalbandian, A.; Llewellyn, K.J.; Kitazawa, M.; Yin, H.Z.; Badadani, M.; Khanlou, N.; Edwards, R.; Nguyen, C.; Mukherjee, J.; Mozaffar, T.; et al. The homozygote VCP(R155H/R155H) mouse model exhibits accelerated human VCP-associated disease pathology. PLoS ONE 2012, 7, e46308. [Google Scholar] [CrossRef]
- Skehel, P.A.; Martin, K.C.; Kandel, E.R.; Bartsch, D. A VAMP-binding protein from Aplysia required for neurotransmitter release. Science 1995, 269, 1580–1583. [Google Scholar] [CrossRef]
- Murphy, S.E.; Levine, T.P. VAP, a Versatile Access Point for the Endoplasmic Reticulum: Review and analysis of FFAT-like motifs in the VAPome. Biochim. Biophys. Acta 2016, 1861, 952–961. [Google Scholar] [CrossRef]
- Lev, S.; Ben Halevy, D.; Peretti, D.; Dahan, N. The VAP protein family: From cellular functions to motor neuron disease. Trends Cell Biol. 2008, 18, 282–290. [Google Scholar] [CrossRef]
- Borgese, N.; Iacomino, N.; Colombo, S.F.; Navone, F. The Link between VAPB Loss of Function and Amyotrophic Lateral Sclerosis. Cells 2021, 10, 1865. [Google Scholar] [CrossRef]
- Dudás, E.F.; Huynen, M.A.; Lesk, A.M.; Pastore, A. Invisible leashes: The tethering VAPs from infectious diseases to neurodegeneration. J. Biol. Chem. 2021, 296, 100421. [Google Scholar] [CrossRef]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.A.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.M.; Gillingwater, T.; Webb, J.; et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef]
- Chen, H.-J.; Anagnostou, G.; Chai, A.; Withers, J.; Morris, A.; Adhikaree, J.; Pennetta, G.; de Belleroche, J.S. Characterization of the properties of a novel mutation in VAPB in familial amyotrophic lateral sclerosis. J. Biol. Chem. 2010, 285, 40266–40281. [Google Scholar] [CrossRef]
- Kabashi, E.; El Oussini, H.; Bercier, V.; Gros-Louis, F.; Valdmanis, P.N.; McDearmid, J.; Mejier, I.A.; Dion, P.A.; Dupre, N.; Hollinger, D.; et al. Investigating the contribution of VAPB/ALS8 loss of function in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 2350–2360. [Google Scholar] [CrossRef]
- Kuijpers, M.; van Dis, V.; Haasdijk, E.D.; Harterink, M.; Vocking, K.; Post, J.A.; Scheper, W.; Hoogenraad, C.C.; Jaarsma, D. Amyotrophic lateral sclerosis (ALS)-associated VAPB-P56S inclusions represent an ER quality control compartment. Acta Neuropathol. Commun. 2013, 1, 24. [Google Scholar] [CrossRef]
- Aliaga, L.; Lai, C.; Yu, J.; Chub, N.; Shim, H.; Sun, L.; Xie, C.; Yang, W.-J.; Lin, X.; O’Donovan, M.J.; et al. Amyotrophic lateral sclerosis-related VAPB P56S mutation differentially affects the function and survival of corticospinal and spinal motor neurons. Hum. Mol. Genet. 2013, 22, 4293–4305. [Google Scholar] [CrossRef]
- Qiu, L.; Qiao, T.; Beers, M.; Tan, W.; Wang, H.; Yang, B.; Xu, Z. Widespread aggregation of mutant VAPB associated with ALS does not cause motor neuron degeneration or modulate mutant SOD1 aggregation and toxicity in mice. Mol. Neurodegener. 2013, 8, 1. [Google Scholar] [CrossRef]
- Larroquette, F.; Seto, L.; Gaub, P.L.; Kamal, B.; Wallis, D.; Larivière, R.; Vallée, J.; Robitaille, R.; Tsuda, H. Vapb/Amyotrophic lateral sclerosis 8 knock-in mice display slowly progressive motor behavior defects accompanying ER stress and autophagic response. Hum. Mol. Genet. 2015, 24, 6515–6529. [Google Scholar] [CrossRef]
- den Hartog Jager, W.A. Experimental amyotrophic lateral sclerosis in the guinea-pig. J. Neurol. Sci. 1985, 67, 133–142. [Google Scholar] [CrossRef]
- Engelhardt, J.I.; Appel, S.H.; Killian, J.M. Experimental autoimmune motoneuron disease. Ann. Neurol. 1989, 26, 368–376. [Google Scholar] [CrossRef]
- Engelhardt, J.I.; Appel, S.H.; Killian, J.M. Motor neuron destruction in guinea pigs immunized with bovine spinal cord ventral horn homogenate: Experimental autoimmune gray matter disease. J. Neuroimmunol. 1990, 27, 21–31. [Google Scholar] [CrossRef]
- Engelhardt, J.I.; Siklós, L.; Appel, S.H. Immunization of guinea pigs with human choline acetyltransferase induces selective lower motoneuron destruction. J. Neuroimmunol. 1997, 78, 57–68. [Google Scholar] [CrossRef]
- Bennett, S.A.; Tanaz, R.; Cobos, S.N.; Torrente, M.P. Epigenetics in amyotrophic lateral sclerosis: A role for histone post-translational modifications in neurodegenerative disease. Transl. Res. 2019, 204, 19–30. [Google Scholar] [CrossRef]
- Nowicka, N.; Juranek, J.; Juranek, J.K.; Wojtkiewicz, J. Risk Factors and Emerging Therapies in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- de Munck, E.; Muñoz-Sáez, E.; Miguel, B.G.; Solas, M.T.; Ojeda, I.; Martínez, A.; Gil, C.; Arahuetes, R.M. β-N-methylamino-l-alanine causes neurological and pathological phenotypes mimicking Amyotrophic Lateral Sclerosis (ALS): The first step towards an experimental model for sporadic ALS. Environ. Toxicol. Pharmacol. 2013, 36, 243–255. [Google Scholar] [CrossRef]
- de Munck, E.; Muñoz-Sáez, E.; Miguel, B.G.; Solas, M.T.; Martínez, A.; Arahuetes, R.M. Morphometric and neurochemical alterations found in l-BMAA treated rats. Environ. Toxicol. Pharmacol. 2015, 39, 1232–1245. [Google Scholar] [CrossRef]
- Yin, H.Z.; Yu, S.; Hsu, C.-I.; Liu, J.; Acab, A.; Wu, R.; Tao, A.; Chiang, B.J.; Weiss, J.H. Intrathecal infusion of BMAA induces selective motor neuron damage and astrogliosis in the ventral horn of the spinal cord. Exp. Neurol. 2014, 261, 1–9. [Google Scholar] [CrossRef]
- Tian, K.-W.; Jiang, H.; Wang, B.-B.; Zhang, F.; Han, S. Intravenous injection of l-BMAA induces a rat model with comprehensive characteristics of amyotrophic lateral sclerosis/Parkinson-dementia complex. Toxicol. Res. 2016, 5, 79–96. [Google Scholar] [CrossRef]
- Bolus, H.; Crocker, K.; Boekhoff-Falk, G.; Chtarbanova, S. Modeling Neurodegenerative Disorders in Drosophila melanogaster. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Lee, I.-S.; Jantrapirom, S.; Suda, K.; Yoshida, H. Drosophila models to study causative genes for human rare intractable neurological diseases. Exp. Cell Res. 2021, 403, 112584. [Google Scholar] [CrossRef]
- Adams, M.D.; Celniker, S.E.; Holt, R.A.; Evans, C.A.; Gocayne, J.D.; Amanatides, P.G.; Scherer, S.E.; Li, P.W.; Hoskins, R.A.; Galle, R.F.; et al. The genome sequence of Drosophila melanogaster. Science 2000, 287, 2185–2195. [Google Scholar] [CrossRef]
- Myers, E.W.; Sutton, G.G.; Delcher, A.L.; Dew, I.M.; Fasulo, D.P.; Flanigan, M.J.; Kravitz, S.A.; Mobarry, C.M.; Reinert, K.H.; Remington, K.A.; et al. A whole-genome assembly of Drosophila. Science 2000, 287, 2196–2204. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Reiter, L.T.; Potocki, L.; Chien, S.; Gribskov, M.; Bier, E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Casci, I.; Pandey, U.B. A fruitful endeavor: Modeling ALS in the fruit fly. Brain Res. 2015, 1607, 47–74. [Google Scholar] [CrossRef]
- Liguori, F.; Amadio, S.; Volonté, C. Fly for ALS: Drosophila modeling on the route to amyotrophic lateral sclerosis modifiers. Cell. Mol. Life Sci. 2021, 78, 6143–6160. [Google Scholar] [CrossRef]
- Braems, E.; Tziortzouda, P.; Van Den Bosch, L. Exploring the alternative: Fish, flies and worms as preclinical models for ALS. Neurosci. Lett. 2021, 759, 136041. [Google Scholar] [CrossRef]
- Marsh, J.L.; Thompson, L.M. Drosophila in the study of neurodegenerative disease. Neuron 2006, 52, 169–178. [Google Scholar] [CrossRef]
- McGurk, L.; Berson, A.; Bonini, N.M. Drosophila as an In Vivo Model for Human Neurodegenerative Disease. Genetics 2015, 201, 377–402. [Google Scholar] [CrossRef]
- Şentürk, M.; Bellen, H.J. Genetic strategies to tackle neurological diseases in fruit flies. Curr. Opin. Neurobiol. 2018, 50, 24–32. [Google Scholar] [CrossRef]
- Layalle, S.; They, L.; Ourghani, S.; Raoul, C.; Soustelle, L. Amyotrophic Lateral Sclerosis Genes in Drosophila melanogaster. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Phillips, J.P.; Campbell, S.D.; Michaud, D.; Charbonneau, M.; Hilliker, A.J. Null mutation of copper/zinc superoxide dismutase in Drosophila confers hypersensitivity to paraquat and reduced longevity. Proc. Natl. Acad. Sci. USA 1989, 86, 2761–2765. [Google Scholar] [CrossRef]
- Staveley, B.E.; Phillips, J.P.; Hilliker, A.J. Phenotypic consequences of copper-zinc superoxide dismutase overexpression in Drosophila melanogaster. Genome 1990, 33, 867–872. [Google Scholar] [CrossRef]
- Phillips, J.P.; Tainer, J.A.; Getzoff, E.D.; Boulianne, G.L.; Kirby, K.; Hilliker, A.J. Subunit-destabilizing mutations in Drosophila copper/zinc superoxide dismutase: Neuropathology and a model of dimer dysequilibrium. Proc. Natl. Acad. Sci. USA 1995, 92, 8574–8578. [Google Scholar] [CrossRef]
- Mockett, R.J.; Radyuk, S.N.; Benes, J.J.; Orr, W.C.; Sohal, R.S. Phenotypic effects of familial amyotrophic lateral sclerosis mutant Sod alleles in transgenic Drosophila. Proc. Natl. Acad. Sci. USA 2003, 100, 301–306. [Google Scholar] [CrossRef]
- Watson, M.R.; Lagow, R.D.; Xu, K.; Zhang, B.; Bonini, N.M. A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J. Biol. Chem. 2008, 283, 24972–24981. [Google Scholar] [CrossRef]
- Islam, R.; Kumimoto, E.L.; Bao, H.; Zhang, B. ALS-linked SOD1 in glial cells enhances ß-N-Methylamino L-Alanine (BMAA)-induced toxicity in Drosophila. F1000Research 2012, 1, 47. [Google Scholar] [CrossRef]
- Şahin, A.; Held, A.; Bredvik, K.; Major, P.; Achilli, T.-M.; Kerson, A.G.; Wharton, K.; Stilwell, G.; Reenan, R. Human SOD1 ALS Mutations in a Drosophila Knock-In Model Cause Severe Phenotypes and Reveal Dosage-Sensitive Gain- and Loss-of-Function Components. Genetics 2017, 205, 707–723. [Google Scholar] [CrossRef]
- Held, A.; Major, P.; Sahin, A.; Reenan, R.A.; Lipscombe, D.; Wharton, K.A. Circuit Dysfunction in SOD1-ALS Model First Detected in Sensory Feedback Prior to Motor Neuron Degeneration Is Alleviated by BMP Signaling. J. Neurosci. 2019, 39, 2347–2364. [Google Scholar] [CrossRef]
- Bahadorani, S.; Mukai, S.T.; Rabie, J.; Beckman, J.S.; Phillips, J.P.; Hilliker, A.J. Expression of zinc-deficient human superoxide dismutase in Drosophila neurons produces a locomotor defect linked to mitochondrial dysfunction. Neurobiol. Aging 2013, 34, 2322–2330. [Google Scholar] [CrossRef]
- Wang, H.-Y.; Wang, I.-F.; Bose, J.; Shen, C.-K.J. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics 2004, 83, 130–139. [Google Scholar] [CrossRef]
- Fiesel, F.C.; Voigt, A.; Weber, S.S.; Van den Haute, C.; Waldenmaier, A.; Görner, K.; Walter, M.; Anderson, M.L.; Kern, J.V.; Rasse, T.M.; et al. Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J. 2010, 29, 209–221. [Google Scholar] [CrossRef]
- Hazelett, D.J.; Chang, J.-C.; Lakeland, D.L.; Morton, D.B. Comparison of parallel high-throughput RNA sequencing between knockout of TDP-43 and its overexpression reveals primarily nonreciprocal and nonoverlapping gene expression changes in the central nervous system of Drosophila. Genes Genomes Genet. 2012, 2, 789–802. [Google Scholar] [CrossRef]
- Lin, M.-J.; Cheng, C.-W.; Shen, C.-K.J. Neuronal function and dysfunction of Drosophila dTDP. PLoS ONE 2011, 6, e20371. [Google Scholar] [CrossRef]
- Feiguin, F.; Godena, V.K.; Romano, G.; D’Ambrogio, A.; Klima, R.; Baralle, F.E. Depletion of TDP-43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett. 2009, 583, 1586–1592. [Google Scholar] [CrossRef]
- Diaper, D.C.; Adachi, Y.; Lazarou, L.; Greenstein, M.; Simoes, F.A.; Di Domenico, A.; Solomon, D.A.; Lowe, S.; Alsubaie, R.; Cheng, D.; et al. Drosophila TDP-43 dysfunction in glia and muscle cells cause cytological and behavioural phenotypes that characterize ALS and FTLD. Hum. Mol. Genet. 2013, 22, 3883–3893. [Google Scholar] [CrossRef]
- Lu, Y.; Ferris, J.; Gao, F.-B. Frontotemporal dementia and amyotrophic lateral sclerosis-associated disease protein TDP-43 promotes dendritic branching. Mol. Brain 2009, 2, 30. [Google Scholar] [CrossRef]
- Baldwin, K.R.; Godena, V.K.; Hewitt, V.L.; Whitworth, A.J. Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum. Mol. Genet. 2016, 25, 2378–2392. [Google Scholar]
- Diaper, D.C.; Adachi, Y.; Sutcliffe, B.; Humphrey, D.M.; Elliott, C.J.H.; Stepto, A.; Ludlow, Z.N.; Vanden Broeck, L.; Callaerts, P.; Dermaut, B.; et al. Loss and gain of Drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum. Mol. Genet. 2013, 22, 1539–1557. [Google Scholar] [CrossRef]
- Magrané, J.; Cortez, C.; Gan, W.-B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef]
- Choksi, D.K.; Roy, B.; Chatterjee, S.; Yusuff, T.; Bakhoum, M.F.; Sengupta, U.; Ambegaokar, S.; Kayed, R.; Jackson, G.R. TDP-43 Phosphorylation by casein kinase Iε promotes oligomerization and enhances toxicity in vivo. Hum. Mol. Genet. 2014, 23, 1025–1035. [Google Scholar] [CrossRef]
- Voigt, A.; Herholz, D.; Fiesel, F.C.; Kaur, K.; Müller, D.; Karsten, P.; Weber, S.S.; Kahle, P.J.; Marquardt, T.; Schulz, J.B. TDP-43-mediated neuron loss in vivo requires RNA-binding activity. PLoS ONE 2010, 5, e12247. [Google Scholar] [CrossRef] [PubMed]
- McGurk, L.; Gomes, E.; Guo, L.; Mojsilovic-Petrovic, J.; Tran, V.; Kalb, R.G.; Shorter, J.; Bonini, N.M. Poly(ADP-Ribose) Prevents Pathological Phase Separation of TDP-43 by Promoting Liquid Demixing and Stress Granule Localization. Mol. Cell 2018, 71, 703–717.e9. [Google Scholar] [CrossRef] [PubMed]
- Krug, L.; Chatterjee, N.; Borges-Monroy, R.; Hearn, S.; Liao, W.-W.; Morrill, K.; Prazak, L.; Rozhkov, N.; Theodorou, D.; Hammell, M.; et al. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet. 2017, 13, e1006635. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.; Cabirol-Pol, M.-J.; Miguel, L.; Whitworth, A.J.; Lecourtois, M.; Liévens, J.-C. Enhancing Mitofusin/Marf ameliorates neuromuscular dysfunction in Drosophila models of TDP-43 proteinopathies. Neurobiol. Aging 2017, 54, 71–83. [Google Scholar] [CrossRef]
- Chang, J.-C.; Hazelett, D.J.; Stewart, J.A.; Morton, D.B. Motor neuron expression of the voltage-gated calcium channel cacophony restores locomotion defects in a Drosophila, TDP-43 loss of function model of ALS. Brain Res. 2014, 1584, 39–51. [Google Scholar] [CrossRef]
- Chang, J.-C.; Morton, D.B. Drosophila lines with mutant and wild type human TDP-43 replacing the endogenous gene reveals phosphorylation and ubiquitination in mutant lines in the absence of viability or lifespan defects. PLoS ONE 2017, 12, e0180828. [Google Scholar] [CrossRef]
- Estes, P.S.; Boehringer, A.; Zwick, R.; Tang, J.E.; Grigsby, B.; Zarnescu, D.C. Wild-type and A315T mutant TDP-43 exert differential neurotoxicity in a Drosophila model of ALS. Hum. Mol. Genet. 2011, 20, 2308–2321. [Google Scholar] [CrossRef]
- Estes, P.S.; Daniel, S.G.; McCallum, A.P.; Boehringer, A.V.; Sukhina, A.S.; Zwick, R.A.; Zarnescu, D.C. Motor neurons and glia exhibit specific individualized responses to TDP-43 expression in a Drosophila model of amyotrophic lateral sclerosis. Dis. Model. Mech. 2013, 6, 721–733. [Google Scholar] [CrossRef]
- Ritson, G.P.; Custer, S.K.; Freibaum, B.D.; Guinto, J.B.; Geffel, D.; Moore, J.; Tang, W.; Winton, M.J.; Neumann, M.; Trojanowski, J.Q.; et al. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J. Neurosci. 2010, 30, 7729–7739. [Google Scholar] [CrossRef]
- Sreedharan, J.; Neukomm, L.J.; Brown, R.H.J.; Freeman, M.R. Age-Dependent TDP-43-Mediated Motor Neuron Degeneration Requires GSK3, hat-trick, and xmas-2. Curr. Biol. 2015, 25, 2130–2136. [Google Scholar] [CrossRef]
- Stolow, D.T.; Haynes, S.R. Cabeza, a Drosophila gene encoding a novel RNA binding protein, shares homology with EWS and TLS, two genes involved in human sarcoma formation. Nucleic Acids Res. 1995, 23, 835–843. [Google Scholar] [CrossRef][Green Version]
- Frickenhaus, M.; Wagner, M.; Mallik, M.; Catinozzi, M.; Storkebaum, E. Highly efficient cell-type-specific gene inactivation reveals a key function for the Drosophila FUS homolog cabeza in neurons. Sci. Rep. 2015, 5, 9107. [Google Scholar] [CrossRef]
- Sasayama, H.; Shimamura, M.; Tokuda, T.; Azuma, Y.; Yoshida, T.; Mizuno, T.; Nakagawa, M.; Fujikake, N.; Nagai, Y.; Yamaguchi, M. Knockdown of the Drosophila fused in sarcoma (FUS) homologue causes deficient locomotive behavior and shortening of motoneuron terminal branches. PLoS ONE 2012, 7, e39483. [Google Scholar] [CrossRef]
- Shimamura, M.; Kyotani, A.; Azuma, Y.; Yoshida, H.; Binh Nguyen, T.; Mizuta, I.; Yoshida, T.; Mizuno, T.; Nakagawa, M.; Tokuda, T.; et al. Genetic link between Cabeza, a Drosophila homologue of Fused in Sarcoma (FUS), and the EGFR signaling pathway. Exp. Cell Res. 2014, 326, 36–45. [Google Scholar] [CrossRef]
- Wang, J.-W.; Brent, J.R.; Tomlinson, A.; Shneider, N.A.; McCabe, B.D. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J. Clin. Investig. 2011, 121, 4118–4126. [Google Scholar] [CrossRef]
- Xia, R.; Liu, Y.; Yang, L.; Gal, J.; Zhu, H.; Jia, J. Motor neuron apoptosis and neuromuscular junction perturbation are prominent features in a Drosophila model of Fus-mediated ALS. Mol. Neurodegener. 2012, 7, 10. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, M.; Deng, J.; Chen, X.; Ye, Y.; Zhu, L.; Liu, J.; Ye, H.; Shen, Y.; Li, Y.; et al. Expression of human FUS protein in Drosophila leads to progressive neurodegeneration. Protein Cell 2011, 2, 477–486. [Google Scholar] [CrossRef]
- Jäckel, S.; Summerer, A.K.; Thömmes, C.M.; Pan, X.; Voigt, A.; Schulz, J.B.; Rasse, T.M.; Dormann, D.; Haass, C.; Kahle, P.J. Nuclear import factor transportin and arginine methyltransferase 1 modify FUS neurotoxicity in Drosophila. Neurobiol. Dis. 2015, 74, 76–88. [Google Scholar] [CrossRef]
- Lanson, N.A.J.; Maltare, A.; King, H.; Smith, R.; Kim, J.H.; Taylor, J.P.; Lloyd, T.E.; Pandey, U.B. A Drosophila model of FUS-related neurodegeneration reveals genetic interaction between FUS and TDP-43. Hum. Mol. Genet. 2011, 20, 2510–2523. [Google Scholar] [CrossRef]
- Machamer, J.B.; Collins, S.E.; Lloyd, T.E. The ALS gene FUS regulates synaptic transmission at the Drosophila neuromuscular junction. Hum. Mol. Genet. 2014, 23, 3810–3822. [Google Scholar] [CrossRef][Green Version]
- Miguel, L.; Avequin, T.; Delarue, M.; Feuillette, S.; Frébourg, T.; Campion, D.; Lecourtois, M. Accumulation of insoluble forms of FUS protein correlates with toxicity in Drosophila. Neurobiol. Aging 2012, 33, e1–e15. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.; Acharya, K.R.; Subramanian, V. A comparative bioinformatic analysis of C9orf72. PeerJ 2018, 6, e4391. [Google Scholar] [CrossRef]
- Yuva-Aydemir, Y.; Almeida, S.; Gao, F.-B. Insights into C9ORF72-Related ALS/FTD from Drosophila and iPSC Models. Trends Neurosci. 2018, 41, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Celona, B.; von Dollen, J.; Vatsavayai, S.C.; Kashima, R.; Johnson, J.R.; Tang, A.A.; Hata, A.; Miller, B.L.; Huang, E.J.; Krogan, N.J.; et al. Suppression of C9orf72 RNA repeat-induced neurotoxicity by the ALS-associated RNA-binding protein Zfp106. eLife 2017, 6, e19032. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.-H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 2015, 525, 129–133. [Google Scholar] [CrossRef]
- Kramer, N.J.; Carlomagno, Y.; Zhang, Y.-J.; Almeida, S.; Cook, C.N.; Gendron, T.F.; Prudencio, M.; Van Blitterswijk, M.; Belzil, V.; Couthouis, J.; et al. Spt4 selectively regulates the expression of C9orf72 sense and antisense mutant transcripts. Science 2016, 353, 708–712. [Google Scholar] [CrossRef]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef]
- Gendron, T.F.; Belzil, V.V.; Zhang, Y.-J.; Petrucelli, L. Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol. 2014, 127, 359–376. [Google Scholar] [CrossRef]
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Lee, K.-H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788.e17. [Google Scholar] [CrossRef]
- Mizielinska, S.; Grönke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef]
- Mizielinska, S.; Ridler, C.E.; Balendra, R.; Thoeng, A.; Woodling, N.S.; Grässer, F.A.; Plagnol, V.; Lashley, T.; Partridge, L.; Isaacs, A.M. Bidirectional nucleolar dysfunction in C9orf72 frontotemporal lobar degeneration. Acta Neuropathol. Commun. 2017, 5, 29. [Google Scholar] [CrossRef]
- Tran, H.; Almeida, S.; Moore, J.; Gendron, T.F.; Chalasani, U.; Lu, Y.; Du, X.; Nickerson, J.A.; Petrucelli, L.; Weng, Z.; et al. Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron 2015, 87, 1207–1214. [Google Scholar] [CrossRef]
- Phillips, J.B.; Westerfield, M. Zebrafish models in translational research: Tipping the scales toward advancements in human health. DMM Dis. Model. Mech. 2014, 7, 739–743. [Google Scholar] [CrossRef]
- Patten, S.A.; Armstrong, G.A.B.; Lissouba, A.; Kabashi, E.; Parker, J.A.; Drapeau, P. Fishing for causes and cures of motor neuron disorders. Dis. Model. Mech. 2014, 7, 799–809. [Google Scholar] [CrossRef]
- Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Modelling amyotrophic lateral sclerosis: Progress and possibilities. Dis. Model. Mech. 2017, 10, 537–549. [Google Scholar] [CrossRef]
- Fortier, G.; Butti, Z.; Patten, S.A. Modelling C9orf72-Related Amyotrophic Lateral Sclerosis in Zebrafish. Biomedicines 2020, 8, e19032. [Google Scholar] [CrossRef]
- Gois, A.M.; Mendonça, D.M.F.; Freire, M.A.M.; Santos, J.R. In vitro and in vivo models of amyotrophic lateral sclerosis: An updated overview. Brain Res. Bull. 2020, 159, 32–43. [Google Scholar] [CrossRef]
- Gerety, S.S.; Wilkinson, D.G. Morpholino artifacts provide pitfalls and reveal a novel role for pro-apoptotic genes in hindbrain boundary development. Dev. Biol. 2011, 350, 279–289. [Google Scholar] [CrossRef]
- Ramesh, T.; Lyon, A.N.; Pineda, R.H.; Wang, C.; Janssen, P.M.L.; Canan, B.D.; Burghes, A.H.M.; Beattie, C.E. A genetic model of amyotrophic lateral sclerosis in zebrafish displays phenotypic hallmarks of motoneuron disease. Dis. Model. Mech. 2010, 3, 652–662. [Google Scholar] [CrossRef]
- Lemmens, R.; Van Hoecke, A.; Hersmus, N.; Geelen, V.; D’Hollander, I.; Thijs, V.; Van Den Bosch, L.; Carmeliet, P.; Robberecht, W. Overexpression of mutant superoxide dismutase 1 causes a motor axonopathy in the zebrafish. Hum. Mol. Genet. 2007, 16, 2359–2365. [Google Scholar] [CrossRef]
- Da Costa, M.M.J.; Allen, C.E.; Higginbottom, A.; Ramesh, T.; Shaw, P.J.; McDermott, C.J. A new zebrafish model produced by TILLING of SOD1-related amyotrophic lateral sclerosis replicates key features of the disease and represents a tool for in vivo therapeutic screening. Dis. Model. Mech. 2014, 7, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, L.; Ghilardi, A.; Rottoli, E.; De Maglie, M.; Prosperi, L.; Perego, C.; Baruscotti, M.; Bucchi, A.; Del Giacco, L.; Francolini, M. INaP selective inhibition reverts precocious inter- and motorneurons hyperexcitability in the Sod1-G93R zebrafish ALS model. Sci. Rep. 2016, 6, 24515. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.; Kwok, S.; Lovejoy, E.; Lavin, T.; Sher, R.B. Editor’s Highlight: Embryonic Exposure to the Environmental Neurotoxin BMAA Negatively Impacts Early Neuronal Development and Progression of Neurodegeneration in the Sod1-G93R Zebrafish Model of Amyotrophic Lateral Sclerosis. Toxicol. Sci. 2017, 157, 129–140. [Google Scholar]
- Choi, S.Y.; Lee, J.-H.; Chung, A.-Y.; Jo, Y.; Shin, J.-H.; Park, H.-C.; Kim, H.; Lopez-Gonzalez, R.; Ryu, J.R.; Sun, W. Prevention of mitochondrial impairment by inhibition of protein phosphatase 1 activity in amyotrophic lateral sclerosis. Cell Death Dis. 2020, 11, 888. [Google Scholar] [CrossRef]
- Goldshtein, H.; Muhire, A.; Petel Légaré, V.; Pushett, A.; Rotkopf, R.; Shefner, J.M.; Peterson, R.T.; Armstrong, G.A.B.; Russek-Blum, N. Efficacy of Ciprofloxacin/Celecoxib combination in zebrafish models of amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 1883–1897. [Google Scholar] [CrossRef]
- Parakh, S.; Shadfar, S.; Perri, E.R.; Ragagnin, A.M.G.; Piattoni, C.V.; Fogolín, M.B.; Yuan, K.C.; Shahheydari, H.; Don, E.K.; Thomas, C.J.; et al. The Redox Activity of Protein Disulfide Isomerase Inhibits ALS Phenotypes in Cellular and Zebrafish Models. iScience 2020, 23, 101097. [Google Scholar] [CrossRef]
- Laird, A.S.; Van Hoecke, A.; De Muynck, L.; Timmers, M.; Van den Bosch, L.; Van Damme, P.; Robberecht, W. Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLoS ONE 2010, 5, e13368. [Google Scholar] [CrossRef]
- Kabashi, E.; Lin, L.; Tradewell, M.L.; Dion, P.A.; Bercier, V.; Bourgouin, P.; Rochefort, D.; Bel Hadj, S.; Durham, H.D.; Vande Velde, C.; et al. Gain and loss of function of ALS-related mutations of TARDBP (TDP-43) cause motor deficits in vivo. Hum. Mol. Genet. 2010, 19, 671–683. [Google Scholar] [CrossRef]
- Vaccaro, A.; Patten, S.A.; Aggad, D.; Julien, C.; Maios, C.; Kabashi, E.; Drapeau, P.; Parker, J.A. Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol. Dis. 2013, 55, 64–75. [Google Scholar] [CrossRef]
- Hewamadduma, C.A.A.; Grierson, A.J.; Ma, T.P.; Pan, L.; Moens, C.B.; Ingham, P.W.; Ramesh, T.; Shaw, P.J. Tardbpl splicing rescues motor neuron and axonal development in a mutant tardbp zebrafish. Hum. Mol. Genet. 2013, 22, 2376–2386. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Brustein, E.; Champagne, N.; Drapeau, P. Zebrafish models for the functional genomics of neurogenetic disorders. Biochim. Biophys. Acta 2011, 1812, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.A.B.; Drapeau, P. Loss and gain of FUS function impair neuromuscular synaptic transmission in a genetic model of ALS. Hum. Mol. Genet. 2013, 22, 4282–4292. [Google Scholar] [CrossRef]
- Bosco, D.A.; Lemay, N.; Ko, H.K.; Zhou, H.; Burke, C.; Kwiatkowski, T.J.J.; Sapp, P.; McKenna-Yasek, D.; Brown, R.H.J.; Hayward, L.J. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum. Mol. Genet. 2010, 19, 4160–4175. [Google Scholar] [CrossRef]
- Acosta, J.R.; Goldsbury, C.; Winnick, C.; Badrock, A.P.; Fraser, S.T.; Laird, A.S.; Hall, T.E.; Don, E.K.; Fifita, J.A.; Blair, I.P.; et al. Mutant human FUS Is ubiquitously mislocalized and generates persistent stress granules in primary cultured transgenic zebrafish cells. PLoS ONE 2014, 9, e90572. [Google Scholar] [CrossRef]
- Armstrong, G.A.B.; Liao, M.; You, Z.; Lissouba, A.; Chen, B.E.; Drapeau, P. Homology Directed Knockin of Point Mutations in the Zebrafish tardbp and fus Genes in ALS Using the CRISPR/Cas9 System. PLoS ONE 2016, 11, e0150188. [Google Scholar] [CrossRef]
- Bourefis, A.-R.; Campanari, M.-L.; Buee-Scherrer, V.; Kabashi, E. Functional characterization of a FUS mutant zebrafish line as a novel genetic model for ALS. Neurobiol. Dis. 2020, 142, 104935. [Google Scholar] [CrossRef]
- Campanari, M.-L.; Bourefis, A.-R.; Buee-Scherrer, V.; Kabashi, E. Freezing activity brief data from a new FUS mutant zebrafish line. Data Br. 2020, 31, 105921. [Google Scholar] [CrossRef]
- Ciura, S.; Lattante, S.; Le Ber, I.; Latouche, M.; Tostivint, H.; Brice, A.; Kabashi, E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 180–187. [Google Scholar] [CrossRef]
- Yeh, T.-H.; Liu, H.-F.; Li, Y.-W.; Lu, C.-S.; Shih, H.-Y.; Chiu, C.-C.; Lin, S.-J.; Huang, Y.-C.; Cheng, Y.-C. C9orf72 is essential for neurodevelopment and motility mediated by Cyclin G1. Exp. Neurol. 2018, 304, 114–124. [Google Scholar] [CrossRef]
- Butti, Z.; Pan, Y.E.; Giacomotto, J.; Patten, S.A. Reduced C9orf72 function leads to defective synaptic vesicle release and neuromuscular dysfunction in zebrafish. Commun. Biol. 2021, 4, 792. [Google Scholar] [CrossRef]
- Lee, Y.-B.; Chen, H.-J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Stalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013, 5, 1178–1186. [Google Scholar] [CrossRef]
- Swinnen, B.; Bento-Abreu, A.; Gendron, T.F.; Boeynaems, S.; Bogaert, E.; Nuyts, R.; Timmers, M.; Scheveneels, W.; Hersmus, N.; Wang, J.; et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. 2018, 135, 427–443. [Google Scholar] [CrossRef]
- Shaw, M.P.; Higginbottom, A.; McGown, A.; Castelli, L.M.; James, E.; Hautbergue, G.M.; Shaw, P.J.; Ramesh, T.M. Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol. Commun. 2018, 6, 125. [Google Scholar] [CrossRef]
- Schludi, M.H.; May, S.; Grässer, F.A.; Rentzsch, K.; Kremmer, E.; Küpper, C.; Klopstock, T.; Arzberger, T.; Edbauer, D. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol. 2015, 130, 537–555. [Google Scholar] [CrossRef]
- Ohki, Y.; Wenninger-Weinzierl, A.; Hruscha, A.; Asakawa, K.; Kawakami, K.; Haass, C.; Edbauer, D.; Schmid, B. Glycine-alanine dipeptide repeat protein contributes to toxicity in a zebrafish model of C9orf72 associated neurodegeneration. Mol. Neurodegener. 2017, 12, 6. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Swaminathan, A.; Bouffard, M.; Liao, M.; Ryan, S.; Callister, J.B.; Pickering-Brown, S.M.; Armstrong, G.A.B.; Drapeau, P. Expression of C9orf72-related dipeptides impairs motor function in a vertebrate model. Hum. Mol. Genet. 2018, 27, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, K.A.; Willicott, C.W.; Caldwell, G.A. Modeling neurodegeneration in Caenorhabditis elegans. Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Patten, S.A.; Parker, J.A.; Wen, X.-Y.; Drapeau, P. Simple animal models for amyotrophic lateral sclerosis drug discovery. Expert Opin. Drug Discov. 2016, 11, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhao, Y.; Chen, Y.; Cheng, B.; Peng, A.; Huang, K. Caenorhabditis elegans as a model system for target identification and drug screening against neurodegenerative diseases. Eur. J. Pharmacol. 2018, 819, 169–180. [Google Scholar] [CrossRef]
- Shaye, D.D.; Greenwald, I. OrthoList: A compendium of C. elegans genes with human orthologs. PLoS ONE 2011, 6, e20085. [Google Scholar] [CrossRef]
- Kim, W.; Underwood, R.S.; Greenwald, I.; Shaye, D.D. OrthoList 2: A New Comparative Genomic Analysis of Human and Caenorhabditis elegans Genes. Genetics 2018, 210, 445–461. [Google Scholar] [CrossRef]
- Wang, J.; Farr, G.W.; Hall, D.H.; Li, F.; Furtak, K.; Dreier, L.; Horwich, A.L. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009, 5, e1000350. [Google Scholar] [CrossRef]
- Li, J.; Li, T.; Zhang, X.; Tang, Y.; Yang, J.; Le, W. Human superoxide dismutase 1 overexpression in motor neurons of Caenorhabditis elegans causes axon guidance defect and neurodegeneration. Neurobiol. Aging 2014, 35, 837–846. [Google Scholar] [CrossRef]
- Baskoylu, S.N.; Yersak, J.; O’Hern, P.; Grosser, S.; Simon, J.; Kim, S.; Schuch, K.; Dimitriadi, M.; Yanagi, K.S.; Lins, J.; et al. Single copy/knock-in models of ALS SOD1 in C. elegans suggest loss and gain of function have different contributions to cholinergic and glutamatergic neurodegeneration. PLoS Genet. 2018, 14, e1007682. [Google Scholar] [CrossRef]
- Gidalevitz, T.; Krupinski, T.; Garcia, S.; Morimoto, R.I. Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 2009, 5, e1000399. [Google Scholar] [CrossRef]
- Ash, P.E.A.; Zhang, Y.-J.; Roberts, C.M.; Saldi, T.; Hutter, H.; Buratti, E.; Petrucelli, L.; Link, C.D. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum. Mol. Genet. 2010, 19, 3206–3218. [Google Scholar] [CrossRef]
- Liachko, N.F.; Guthrie, C.R.; Kraemer, B.C. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J. Neurosci. 2010, 30, 16208–16219. [Google Scholar] [CrossRef]
- Vaccaro, A.; Tauffenberger, A.; Ash, P.E.A.; Carlomagno, Y.; Petrucelli, L.; Parker, J.A. TDP-1/TDP-43 regulates stress signaling and age-dependent proteotoxicity in Caenorhabditis elegans. PLoS Genet. 2012, 8, e1002806. [Google Scholar] [CrossRef]
- Zhang, T.; Hwang, H.-Y.; Hao, H.; Talbot, C.J.; Wang, J. Caenorhabditis elegans RNA-processing protein TDP-1 regulates protein homeostasis and life span. J. Biol. Chem. 2012, 287, 8371–8382. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, A.; Tauffenberger, A.; Aggad, D.; Rouleau, G.; Drapeau, P.; Parker, J.A. Mutant TDP-43 and FUS cause age-dependent paralysis and neurodegeneration in C. elegans. PLoS ONE 2012, 7, e31321. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Yang, S.-P.; Xie, L.; Kawano, T.; Fu, D.; Mukai, A.; Bohm, C.; Chen, F.; Robertson, J.; Suzuki, H.; et al. ALS mutations in FUS cause neuronal dysfunction and death in Caenorhabditis elegans by a dominant gain-of-function mechanism. Hum. Mol. Genet. 2012, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Therrien, M.; Rouleau, G.A.; Dion, P.A.; Parker, J.A. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS ONE 2013, 8, e83450. [Google Scholar] [CrossRef]
- Corrionero, A.; Horvitz, H.R. A C9orf72 ALS/FTD Ortholog Acts in Endolysosomal Degradation and Lysosomal Homeostasis. Curr. Biol. 2018, 28, 1522–1535.e5. [Google Scholar] [CrossRef]
- Wang, X.; Hao, L.; Saur, T.; Joyal, K.; Zhao, Y.; Zhai, D.; Li, J.; Pribadi, M.; Coppola, G.; Cohen, B.M.; et al. Forward Genetic Screen in Caenorhabditis elegans Suggests F57A10.2 and acp-4 As Suppressors of C9ORF72 Related Phenotypes. Front. Mol. Neurosci. 2016, 9, 113. [Google Scholar] [CrossRef]
- Cherry, J.M.; Hong, E.L.; Amundsen, C.; Balakrishnan, R.; Binkley, G.; Chan, E.T.; Christie, K.R.; Costanzo, M.C.; Dwight, S.S.; Engel, S.R.; et al. Saccharomyces Genome Database: The genomics resource of budding yeast. Nucleic Acids Res. 2012, 40, D700–D705. [Google Scholar] [CrossRef]
- Kryndushkin, D.; Shewmaker, F. Modeling ALS and FTLD proteinopathies in yeast: An efficient approach for studying protein aggregation and toxicity. Prion 2011, 5, 250–257. [Google Scholar] [CrossRef][Green Version]
- Tenreiro, S.; Munder, M.C.; Alberti, S.; Outeiro, T.F. Harnessing the power of yeast to unravel the molecular basis of neurodegeneration. J. Neurochem. 2013, 127, 438–452. [Google Scholar] [CrossRef]
- Monahan, Z.T.; Rhoads, S.N.; Yee, D.S.; Shewmaker, F.P. Yeast Models of Prion-Like Proteins That Cause Amyotrophic Lateral Sclerosis Reveal Pathogenic Mechanisms. Front. Mol. Neurosci. 2018, 11, 453. [Google Scholar] [CrossRef]
- Rabizadeh, S.; Gralla, E.B.; Borchelt, D.R.; Gwinn, R.; Valentine, J.S.; Sisodia, S.; Wong, P.; Lee, M.; Hahn, H.; Bredesen, D.E. Mutations associated with amyotrophic lateral sclerosis convert superoxide dismutase from an antiapoptotic gene to a proapoptotic gene: Studies in yeast and neural cells. Proc. Natl. Acad. Sci. USA 1995, 92, 3024–3028. [Google Scholar] [CrossRef]
- Kryndushkin, D.; Ihrke, G.; Piermartiri, T.C.; Shewmaker, F. A yeast model of optineurin proteinopathy reveals a unique aggregation pattern associated with cellular toxicity. Mol. Microbiol. 2012, 86, 1531–1547. [Google Scholar] [CrossRef]
- Armakola, M.; Hart, M.P.; Gitler, A.D. TDP-43 toxicity in yeast. Methods 2011, 53, 238–245. [Google Scholar] [CrossRef]
- Gurney, M.E. The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. J. Neurol. Sci. 1997, 152, S67–S73. [Google Scholar] [CrossRef]
- Reaume, A.G.; Elliott, J.L.; Hoffman, E.K.; Kowall, N.W.; Ferrante, R.J.; Siwek, D.F.; Wilcox, H.M.; Flood, D.G.; Beal, M.F.; Brown, R.H.J.; et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 1996, 13, 43–47. [Google Scholar] [CrossRef]
- Klöppel, C.; Michels, C.; Zimmer, J.; Herrmann, J.M.; Riemer, J. In yeast redistribution of Sod1 to the mitochondrial intermembrane space provides protection against respiration derived oxidative stress. Biochem. Biophys. Res. Commun. 2010, 403, 114–119. [Google Scholar] [CrossRef]
- Sturtz, L.A.; Diekert, K.; Jensen, L.T.; Lill, R.; Culotta, V.C. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J. Biol. Chem. 2001, 276, 38084–38089. [Google Scholar] [CrossRef]
- Bastow, E.L.; Peswani, A.R.; Tarrant, D.S.J.; Pentland, D.R.; Chen, X.; Morgan, A.; Staniforth, G.L.; Tullet, J.M.; Rowe, M.L.; Howard, M.J.; et al. New links between SOD1 and metabolic dysfunction from a yeast model of amyotrophic lateral sclerosis. J. Cell Sci. 2016, 129, 4118–4129. [Google Scholar] [CrossRef]
- Armakola, M.; Higgins, M.J.; Figley, M.D.; Barmada, S.J.; Scarborough, E.A.; Diaz, Z.; Fang, X.; Shorter, J.; Krogan, N.J.; Finkbeiner, S.; et al. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat. Genet. 2012, 44, 1302–1309. [Google Scholar] [CrossRef]
- Johnson, B.S.; McCaffery, J.M.; Lindquist, S.; Gitler, A.D. A yeast TDP-43 proteinopathy model: Exploring the molecular determinants of TDP-43 aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 6439–6444. [Google Scholar] [CrossRef]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef]
- Figley, M.D.; Gitler, A.D. Yeast genetic screen reveals novel therapeutic strategy for ALS. Rare Dis. 2013, 1, e24420. [Google Scholar] [CrossRef]
- Bharathi, V.; Girdhar, A.; Patel, B.K. Role of CNC1 gene in TDP-43 aggregation-induced oxidative stress-mediated cell death in S. cerevisiae model of ALS. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118993. [Google Scholar] [CrossRef]
- Fushimi, K.; Long, C.; Jayaram, N.; Chen, X.; Li, L.; Wu, J.Y. Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy. Protein Cell 2011, 2, 141–149. [Google Scholar] [CrossRef]
- Ju, S.; Tardiff, D.F.; Han, H.; Divya, K.; Zhong, Q.; Maquat, L.E.; Bosco, D.A.; Hayward, L.J.; Brown, R.H.J.; Lindquist, S.; et al. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 2011, 9, e1001052. [Google Scholar] [CrossRef]
- Sun, Z.; Diaz, Z.; Fang, X.; Hart, M.P.; Chesi, A.; Shorter, J.; Gitler, A.D. Molecular determinants and genetic modifiers of aggregation and toxicity for the ALS disease protein FUS/TLS. PLoS Biol. 2011, 9, e1000614. [Google Scholar] [CrossRef]
- Gal, J.; Zhang, J.; Kwinter, D.M.; Zhai, J.; Jia, H.; Jia, J.; Zhu, H. Nuclear localization sequence of FUS and induction of stress granules by ALS mutants. Neurobiol. Aging 2011, 32, e27–e40. [Google Scholar] [CrossRef]
- Park, S.-K.; Arslan, F.; Kanneganti, V.; Barmada, S.J.; Purushothaman, P.; Verma, S.C.; Liebman, S.W. Overexpression of a conserved HSP40 chaperone reduces toxicity of several neurodegenerative disease proteins. Prion 2018, 12, 16–22. [Google Scholar] [CrossRef]
- Hayden, E.; Chen, S.; Chumley, A.; Xia, C.; Zhong, Q.; Ju, S. A Genetic Screen for Human Genes Suppressing FUS Induced Toxicity in Yeast. G3 Bethesda 2020, 10, 1843–1852. [Google Scholar] [CrossRef]
- Averill, D.R.J. Degenerative myelopathy in the aging German Shepherd dog: Clinical and pathologic findings. J. Am. Vet. Med. Assoc. 1973, 162, 1045–1051. [Google Scholar]
- Nardone, R.; Höller, Y.; Taylor, A.C.; Lochner, P.; Tezzon, F.; Golaszewski, S.; Brigo, F.; Trinka, E. Canine degenerative myelopathy: A model of human amyotrophic lateral sclerosis. Zoology 2016, 119, 64–73. [Google Scholar] [CrossRef] [PubMed]
- ENGEL, W.K.; KURLAND, L.T.; KLATZO, I. An inherited disease similar to amyotrophic lateral sclerosis with a pattern of posterior column involvement. An intermediate form? Brain 1959, 82, 203–220. [Google Scholar] [CrossRef] [PubMed]
- Hirano, A.; Kurland, L.T.; Sayre, G.P. Familial amyotrophic lateral sclerosis. A subgroup characterized by posterior and spinocerebellar tract involvement and hyaline inclusions in the anterior horn cells. Arch. Neurol. 1967, 16, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Awano, T.; Johnson, G.S.; Wade, C.M.; Katz, M.L.; Johnson, G.C.; Taylor, J.F.; Perloski, M.; Biagi, T.; Baranowska, I.; Long, S.; et al. Genome-wide association analysis reveals a SOD1 mutation in canine degenerative myelopathy that resembles amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 2794–2799. [Google Scholar] [CrossRef]
- Wininger, F.A.; Zeng, R.; Johnson, G.S.; Katz, M.L.; Johnson, G.C.; Bush, W.W.; Jarboe, J.M.; Coates, J.R. Degenerative myelopathy in a Bernese Mountain Dog with a novel SOD1 missense mutation. J. Vet. Intern. Med. 2011, 25, 1166–1170. [Google Scholar] [CrossRef]
- Crisp, M.J.; Beckett, J.; Coates, J.R.; Miller, T.M. Canine degenerative myelopathy: Biochemical characterization of superoxide dismutase 1 in the first naturally occurring non-human amyotrophic lateral sclerosis model. Exp. Neurol. 2013, 248, 1–9. [Google Scholar] [CrossRef]
- Kimura, S.; Kamatari, Y.O.; Kuwahara, Y.; Hara, H.; Yamato, O.; Maeda, S.; Kamishina, H.; Honda, R. Canine SOD1 harboring E40K or T18S mutations promotes protein aggregation without reducing the global structural stability. PeerJ 2020, 8, e9512. [Google Scholar] [CrossRef]
- Golubczyk, D.; Malysz-Cymborska, I.; Kalkowski, L.; Janowski, M.; Coates, J.R.; Wojtkiewicz, J.; Maksymowicz, W.; Walczak, P. The Role of Glia in Canine Degenerative Myelopathy: Relevance to Human Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2019, 56, 5740–5748. [Google Scholar] [CrossRef]
- Toedebusch, C.M.; Snyder, J.C.; Jones, M.R.; Garcia, V.B.; Johnson, G.C.; Villalón, E.L.; Coates, J.R.; Garcia, M.L. Arginase-1 expressing microglia in close proximity to motor neurons were increased early in disease progression in canine degenerative myelopathy, a model of amyotrophic lateral sclerosis. Mol. Cell. Neurosci. 2018, 88, 148–157. [Google Scholar] [CrossRef]
- Fernández-Trapero, M.; Espejo-Porras, F.; Rodríguez-Cueto, C.; Coates, J.R.; Pérez-Díaz, C.; de Lago, E.; Fernández-Ruiz, J. Upregulation of CB(2) receptors in reactive astrocytes in canine degenerative myelopathy, a disease model of amyotrophic lateral sclerosis. Dis. Model. Mech. 2017, 10, 551–558. [Google Scholar]
- Bendixen, E.; Danielsen, M.; Larsen, K.; Bendixen, C. Advances in porcine genomics and proteomics--a toolbox for developing the pig as a model organism for molecular biomedical research. Brief. Funct. Genom. 2010, 9, 208–219. [Google Scholar] [CrossRef]
- Lind, N.M.; Moustgaard, A.; Jelsing, J.; Vajta, G.; Cumming, P.; Hansen, A.K. The use of pigs in neuroscience: Modeling brain disorders. Neurosci. Biobehav. Rev. 2007, 31, 728–751. [Google Scholar] [CrossRef]
- Bjarkam, C.R.; Nielsen, M.S.; Glud, A.N.; Rosendal, F.; Mogensen, P.; Bender, D.; Doudet, D.; Møller, A.; Sørensen, J.C. Neuromodulation in a minipig MPTP model of Parkinson disease. Br. J. Neurosurg. 2008, 22 (Suppl. 1), S9–S12. [Google Scholar] [CrossRef]
- Kragh, P.M.; Nielsen, A.L.; Li, J.; Du, Y.; Lin, L.; Schmidt, M.; Bøgh, I.B.; Holm, I.E.; Jakobsen, J.E.; Johansen, M.G.; et al. Hemizygous minipigs produced by random gene insertion and handmade cloning express the Alzheimer’s disease-causing dominant mutation APPsw. Transgenic Res. 2009, 18, 545–558. [Google Scholar] [CrossRef]
- Lorson, M.A.; Spate, L.D.; Samuel, M.S.; Murphy, C.N.; Lorson, C.L.; Prather, R.S.; Wells, K.D. Disruption of the Survival Motor Neuron (SMN) gene in pigs using ssDNA. Transgenic Res. 2011, 20, 1293–1304. [Google Scholar] [CrossRef][Green Version]
- Uchida, M.; Shimatsu, Y.; Onoe, K.; Matsuyama, N.; Niki, R.; Ikeda, J.E.; Imai, H. Production of transgenic miniature pigs by pronuclear microinjection. Transgenic Res. 2001, 10, 577–582. [Google Scholar] [CrossRef]
- Dolezalova, D.; Hruska-Plochan, M.; Bjarkam, C.R.; Sørensen, J.C.H.; Cunningham, M.; Weingarten, D.; Ciacci, J.D.; Juhas, S.; Juhasova, J.; Motlik, J.; et al. Pig models of neurodegenerative disorders: Utilization in cell replacement-based preclinical safety and efficacy studies. J. Comp. Neurol. 2014, 522, 2784–2801. [Google Scholar] [CrossRef]
- Holm, I.E.; Alstrup, A.K.O.; Luo, Y. Genetically modified pig models for neurodegenerative disorders. J. Pathol. 2016, 238, 267–287. [Google Scholar] [CrossRef]
- Chieppa, M.N.; Perota, A.; Corona, C.; Grindatto, A.; Lagutina, I.; Vallino Costassa, E.; Lazzari, G.; Colleoni, S.; Duchi, R.; Lucchini, F.; et al. Modeling amyotrophic lateral sclerosis in hSOD1 transgenic swine. Neurodegener. Dis. 2014, 13, 246–254. [Google Scholar] [CrossRef]
- Yang, H.; Wang, G.; Sun, H.; Shu, R.; Liu, T.; Wang, C.-E.; Liu, Z.; Zhao, Y.; Zhao, B.; Ouyang, Z.; et al. Species-dependent neuropathology in transgenic SOD1 pigs. Cell Res. 2014, 24, 464–481. [Google Scholar] [CrossRef]
- Crociara, P.; Chieppa, M.N.; Vallino Costassa, E.; Berrone, E.; Gallo, M.; Lo Faro, M.; Pintore, M.D.; Iulini, B.; D’Angelo, A.; Perona, G.; et al. Motor neuron degeneration, severe myopathy and TDP-43 increase in a transgenic pig model of SOD1-linked familiar ALS. Neurobiol. Dis. 2019, 124, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yang, H.; Yan, S.; Wang, C.-E.; Liu, X.; Zhao, B.; Ouyang, Z.; Yin, P.; Liu, Z.; Zhao, Y.; et al. Cytoplasmic mislocalization of RNA splicing factors and aberrant neuronal gene splicing in TDP-43 transgenic pig brain. Mol. Neurodegener. 2015, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Hernandez, M.; Tadokoro, T.; Navarro, M.R.; Platoshyn, O.; Kobayashi, Y.; Marsala, S.; Miyanohara, A.; Juhas, S.; Juhasova, J.; Skalnikova, H.; et al. Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat. Med. 2020, 26, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Harding, J.D. Nonhuman Primates and Translational Research: Progress, Opportunities, and Challenges. ILAR J. 2017, 58, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Teil, M.; Arotcarena, M.-L.; Dehay, B. A New Rise of Non-Human Primate Models of Synucleinopathies. Biomedicines 2021, 9, 671–683. [Google Scholar] [CrossRef]
- Arnsten, A.F.T.; Datta, D.; Preuss, T.M. Studies of aging nonhuman primates illuminate the etiology of early-stage Alzheimer’s-like neuropathology: An evolutionary perspective. Am. J. Primatol. 2021, e23254. [Google Scholar] [CrossRef]
- Uchida, A.; Sasaguri, H.; Kimura, N.; Tajiri, M.; Ohkubo, T.; Ono, F.; Sakaue, F.; Kanai, K.; Hirai, T.; Sano, T.; et al. Non-human primate model of amyotrophic lateral sclerosis with cytoplasmic mislocalization of TDP-43. Brain 2012, 135, 833–846. [Google Scholar] [CrossRef]
- Borel, F.; Gernoux, G.; Cardozo, B.; Metterville, J.P.; Toro Cabrera, G.C.; Song, L.; Su, Q.; Gao, G.P.; Elmallah, M.K.; Brown, R.H.J.; et al. Therapeutic rAAVrh10 Mediated SOD1 Silencing in Adult SOD1(G93A) Mice and Nonhuman Primates. Hum. Gene Ther. 2016, 27, 19–31. [Google Scholar] [CrossRef]
- Borel, F.; Gernoux, G.; Sun, H.; Stock, R.; Blackwood, M.; Brown, R.H.J.; Mueller, C. Safe and effective superoxide dismutase 1 silencing using artificial microRNA in macaques. Sci. Transl. Med. 2018, 10, eaau6414. [Google Scholar] [CrossRef]
- Johnson, J.O.; Pioro, E.P.; Boehringer, A.; Chia, R.; Feit, H.; Renton, A.E.; Pliner, H.A.; Abramzon, Y.; Marangi, G.; Winborn, B.J.; et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat. Neurosci. 2014, 17, 664–666. [Google Scholar] [CrossRef]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.-F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef]
- Smith, B.N.; Ticozzi, N.; Fallini, C.; Gkazi, A.S.; Topp, S.; Kenna, K.P.; Scotter, E.L.; Kost, J.; Keagle, P.; Miller, J.W.; et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron 2014, 84, 324–331. [Google Scholar] [CrossRef]
- Kenna, K.P.; van Doormaal, P.T.C.; Dekker, A.M.; Ticozzi, N.; Kenna, B.J.; Diekstra, F.P.; van Rheenen, W.; van Eijk, K.R.; Jones, A.R.; Keagle, P.; et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1037–1042. [Google Scholar] [CrossRef]
- Brenner, D.; Müller, K.; Wieland, T.; Weydt, P.; Böhm, S.; Lulé, D.; Hübers, A.; Neuwirth, C.; Weber, M.; Borck, G.; et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 2016, 139, e28. [Google Scholar] [CrossRef]
- Smith, B.N.; Topp, S.D.; Fallini, C.; Shibata, H.; Chen, H.-J.; Troakes, C.; King, A.; Ticozzi, N.; Kenna, K.P.; Soragia-Gkazi, A.; et al. Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9, aad9157. [Google Scholar] [CrossRef]
- van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.A.; Võsa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef]
- Williams, K.L.; Topp, S.; Yang, S.; Smith, B.; Fifita, J.A.; Warraich, S.T.; Zhang, K.Y.; Farrawell, N.; Vance, C.; Hu, X.; et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 2016, 7, 11253. [Google Scholar] [CrossRef]
- Nakayasu, H.; Berezney, R. Nuclear matrins: Identification of the major nuclear matrix proteins. Proc. Natl. Acad. Sci. USA 1991, 88, 10312–10316. [Google Scholar] [CrossRef]
- Malik, A.M.; Barmada, S.J. Matrin 3 in neuromuscular disease: Physiology and pathophysiology. JCI Insight 2021, 6, e143948. [Google Scholar] [CrossRef] [PubMed]
- Moloney, C.; Rayaprolu, S.; Howard, J.; Fromholt, S.; Brown, H.; Collins, M.; Cabrera, M.; Duffy, C.; Siemienski, Z.; Miller, D.; et al. Transgenic mice overexpressing the ALS-linked protein Matrin 3 develop a profound muscle phenotype. Acta Neuropathol. Commun. 2016, 4, 122. [Google Scholar] [CrossRef] [PubMed]
- Moloney, C.; Rayaprolu, S.; Howard, J.; Fromholt, S.; Brown, H.; Collins, M.; Cabrera, M.; Duffy, C.; Siemienski, Z.; Miller, D.; et al. Analysis of spinal and muscle pathology in transgenic mice overexpressing wild-type and ALS-linked mutant MATR3. Acta Neuropathol. Commun. 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yamashita, S.; Hara, K.; Doki, T.; Tawara, N.; Ikeda, T.; Misumi, Y.; Zhang, Z.; Matsuo, Y.; Nagai, M.; et al. A mutant MATR3 mouse model to explain multisystem proteinopathy. J. Pathol. 2019, 249, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.S.; van Bruggen, R.; Kim, J.R.; Chen, X.X.L.; Chan, C.; Lee, J.; Cho, W.I.; Zhao, M.; Arndt, C.; Maksimovic, K.; et al. Selective neuronal degeneration in MATR3 S85C knock-in mouse model of early-stage ALS. Nat. Commun. 2020, 11, 5304. [Google Scholar] [CrossRef]
- Zhao, M.; Kao, C.S.; Arndt, C.; Tran, D.D.; Cho, W.I.; Maksimovic, K.; Chen, X.X.L.; Khan, M.; Zhu, H.; Qiao, J.; et al. Knockdown of genes involved in axonal transport enhances the toxicity of human neuromuscular disease-linked MATR3 mutations in Drosophila. FEBS Lett. 2020, 594, 2800–2818. [Google Scholar] [CrossRef]
- Ramesh, N.; Kour, S.; Anderson, E.N.; Rajasundaram, D.; Pandey, U.B. RNA-recognition motif in Matrin-3 mediates neurodegeneration through interaction with hnRNPM. Acta Neuropathol. Commun. 2020, 8, 138. [Google Scholar] [CrossRef]
- Imai, Y.; Meng, H.; Shiba-Fukushima, K.; Hattori, N. Twin CHCH Proteins, CHCHD2, and CHCHD10: Key Molecules of Parkinson’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Int. J. Mol. Sci. 2019, 20, 908. [Google Scholar] [CrossRef]
- Burstein, S.R.; Valsecchi, F.; Kawamata, H.; Bourens, M.; Zeng, R.; Zuberi, A.; Milner, T.A.; Cloonan, S.M.; Lutz, C.; Barrientos, A.; et al. In vitro and in vivo studies of the ALS-FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Hum. Mol. Genet. 2018, 27, 160–177. [Google Scholar] [CrossRef]
- Genin, E.C.; Madji Hounoum, B.; Bannwarth, S.; Fragaki, K.; Lacas-Gervais, S.; Mauri-Crouzet, A.; Lespinasse, F.; Neveu, J.; Ropert, B.; Augé, G.; et al. Mitochondrial defect in muscle precedes neuromuscular junction degeneration and motor neuron death in CHCHD10(S59L/+) mouse. Acta Neuropathol. 2019, 138, 123–145. [Google Scholar] [CrossRef]
- Anderson, C.J.; Bredvik, K.; Burstein, S.R.; Davis, C.; Meadows, S.M.; Dash, J.; Case, L.; Milner, T.A.; Kawamata, H.; Zuberi, A.; et al. ALS/FTD mutant CHCHD10 mice reveal a tissue-specific toxic gain-of-function and mitochondrial stress response. Acta Neuropathol. 2019, 138, 103–121. [Google Scholar] [CrossRef]
- Ryan, É.B.; Yan, J.; Miller, N.; Dayanidhi, S.; Ma, Y.C.; Deng, H.-X.; Siddique, T. Early death of ALS-linked CHCHD10-R15L transgenic mice with central nervous system, skeletal muscle, and cardiac pathology. iScience 2021, 24, 102061. [Google Scholar] [CrossRef]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef]
- Pottier, C.; Bieniek, K.F.; Finch, N.; van de Vorst, M.; Baker, M.; Perkersen, R.; Brown, P.; Ravenscroft, T.; van Blitterswijk, M.; Nicholson, A.M.; et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015, 130, 77–92. [Google Scholar] [CrossRef]
- Catanese, A.; Olde Heuvel, F.; Mulaw, M.; Demestre, M.; Higelin, J.; Barbi, G.; Freischmidt, A.; Weishaupt, J.H.; Ludolph, A.C.; Roselli, F.; et al. Retinoic acid worsens ATG10-dependent autophagy impairment in TBK1-mutant hiPSC-derived motoneurons through SQSTM1/p62 accumulation. Autophagy 2019, 15, 1719–1737. [Google Scholar] [CrossRef]
- Bonnard, M.; Mirtsos, C.; Suzuki, S.; Graham, K.; Huang, J.; Ng, M.; Itié, A.; Wakeham, A.; Shahinian, A.; Henzel, W.J.; et al. Deficiency of T2K leads to apoptotic liver degeneration and impaired NF-kappaB-dependent gene transcription. EMBO J. 2000, 19, 4976–4985. [Google Scholar] [CrossRef]
- Marchlik, E.; Thakker, P.; Carlson, T.; Jiang, Z.; Ryan, M.; Marusic, S.; Goutagny, N.; Kuang, W.; Askew, G.R.; Roberts, V.; et al. Mice lacking Tbk1 activity exhibit immune cell infiltrates in multiple tissues and increased susceptibility to LPS-induced lethality. J. Leukoc. Biol. 2010, 88, 1171–1180. [Google Scholar] [CrossRef]
- Gerbino, V.; Kaunga, E.; Ye, J.; Canzio, D.; O’Keeffe, S.; Rudnick, N.D.; Guarnieri, P.; Lutz, C.M.; Maniatis, T. The Loss of TBK1 Kinase Activity in Motor Neurons or in All Cell Types Differentially Impacts ALS Disease Progression in SOD1 Mice. Neuron 2020, 106, 789–805.e5. [Google Scholar] [CrossRef]
- Sieverding, K.; Ulmer, J.; Bruno, C.; Satoh, T.; Tsao, W.; Freischmidt, A.; Akira, S.; Wong, P.C.; Ludolph, A.C.; Danzer, K.M.; et al. Hemizygous deletion of Tbk1 worsens neuromuscular junction pathology in TDP-43(G298S) transgenic mice. Exp. Neurol. 2021, 335, 113496. [Google Scholar] [CrossRef]
- Hogan, A.L.; Don, E.K.; Rayner, S.L.; Lee, A.; Laird, A.S.; Watchon, M.; Winnick, C.; Tarr, I.S.; Morsch, M.; Fifita, J.A.; et al. Expression of ALS/FTD-linked mutant CCNF in zebrafish leads to increased cell death in the spinal cord and an aberrant motor phenotype. Hum. Mol. Genet. 2017, 26, 2616–2626. [Google Scholar] [CrossRef]
- Li, J.; He, J.; Tang, L.; Chen, L.; Ma, Y.; Fan, D. Screening for TUBA4A mutations in a large Chinese cohort of patients with ALS: Re-evaluating the pathogenesis of TUBA4A in ALS. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1350–1352. [Google Scholar] [CrossRef]
- Clark, J.A.; Yeaman, E.J.; Blizzard, C.A.; Chuckowree, J.A.; Dickson, T.C. A Case for Microtubule Vulnerability in Amyotrophic Lateral Sclerosis: Altered Dynamics During Disease. Front. Cell. Neurosci. 2016, 10, 204. [Google Scholar] [CrossRef]
- Helferich, A.M.; Brockmann, S.J.; Reinders, J.; Deshpande, D.; Holzmann, K.; Brenner, D.; Andersen, P.M.; Petri, S.; Thal, D.R.; Michaelis, J.; et al. Dysregulation of a novel miR-1825/TBCB/TUBA4A pathway in sporadic and familial ALS. Cell. Mol. Life Sci. 2018, 75, 4301–4319. [Google Scholar] [CrossRef]
- Maraldi, T.; Beretti, F.; Anselmi, L.; Franchin, C.; Arrigoni, G.; Braglia, L.; Mandrioli, J.; Vinceti, M.; Marmiroli, S. Influence of selenium on the emergence of neuro tubule defects in a neuron-like cell line and its implications for amyotrophic lateral sclerosis. Neurotoxicology 2019, 75, 209–220. [Google Scholar] [CrossRef]
- Teyssou, E.; Muratet, F.; Amador, M.-D.-M.; Ferrien, M.; Lautrette, G.; Machat, S.; Boillée, S.; Larmonier, T.; Saker, S.; Leguern, E.; et al. Genetic screening of ANXA11 revealed novel mutations linked to amyotrophic lateral sclerosis. Neurobiol. Aging 2021, 99, 102.e11–102.e20. [Google Scholar] [CrossRef]
- Iyer, S.; Acharya, K.R.; Subramanian, V. Prediction of structural consequences for disease causing variants in C21orf2 protein using computational approaches. J. Biomol. Struct. Dyn. 2019, 37, 465–480. [Google Scholar] [CrossRef]
- Lu, H.; Le, W.D.; Xie, Y.-Y.; Wang, X.-P. Current Therapy of Drugs in Amyotrophic Lateral Sclerosis. Curr. Neuropharmacol. 2016, 14, 314–321. [Google Scholar] [CrossRef]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Verber, N.S.; Shepheard, S.R.; Sassani, M.; McDonough, H.E.; Moore, S.A.; Alix, J.J.P.; Wilkinson, I.D.; Jenkins, T.M.; Shaw, P.J. Biomarkers in Motor Neuron Disease: A State of the Art Review. Front. Neurol. 2019, 10, 291. [Google Scholar] [CrossRef]
- Tadenev, A.L.D.; Burgess, R.W. Model validity for preclinical studies in precision medicine: Precisely how precise do we need to be? Mamm. Genome 2019, 30, 111–122. [Google Scholar] [CrossRef]
- Jankovska, N.; Matej, R. Molecular Pathology of ALS: What We Currently Know and What Important Information Is Still Missing. Diagnostics 2021, 11, 1365. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.C.; McLachlan, E.M.; Zeller, R. Transparency in Research involving Animals: The Basel Declaration and new principles for reporting research in BJP manuscripts. Br. J. Pharmacol. 2015, 172, 2427–2432. [Google Scholar] [CrossRef] [PubMed]
- Ludolph, A.C.; Bendotti, C.; Blaugrund, E.; Hengerer, B.; Löffler, J.-P.; Martin, J.; Meininger, V.; Meyer, T.; Moussaoui, S.; Robberecht, W.; et al. Guidelines for the preclinical in vivo evaluation of pharmacological active drugs for ALS/MND: Report on the 142nd ENMC international workshop. Amyotroph. Lateral Scler. 2007, 8, 217–223. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonifacino, T.; Zerbo, R.A.; Balbi, M.; Torazza, C.; Frumento, G.; Fedele, E.; Bonanno, G.; Milanese, M. Nearly 30 Years of Animal Models to Study Amyotrophic Lateral Sclerosis: A Historical Overview and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 12236. https://doi.org/10.3390/ijms222212236
Bonifacino T, Zerbo RA, Balbi M, Torazza C, Frumento G, Fedele E, Bonanno G, Milanese M. Nearly 30 Years of Animal Models to Study Amyotrophic Lateral Sclerosis: A Historical Overview and Future Perspectives. International Journal of Molecular Sciences. 2021; 22(22):12236. https://doi.org/10.3390/ijms222212236
Chicago/Turabian StyleBonifacino, Tiziana, Roberta Arianna Zerbo, Matilde Balbi, Carola Torazza, Giulia Frumento, Ernesto Fedele, Giambattista Bonanno, and Marco Milanese. 2021. "Nearly 30 Years of Animal Models to Study Amyotrophic Lateral Sclerosis: A Historical Overview and Future Perspectives" International Journal of Molecular Sciences 22, no. 22: 12236. https://doi.org/10.3390/ijms222212236
APA StyleBonifacino, T., Zerbo, R. A., Balbi, M., Torazza, C., Frumento, G., Fedele, E., Bonanno, G., & Milanese, M. (2021). Nearly 30 Years of Animal Models to Study Amyotrophic Lateral Sclerosis: A Historical Overview and Future Perspectives. International Journal of Molecular Sciences, 22(22), 12236. https://doi.org/10.3390/ijms222212236