
SARS-CoV-2 Variants, RBD Mutations, Binding Affinity, and Antibody Escape


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Theory of the Spontaneous Nature of Protein–Protein Docking
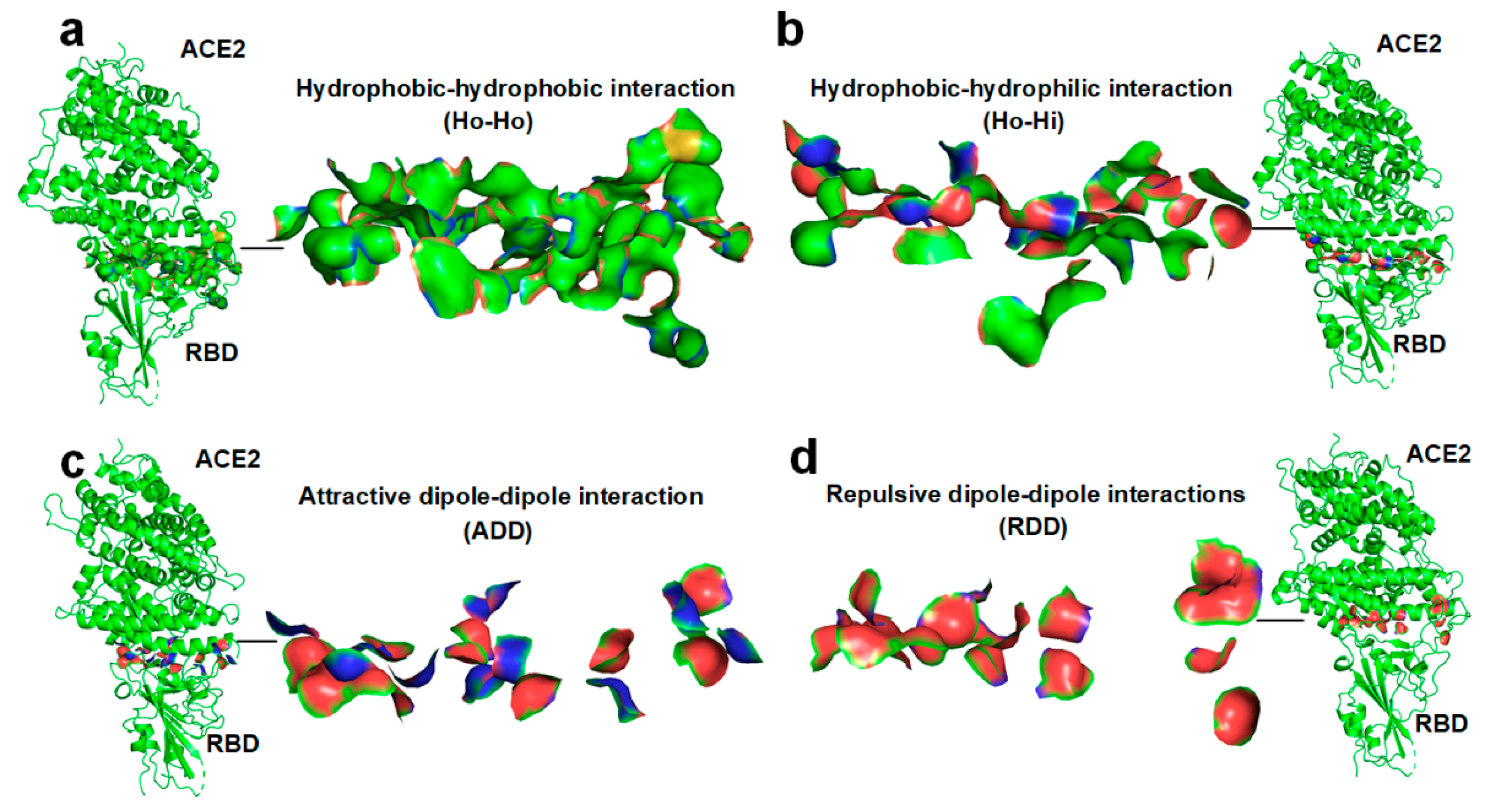
2.2. Docking between Hydrophobic Surface Areas
2.3. Docking between the Hydrophobic Surface Area and the Hydrophilic Surface Area
2.4. Docking-Induced Attractive Dipole–Dipole Interaction
2.5. Docking-Induced Repulsive Dipole–Dipole Interaction
3. Discussion
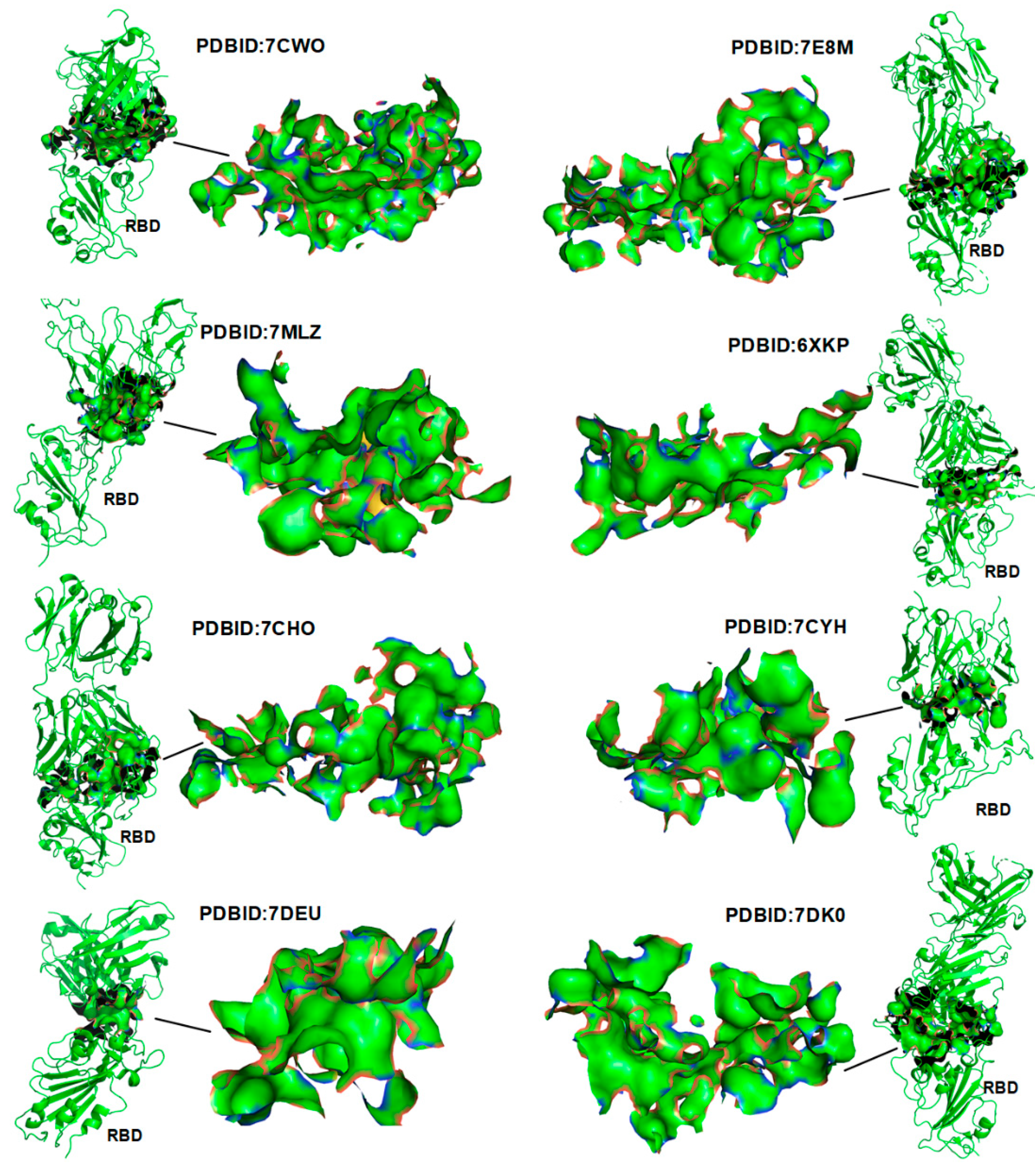
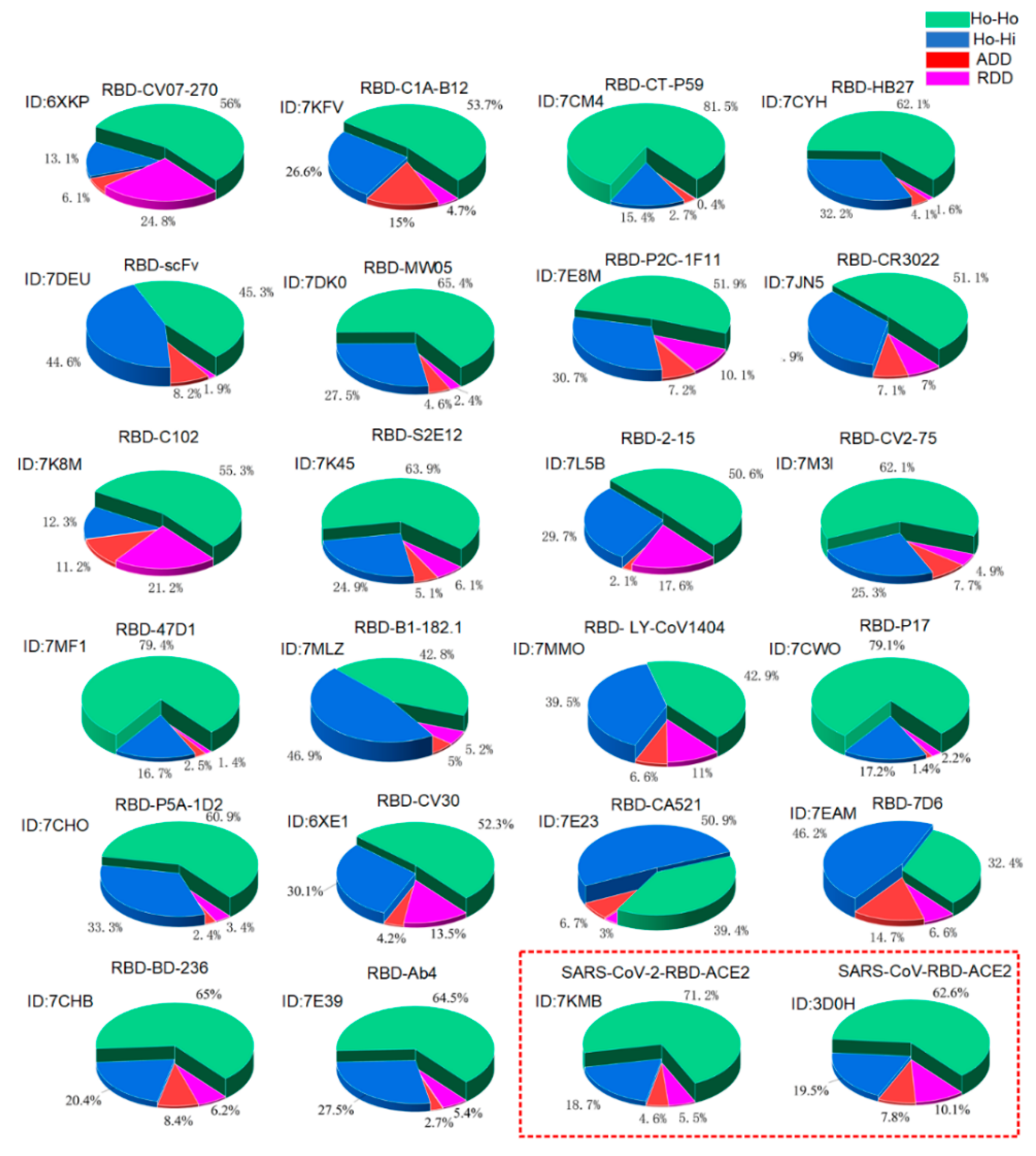
3.1. Verification of the Docking Theory
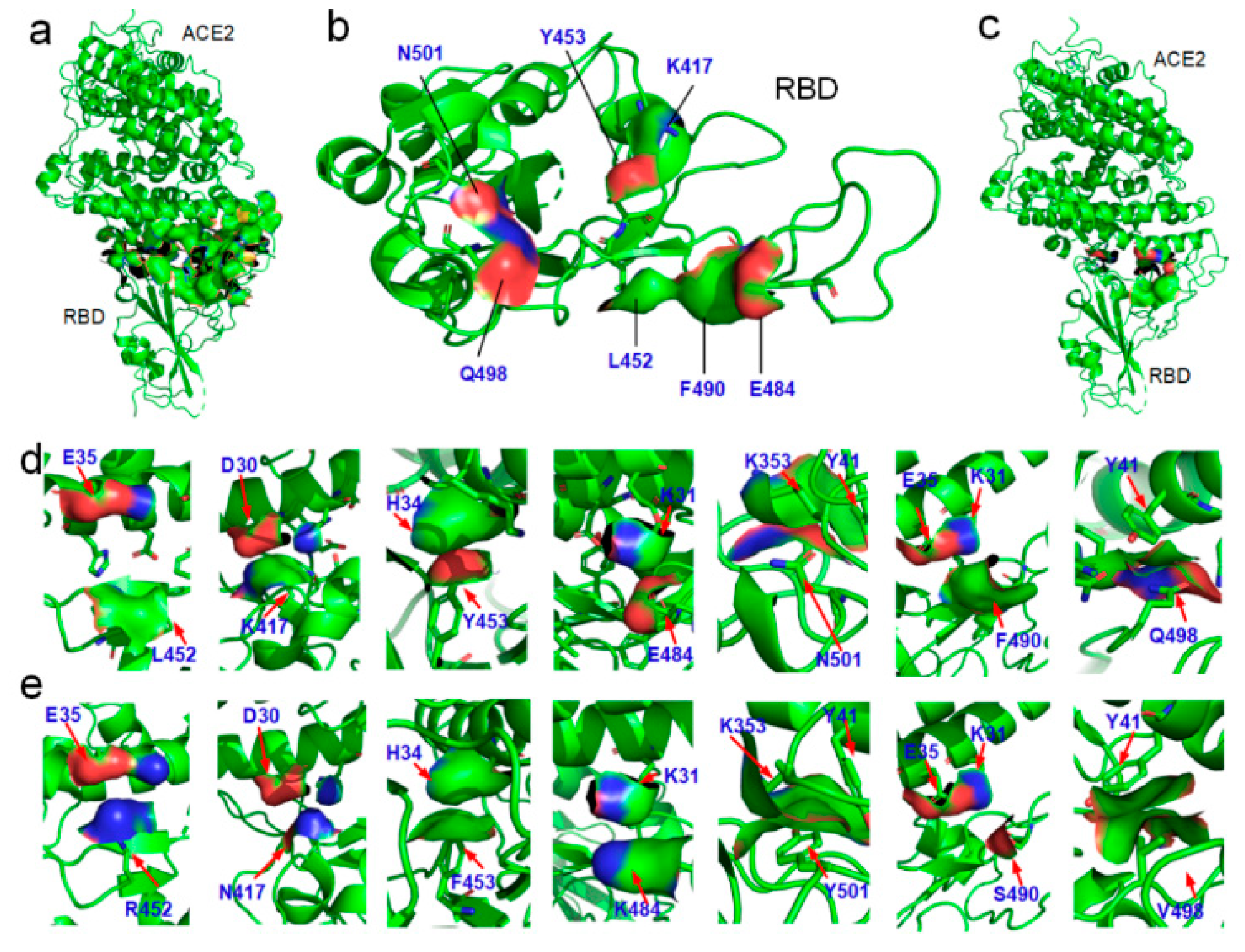
3.2. Variations of the Binding Affinities of SARS-CoV-2 Due to Mutations
4. Materials and Methods
4.1. Protein Structures
4.2. Calculation of Hydrophobic or Hydrophilic Surface Area
4.3. The Parameters of the Equation (1)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Le, T.T.; Cramer, J.P.; Chen, R.; Mayhew, S. Evolution of the COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 667–668. [Google Scholar] [CrossRef]
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Lambson, B.E.; Vermeulen, M.; van den Berg, K.; Rossouw, T.; Boswell, M.; et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 2021, 27, 622–625. [Google Scholar] [CrossRef]
- Jon, C. South Africa suspends use of AstraZeneca’s COVID-19 vaccine after it fails to clearly stop virus variant. Science 2021, 1–7. [Google Scholar]
- Meredith Wadman, J.C. Novavax vaccine delivers 89% efficacy against COVID-19 in U.K.—But is less potent in South Africa. Science 2021. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 1–17. [Google Scholar]
- Wang, R.; Chen, J.; Gao, K.; Wei, G.-W. Vaccine-escape and fast-growing mutations in the United Kingdom, the United States, Singapore, Spain, India, and other COVID-19-devastated countries. Genomics 2021, 113, 2158–2170. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv 2021. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020. [Google Scholar] [CrossRef]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.; Kopp, M.; Hsu, A.; Borgnia, M.; Parks, R.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wang, Q.; Zhou, H.; Yan, J.; Qi, J. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e899. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, C.O.; Jette, C.A.; Abernathy, M.E.; Dam, K.-M.A.; Esswein, S.R.; Gristick, H.B.; Malyutin, A.G.; Sharaf, N.G.; Huey-Tubman, K.E.; Lee, Y.E.; et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature 2020, 588, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Beltramello, M.; Lempp, F.A.; Pinto, D.; Veesler, D.J.S. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science 2020, 370, 950–957. [Google Scholar] [CrossRef]
- Du, S.; Cao, Y.; Zhu, Q.; Yu, P.; Qi, F.; Wang, G.; Du, X.; Bao, L.; Deng, W.; Zhu, H.; et al. Structurally resolved SARS-CoV-2 Antibody shows high efficacy in severely infected hamsters and provides a potent cocktail pairing strategy. Cell 2020, 183, 1013–1023. [Google Scholar] [CrossRef]
- Hurlburt, N.K.; Seydoux, E.; Wan, Y.H.; Edara, V.V.; Stuart, A.B.; Feng, J.; Suthar, M.S.; McGuire, A.T.; Stamatatos, L.; Pancera, M. Structural basis for potent neutralization of SARS-CoV-2 and role of antibody affinity maturation. Nat. Commun. 2020, 11, 5413. [Google Scholar] [CrossRef]
- Kreye, J.; Reincke, M.; Korn Au, H.C.; Sánchez-Sendin, E.; Cell, H.P.J. A therapeutic non-self-reactive SARS-CoV-2 antibody protects from lung pathology in a COVID-19 hamster model. Cell 2020, 183, 1058–1069. [Google Scholar] [CrossRef]
- Kim, C.; Ryu, D.K.; Lee, J.; Kim, Y.I.; Lee, S.Y.J.N.C. A therapeutic neutralizing antibody targeting receptor binding domain of SARS-CoV-2 spike protein. Nat. Commun. 2021, 12, 1–10. [Google Scholar]
- Yao, H.; Sun, Y.; Deng, Y.-Q.; Wang, N.; Tan, Y.; Zhang, N.-N.; Li, X.-F.; Kong, C.; Xu, Y.-P.; Chen, Q.; et al. Rational development of a human antibody cocktail that deploys multiple functions to confer Pan-SARS-CoVs protection. from the Chinese Academy of Medical Sciences. Cell Res. 2021, 31, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Xie, J.; Liu, S.; Wu, J.; Liu, C.; Li, J.; Liu, Y.; Wang, M.; Zhao, H.; Zhang, Y.; et al. Three epitope-distinct human antibodies from RenMab mice neutralize SARS-CoV-2 and cooperatively minimize the escape of mutants. Cell Discov. 2021, 7, 1–11. [Google Scholar] [CrossRef]
- Jennewein, M.; Maccamey, A.; Akins, N.; Feng, J.; Stamatatos, L.J.S.E.J. Isolation and characterization of cross-neutralizing coronavirus antibodies from COVID-19+ subjects. Cell Rep. 2021, 109353. [Google Scholar] [CrossRef]
- Zhou, X.; Ma, F.; Xie, J.; Yuan, M.; Li, Y.; Shaabani, N.; Zhao, F.; Huang, D.; Wu, N.C.; Lee, C.-C.D.; et al. Diverse immunoglobulin gene usage and convergent epitope targeting in neutralizing antibody responses to SARS-CoV-2. Cell Rep. 2021, 35, 109109. [Google Scholar] [CrossRef] [PubMed]
- Rapp, M.; Guo, Y.; Reddem, E.R.; Liu, L.; Sheng, Z.J.C.R. Modular basis for potent SARS-CoV-2 neutralization by a prevalent VH1-2-derived antibody class. Cell Rep. 2021, 35, 108950. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Zhang, G.; Wang, Y.; Zhang, Z.; Guo, Y.J.P.B. Structural basis for SARS-CoV-2 neutralizing antibodies with novel binding epitopes. PLoS Biol. 2021, 19, e3001209. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Yuan, M.; Bangaru, S.; Huang, D.; Wilson, I.A.J.P.P. A natural mutation between SARS-CoV-2 and SARS-CoV determines neutralization by a cross-reactive antibody. PLoS Pathog. 2020, 16, e1009089. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into protein-ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Vakser, Y.A. Protein-protein docking: From interaction to interactome. Biophys. J. 2014, 107, 1785–1793. [Google Scholar] [CrossRef] [Green Version]
- American Association for the Advancement of Science. So much more to know…. Science 2005, 309, 78. [Google Scholar]
- Wang, L.-S. Entropy Growth Is the Manifestation of Spontaneity. J. Thermodyn. 2014, 2014, 387698. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, L.; Kao, Y.-T.; Qiu, W.; Yang, Y.; Okobiah, O.; Zhong, D. Mapping hydration dynamics around a protein surface. Proc. Natl. Acad. Sci. USA 2007, 104, 18461–18466. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Shuai, G.; Xiao-liang, M.; Cheng-yu, H.; Li-ping, S.; Jia-cheng, L.; Xiao-dong, H. Universal initial thermodynamic metastable state of unfolded proteins. Prog. Biochem. Biophys. 2019, 46, 8. [Google Scholar]
- Qiao, B.; Jiménez-Ángeles, F.; Nguyen, T.D.; Olvera de la Cruz, M. Water follows polar and nonpolar protein surface domains. Proc. Natl. Acad. Sci. USA 2019, 116, 19274–19281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Yang, L.; et al. Structure relaxation via long trajectories made stable. Phys. Chem. Chem. Phys. 2017, 19, 24478–24484. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring nature and predicting strength of hydrogen bonds: A correlation analysis between atoms-in-molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Lewis, D.F.V. Hydrogen bonding in human p450-substrate interactions: A major contribution to binding affinity. Sci. World J. 2004, 4, 1074–1082. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, S.J. Hydrogen bonding strength—Measures based on geometric and topological parameters. J. Phys. Organic Chem. 2004, 17, 18–31. [Google Scholar] [CrossRef]
- Grabowski, S.J. A new measure of hydrogen bonding strength—Ab initio and atoms in molecules studies. Chem. Phys. Lett. 2001, 338, 361–366. [Google Scholar] [CrossRef]
- Urban, J.J.; Tillman, B.G.; Cronin, W.A. Fluoroolefins as peptide mimetics: A computational study of structure, charge distribution, hydration, and hydrogen bonding. J. Phys. Chem. A 2006, 110, 11120–11129. [Google Scholar] [CrossRef]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, C.M.; Teixeira, V.H.; Baptista, A.M. Protein structure and dynamics in nonaqueous solvents: Insights from molecular dynamics simulation studies. Biophys. J. 2003, 84, 1628–1641. [Google Scholar] [CrossRef] [Green Version]
- McPherson, A.; Gavira, J.A. Introduction to protein crystallization. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 2–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ma, X.; Guo, S.; Hou, C.; Shi, L.; Zhang, H.; Zheng, B.; Liao, C.; Yang, L.; Ye, L.; He, X. A hydrophobic-interaction-based mechanism triggers docking between the SARS-CoV-2 spike and angiotensin-converting enzyme 2. Glob. Chall. 2020, 4, 2000067. [Google Scholar] [CrossRef] [PubMed]
- Berchanski, A.; Shapira, B.; Eisenstein, M. Hydrophobic complementarity in protein-protein docking. Proteins 2004, 56, 130–142. [Google Scholar] [CrossRef]
- Meyer, E.E.; Rosenberg, K.J.; Israelachvili, J. Recent progress in understanding hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2006, 103, 15739–15746. [Google Scholar] [CrossRef] [Green Version]
- Chothia, C.; Janin, J. Principles of protein-protein recognition. Nature 1975, 256, 705–708. [Google Scholar] [CrossRef]
- Li, J.; Hou, C.; Ma, X.; Guo, S.; Zhang, H.; Shi, L.; Liao, C.; Zheng, B.; Ye, L.; Yang, L.; et al. Entropy-enthalpy compensations fold proteins in precise ways. Int. J. Mol. Sci. 2021, 22, 9653. [Google Scholar] [CrossRef]
- Du, Y.; Wang, H.; Chen, L.; Fang, Q.; Zhang, B.; Jiang, L.; Wu, Z.; Yang, Y.; Zhou, Y.; Chen, B.; et al. Non-RBM mutations impaired SARS-CoV-2 spike protein regulated to the ACE2 receptor based on molecular dynamic simulation. Front. Mol. Biosci. 2021, 8, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef] [PubMed]
- Guruprasad, L. Human SARS CoV-2 spike protein mutations. Proteins Struct. Funct. Bioinform. 2021, 89, 569–576. [Google Scholar] [CrossRef]
- Cui, D.; Ou, S.; Patel, S. Protein-spanning water networks and implications for prediction of protein–protein interactions mediated through hydrophobic effects. Proteins Struct. Funct. Bioinform. 2014, 82, 3312–3326. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Smith, C. Molecular biology genes to proteins, 3rd edition by B.E. Tropp. Biochem. Mol. Biol. Educ. 2008, 36, 318–319. [Google Scholar] [CrossRef]
- Grdadolnik, J.; Merzel, F.; Avbelj, F. Origin of hydrophobicity and enhanced water hydrogen bond strength near purely hydrophobic solutes. Proc. Natl. Acad. Sci. USA 2017, 114, 322–327. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q. The physical origin of hydrophobic effects. Chem. Phys. Lett. 2017, 672, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Wagman, D.; Evans, W.; Parker, V.; Schumm, R.; Nuttall, R.J. The NBS Tables of Chemical Thermodynamic Properties. J. Phys. Chem. Ref. Data. USA 1982, 18, 1807–1812. [Google Scholar]
- Wang, Y.; Liu, M.; Gao, J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc. Natl. Acad. Sci. USA 2020, 117, 13967–13974. [Google Scholar] [CrossRef]
- Newberry, R.W.; Raines, R.T. A prevalent intraresidue hydrogen bond stabilizes proteins. Nat. Chem. Biol. 2016, 12, 1084–1088. [Google Scholar] [CrossRef]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; et al. Structural basis of a shared antibody response to SARS-CoV-2. Science 2020, 369, 1119–1123. [Google Scholar] [CrossRef]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, F.; Shen, C.; Peng, W.; Li, D.; Zhao, C.; Li, Z.; Li, S.; Bi, Y.; Yang, Y.; et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 2020, 368, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor binding site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.C.; Yuan, M.; Liu, H.; Lee, C.-C.D.; Zhu, X.; Bangaru, S.; Torres, J.L.; Caniels, T.G.; Brouwer, P.J.M.; van Gils, M.J.; et al. An alternative binding mode of IGHV3-53 antibodies to the SARS-CoV-2 receptor binding domain. Cell Rep. 2020, 33, 108274. [Google Scholar] [CrossRef]
- Moreira, R.A.; Chwastyk, M.; Baker, J.L.; Guzman, H.V.; Poma, A.B. Quantitative determination of mechanical stability in the novel coronavirus spike protein. Nanoscale 2020, 12, 16409–16413. [Google Scholar] [CrossRef]
- Moreira, R.A.; Guzman, H.V.; Boopathi, S.; Baker, J.L.; Poma, A.B. Characterization of structural and energetic differences between conformations of the SARS-CoV-2 spike. Protein 2020, 13, 5362. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, L.; Li, J.; Guo, S.; Hou, C.; Liao, C.; Shi, L.; Ma, X.; Jiang, S.; Zheng, B.; Fang, Y.; et al. SARS-CoV-2 Variants, RBD Mutations, Binding Affinity, and Antibody Escape. Int. J. Mol. Sci. 2021, 22, 12114. https://doi.org/10.3390/ijms222212114
Yang L, Li J, Guo S, Hou C, Liao C, Shi L, Ma X, Jiang S, Zheng B, Fang Y, et al. SARS-CoV-2 Variants, RBD Mutations, Binding Affinity, and Antibody Escape. International Journal of Molecular Sciences. 2021; 22(22):12114. https://doi.org/10.3390/ijms222212114
Chicago/Turabian StyleYang, Lin, Jiacheng Li, Shuai Guo, Chengyu Hou, Chenchen Liao, Liping Shi, Xiaoliang Ma, Shenda Jiang, Bing Zheng, Yi Fang, and et al. 2021. "SARS-CoV-2 Variants, RBD Mutations, Binding Affinity, and Antibody Escape" International Journal of Molecular Sciences 22, no. 22: 12114. https://doi.org/10.3390/ijms222212114
APA StyleYang, L., Li, J., Guo, S., Hou, C., Liao, C., Shi, L., Ma, X., Jiang, S., Zheng, B., Fang, Y., Ye, L., & He, X. (2021). SARS-CoV-2 Variants, RBD Mutations, Binding Affinity, and Antibody Escape. International Journal of Molecular Sciences, 22(22), 12114. https://doi.org/10.3390/ijms222212114