
RNA Modifications and RNA Metabolism in Neurological Disease Pathogenesis


Abstract
1. Introduction
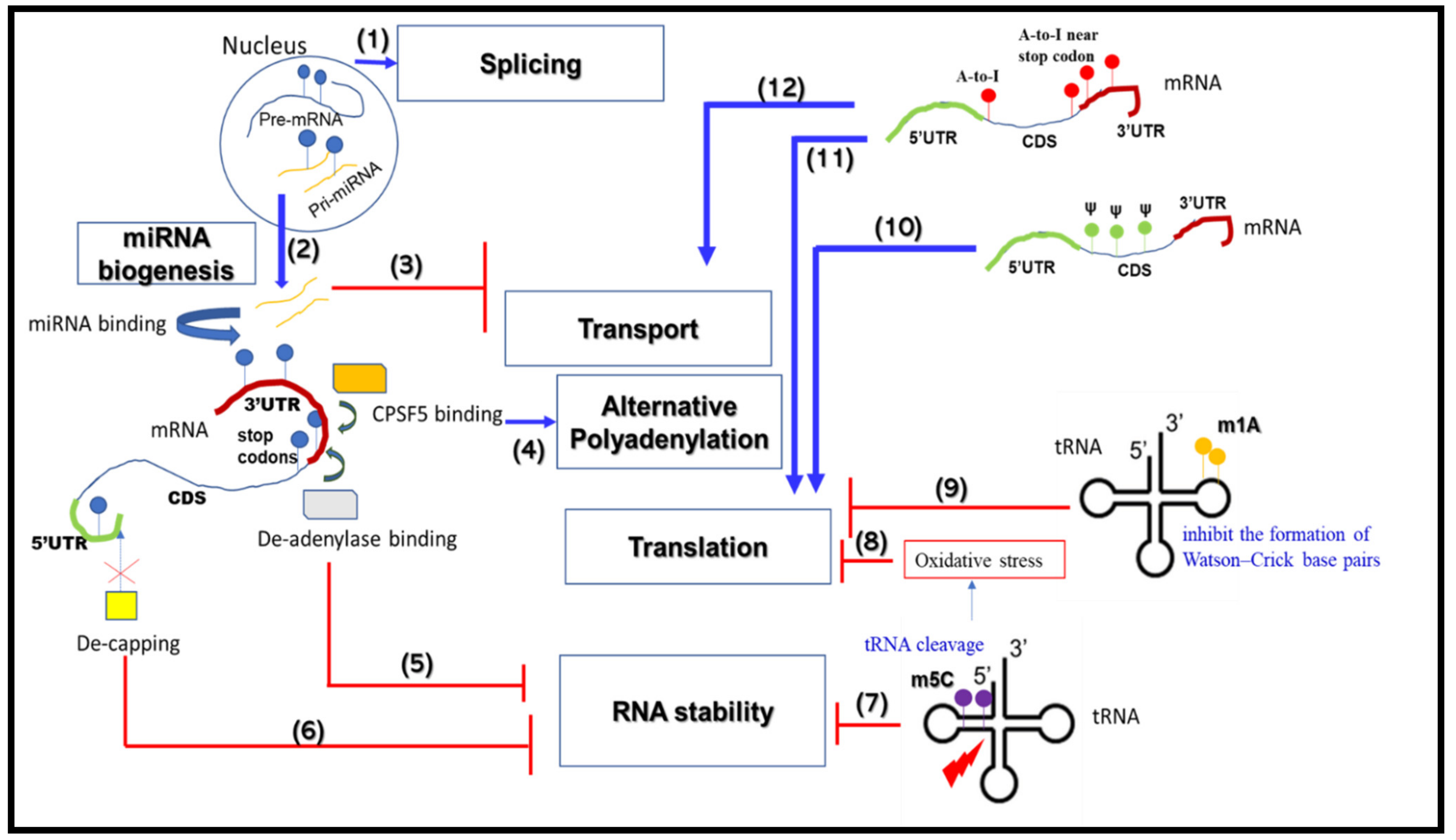
2. RNA Metabolism-Associated Neurological Disease Mechanisms
2.1. mRNA Splicing
2.2. mRNA Alternative Polyadenylation
2.3. mRNA Transport and Translation
2.4. mRNA Stability
2.5. miRNA Biogenesis
2.6. Roles for RBPs in RNA Metabolism and Neurological Diseases
3. RNA Modifications that Change RNA Metabolic Processes
3.1. m6A
3.2. m1A

{kind=link}
| RNA Modification | Effect on RNA Metabolism and/or Protein Function | Reader/Writer/Eraser | Mechanism | Neurological Functions/Diseases | References |
|---|---|---|---|---|---|
| m6A | mRNA stability | Writer: METTL3, METTL14 Reader: YTHDF2, YTHDF3, YTHDC2 | Readers selectively recognize and bind G-(m6 A)-C-containing mRNAs via their CTD, whereas the NTD localizes mRNP complexes at the cellular RNA degradation machinery | Neurogenesis | [135] |
| Splicing, Transport | Reader: YTHDC1 | In the nucleus, this reader recognizes and binds pre-mRNAs with m6A methylation marks and selectively recruits SRSF3 to promote exon inclusion and nuclear-to-cytoplasmic transport of target mRNAs. | Facilitates neuron survival after brain injury and ischemic stroke | [120] | |
| Transport, Translation | Reader: YTHDF1 | This reader recruits the mRNP complex to the cellular transport and translation machinery and activates protein translation | Facilitates learning and memory development | [116] | |
| Translation | Eraser: FTO | In diseased neurons, FTO is translated and accumulates at axons, increasing m6A demethylation and NMDAR1 expression followed by neuronal apoptosis | PD | [115] | |
| Translation | Writer: METTL3 Eraser: FTO | mRNA methylation controls expression of AD-related transcripts, but the underlying mechanism remains obscure | AD | [122] | |
| m1A | tRNA stability/Translation | Writer: MRPP1 | Mitochondrial tRNA methylation causes stabilization of the tRNA to facilitate translational initiation. In disease conditions, improper processing of tRNAs results in reduced mitochondrial protein synthesis | HSD10 disease | [134] |
| m5C | Translation | Writer: NSUN2 | In the absence of tRNA methylation, angiogenin-mediated tRNA cleavage causes an accumulation of tRNA fragments that activate stress-response pathways and impair translation | Dubowitz-like syndrome, Noonan like syndrome | [133] |
| tRNA stability/Translation | Writer: DNMT2 | Methylation of tRNAs enhance their stability and facilitate their translation tRNA cleavage as a result of impaired m5C methylation limits translation in diseased neurons | Brain development and neurogenesis, embryogenesis | [136,137,138] | |
| Pseudoeuridine | mRNA stability, Translation | Writer: PUS1 | Exact mechanism not yet known. It is likely the presence of pseudoeuridine reduces mRNA stability and impairs translation | AD | [139] |
| RNA editing | Transport/Translation | Writer: ADAR2 | AMPA receptor pre-mRNA is edited by ADAR2 to regulate its function. Downregulation of ADAR2 causes reduced editing accompanied with functional defects of AMPAR under disease conditions | Schizophrenia, mood disorders | [140,141] |
3.3. m5C
3.4. Pseudouridine (Ψ)
3.5. RNA Editing
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, E.Y.; Cali, C.P.; Lee, E.B. RNA metabolism in neurodegenerative disease. Dis. Model Mech. 2017, 10, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Prashad, S.; Gopal, P.P. RNA-binding proteins in neurological development and disease. RNA Biol. 2021, 18, 972–987. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Hayakawa-Yano, Y.; Okano, H. RNA regulation went wrong in neurodevelopmental disorders: The example of Msi/Elavl RNA binding proteins. Int. J. Dev. Neurosci. 2016, 55, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Nussbacher, J.K.; Tabet, R.; Yeo, G.W.; Lagier-Tourenne, C. Disruption of RNA Metabolism in Neurological Diseases and Emerging Therapeutic Interventions. Neuron 2019, 102, 294–320. [Google Scholar] [CrossRef] [PubMed]
- Schieweck, R.; Ninkovic, J.; Kiebler, M.A. RNA-binding proteins balance brain function in health and disease. Physiol. Rev. 2021, 101, 1309–1370. [Google Scholar] [CrossRef] [PubMed]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crecy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Esteve-Puig, R.; Bueno-Costa, A.; Esteller, M. Writers, readers and erasers of RNA modifications in cancer. Cancer Lett. 2020, 474, 127–137. [Google Scholar] [CrossRef]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef]
- Vuong, C.K.; Black, D.L.; Zheng, S. The neurogenetics of alternative splicing. Nat. Rev. Neurosci. 2016, 17, 265–281. [Google Scholar] [CrossRef]
- Chabot, B.; Shkreta, L. Defective control of pre-messenger RNA splicing in human disease. J. Cell Biol. 2016, 212, 13–27. [Google Scholar] [CrossRef]
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef]
- Mills, J.D.; Janitz, M. Alternative splicing of mRNA in the molecular pathology of neurodegenerative diseases. Neurobiol. Aging 2012, 33, 1012.e11–1012.e24. [Google Scholar] [CrossRef]
- Li, D.; McIntosh, C.S.; Mastaglia, F.L.; Wilton, S.D.; Aung-Htut, M.T. Neurodegenerative diseases: A hotbed for splicing defects and the potential therapies. Transl. Neurodegener. 2021, 10, 16. [Google Scholar] [CrossRef]
- Sanders, S.J.; Schwartz, G.B.; Farh, K.K. Clinical impact of splicing in neurodevelopmental disorders. Genome Med. 2020, 12, 36. [Google Scholar] [CrossRef]
- Thacker, S.; Sefyi, M.; Eng, C. Alternative splicing landscape of the neural transcriptome in a cytoplasmic-predominant Pten expression murine model of autism-like Behavior. Transl. Psychiatry 2020, 10, 380. [Google Scholar] [CrossRef]
- Dick, F.; Nido, G.S.; Alves, G.W.; Tysnes, O.B.; Nilsen, G.H.; Dolle, C.; Tzoulis, C. Differential transcript usage in the Parkinson’s disease brain. PLoS Genet. 2020, 16, e1009182. [Google Scholar] [CrossRef]
- Perrone, B.; La Cognata, V.; Sprovieri, T.; Ungaro, C.; Conforti, F.L.; Ando, S.; Cavallaro, S. Alternative Splicing of ALS Genes: Misregulation and Potential Therapies. Cell Mol. Neurobiol. 2020, 40, 1–14. [Google Scholar] [CrossRef]
- Arnold, E.S.; Ling, S.C.; Huelga, S.C.; Lagier-Tourenne, C.; Polymenidou, M.; Ditsworth, D.; Kordasiewicz, H.B.; McAlonis-Downes, M.; Platoshyn, O.; Parone, P.A.; et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc. Natl. Acad. Sci. USA 2013, 110, E736–E745. [Google Scholar] [CrossRef]
- Schilling, J.; Broemer, M.; Atanassov, I.; Duernberger, Y.; Vorberg, I.; Dieterich, C.; Dagane, A.; Dittmar, G.; Wanker, E.; van Roon-Mom, W.; et al. Deregulated Splicing Is a Major Mechanism of RNA-Induced Toxicity in Huntington’s Disease. J. Mol. Biol. 2019, 431, 1869–1877. [Google Scholar] [CrossRef]
- Aikawa, T.; Watanabe, T.; Miyazaki, T.; Mikuni, T.; Wakamori, M.; Sakurai, M.; Aizawa, H.; Ishizu, N.; Watanabe, M.; Kano, M.; et al. Alternative splicing in the C-terminal tail of Cav2.1 is essential for preventing a neurological disease in mice. Hum. Mol. Genet. 2017, 26, 3094–3104. [Google Scholar] [CrossRef]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef]
- Ren, F.; Zhang, N.; Zhang, L.; Miller, E.; Pu, J.J. Alternative Polyadenylation: A new frontier in post transcriptional regulation. Biomark. Res. 2020, 8, 67. [Google Scholar] [CrossRef]
- Di Giammartino, D.C.; Nishida, K.; Manley, J.L. Mechanisms and consequences of alternative polyadenylation. Mol. Cell 2011, 43, 853–866. [Google Scholar] [CrossRef]
- Tian, B.; Manley, J.L. Alternative cleavage and polyadenylation: The long and short of it. Trends Biochem. Sci. 2013, 38, 312–320. [Google Scholar] [CrossRef]
- Derti, A.; Garrett-Engele, P.; Macisaac, K.D.; Stevens, R.C.; Sriram, S.; Chen, R.; Rohl, C.A.; Johnson, J.M.; Babak, T. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012, 22, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.; Ji, Z.; Zheng, D.; Luo, W.; Li, W.; You, B.; Park, J.Y.; Yehia, G.; Tian, B. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat. Methods 2013, 10, 133–139. [Google Scholar] [CrossRef]
- Shi, Y. Alternative polyadenylation: New insights from global analyses. RNA 2012, 18, 2105–2117. [Google Scholar] [CrossRef]
- Zhang, H.; Lee, J.Y.; Tian, B. Biased alternative polyadenylation in human tissues. Genome Biol. 2005, 6, R100. [Google Scholar] [CrossRef]
- Liu, D.; Brockman, J.M.; Dass, B.; Hutchins, L.N.; Singh, P.; McCarrey, J.R.; MacDonald, C.C.; Graber, J.H. Systematic variation in mRNA 3′-processing signals during mouse spermatogenesis. Nucleic Acids Res. 2007, 35, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.W.; Rissland, O.S.; Koppstein, D.; Abreu-Goodger, C.; Jan, C.H.; Agarwal, V.; Yildirim, M.A.; Rodriguez, A.; Bartel, D.P. Global analyses of the effect of different cellular contexts on microRNA targeting. Mol. Cell 2014, 53, 1031–1043. [Google Scholar] [CrossRef]
- An, J.J.; Gharami, K.; Liao, G.Y.; Woo, N.H.; Lau, A.G.; Vanevski, F.; Torre, E.R.; Jones, K.R.; Feng, Y.; Lu, B.; et al. Distinct role of long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell 2008, 134, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Berkovits, B.D.; Mayr, C. Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Ji, Z.; Pan, Z.; You, B.; Hoque, M.; Li, W.; Gunderson, S.I.; Tian, B. The conserved intronic cleavage and polyadenylation site of CstF-77 gene imparts control of 3′ end processing activity through feedback autoregulation and by U1 snRNP. PLoS Genet. 2013, 9, e1003613. [Google Scholar] [CrossRef]
- Braz, S.O.; Cruz, A.; Lobo, A.; Bravo, J.; Moreira-Ribeiro, J.; Pereira-Castro, I.; Freitas, J.; Relvas, J.B.; Summavielle, T.; Moreira, A. Expression of Rac1 alternative 3′ UTRs is a cell specific mechanism with a function in dendrite outgrowth in cortical neurons. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 685–694. [Google Scholar] [CrossRef]
- Jereb, S.; Hwang, H.W.; Van Otterloo, E.; Govek, E.E.; Fak, J.J.; Yuan, Y.; Hatten, M.E.; Darnell, R.B. Differential 3′ Processing of Specific Transcripts Expands Regulatory and Protein Diversity Across Neuronal Cell Types. Elife 2018, 7, e34042. [Google Scholar] [CrossRef]
- Yang, Y.; Paul, A.; Bach, T.N.; Huang, Z.J.; Zhang, M.Q. Single-cell alternative polyadenylation analysis delineates GABAergic neuron types. BMC Biol. 2021, 19, 144. [Google Scholar] [CrossRef]
- Grassi, E.; Santoro, R.; Umbach, A.; Grosso, A.; Oliviero, S.; Neri, F.; Conti, L.; Ala, U.; Provero, P.; DiCunto, F.; et al. Choice of Alternative Polyadenylation Sites, Mediated by the RNA-Binding Protein Elavl3, Plays a Role in Differentiation of Inhibitory Neuronal Progenitors. Front. Cell Neurosci. 2018, 12, 518. [Google Scholar] [CrossRef]
- Coutinho, A.M.; Oliveira, G.; Katz, C.; Feng, J.; Yan, J.; Yang, C.; Marques, C.; Ataide, A.; Miguel, T.S.; Borges, L.; et al. MECP2 coding sequence and 3′UTR variation in 172 unrelated autistic patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2007, 144, 475–483. [Google Scholar] [CrossRef]
- Hoffbuhr, K.; Devaney, J.M.; LaFleur, B.; Sirianni, N.; Scacheri, C.; Giron, J.; Schuette, J.; Innis, J.; Marino, M.; Philippart, M.; et al. MeCP2 mutations in children with and without the phenotype of Rett syndrome. Neurology 2001, 56, 1486–1495. [Google Scholar] [CrossRef]
- Newnham, C.M.; Hall-Pogar, T.; Liang, S.; Wu, J.; Tian, B.; Hu, J.; Lutz, C.S. Alternative polyadenylation of MeCP2: Influence of cis-acting elements and trans-acting factors. RNA Biol. 2010, 7, 361–372. [Google Scholar] [CrossRef][Green Version]
- Shibayama, A.; Cook, E.H., Jr.; Feng, J.; Glanzmann, C.; Yan, J.; Craddock, N.; Jones, I.R.; Goldman, D.; Heston, L.L.; Sommer, S.S. MECP2 structural and 3′-UTR variants in schizophrenia, autism and other psychiatric diseases: A possible association with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2004, 128, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; De Rubeis, S.; Carosi, C.; La Fata, G.; Serpa, G.; Raske, C.; Willemsen, R.; Hagerman, P.J.; Bagni, C. Differential usage of transcriptional start sites and polyadenylation sites in FMR1 premutation alleles. Nucleic Acids Res. 2011, 39, 6172–6185. [Google Scholar] [CrossRef] [PubMed]
- Szkop, K.J.; Cooke, P.I.C.; Humphries, J.A.; Kalna, V.; Moss, D.S.; Schuster, E.F.; Nobeli, I. Dysregulation of Alternative Poly-adenylation as a Potential Player in Autism Spectrum Disorder. Front. Mol. Neurosci. 2017, 10, 279. [Google Scholar] [CrossRef] [PubMed]
- Gennarino, V.A.; Alcott, C.E.; Chen, C.A.; Chaudhury, A.; Gillentine, M.A.; Rosenfeld, J.A.; Parikh, S.; Wheless, J.W.; Roeder, E.R.; Horovitz, D.D.; et al. NUDT21-spanning CNVs lead to neuropsychiatric disease and altered MeCP2 abundance via alternative polyadenylation. Elife 2015, 4, e10782. [Google Scholar] [CrossRef]
- Habib, R.; Noureen, N.; Nadeem, N. Decoding Common Features of Neurodegenerative Disorders: From Differentially Expressed Genes to Pathways. Curr. Genomics 2018, 19, 300–312. [Google Scholar] [CrossRef]
- Noori, A.; Mezlini, A.M.; Hyman, B.T.; Serrano-Pozo, A.; Das, S. Systematic review and meta-analysis of human transcriptomics reveals neuroinflammation, deficient energy metabolism, and proteostasis failure across neurodegeneration. Neurobiol. Dis. 2021, 149, 105225. [Google Scholar] [CrossRef]
- Patel, R.; Brophy, C.; Hickling, M.; Neve, J.; Furger, A. Alternative cleavage and polyadenylation of genes associated with protein turnover and mitochondrial function are deregulated in Parkinson’s, Alzheimer’s and ALS disease. BMC Med. Genomics 2019, 12, 60. [Google Scholar] [CrossRef]
- Rhinn, H.; Qiang, L.; Yamashita, T.; Rhee, D.; Zolin, A.; Vanti, W.; Abeliovich, A. Alternative alpha-synuclein transcript usage as a convergent mechanism in Parkinson’s disease pathology. Nat. Commun. 2012, 3, 1084. [Google Scholar] [CrossRef]
- Jenal, M.; Elkon, R.; Loayza-Puch, F.; van Haaften, G.; Kuhn, U.; Menzies, F.M.; Oude Vrielink, J.A.; Bos, A.J.; Drost, J.; Rooijers, K.; et al. The poly(A)-binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell 2012, 149, 538–553. [Google Scholar] [CrossRef]
- Romo, L.; Ashar-Patel, A.; Pfister, E.; Aronin, N. Alterations in mRNA 3′ UTR Isoform Abundance Accompany Gene Expression Changes in Human Huntington’s Disease Brains. Cell Rep. 2017, 20, 3057–3070. [Google Scholar] [CrossRef]
- Melamed, Z.; Lopez-Erauskin, J.; Baughn, M.W.; Zhang, O.; Drenner, K.; Sun, Y.; Freyermuth, F.; McMahon, M.A.; Beccari, M.S.; Artates, J.W.; et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 2019, 22, 180–190. [Google Scholar] [CrossRef]
- Dickson, J.R.; Kruse, C.; Montagna, D.R.; Finsen, B.; Wolfe, M.S. Alternative polyadenylation and miR-34 family members regulate tau expression. J. Neurochem. 2013, 127, 739–749. [Google Scholar] [CrossRef]
- Akbalik, G.; Schuman, E.M. Molecular biology. mRNA, live and unmasked. Science 2014, 343, 375–376. [Google Scholar] [CrossRef]
- Nagano, S.; Jinno, J.; Abdelhamid, R.F.; Jin, Y.; Shibata, M.; Watanabe, S.; Hirokawa, S.; Nishizawa, M.; Sakimura, K.; Onodera, O.; et al. TDP-43 transports ribosomal protein mRNA to regulate axonal local translation in neuronal axons. Acta Neuropathol. 2020, 140, 695–713. [Google Scholar] [CrossRef]
- Shigeoka, T.; Koppers, M.; Wong, H.H.; Lin, J.Q.; Cagnetta, R.; Dwivedy, A.; de Freitas Nascimento, J.; van Tartwijk, F.W.; Strohl, F.; Cioni, J.M.; et al. On-Site Ribosome Remodeling by Locally Synthesized Ribosomal Proteins in Axons. Cell Rep. 2019, 29, 3605–3619.e10. [Google Scholar] [CrossRef]
- Richter, J.D.; Bassell, G.J.; Klann, E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat. Rev. Neurosci. 2015, 16, 595–605. [Google Scholar] [CrossRef]
- Guedes-Dias, P.; Holzbaur, E.L.F. Axonal transport: Driving synaptic function. Science 2019, 366, eaaw9997. [Google Scholar] [CrossRef]
- Nagano, S.; Araki, T. Axonal Transport and Local Translation of mRNA in Neurodegenerative Diseases. Front. Mol. Neurosci. 2021, 14, 697973. [Google Scholar] [CrossRef]
- Banez-Coronel, M.; Ranum, L.P.W. Repeat-associated non-AUG (RAN) translation: Insights from pathology. Lab. Investig. 2019, 99, 929–942. [Google Scholar] [CrossRef]
- Cleary, J.D.; Pattamatta, A.; Ranum, L.P.W. Repeat-associated non-ATG (RAN) translation. J. Biol. Chem. 2018, 293, 16127–16141. [Google Scholar] [CrossRef]
- Bagni, C.; Zukin, R.S. A Synaptic Perspective of Fragile X Syndrome and Autism Spectrum Disorders. Neuron 2019, 101, 1070–1088. [Google Scholar] [CrossRef]
- Chu, J.F.; Majumder, P.; Chatterjee, B.; Huang, S.L.; Shen, C.J. TDP-43 Regulates Coupled Dendritic mRNA Transport-Translation Processes in Co-operation with FMRP and Staufen1. Cell Rep. 2019, 29, 3118–3133.e6. [Google Scholar] [CrossRef]
- Majumder, P.; Chu, J.F.; Chatterjee, B.; Swamy, K.B.; Shen, C.J. Co-regulation of mRNA translation by TDP-43 and Fragile X Syndrome protein FMRP. Acta Neuropathol. 2016, 132, 721–738. [Google Scholar] [CrossRef]
- Wang, E.T.; Taliaferro, J.M.; Lee, J.A.; Sudhakaran, I.P.; Rossoll, W.; Gross, C.; Moss, K.R.; Bassell, G.J. Dysregulation of mRNA Localization and Translation in Genetic Disease. J. Neurosci. 2016, 36, 11418–11426. [Google Scholar] [CrossRef]
- Shukla, S.; Parker, R. Hypo- and Hyper-Assembly Diseases of RNA-Protein Complexes. Trends Mol. Med. 2016, 22, 615–628. [Google Scholar] [CrossRef]
- Buchan, J.R.; Parker, R. Eukaryotic stress granules: The ins and outs of translation. Mol. Cell 2009, 36, 932–941. [Google Scholar] [CrossRef]
- Wang, P.Y.; Lin, Y.M.; Wang, L.H.; Kuo, T.Y.; Cheng, S.J.; Wang, G.S. Reduced cytoplasmic MBNL1 is an early event in a brain-specific mouse model of myotonic dystrophy. Hum. Mol. Genet. 2017, 26, 2247–2257. [Google Scholar] [CrossRef][Green Version]
- Goodman, L.D.; Bonini, N.M. Repeat-associated non-AUG (RAN) translation mechanisms are running into focus for GGGGCC-repeat associated ALS/FTD. Prog. Neurobiol. 2019, 183, 101697. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Nagai, Y.; Ishikawa, K. Insight Into Spinocerebellar Ataxia Type 31 (SCA31) From Drosophila Model. Front. Neurosci. 2021, 15, 472. [Google Scholar] [CrossRef] [PubMed]
- Banez-Coronel, M.; Ayhan, F.; Tarabochia, A.D.; Zu, T.; Perez, B.A.; Tusi, S.K.; Pletnikova, O.; Borchelt, D.R.; Ross, C.A.; Margolis, R.L.; et al. RAN Translation in Huntington Disease. Neuron 2015, 88, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef]
- Eshraghi, M.; Karunadharma, P.P.; Blin, J.; Shahani, N.; Ricci, E.P.; Michel, A.; Urban, N.T.; Galli, N.; Sharma, M.; Ramirez-Jarquin, U.N.; et al. Mutant Huntingtin stalls ribosomes and represses protein synthesis in a cellular model of Huntington disease. Nat. Commun. 2021, 12, 1461. [Google Scholar] [CrossRef]
- Akiyama, T.; Suzuki, N.; Ishikawa, M.; Fujimori, K.; Sone, T.; Kawada, J.; Funayama, R.; Fujishima, F.; Mitsuzawa, S.; Ikeda, K.; et al. Aberrant axon branching via Fos-B dysregulation in FUS-ALS motor neurons. EBioMedicine 2019, 45, 362–378. [Google Scholar] [CrossRef]
- Imperatore, J.A.; McAninch, D.S.; Valdez-Sinon, A.N.; Bassell, G.J.; Mihailescu, M.R. FUS Recognizes G Quadruplex Structures Within Neuronal mRNAs. Front. Mol. Biosci. 2020, 7, 6. [Google Scholar] [CrossRef]
- Inagaki, H.; Hosoda, N.; Tsuiji, H.; Hoshino, S.I. Direct evidence that Ataxin-2 is a translational activator mediating cytoplasmic polyadenylation. J. Biol. Chem. 2020, 295, 15810–15825. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Cascio, F.L.; Sengupta, U.; Garcia, S.; Bhatt, N.; Ellsworth, A.; Heidelman, E.A.; Johnson, O.D.; Doskocil, S.; et al. TDP-43 and Tau Oligomers in Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Neurobiol. Dis. 2020, 146, 105130. [Google Scholar] [CrossRef]
- Houseley, J.; Tollervey, D. The many pathways of RNA degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef]
- Chen, C.Y.; Ezzeddine, N.; Shyu, A.B. Messenger RNA half-life measurements in mammalian cells. Methods Enzymol. 2008, 448, 335–357. [Google Scholar]
- Burow, D.A.; Umeh-Garcia, M.C.; True, M.B.; Bakhaj, C.D.; Ardell, D.H.; Cleary, M.D. Dynamic regulation of mRNA decay during neural development. Neural Dev. 2015, 10, 11. [Google Scholar] [CrossRef]
- Porter, R.S.; Jaamour, F.; Iwase, S. Neuron-specific alternative splicing of transcriptional machineries: Implications for neurodevelopmental disorders. Mol. Cell Neurosci. 2018, 87, 35–45. [Google Scholar] [CrossRef]
- Lee, S.; Wei, L.; Zhang, B.; Goering, R.; Majumdar, S.; Wen, J.; Taliaferro, J.M.; Lai, E.C. ELAV/Hu RNA binding proteins determine multiple programs of neural alternative splicing. PLoS Genet. 2021, 17, e1009439. [Google Scholar] [CrossRef]
- DeBoer, E.M.; Azevedo, R.; Vega, T.A.; Brodkin, J.; Akamatsu, W.; Okano, H.; Wagner, G.C.; Rasin, M.R. Prenatal deletion of the RNA-binding protein HuD disrupts postnatal cortical circuit maturation and behavior. J. Neurosci. 2014, 34, 3674–3686. [Google Scholar] [CrossRef]
- Berto, S.; Usui, N.; Konopka, G.; Fogel, B.L. ELAVL2-regulated transcriptional and splicing networks in human neurons link neurodevelopment and autism. Hum. Mol. Genet. 2016, 25, 2451–2464. [Google Scholar] [CrossRef]
- Cha, I.J.; Lee, D.; Park, S.S.; Chung, C.G.; Kim, S.Y.; Jo, M.G.; Kim, S.Y.; Lee, B.H.; Lee, Y.S.; Lee, S.B. Ataxin-2 Dysregulation Triggers a Compensatory Fragile X Mental Retardation Protein Decrease in Drosophila C4da Neurons. Mol. Cells 2020, 43, 870–879. [Google Scholar]
- Scheckel, C.; Drapeau, E.; Frias, M.A.; Park, C.Y.; Fak, J.; Zucker-Scharff, I.; Kou, Y.; Haroutunian, V.; Ma’ayan, A.; Buxbaum, J.D.; et al. Regulatory consequences of neuronal ELAV-like protein binding to coding and non-coding RNAs in human brain. Elife 2016, 5, e10421. [Google Scholar] [CrossRef]
- Ostrowski, L.A.; Hall, A.C.; Mekhail, K. Ataxin-2: From RNA Control to Human Health and Disease. Genes 2017, 8, 157. [Google Scholar] [CrossRef]
- Alkallas, R.; Fish, L.; Goodarzi, H.; Najafabadi, H.S. Inference of RNA decay rate from transcriptional profiling highlights the regulatory programs of Alzheimer’s disease. Nat. Commun. 2017, 8, 909. [Google Scholar] [CrossRef]
- Costessi, L.; Porro, F.; Iaconcig, A.; Muro, A.F. TDP-43 regulates beta-adducin (Add2) transcript stability. RNA Biol. 2014, 11, 1280–1290. [Google Scholar] [CrossRef]
- Wang, H.; Taguchi, Y.H.; Liu, X. Editorial: miRNAs and Neurological Diseases. Front. Neurol. 2021, 12, 662373. [Google Scholar] [CrossRef]
- Guo, L.; Yin, M.; Wang, Y. CREB1, a direct target of miR-122, promotes cell proliferation and invasion in bladder cancer. Oncol. Lett. 2018, 16, 3842–3848. [Google Scholar] [CrossRef]
- Jasinska, M.; Milek, J.; Cymerman, I.A.; Leski, S.; Kaczmarek, L.; Dziembowska, M. miR-132 Regulates Dendritic Spine Structure by Direct Targeting of Matrix Metalloproteinase 9 mRNA. Mol. Neurobiol. 2016, 53, 4701–4712. [Google Scholar] [CrossRef] [PubMed]
- Rey, R.; Suaud-Chagny, M.F.; Dorey, J.M.; Teyssier, J.R.; d’Amato, T. Widespread transcriptional disruption of the microRNA biogenesis machinery in brain and peripheral tissues of individuals with schizophrenia. Transl. Psychiatry 2020, 10, 376. [Google Scholar] [CrossRef] [PubMed]
- McKeever, P.M.; Schneider, R.; Taghdiri, F.; Weichert, A.; Multani, N.; Brown, R.A.; Boxer, A.L.; Karydas, A.; Miller, B.; Robertson, J.; et al. MicroRNA Expression Levels Are Altered in the Cerebrospinal Fluid of Patients with Young-Onset Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 8826–8841. [Google Scholar] [CrossRef] [PubMed]
- Riancho, J.; Vazquez-Higuera, J.L.; Pozueta, A.; Lage, C.; Kazimierczak, M.; Bravo, M.; Calero, M.; Gonalezalez, A.; Rodriguez, E.; Lleo, A.; et al. MicroRNA Profile in Patients with Alzheimer’s Disease: Analysis of miR-9-5p and miR-598 in Raw and Exosome Enriched Cerebrospinal Fluid Samples. J. Alzheimers Dis. 2017, 57, 483–491. [Google Scholar] [CrossRef]
- Rajgor, D. Macro roles for microRNAs in neurodegenerative diseases. Noncoding RNA Res. 2018, 3, 154–159. [Google Scholar] [CrossRef]
- Rinchetti, P.; Rizzuti, M.; Faravelli, I.; Corti, S. MicroRNA Metabolism and Dysregulation in Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 2617–2630. [Google Scholar] [CrossRef]
- Dong, X.; Cong, S. The Emerging Role of microRNAs in Polyglutamine Diseases. Front. Mol. Neurosci. 2019, 12, 156. [Google Scholar] [CrossRef]
- Lopez Castel, A.; Overby, S.J.; Artero, R. MicroRNA-Based Therapeutic Perspectives in Myotonic Dystrophy. Int. J. Mol. Sci. 2019, 20, 5600. [Google Scholar] [CrossRef]
- Parras, A.; Anta, H.; Santos-Galindo, M.; Swarup, V.; Elorza, A.; Nieto-Gonzalez, J.L.; Pico, S.; Hernandez, I.H.; Diaz-Hernandez, J.I.; Belloc, E.; et al. Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing. Nature 2018, 560, 441–446. [Google Scholar] [CrossRef]
- Carreira-Rosario, A.; Bhargava, V.; Hillebrand, J.; Kollipara, R.K.; Ramaswami, M.; Buszczak, M. Repression of Pumilio Protein Expression by Rbfox1 Promotes Germ Cell Differentiation. Dev. Cell 2016, 36, 562–571. [Google Scholar] [CrossRef]
- Lee, J.A.; Damianov, A.; Lin, C.H.; Fontes, M.; Parikshak, N.N.; Anderson, E.S.; Geschwind, D.H.; Black, D.L.; Martin, K.C. Cytoplasmic Rbfox1 Regulates the Expression of Synaptic and Autism-Related Genes. Neuron 2016, 89, 113–128. [Google Scholar] [CrossRef]
- Wei, C.; Xiao, R.; Chen, L.; Cui, H.; Zhou, Y.; Xue, Y.; Hu, J.; Zhou, B.; Tsutsui, T.; Qiu, J.; et al. RBFox2 Binds Nascent RNA to Globally Regulate Polycomb Complex 2 Targeting in Mammalian Genomes. Mol. Cell 2016, 62, 982. [Google Scholar] [CrossRef]
- Lee, J.E.; Cooper, T.A. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 2009, 37 Pt 6, 1281–1286. [Google Scholar] [CrossRef]
- Ramesh, N.; Daley, E.L.; Gleixner, A.M.; Mann, J.R.; Kour, S.; Mawrie, D.; Anderson, E.N.; Kofler, J.; Donnelly, C.J.; Kiskinis, E.; et al. RNA dependent suppression of C9orf72 ALS/FTD associated neurodegeneration by Matrin-3. Acta Neuropathol. Commun. 2020, 8, 177. [Google Scholar] [CrossRef]
- Tan, M.S.; Yu, J.T.; Tan, L. Bridging integrator 1 (BIN1): Form, function, and Alzheimer’s disease. Trends Mol. Med. 2013, 19, 594–603. [Google Scholar] [CrossRef]
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.W.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A.; et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014, 81, 536–543. [Google Scholar] [CrossRef]
- Coyne, A.N.; Yamada, S.B.; Siddegowda, B.B.; Estes, P.S.; Zaepfel, B.L.; Johannesmeyer, J.S.; Lockwood, D.B.; Pham, L.T.; Hart, M.P.; Cassel, J.A.; et al. Fragile X protein mitigates TDP-43 toxicity by remodeling RNA granules and restoring translation. Hum. Mol. Genet. 2015, 24, 6886–6898. [Google Scholar] [CrossRef]
- Ling, S.C.; Albuquerque, C.P.; Han, J.S.; Lagier-Tourenne, C.; Tokunaga, S.; Zhou, H.; Cleveland, D.W. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc. Natl. Acad. Sci. USA 2010, 107, 13318–13323. [Google Scholar] [CrossRef]
- Lopez-Erauskin, J.; Tadokoro, T.; Baughn, M.W.; Myers, B.; McAlonis-Downes, M.; Chillon-Marinas, C.; Asiaban, J.N.; Artates, J.; Bui, A.T.; Vetto, A.P.; et al. ALS/FTD-Linked Mutation in FUS Suppresses Intra-axonal Protein Synthesis and Drives Disease Without Nuclear Loss-of-Function of FUS. Neuron 2018, 100, 816–830.e7. [Google Scholar] [CrossRef]
- Sellier, C.; Rau, F.; Liu, Y.; Tassone, F.; Hukema, R.K.; Gattoni, R.; Schneider, A.; Richard, S.; Willemsen, R.; Elliott, D.J.; et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010, 29, 1248–1261. [Google Scholar] [CrossRef]
- Conlon, E.G.; Manley, J.L. RNA-binding proteins in neurodegeneration: Mechanisms in aggregate. Genes Dev. 2017, 31, 1509–1528. [Google Scholar] [CrossRef]
- Kelaini, S.; Chan, C.; Cornelius, V.A.; Margariti, A. RNA-Binding Proteins Hold Key Roles in Function, Dysfunction, and Disease. Biology 2021, 10, 366. [Google Scholar] [CrossRef]
- Jung, Y.; Goldman, D. Role of RNA modifications in brain and behavior. Genes Brain Behav. 2018, 17, e12444. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vasquez, E.; Alata Jimenez, N.; Vazquez, N.A.; Strobl-Mazzulla, P.H. Emerging role of dynamic RNA modifications during animal development. Mech. Dev. 2018, 154, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Yen, Y.P.; Chen, J.A. The m(6)A epitranscriptome on neural development and degeneration. J. Biomed. Sci. 2021, 28, 40. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, M.; Huang, H.; Zhu, J.; Song, H.; Zhu, J.; Park, J.; Ji, S.J. Dynamic m6A modification regulates local translation of mRNA in axons. Nucleic Acids Res. 2018, 46, 1412–1423. [Google Scholar] [CrossRef] [PubMed]
- Alarcon, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-methyladenosine marks primary microRNAs for processing. Nature 2015, 519, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Weng, H.; Sun, W.; Qin, X.; Shi, H.; Wu, H.; Zhao, B.S.; Mesquita, A.; Liu, C.; Yuan, C.L.; et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 2018, 20, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.I.; Pan, T. An additional class of m(6)A readers. Nat. Cell Biol. 2018, 20, 230–232. [Google Scholar] [CrossRef]
- Sokpor, G.; Xie, Y.; Nguyen, H.P.; Tuoc, T. Emerging Role of m(6) A Methylome in Brain Development: Implications for Neurological Disorders and Potential Treatment. Front. Cell Dev. Biol. 2021, 9, 656849. [Google Scholar] [CrossRef]
- Livneh, I.; Moshitch-Moshkovitz, S.; Amariglio, N.; Rechavi, G.; Dominissini, D. The m(6)A epitranscriptome: Transcriptome plasticity in brain development and function. Nat. Rev. Neurosci. 2020, 21, 36–51. [Google Scholar] [CrossRef]
- Han, M.; Liu, Z.; Xu, Y.; Liu, X.; Wang, D.; Li, F.; Wang, Y.; Bi, J. Abnormality of m6A mRNA Methylation Is Involved in Alzheimer’s Disease. Front. Neurosci. 2020, 14, 98. [Google Scholar] [CrossRef]
- Dermentzaki, G.; Lotti, F. New Insights on the Role of N (6)-Methyladenosine RNA Methylation in the Physiology and Pathology of the Nervous System. Front. Mol. Biosci. 2020, 7, 555372. [Google Scholar] [CrossRef]
- Mendel, M.; Delaney, K.; Pandey, R.R.; Chen, K.M.; Wenda, J.M.; Vagbo, C.B.; Steiner, F.A.; Homolka, D.; Pillai, R.S. Splice site m(6)A methylation prevents binding of U2AF35 to inhibit RNA splicing. Cell 2021, 184, 3125–3142.e25. [Google Scholar] [CrossRef]
- Shima, H.; Igarashi, K. N 1-methyladenosine (m1A) RNA modification: The key to ribosome control. J. Biochem. 2020, 167, 535–539. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef]
- Zhang, C.; Jia, G. Reversible RNA Modification N(1)-methyladenosine (m(1)A) in mRNA and tRNA. Genomics Proteomics Bioinform. 2018, 16, 155–161. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Zhang, M.; Wang, K.; Chen, Y.; Zhou, J.; Mao, Y.; Lv, J.; Yi, D.; Chen, X.W.; et al. Base-Resolution Mapping Reveals Distinct m(1)A Methylome in Nuclear- and Mitochondrial-Encoded Transcripts. Mol. Cell 2017, 68, 993–1005.e9. [Google Scholar] [CrossRef]
- Mathlin, J.; Le Pera, L.; Colombo, T. A Census and Categorization Method of Epitranscriptomic Marks. Int. J. Mol. Sci. 2020, 21, 4684. [Google Scholar] [CrossRef]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 2017, 551, 251–255. [Google Scholar] [CrossRef]
- Liu, F.; Clark, W.; Luo, G.; Wang, X.; Fu, Y.; Wei, J.; Wang, X.; Hao, Z.; Dai, Q.; Zheng, G.; et al. ALKBH1-Mediated tRNA Demethylation Regulates Translation. Cell 2016, 167, 816–828.e16. [Google Scholar] [CrossRef]
- Zheng, Q.; Gan, H.; Yang, F.; Yao, Y.; Hao, F.; Hong, L.; Jin, L. Cytoplasmic m(1)A reader YTHDF3 inhibits trophoblast invasion by downregulation of m(1)A-methylated IGF1R. Cell Discov. 2020, 6, 12. [Google Scholar] [CrossRef]
- Satterlee, J.S.; Basanta-Sanchez, M.; Blanco, S.; Li, J.B.; Meyer, K.; Pollock, J.; Sadri-Vakili, G.; Rybak-Wolf, A. Novel RNA modifications in the nervous system: Form and function. J. Neurosci. 2014, 34, 15170–15177. [Google Scholar] [CrossRef]
- Bohnsack, M.T.; Sloan, K.E. The mitochondrial epitranscriptome: The roles of RNA modifications in mitochondrial translation and human disease. Cell Mol. Life Sci. 2018, 75, 241–260. [Google Scholar] [CrossRef]
- Kretschmer, J.; Rao, H.; Hackert, P.; Sloan, K.E.; Hobartner, C.; Bohnsack, M.T. The m(6)A reader protein YTHDC2 interacts with the small ribosomal subunit and the 5′-3′ exoribonuclease XRN1. RNA 2018, 24, 1339–1350. [Google Scholar] [CrossRef]
- Chan, C.T.Y.; Pang, Y.L.J.; Deng, W.J.; Babu, I.R.; Dyavaiah, M.; Begley, T.J.; Dedon, P.C. Reprogramming of tRNA modifications controls the oxidative stress response by codon-biased translation of proteins. Nat. Commun. 2012, 3, 1–9. [Google Scholar] [CrossRef]
- Shanmugam, R.; Fierer, J.; Kaiser, S.; Helm, M.; Jurkowski, T.P.; Jeltsch, A. Cytosine methylation of tRNA-Asp by DNMT2 has a role in translation of proteins containing poly-Asp sequences. Cell Discov. 2015, 1, 1–10. [Google Scholar] [CrossRef]
- Rai, K.; Chidester, S.; Zavala, C.V.; Manos, E.J.; James, S.R.; Karpf, A.R.; Jones, D.A.; Cairns, B.R. Dnmt2 functions in the cytoplasm to promote liver, brain, and retina development in zebrafish. Genes Dev. 2007, 21, 261–266. [Google Scholar] [CrossRef]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Kubota-Sakashita, M.; Iwamoto, K.; Bundo, M.; Kato, T. A role of ADAR2 and RNA editing of glutamate receptors in mood disorders and schizophrenia. Mol. Brain 2014, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- La Via, L.; Bonini, D.; Russo, I.; Orlandi, C.; Barlati, S.; Barbon, A. Modulation of dendritic AMPA receptor mRNA trafficking by RNA splicing and editing. Nucleic Acids Res. 2013, 41, 617–631. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Sun, B.F.; Chen, Y.S.; Xu, J.W.; Lai, W.Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-methylcytosine promotes mRNA export-NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 2017, 27, 606–625. [Google Scholar] [CrossRef] [PubMed]
- Schumann, U.; Zhang, H.N.; Sibbritt, T.; Pan, A.Y.; Horvath, A.; Gross, S.; Clark, S.J.; Yang, L.; Preiss, T. Multiple links between 5-methylcytosine content of mRNA and translation. BMC Biol. 2020, 18, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, L.; Han, X.; Yang, W.L.; Zhang, M.M.; Ma, H.L.; Sun, B.F.; Li, A.; Xia, J.; Chen, J.; et al. RNA 5-Methylcytosine Facilitates the Maternal-to-Zygotic Transition by Preventing Maternal mRNA Decay. Mol. Cell 2019, 75, 1188–1202. [Google Scholar] [CrossRef] [PubMed]
- David, R.; Burgess, A.; Parker, B.; Li, J.; Pulsford, K.; Sibbritt, T.; Preiss, T.; Searle, I.R. Transcriptome-Wide Mapping of RNA 5-Methylcytosine in Arabidopsis mRNAs and Noncoding RNAs. Plant Cell 2017, 29, 445–460. [Google Scholar] [CrossRef]
- Xue, S.L.; Xu, H.; Sun, Z.; Shen, H.; Chen, S.H.; Ouyang, J.; Zhou, Q.Q.; Hu, X.M.; Cui, H.M. Depletion of TRDMT1 affects 5-methylcytosine modification of mRNA and inhibits HEK293 cell proliferation and migration. Biochem. Biophys. Res. Commun. 2019, 520, 60–66. [Google Scholar] [CrossRef]
- Hussain, S.; Sajini, A.A.; Blanco, S.; Dietmann, S.; Lombard, P.; Sugimoto, Y.; Paramor, M.; Gleeson, J.G.; Odom, D.T.; Ule, J.; et al. NSun2-Mediated Cytosine-5 Methylation of Vault Noncoding RNA Determines Its Processing into Regulatory Small RNAs. Cell Rep. 2013, 4, 255–261. [Google Scholar] [CrossRef]
- Reid, R.; Greene, P.J.; Santi, D.V. Exposition of a family of RNA m(5)C methyltransferases from searching genomic and proteomic sequences. Nucleic Acids Res. 1999, 27, 3138–3145. [Google Scholar] [CrossRef]
- Bohnsack, K.E.; Hobartner, C.; Bohnsack, M.T. Eukaryotic 5-methylcytosine (m(5)C) RNA Methyltransferases: Mechanisms, Cellular Functions, and Links to Disease. Genes 2019, 10, 102. [Google Scholar] [CrossRef]
- Van Haute, L.; Lee, S.Y.; McCann, B.J.; Powell, C.A.; Bansal, D.; Vasiliauskaite, L.; Garone, C.; Shin, S.; Kim, J.S.; Frye, M.; et al. NSUN2 introduces 5-methylcytosines in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2019, 47, 8720–8733. [Google Scholar] [CrossRef]
- Aguilo, F.; Li, S.D.; Balasubramaniyan, N.; Sancho, A.; Benko, S.; Zhang, F.; Vashisht, A.; Rengasamy, M.; Andino, B.; Chen, C.H.; et al. Deposition of 5-Methylcytosine on Enhancer RNAs Enables the Coactivator Function of PGC-1 alpha. Cell Rep. 2016, 14, 479–492. [Google Scholar] [CrossRef]
- Liu, Y.Q.; Santi, D.V. m(5)C RNA and m(5)C DNA methyl transferases use different cysteine residues as catalysts. Proc. Natl. Acad. Sci. USA 2000, 97, 8263–8265. [Google Scholar] [CrossRef]
- King, M.Y.; Redman, K.L. RNA Methyltransferases utilize two cysteine residues in the formation of 5-methylcytosine. Biochemistry 2002, 41, 11218–11225. [Google Scholar] [CrossRef]
- Huang, W.; Lan, M.D.; Qi, C.B.; Zheng, S.J.; Wei, S.Z.; Yuan, B.F.; Feng, Y.Q. Formation and determination of the oxidation products of 5-methylcytosine in RNA. Chem. Sci. 2016, 7, 5495–5502. [Google Scholar] [CrossRef]
- Schosserer, M.; Minois, N.; Angerer, T.B.; Amring, M.; Dellago, H.; Harreither, E.; Calle-Perez, A.; Pircher, A.; Gerstl, M.P.; Pfeifenberger, S.; et al. Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat. Commun. 2015, 6, 1–7. [Google Scholar] [CrossRef]
- Tuorto, F.; Herbst, F.; Alerasool, N.; Bender, S.; Popp, O.; Federico, G.; Reitter, S.; Liebers, R.; Stoecklin, G.; Grone, H.J.; et al. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J. 2015, 34, 2350–2362. [Google Scholar] [CrossRef]
- Cui, X.A.; Liang, Z.; Shen, L.S.; Zhang, Q.; Bao, S.J.; Geng, Y.K.; Zhang, B.; Leo, V.; Vardy, L.A.; Lu, T.G.; et al. 5-Methylcytosine RNA Methylation in &ITArabidopsis Thaliana&IT. Mol. Plant. 2017, 10, 1387–1399. [Google Scholar]
- Huang, T.; Chen, W.; Liu, J.; Gu, N.; Zhang, R. Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat. Struct. Mol. Biol. 2019, 26, 380–388. [Google Scholar] [CrossRef]
- Li, Q.; Li, X.; Tang, H.; Jiang, B.; Dou, Y.; Gorospe, M.; Wang, W. NSUN2-Mediated m5C Methylation and METTL3/METTL14-Mediated m6A Methylation Cooperatively Enhance p21 Translation. J. Cell Biochem. 2017, 118, 2587–2598. [Google Scholar] [CrossRef]
- Blanco, S.; Frye, M. Role of RNA methyltransferases in tissue renewal and pathology. Curr. Opin. Cell Biol. 2014, 31, 1–7. [Google Scholar] [CrossRef]
- Blanco, S.; Kurowski, A.; Nichols, J.; Watt, F.M.; Benitah, S.A.; Frye, M. The RNA-Methyltransferase Misu (NSun2) Poises Epidermal Stem Cells to Differentiate. PLoS Genet. 2011, 7, e1002403. [Google Scholar] [CrossRef]
- Hussain, S.; Tuorto, F.; Menon, S.; Blanco, S.; Cox, C.; Flores, J.V.; Watt, S.; Kudo, N.R.; Lyko, F.; Frye, M. The Mouse Cytosine-5 RNA Methyltransferase NSun2 Is a Component of the Chromatoid Body and Required for Testis Differentiation. Mol. Cell Biol. 2013, 33, 1561–1570. [Google Scholar] [CrossRef]
- Khan, M.A.; Rafiq, M.A.; Noor, A.; Hussain, S.; Flores, J.V.; Rupp, V.; Vincent, A.K.; Malli, R.; Ali, G.; Khan, F.S.; et al. Mutation in NSUN2, which Encodes an RNA Methyltransferase, Causes Autosomal-Recessive Intellectual Disability. Am. J. Hum. Genet. 2012, 90, 856–863. [Google Scholar] [CrossRef]
- Abbasi-Moheb, L.; Mertel, S.; Gonsior, M.; Nouri-Vahid, L.; Kahrizi, K.; Cirak, S.; Wieczorek, D.; Motazacker, M.M.; Esmaeeli-Nieh, S.; Cremer, K.; et al. Mutations in NSUN2 Cause Autosomal-Recessive Intellectual Disability. Am. J. Hum. Genet. 2012, 90, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Lee, J.H.; Lee, J.E.; Blanco, S.; Nickerson, E.; Gabriel, S.; Frye, M.; Al-Gazali, L.; Gleeson, J.G. Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz-like syndrome. J. Med. Genet. 2012, 49, 380–385. [Google Scholar] [CrossRef]
- Blaze, J.; Navickas, A.; Phillips, H.L.; Heissel, S.; Plaza-Jennings, A.; Miglani, S.; Asgharian, H.; Foo, M.; Katanski, C.D.; Watkins, C.P.; et al. Neuronal Nsun2 deficiency produces tRNA epitranscriptomic alterations and proteomic shifts impacting synaptic signaling and behavior. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Flores, J.V.; Cordero-Espinoza, L.; Oeztuerk-Winder, F.; Andersson-Rolf, A.; Selmi, T.; Blanco, S.; Tailor, J.; Dietmann, S.; Frye, M. Cytosine-5 RNA Methylation Regulates Neural Stem Cell Differentiation and Motility. Stem Cell Rep. 2017, 8, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Van Haute, L.; Dietmann, S.; Kremer, L.; Hussain, S.; Pearce, S.F.; Powell, C.A.; Rorbach, J.; Lantaff, R.; Blanco, S.; Sauer, S.; et al. Deficient methylation and formylation of mt- tRNA(Met) wobble cytosine in a patient carrying mutations in NSUN3. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Borchardt, E.K.; Martinez, N.M.; Gilbert, W.V. Regulation and Function of RNA Pseudouridylation in Human Cells. Annu. Rev. Genet. 2020, 54, 309–336. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.T.; Dimitrova, D.G.; Dinges, N.; Lence, T.; Worpenberg, L.; Carre, C.; Roignant, J.Y. The Emerging Field of Epitranscriptomics in Neurodevelopmental and Neuronal Disorders. Front. Bioeng. Biotechnol. 2018, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Nainar, S.; Marshall, P.R.; Tyler, C.R.; Spitale, R.C.; Bredy, T.W. Evolving insights into RNA modifications and their functional diversity in the brain. Nat. Neurosci. 2016, 19, 1292–1298. [Google Scholar] [CrossRef]
- Song, W.; Ressl, S.; Tracey, W.D. Loss of Pseudouridine Synthases in the RluA Family Causes Hypersensitive Nociception in Drosophila. G3-Genes Genomes Genet. 2020, 10, 4425–4438. [Google Scholar] [CrossRef]
- Lee, S.H.; Kim, I.; Chung, B.C. Increased urinary level of oxidized nucleosides in patients with mild-to-moderate Alzheimer’s disease. Clin. Biochem. 2007, 40, 936–938. [Google Scholar] [CrossRef]
- Delorimier, E.; Hinman, M.N.; Copperman, J.; Datta, K.; Guenza, M.; Berglund, J.A. Pseudouridine Modification Inhibits Muscleblind-like 1 (MBNL1) Binding to CCUG Repeats and Minimally Structured RNA through Reduced RNA Flexibility. J. Biol. Chem. 2017, 292, 4350–4357. [Google Scholar] [CrossRef]
- Cao, M.; Dona, M.; Valentino, L.; Semplicini, C.; Maresca, A.; Cassina, M.; Torraco, A.; Galletta, E.; Manfioli, V.; Soraru, G.; et al. Clinical and molecular study in a long-surviving patient with MLASA syndrome due to novel PUS1 mutations. Neurogenetics 2016, 17, 65–70. [Google Scholar] [CrossRef]
- Diez-Roux, G.; Banfi, S.; Sultan, M.; Geffers, L.; Anand, S.; Rozado, D.; Magen, A.; Canidio, E.; Pagani, M.; Peluso, I.; et al. A High-Resolution Anatomical Atlas of the Transcriptome in the Mouse Embryo. PLoS Biol. 2011, 9, e1000582. [Google Scholar] [CrossRef]
- Shaheen, R.; Han, L.; Faqeih, E.; Ewida, N.; Alobeid, E.; Phizicky, E.M.; Alkuraya, F.S. A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum. Genet. 2016, 135, 707–713. [Google Scholar] [CrossRef]
- De Brouwer, A.P.M.; Abou Jamra, R.; Kortel, N.; Soyris, C.; Polla, D.L.; Safra, M.; Zisso, A.; Powell, C.A.; Rebelo-Guiomar, P.; Dinges, N.; et al. Variants in PUS7 Cause Intellectual Disability with Speech Delay, Microcephaly, Short Stature, and Aggressive Behavior. Am. J. Hum. Genet. 2018, 103, 1045–1052. [Google Scholar] [CrossRef]
- Shaheen, R.; Tasak, M.; Maddirevula, S.; Abdel-Salam, G.M.H.; Sayed, I.S.M.; Alazami, A.M.; Al-Sheddi, T.; Alobeid, E.; Phizicky, E.M.; Alkuraya, F.S. PUS7 mutations impair pseudouridylation in humans and cause intellectual disability and microcephaly. Hum. Genet. 2019, 138, 231–239. [Google Scholar] [CrossRef]
- Heiss, N.S.; Bachner, D.; Salowsky, R.; Kolb, A.; Kioschis, P.; Poustka, A. Gene structure and expression of the mouse dyskeratosis congenita gene, Dkc1. Genomics 2000, 67, 153–163. [Google Scholar] [CrossRef]
- Christofi, T.; Zaravinos, A. RNA editing in the forefront of epitranscriptomics and human health. J. Transl. Med. 2019, 17, 319. [Google Scholar] [CrossRef] [PubMed]
- Bar-Yaacov, D.; Mordret, E.; Towers, R.; Biniashvili, T.; Soyris, C.; Schwartz, S.; Dahan, O.; Pilpel, Y. RNA editing in bacteria recodes multiple proteins and regulates an evolutionarily conserved toxin-antitoxin system. Genome Res. 2017, 27, 1696–1703. [Google Scholar] [CrossRef] [PubMed]
- Grice, L.F.; Degnan, B.M. The origin of the ADAR gene family and animal RNA editing. BMC Evol. Biol. 2015, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- Keegan, L.P.; McGurk, L.; Palavicini, J.P.; Brindle, J.; Paro, S.; Li, X.; Rosenthal, J.J.; O’Connell, M.A. Functional conservation in human and Drosophila of Metazoan ADAR2 involved in RNA editing: Loss of ADAR1 in insects. Nucleic Acids Res. 2011, 39, 7249–7262. [Google Scholar] [CrossRef] [PubMed]
- Slavov, D.; Crnogorac-Jurcevic, T.; Clark, M.; Gardiner, K. Comparative analysis of the DRADA A-to-I RNA editing gene from mammals, pufferfish and zebrafish. Gene 2000, 250, 53–60. [Google Scholar] [CrossRef]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004, 2, e391. [Google Scholar] [CrossRef] [PubMed]
- Jinnah, H.; Ulbricht, R.J. Using mouse models to unlock the secrets of non-synonymous RNA editing. Methods 2019, 156, 40–45. [Google Scholar] [CrossRef]
- Kawahara, Y.; Megraw, M.; Kreider, E.; Iizasa, H.; Valente, L.; Hatzigeorgiou, A.G.; Nishikura, K. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 2008, 36, 5270–5280. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef]
- Tonkin, L.A.; Saccomanno, L.; Morse, D.P.; Brodigan, T.; Krause, M.; Bass, B.L. RNA editing by ADARs is important for normal behavior in Caenorhabditis elegans. EMBO J. 2002, 21, 6025–6035. [Google Scholar] [CrossRef]
- Palladino, M.J.; Keegan, L.P.; O’Connell, M.A.; Reenan, R.A. dADAR, a Drosophila double-stranded RNA-specific adenosine deaminase is highly developmentally regulated and is itself a target for RNA editing. RNA 2000, 6, 1004–1018. [Google Scholar] [CrossRef]
- Torres, A.G.; Pineyro, D.; Filonava, L.; Stracker, T.H.; Batlle, E.; Ribas de Pouplana, L. A-to-I editing on tRNAs: Biochemical, biological and evolutionary implications. FEBS Lett. 2014, 588, 4279–4286. [Google Scholar] [CrossRef]
- Wolf, J.; Gerber, A.P.; Keller, W. tadA, an essential tRNA-specific adenosine deaminase from Escherichia coli. EMBO J. 2002, 21, 3841–3851. [Google Scholar] [CrossRef]
- Knisbacher, B.A.; Gerber, D.; Levanon, E.Y. DNA Editing by APOBECs: A Genomic Preserver and Transformer. Trends Genet. 2016, 32, 16–28. [Google Scholar] [CrossRef]
- Smith, H.C.; Bennett, R.P.; Kizilyer, A.; McDougall, W.M.; Prohaska, K.M. Functions and regulation of the APOBEC family of proteins. Semin. Cell Dev. Biol. 2012, 23, 258–268. [Google Scholar] [CrossRef]
- Krishnan, A.; Iyer, L.M.; Holland, S.J.; Boehm, T.; Aravind, L. Diversification of AID/APOBEC-like deaminases in metazoa: Multiplicity of clades and widespread roles in immunity. Proc. Natl. Acad. Sci. USA 2018, 115, E3201–E3210. [Google Scholar] [CrossRef]
- Iyer, L.M.; Zhang, D.; Rogozin, I.B.; Aravind, L. Evolution of the deaminase fold and multiple origins of eukaryotic editing and mutagenic nucleic acid deaminases from bacterial toxin systems. Nucleic Acids Res. 2011, 39, 9473–9497. [Google Scholar] [CrossRef]
- Wang, I.X.; So, E.; Devlin, J.L.; Zhao, Y.; Wu, M.; Cheung, V.G. ADAR regulates RNA editing, transcript stability, and gene expression. Cell Rep. 2013, 5, 849–860. [Google Scholar] [CrossRef]
- Solomon, O.; Di Segni, A.; Cesarkas, K.; Porath, H.T.; Marcu-Malina, V.; Mizrahi, O.; Stern-Ginossar, N.; Kol, N.; Farage-Barhom, S.; Glick-Saar, E.; et al. RNA editing by ADAR1 leads to context-dependent transcriptome-wide changes in RNA secondary structure. Nat. Commun. 2017, 8, 1440. [Google Scholar] [CrossRef]
- Correia de Sousa, M.; Gjorgjieva, M.; Dolicka, D.; Sobolewski, C.; Foti, M. Deciphering miRNAs’ Action through miRNA Editing. Int. J. Mol. Sci. 2019, 20, 6249. [Google Scholar] [CrossRef]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef]
- Gassner, F.J.; Zaborsky, N.; Buchumenski, I.; Levanon, E.Y.; Gatterbauer, M.; Schubert, M.; Rauscher, S.; Hebenstreit, D.; Nadeu, F.; Campo, E.; et al. RNA editing contributes to epitranscriptome diversity in chronic lymphocytic leukemia. Leukemia 2021, 35, 1053–1063. [Google Scholar] [CrossRef]
- Peng, X.; Xu, X.; Wang, Y.; Hawke, D.H.; Yu, S.; Han, L.; Zhou, Z.; Mojumdar, K.; Jeong, K.J.; Labrie, M.; et al. A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer Cell 2018, 33, 817–828.e7. [Google Scholar] [CrossRef]
- Konen, L.M.; Wright, A.L.; Royle, G.A.; Morris, G.P.; Lau, B.K.; Seow, P.W.; Zinn, R.; Milham, L.T.; Vaughan, C.W.; Vissel, B. A new mouse line with reduced GluA2 Q/R site RNA editing exhibits loss of dendritic spines, hippocampal CA1-neuron loss, learning and memory impairments and NMDA receptor-independent seizure vulnerability. Mol. Brain 2020, 13, 27. [Google Scholar] [CrossRef]
- Behm, M.; Ohman, M. RNA Editing: A Contributor to Neuronal Dynamics in the Mammalian Brain. Trends Genet. 2016, 32, 165–175. [Google Scholar] [CrossRef]
- Sapiro, A.L.; Shmueli, A.; Henry, G.L.; Li, Q.; Shalit, T.; Yaron, O.; Paas, Y.; Billy Li, J.; Shohat-Ophir, G. Illuminating spatial A-to-I RNA editing signatures within the Drosophila brain. Proc. Natl. Acad. Sci. USA 2019, 116, 2318–2327. [Google Scholar] [CrossRef]
- Shimokawa, T.; Rahman, M.F.; Tostar, U.; Sonkoly, E.; Stahle, M.; Pivarcsi, A.; Palaniswamy, R.; Zaphiropoulos, P.G. RNA editing of the GLI1 transcription factor modulates the output of Hedgehog signaling. RNA Biol. 2013, 10, 321–333. [Google Scholar] [CrossRef]
- Zhang, C.; Frias, M.A.; Mele, A.; Ruggiu, M.; Eom, T.; Marney, C.B.; Wang, H.; Licatalosi, D.D.; Fak, J.J.; Darnell, R.B. Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science 2010, 329, 439–443. [Google Scholar] [CrossRef]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E.; et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014, 24, 365–376. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K.; et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Costa Cruz, P.H.; Kawahara, Y. RNA Editing in Neurological and Neurodegenerative Disorders. Methods Mol. Biol. 2021, 2181, 309–330. [Google Scholar] [PubMed]
- Khermesh, K.; D’Erchia, A.M.; Barak, M.; Annese, A.; Wachtel, C.; Levanon, E.Y.; Picardi, E.; Eisenberg, E. Reduced levels of protein recoding by A-to-I RNA editing in Alzheimer’s disease. RNA 2016, 22, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Bhogal, B.; Jepson, J.E.; Savva, Y.A.; Pepper, A.S.; Reenan, R.A.; Jongens, T.A. Modulation of dADAR-dependent RNA editing by the Drosophila fragile X mental retardation protein. Nat. Neurosci. 2011, 14, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Shamay-Ramot, A.; Khermesh, K.; Porath, H.T.; Barak, M.; Pinto, Y.; Wachtel, C.; Zilberberg, A.; Lerer-Goldshtein, T.; Efroni, S.; Levanon, E.Y.; et al. Fmrp Interacts with Adar and Regulates RNA Editing, Synaptic Density and Locomotor Activity in Zebrafish. PLoS Genet. 2015, 11, e1005702. [Google Scholar] [CrossRef]
- Filippini, A.; Bonini, D.; Lacoux, C.; Pacini, L.; Zingariello, M.; Sancillo, L.; Bosisio, D.; Salvi, V.; Mingardi, J.; La Via, L.; et al. Absence of the Fragile X Mental Retardation Protein results in defects of RNA editing of neuronal mRNAs in mouse. RNA Biol. 2017, 14, 1580–1591. [Google Scholar] [CrossRef]
- Tran, S.S.; Jun, H.I.; Bahn, J.H.; Azghadi, A.; Ramaswami, G.; Van Nostrand, E.L.; Nguyen, T.B.; Hsiao, Y.E.; Lee, C.; Pratt, G.A.; et al. Widespread RNA editing dysregulation in brains from autistic individuals. Nat. Neurosci. 2019, 22, 25–36. [Google Scholar] [CrossRef]

| Disease Type | Altered RNA Metabolism Pathway | RBP(s) Involved | Mechanisms | Neurological Disease(s) | References |
|---|---|---|---|---|---|
| Neuro developmental diseases | Splicing, Translation | CPEB4 | Missplicing of CPEB4 causes reduced inclusion of a neuron-specific microexon, leading to diminished expression of the Cpeb4 transcript that activates translation of mRNAs via polyadenylation under normal conditions | ASD | [99] |
| Splicing Translation, mRNA stability, miRNA biogenesis | RBFOX1, RBFOX2 (RBM9), RBFOX3 (Neun) | RBFOX1 binds to the 3′-UTR of its target mRNAs and regulates:
| ASD | [23,100,101,102] | |
| Transport, Translation | FMRP | CGG repeat expansion beyond 200 (>200) at the 5′-UTR of FMR1 affects protein expression, resulting in dysregulated spatio-temporal transport/translation of dendritic mRNAs | FXS | [64] | |
| APA | NUDT21 | Elevated amount of NUDT21, a subunit of pre-mRNA cleavage factor Im, due to copy number variation causes abnormal usage of polyadenylation sites, resulting in the generation of an inefficiently translated long isoform of MeCP2 protein. | Neuropsychia tric disease | [44] | |
| Neuro degenerative diseases | Splicing | PRPF8 | Mutated Huntingtin (HTT) traps PRPF8 (a splicing factor) to cause CREB1 mis-splicing | HD | [18] |
| Translation | HTT | Mutant HTT stalls ribosomes | HD | [72] | |
| Splicing | MBNL family proteins | RNA corresponding to expanded microsatellite repeats in DMPK traps MBNL-family proteins, impairing their normal function in splicing | DM | [103] | |
| Translation | ATAXIN-2 | CAG expansion in the reading frame of ATAXIN-2 causes loss of protein function that, under normal conditions, acts as an mRNA translation activator via polyadenylation | SCA2, ALS | [75] | |
| RAN Translation, Abnormal RNA structure (RNA foci) | Matrin-3 | GGGGCC repeat expansion mutation in the C9orf72 gene causes sequestration of Matrin-3 at the RNA foci and RAN translated peptides and loss of function of Matrin-3 | FTLD, ALS | [104] | |
| mRNA stability, Splicing, Translation | nELAVL | nELAVL regulates disease-specific splicing of the pre-mRNAs Picalm and Bin1 by incorporating exons 13 and 6a, respectively. The proteins corresponding to these spliced isoforms have been implicated in trafficking of amyloid precursor protein | AD | [105] | |
| Transport, Translation, miRNA biogenesis | TDP-43 |
| FTLD, ALS | [63,106,107,108] | |
| Transport, Translation | FUS | Normal FUS functions such as axonal transport/translation of mRNAs are adversely impacted in diseased neurons. Under disease conditions, the altered intracellular localization of FUS disrupts its functions as an RBP | FTLD, ALS | [109] | |
| Splicing, miRNA biogenesis | hnRNPs, MBNL1 | mRNA corresponding to shorter CGG repeat expansions (<200) in the 5′UTR of FMR1 sequester many RBPs, e.g., hnRNPs and MBNL1 | Fragile X-associated tremor/ataxia syndrome (FXTAS) | [110] | |
| APA | α-synuclein | Presence of an extended 3′-UTR region in α-synuclein transcript impacts accumulation of α-synuclein protein that is redirected away from synaptic terminals towards mitochondria | PD | [48] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatterjee, B.; Shen, C.-K.J.; Majumder, P. RNA Modifications and RNA Metabolism in Neurological Disease Pathogenesis. Int. J. Mol. Sci. 2021, 22, 11870. https://doi.org/10.3390/ijms222111870
Chatterjee B, Shen C-KJ, Majumder P. RNA Modifications and RNA Metabolism in Neurological Disease Pathogenesis. International Journal of Molecular Sciences. 2021; 22(21):11870. https://doi.org/10.3390/ijms222111870
Chicago/Turabian StyleChatterjee, Biswanath, Che-Kun James Shen, and Pritha Majumder. 2021. "RNA Modifications and RNA Metabolism in Neurological Disease Pathogenesis" International Journal of Molecular Sciences 22, no. 21: 11870. https://doi.org/10.3390/ijms222111870
APA StyleChatterjee, B., Shen, C.-K. J., & Majumder, P. (2021). RNA Modifications and RNA Metabolism in Neurological Disease Pathogenesis. International Journal of Molecular Sciences, 22(21), 11870. https://doi.org/10.3390/ijms222111870