
RNA Editing: A New Therapeutic Target in Amyotrophic Lateral Sclerosis and Other Neurological Diseases

Abstract
1. Introduction
2. RNA Editing and Neurological Diseases
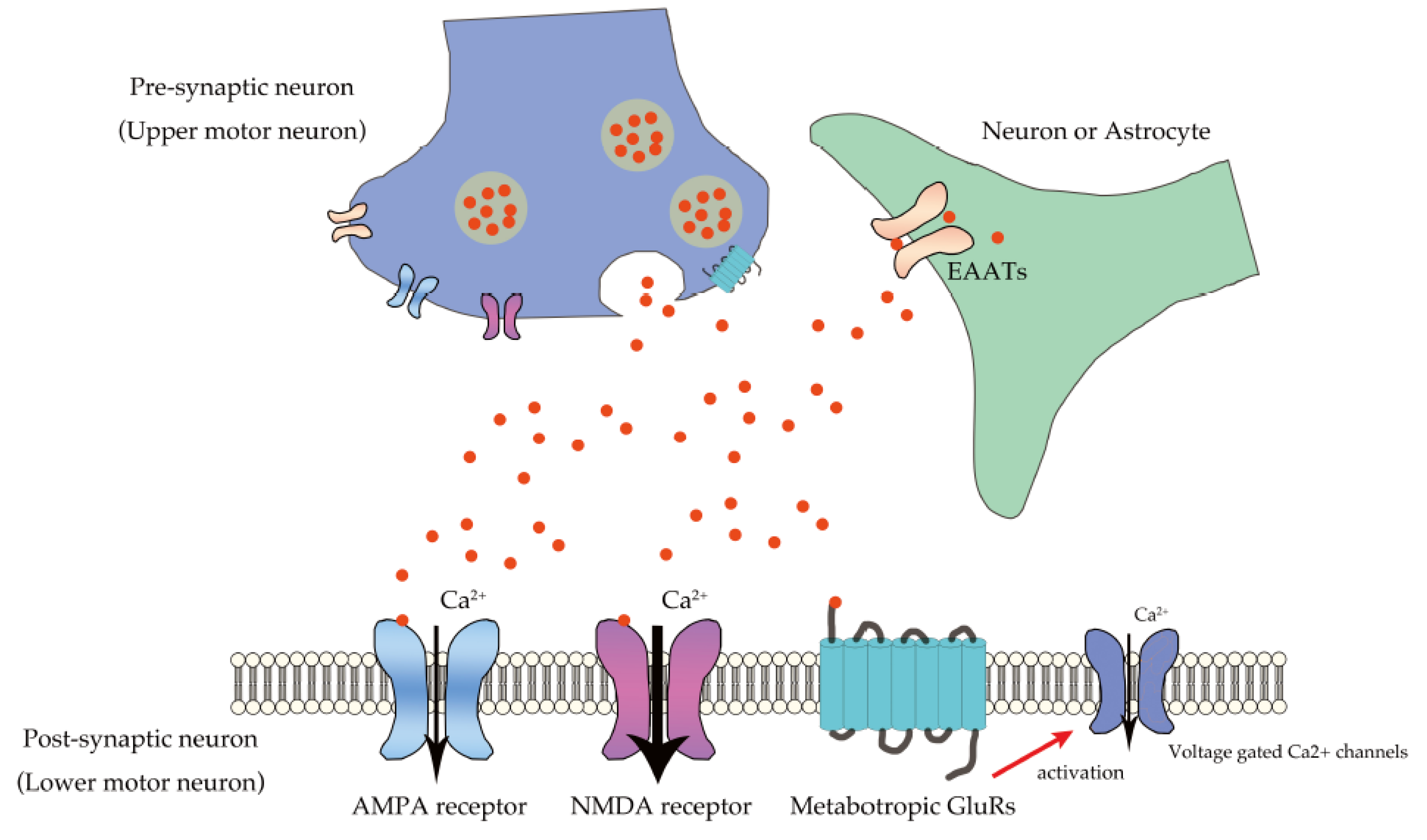
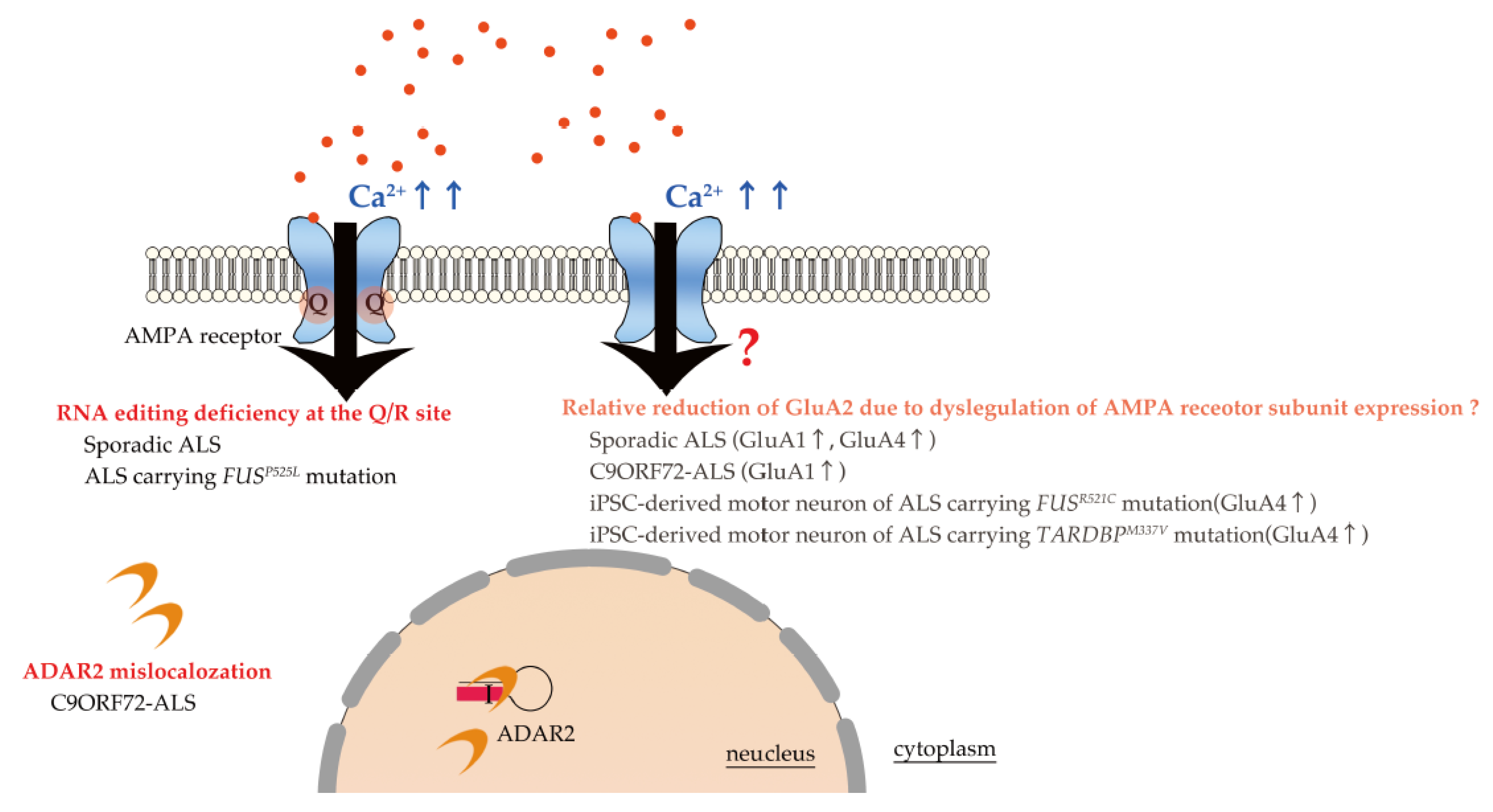
3. Excitotoxicity in ALS Due to Excessive Ca2+ Influx through Unedited GluA2-Conaining AMPA Receptors
4. Promising Therapy Targeting RNA Editing Dysregulation in Neurological Diseases
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Van Damme, P.; Dewil, M.; Robberecht, W.; Van Den Bosch, L. Excitotoxicity and amyotrophic lateral sclerosis. Neurodegener. Dis. 2005, 2, 147–159. [Google Scholar] [CrossRef] [PubMed]
- King, A.E.; Woodhouse, A.; Kirkcaldie, M.T.; Vickers, J.C. Excitotoxicity in ALS: Overstimulation, or overreaction? Exp. Neurol. 2016, 275, 162–171. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Rodriguez-Campuzano, A.G.; Ortega, A. Glutamate transporters: Critical components of glutamatergic transmission. Neuropharmacology 2021, 192, 108602. [Google Scholar] [CrossRef]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Kerchner, G.A.; Nicoll, R.A. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat. Rev. Neurosci. 2008, 9, 813–825. [Google Scholar] [CrossRef]
- Mayer, M.L.; Westbrook, G.L.; Guthrie, P.B. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature 1984, 309, 261–263. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Constantakakis, E.; Smith, J. The neuroexcitotoxic amino acids glutamate and aspartate are altered in the spinal cord and brain in amyotrophic lateral sclerosis. Ann. Neurol. 1988, 24, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Tsai, G.; Kuncl, R.W.; Clawson, L.; Cornblath, D.R.; Drachman, D.B.; Pestronk, A.; Stauch, B.L.; Coyle, J.T. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann. Neurol. 1990, 28, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.J.; Forrest, V.; Ince, P.G.; Richardson, J.P.; Wastell, H.J. CSF and plasma amino acid levels in motor neuron disease: Elevation of CSF glutamate in a subset of patients. Neurodegeneration 1995, 4, 209–216. [Google Scholar] [CrossRef]
- Spreux-Varoquaux, O.; Bensimon, G.; Lacomblez, L.; Salachas, F.; Pradat, P.F.; Le Forestier, N.; Marouan, A.; Dib, M.; Meininger, V. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis: A reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J. Neurol. Sci. 2002, 193, 73–78. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef]
- Plaitakis, A.; Smith, J.; Mandeli, J.; Yahr, M.D. Pilot trial of branched-chain aminoacids in amyotrophic lateral sclerosis. Lancet 1988, 1, 1015–1018. [Google Scholar] [CrossRef]
- Martin, D.; Thompson, M.A.; Nadler, J.V. The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur. J. Pharmacol. 1993, 250, 473–476. [Google Scholar] [CrossRef]
- Bensimon, G.; Lacomblez, L.; Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 1994, 330, 585–591. [Google Scholar] [CrossRef]
- Lacomblez, L.; Bensimon, G.; Leigh, P.N.; Guillet, P.; Meininger, V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 1996, 347, 1425–1431. [Google Scholar] [CrossRef]
- Fang, T.; Al Khleifat, A.; Meurgey, J.-H.; Jones, A.; Leigh, P.N.; Bensimon, G.; Al-Chalabi, A. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: A retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018, 17, 416–422. [Google Scholar] [CrossRef]
- Miller, R.G.; Mitchell, J.D.; Moore, D.H. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst. Rev. 2012, 65, CD001447. [Google Scholar] [CrossRef]
- van Zundert, B.; Izaurieta, P.; Fritz, E.; Alvarez, F.J. Early pathogenesis in the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J. Cell. Biochem. 2012, 113, 3301–3312. [Google Scholar] [CrossRef]
- Vucic, S.; Nicholson, G.A.; Kiernan, M.C. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008, 131, 1540–1550. [Google Scholar] [CrossRef]
- Menon, P.; Geevasinga, N.; Yiannikas, C.; Howells, J.; Kiernan, M.C.; Vucic, S. Sensitivity and specificity of threshold tracking transcranial magnetic stimulation for diagnosis of amyotrophic lateral sclerosis: A prospective study. Lancet Neurol. 2015, 14, 478–484. [Google Scholar] [CrossRef]
- Schanz, O.; Bageac, D.; Braun, L.; Traynor, B.J.; Lehky, T.J.; Floeter, M.K. Cortical hyperexcitability in patients with C9ORF72 mutations: Relationship to phenotype. Muscle Nerve 2016, 54, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Wainger, B.J.; Kiskinis, E.; Mellin, C.; Wiskow, O.; Han, S.S.; Sandoe, J.; Perez, N.P.; Williams, L.A.; Lee, S.; Boulting, G.; et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell. Rep. 2014, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wainger, B.J.; Macklin, E.A.; Vucic, S.; McIlduff, C.E.; Paganoni, S.; Maragakis, N.J.; Bedlack, R.; Goyal, N.A.; Rutkove, S.B.; Lange, D.J.; et al. Effect of Ezogabine on Cortical and Spinal Motor Neuron Excitability in Amyotrophic Lateral Sclerosis: A Randomized Clinical Trial. JAMA Neurol. 2021, 78, 186–196. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, M.; Pinto, S.; Costa, J.; Evangelista, T.; Ohana, B.; Pinto, A. A randomized, placebo-controlled trial of memantine for functional disability in amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler. 2010, 11, 456–460. [Google Scholar] [CrossRef]
- Ryberg, H.; Askmark, H.; Persson, L.I. A double-blind randomized clinical trial in amyotrophic lateral sclerosis using lamotrigine: Effects on CSF glutamate, aspartate, branched-chain amino acid levels and clinical parameters. Acta Neurol. Scand. 2003, 108, 1–8. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Jin, L.; Dykes-Hoberg, M.; Kuncl, R.W. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc. Natl. Acad. Sci. USA 1993, 90, 6591–6595. [Google Scholar] [CrossRef]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2006, 1762, 1068–1082. [Google Scholar] [CrossRef]
- Kwak, S.; Weiss, J.H. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr. Opin. Neurobiol. 2006, 16, 281–287. [Google Scholar] [CrossRef]
- Rosenthal, J.J.; Seeburg, P.H. A-to-I RNA editing: Effects on proteins key to neural excitability. Neuron 2012, 74, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Sommer, B.; Köhler, M.; Sprengel, R.; Seeburg, P.H. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 1991, 67, 11–19. [Google Scholar] [CrossRef]
- Hideyama, T.; Yamashita, T.; Aizawa, H.; Tsuji, S.; Kakita, A.; Takahashi, H.; Kwak, S. Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol. Dis. 2012, 45, 1121–1128. [Google Scholar] [CrossRef]
- Henley, J.M.; Wilkinson, K.A. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 2016, 17, 337–350. [Google Scholar] [CrossRef]
- Greger, I.H.; Khatri, L.; Ziff, E.B. RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron 2002, 34, 759–772. [Google Scholar] [CrossRef]
- Herguedas, B.; Watson, J.F.; Ho, H.; Cais, O.; Garcia-Nafria, J.; Greger, I.H. Architecture of the heteromeric GluA1/2 AMPA receptor in complex with the auxiliary subunit TARP gamma8. Science 2019, 364, 9011. [Google Scholar] [CrossRef]
- Zhang, D.; Watson, J.F.; Matthews, P.M.; Cais, O.; Greger, I.H. Gating and modulation of a hetero-octameric AMPA glutamate receptor. Nature 2021, 594, 454–458. [Google Scholar] [CrossRef]
- Singh, M. Dysregulated A to I RNA editing and non-coding RNAs in neurodegeneration. Front. Genet. 2012, 3, 326. [Google Scholar] [CrossRef]
- Costa Cruz, P.H.; Kawahara, Y. RNA Editing in Neurological and Neurodegenerative Disorders. Methods Mol. Biol. 2021, 2181, 309–330. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, Y.; Yang, X.; Hao, C.; Duan, H. Gene Therapy for Neurodegenerative Disease: Clinical Potential and Directions. Front. Mol. Neurosci. 2021, 14, 618171. [Google Scholar] [CrossRef]
- Sun, J.; Roy, S. Gene-based therapies for neurodegenerative diseases. Nat. Neurosci. 2021, 24, 297–311. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell. Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Elbarbary, R.A.; Lucas, B.A.; Maquat, L.E. Retrotransposons as regulators of gene expression. Science 2016, 351, aac7247. [Google Scholar] [CrossRef] [PubMed]
- Picardi, E.; Manzari, C.; Mastropasqua, F.; Aiello, I.; D’Erchia, A.M.; Pesole, G. Profiling RNA editing in human tissues: Towards the inosinome Atlas. Sci. Rep. 2015, 5, 14941. [Google Scholar] [CrossRef] [PubMed]
- Picardi, E.; Horner, D.S.; Pesole, G. Single-cell transcriptomics reveals specific RNA editing signatures in the human brain. RNA 2017, 23, 860–865. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K.; et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249–254. [Google Scholar] [CrossRef]
- Heraud-Farlow, J.E.; Walkley, C.R. What do editors do? Understanding the physiological functions of A-to-I RNA editing by adenosine deaminase acting on RNAs. Open Biol. 2020, 10, 200085. [Google Scholar] [CrossRef]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Memczak, S.; Wyler, E.; Torti, F.; Porath, H.T.; Orejuela, M.R.; Piechotta, M.; Levanon, E.Y.; Landthaler, M.; Dieterich, C.; et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell. Rep. 2015, 10, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Rueter, S.M.; Dawson, T.R.; Emeson, R.B. Regulation of alternative splicing by RNA editing. Nature 1999, 399, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Solomon, O.; Di Segni, A.; Cesarkas, K.; Porath, H.T.; Marcu-Malina, V.; Mizrahi, O.; Stern-Ginossar, N.; Kol, N.; Farage-Barhom, S.; Glick-Saar, E.; et al. RNA editing by ADAR1 leads to context-dependent transcriptome-wide changes in RNA secondary structure. Nat. Commun. 2017, 8, 1440. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, E.; Levanon, E.Y. A-to-I RNA editing-immune protector and transcriptome diversifier. Nat. Rev. Genet. 2018, 19, 473–490. [Google Scholar] [CrossRef]
- Lorenzini, I.; Moore, S.; Sattler, R. RNA Editing Deficiency in Neurodegeneration. Adv. Neurobiol. 2018, 20, 63–83. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef]
- Kawahara, Y.; Ito, K.; Sun, H.; Kanazawa, I.; Kwak, S. Low editing efficiency of GluR2 mRNA is associated with a low relative abundance of ADAR2 mRNA in white matter of normal human brain. Eur. J. Neurosci. 2003, 18, 23–33. [Google Scholar] [CrossRef]
- Hartner, J.C.; Schmittwolf, C.; Kispert, A.; Muller, A.M.; Higuchi, M.; Seeburg, P.H. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem. 2004, 279, 4894–4902. [Google Scholar] [CrossRef]
- Miyamura, Y.; Suzuki, T.; Kono, M.; Inagaki, K.; Ito, S.; Suzuki, N.; Tomita, Y. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am. J. Hum. Genet. 2003, 73, 693–699. [Google Scholar] [CrossRef]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.Y.; Sedmik, J.; Fitzgerald, M.P.; Halevy, R.S.; Keegan, L.P.; Helbig, I.; Basel-Salmon, L.; Cohen, L.; Straussberg, R.; Chung, W.K.; et al. Bi-allelic ADARB1 Variants Associated with Microcephaly, Intellectual Disability, and Seizures. Am. J. Hum. Genet. 2020, 106, 467–483. [Google Scholar] [CrossRef] [PubMed]
- Maroofian, R.; Sedmik, J.; Mazaheri, N.; Scala, M.; Zaki, M.S.; Keegan, L.P.; Azizimalamiri, R.; Issa, M.; Shariati, G.; Sedaghat, A.; et al. Biallelic variants in ADARB1, encoding a dsRNA-specific adenosine deaminase, cause a severe developmental and epileptic encephalopathy. J. Med. Genet. 2020, 11, 36. [Google Scholar] [CrossRef]
- Hideyama, T.; Yamashita, T.; Suzuki, T.; Tsuji, S.; Higuchi, M.; Seeburg, P.H.; Takahashi, R.; Misawa, H.; Kwak, S. Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J. Neurosci. 2010, 30, 11917–11925. [Google Scholar] [CrossRef]
- Hwang, T.; Park, C.K.; Leung, A.K.; Gao, Y.; Hyde, T.M.; Kleinman, J.E.; Rajpurohit, A.; Tao, R.; Shin, J.H.; Weinberger, D.R. Dynamic regulation of RNA editing in human brain development and disease. Nat. Neurosci. 2016, 19, 1093–1099. [Google Scholar] [CrossRef]
- Tariq, A.; Jantsch, M.F. Transcript diversification in the nervous system: A to I RNA editing in CNS function and disease development. Front. Neurosci. 2012, 6, 99. [Google Scholar] [CrossRef]
- Gal-Mark, N.; Shallev, L.; Sweetat, S.; Barak, M.; Billy Li, J.; Levanon, E.Y.; Eisenberg, E.; Behar, O. Abnormalities in A-to-I RNA editing patterns in CNS injuries correlate with dynamic changes in cell type composition. Sci. Rep. 2017, 7, 43421. [Google Scholar] [CrossRef]
- Tran, S.S.; Jun, H.I.; Bahn, J.H.; Azghadi, A.; Ramaswami, G.; Van Nostrand, E.L.; Nguyen, T.B.; Hsiao, Y.E.; Lee, C.; Pratt, G.A.; et al. Widespread RNA editing dysregulation in brains from autistic individuals. Nat. Neurosci. 2019, 22, 25–36. [Google Scholar] [CrossRef]
- Breen, M.S.; Dobbyn, A.; Li, Q.; Roussos, P.; Hoffman, G.E.; Stahl, E.; Chess, A.; Sklar, P.; Li, J.B.; Devlin, B.; et al. Global landscape and genetic regulation of RNA editing in cortical samples from individuals with schizophrenia. Nat. Neurosci. 2019, 22, 1402–1412. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Ito, K.; Sun, H.; Aizawa, H.; Kanazawa, I.; Kwak, S. Glutamate receptors: RNA editing and death of motor neurons. Nature 2004, 427, 801. [Google Scholar] [CrossRef] [PubMed]
- Khermesh, K.; D’Erchia, A.M.; Barak, M.; Annese, A.; Wachtel, C.; Levanon, E.Y.; Picardi, E.; Eisenberg, E. Reduced levels of protein recoding by A-to-I RNA editing in Alzheimer’s disease. RNA 2016, 22, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Akbarian, S.; Smith, M.A.; Jones, E.G. Editing for an AMPA receptor subunit RNA in prefrontal cortex and striatum in Alzheimer’s disease, Huntington’s disease and schizophrenia. Brain Res. 1995, 699, 297–304. [Google Scholar] [CrossRef]
- Gaisler-Salomon, I.; Kravitz, E.; Feiler, Y.; Safran, M.; Biegon, A.; Amariglio, N.; Rechavi, G. Hippocampus-specific deficiency in RNA editing of GluA2 in Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1785–1791. [Google Scholar] [CrossRef]
- Maas, S.; Patt, S.; Schrey, M.; Rich, A. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc. Natl. Acad. Sci. USA 2001, 98, 14687–14692. [Google Scholar] [CrossRef]
- Ishiuchi, S.; Yoshida, Y.; Sugawara, K.; Aihara, M.; Ohtani, T.; Watanabe, T.; Saito, N.; Tsuzuki, K.; Okado, H.; Miwa, A.; et al. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J. Neurosci. 2007, 27, 7987–8001. [Google Scholar] [CrossRef]
- Cenci, C.; Barzotti, R.; Galeano, F.; Corbelli, S.; Rota, R.; Massimi, L.; Di Rocco, C.; O’Connell, M.A.; Gallo, A. Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J. Biol. Chem. 2008, 283, 7251–7260. [Google Scholar] [CrossRef]
- Patil, V.; Pal, J.; Mahalingam, K.; Somasundaram, K. Global RNA editome landscape discovers reduced RNA editing in glioma: Loss of editing of gamma-amino butyric acid receptor alpha subunit 3 (GABRA3) favors glioma migration and invasion. PeerJ 2020, 8, e9755. [Google Scholar] [CrossRef]
- Galeano, F.; Rossetti, C.; Tomaselli, S.; Cifaldi, L.; Lezzerini, M.; Pezzullo, M.; Boldrini, R.; Massimi, L.; Di Rocco, C.M.; Locatelli, F.; et al. ADAR2-editing activity inhibits glioblastoma growth through the modulation of the CDC14B/Skp2/p21/p27 axis. Oncogene 2013, 32, 998–1009. [Google Scholar] [CrossRef]
- Choudhury, Y.; Tay, F.C.; Lam, D.H.; Sandanaraj, E.; Tang, C.; Ang, B.T.; Wang, S. Attenuated adenosine-to-inosine editing of microRNA-376a* promotes invasiveness of glioblastoma cells. J. Clin. Investig. 2012, 122, 4059–4076. [Google Scholar] [CrossRef]
- Cesarini, V.; Silvestris, D.A.; Tassinari, V.; Tomaselli, S.; Alon, S.; Eisenberg, E.; Locatelli, F.; Gallo, A. ADAR2/miR-589-3p axis controls glioblastoma cell migration/invasion. Nucleic Acids. Res. 2018, 46, 2045–2059. [Google Scholar] [CrossRef]
- Liu, S.; Lau, L.; Wei, J.; Zhu, D.; Zou, S.; Sun, H.S.; Fu, Y.; Liu, F.; Lu, Y. Expression of Ca(2+)-permeable AMPA receptor channels primes cell death in transient forebrain ischemia. Neuron 2004, 43, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.L.; Zhong, X.; Tu, W.; Soundarapandian, M.M.; Molner, P.; Zhu, D.; Lau, L.; Liu, S.; Liu, F.; Lu, Y. ADAR2-dependent RNA editing of AMPA receptor subunit GluR2 determines vulnerability of neurons in forebrain ischemia. Neuron 2006, 49, 719–733. [Google Scholar] [CrossRef]
- Barbon, A.; Fumagalli, F.; Caracciolo, L.; Madaschi, L.; Lesma, E.; Mora, C.; Carelli, S.; Slotkin, T.A.; Racagni, G.; Di Giulio, A.M.; et al. Acute spinal cord injury persistently reduces R/G RNA editing of AMPA receptors. J. Neurochem. 2010, 114, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Di Narzo, A.F.; Kozlenkov, A.; Ge, Y.; Zhang, B.; Sanelli, L.; May, Z.; Li, Y.; Fouad, K.; Cardozo, C.; Koonin, E.V.; et al. Decrease of mRNA Editing after Spinal Cord Injury is Caused by Down-regulation of ADAR2 that is Triggered by Inflammatory Response. Sci. Rep. 2015, 5, 12615. [Google Scholar] [CrossRef] [PubMed]
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef]
- Kortenbruck, G.; Berger, E.; Speckmann, E.J.; Musshoff, U. RNA editing at the Q/R site for the glutamate receptor subunits GLUR2, GLUR5, and GLUR6 in hippocampus and temporal cortex from epileptic patients. Neurobiol. Dis. 2001, 8, 459–468. [Google Scholar] [CrossRef]
- Vollmar, W.; Gloger, J.; Berger, E.; Kortenbruck, G.; Kohling, R.; Speckmann, E.J.; Musshoff, U. RNA editing (R/G site) and flip-flop splicing of the AMPA receptor subunit GluR2 in nervous tissue of epilepsy patients. Neurobiol. Dis. 2004, 15, 371–379. [Google Scholar] [CrossRef]
- Krestel, H.; Raffel, S.; von Lehe, M.; Jagella, C.; Moskau-Hartmann, S.; Becker, A.; Elger, C.E.; Seeburg, P.H.; Nirkko, A. Differences between RNA and DNA due to RNA editing in temporal lobe epilepsy. Neurobiol. Dis. 2013, 56, 66–73. [Google Scholar] [CrossRef]
- Bernard, A.; Ferhat, L.; Dessi, F.; Charton, G.; Represa, A.; Ben-Ari, Y.; Khrestchatisky, M. Q/R editing of the rat GluR5 and GluR6 kainate receptors in vivo and in vitro: Evidence for independent developmental, pathological and cellular regulation. Eur. J. Neurosci. 1999, 11, 604–616. [Google Scholar] [CrossRef]
- Caracciolo, L.; Barbon, A.; Palumbo, S.; Mora, C.; Toscano, C.D.; Bosetti, F.; Barlati, S. Altered mRNA editing and expression of ionotropic glutamate receptors after kainic acid exposure in cyclooxygenase-2 deficient mice. PLoS ONE 2011, 6, e19398. [Google Scholar] [CrossRef] [PubMed]
- Malhi, G.S.; Mann, J.J. Depression. Lancet 2018, 392, 2299–2312. [Google Scholar] [CrossRef]
- Niswender, C.M.; Herrick-Davis, K.; Dilley, G.E.; Meltzer, H.Y.; Overholser, J.C.; Stockmeier, C.A.; Emeson, R.B.; Sanders-Bush, E. RNA editing of the human serotonin 5-HT2C receptor. alterations in suicide and implications for serotonergic pharmacotherapy. Neuropsychopharmacology 2001, 24, 478–491. [Google Scholar] [CrossRef]
- Gurevich, I.; Tamir, H.; Arango, V.; Dwork, A.J.; Mann, J.J.; Schmauss, C. Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron 2002, 34, 349–356. [Google Scholar] [CrossRef]
- Iwamoto, K.; Kato, T. RNA editing of serotonin 2C receptor in human postmortem brains of major mental disorders. Neurosci. Lett. 2003, 346, 169–172. [Google Scholar] [CrossRef]
- Zhu, H.; Urban, D.J.; Blashka, J.; McPheeters, M.T.; Kroeze, W.K.; Mieczkowski, P.; Overholser, J.C.; Jurjus, G.J.; Dieter, L.; Mahajan, G.J.; et al. Quantitative analysis of focused a-to-I RNA editing sites by ultra-high-throughput sequencing in psychiatric disorders. PLoS ONE 2012, 7, e43227. [Google Scholar] [CrossRef]
- Weissmann, D.; van der Laan, S.; Underwood, M.D.; Salvetat, N.; Cavarec, L.; Vincent, L.; Molina, F.; Mann, J.J.; Arango, V.; Pujol, J.F. Region-specific alterations of A-to-I RNA editing of serotonin 2c receptor in the cortex of suicides with major depression. Transl. Psychiatry 2016, 6, e878. [Google Scholar] [CrossRef]
- Sodhi, M.S.; Burnet, P.W.; Makoff, A.J.; Kerwin, R.W.; Harrison, P.J. RNA editing of the 5-HT(2C) receptor is reduced in schizophrenia. Mol. Psychiatry 2001, 6, 373–379. [Google Scholar] [CrossRef][Green Version]
- Dracheva, S.; Patel, N.; Woo, D.A.; Marcus, S.M.; Siever, L.J.; Haroutunian, V. Increased serotonin 2C receptor mRNA editing: A possible risk factor for suicide. Mol. Psychiatry 2008, 13, 1001–1010. [Google Scholar] [CrossRef]
- Chimienti, F.; Cavarec, L.; Vincent, L.; Salvetat, N.; Arango, V.; Underwood, M.D.; Mann, J.J.; Pujol, J.F.; Weissmann, D. Brain region-specific alterations of RNA editing in PDE8A mRNA in suicide decedents. Transl. Psychiatry 2019, 9, 91. [Google Scholar] [CrossRef]
- Salvetat, N.; Chimienti, F.; Cayzac, C.; Dubuc, B.; Checa-Robles, F.; Dupre, P.; Mereuze, S.; Patel, V.; Genty, C.; Lang, J.P.; et al. Phosphodiesterase 8A to discriminate in blood samples depressed patients and suicide attempters from healthy controls based on A-to-I RNA editing modifications. Transl. Psychiatry 2021, 11, 255. [Google Scholar] [CrossRef]
- Lord, C.; Elsabbagh, M.; Baird, G.; Veenstra-Vanderweele, J. Autism spectrum disorder. Lancet 2018, 392, 508–520. [Google Scholar] [CrossRef]
- Eran, A.; Li, J.B.; Vatalaro, K.; McCarthy, J.; Rahimov, F.; Collins, C.; Markianos, K.; Margulies, D.M.; Brown, E.N.; Calvo, S.E.; et al. Comparative RNA editing in autistic and neurotypical cerebella. Mol. Psychiatry 2013, 18, 1041–1048. [Google Scholar] [CrossRef][Green Version]
- Schmidt, H.D.; McFarland, K.N.; Darnell, S.B.; Huizenga, M.N.; Sangrey, G.R.; Cha, J.H.; Pierce, R.C.; Sadri-Vakili, G. ADAR2-dependent GluA2 editing regulates cocaine seeking. Mol. Psychiatry 2015, 20, 1460–1466. [Google Scholar] [CrossRef]
- Tanaka, M.; Watanabe, Y. RNA Editing of Serotonin 2C Receptor and Alcohol Intake. Front. Neurosci 2019, 13, 1390. [Google Scholar] [CrossRef]
- Starr, A.; Sattler, R. Synaptic dysfunction and altered excitability in C9ORF72 ALS/FTD. Brain Res. 2018, 1693, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, T.; Yamashita, T.; Tamaoka, A.; Kwak, S. Extracellular RNAs as Biomarkers of Sporadic Amyotrophic Lateral Sclerosis and Other Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 3148. [Google Scholar] [CrossRef] [PubMed]
- Burnashev, N.; Monyer, H.; Seeburg, P.H.; Sakmann, B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron 1992, 8, 189–198. [Google Scholar] [CrossRef]
- Feldmeyer, D.; Kask, K.; Brusa, R.; Kornau, H.C.; Kolhekar, R.; Rozov, A.; Burnashev, N.; Jensen, V.; Hvalby, O.; Sprengel, R.; et al. Neurological dysfunctions in mice expressing different levels of the Q/R site-unedited AMPAR subunit GluR-B. Nat. Neurosci. 1999, 2, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Seeburg, P.H. A-to-I editing: New and old sites, functions and speculations. Neuron 2002, 35, 17–20. [Google Scholar] [CrossRef]
- Jia, Z.; Agopyan, N.; Miu, P.; Xiong, Z.; Henderson, J.; Gerlai, R.; Taverna, F.A.; Velumian, A.; MacDonald, J.; Carlen, P.; et al. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron 1996, 17, 945–956. [Google Scholar] [CrossRef]
- D’Erchia, A.M.; Gallo, A.; Manzari, C.; Raho, S.; Horner, D.S.; Chiara, M.; Valletti, A.; Aiello, I.; Mastropasqua, F.; Ciaccia, L.; et al. Massive transcriptome sequencing of human spinal cord tissues provides new insights into motor neuron degeneration in ALS. Sci. Rep. 2017, 7, 10046. [Google Scholar] [CrossRef]
- Ichiyanagi, N.; Fujimori, K.; Yano, M.; Ishihara-Fujisaki, C.; Sone, T.; Akiyama, T.; Okada, Y.; Akamatsu, W.; Matsumoto, T.; Ishikawa, M.; et al. Establishment of In Vitro FUS-Associated Familial Amyotrophic Lateral Sclerosis Model Using Human Induced Pluripotent Stem Cells. Stem Cell Reports 2016, 6, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Gregory, J.M.; Livesey, M.R.; McDade, K.; Selvaraj, B.T.; Barton, S.K.; Chandran, S.; Smith, C. Dysregulation of AMPA receptor subunit expression in sporadic ALS post-mortem brain. J. Pathol. 2020, 250, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, H.; Sawada, J.; Hideyama, T.; Yamashita, T.; Katayama, T.; Hasebe, N.; Kimura, T.; Yahara, O.; Kwak, S. TDP-43 pathology in sporadic ALS occurs in motor neurons lacking the RNA editing enzyme ADAR2. Acta Neuropathol. 2010, 120, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Kwak, S. The molecular link between inefficient GluA2 Q/R site-RNA editing and TDP-43 pathology in motor neurons of sporadic amyotrophic lateral sclerosis patients. Brain Res. 2014, 1584, 28–38. [Google Scholar] [CrossRef]
- Yamashita, T.; Hideyama, T.; Hachiga, K.; Teramoto, S.; Takano, J.; Iwata, N.; Saido, T.C.; Kwak, S. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat. Commun 2012, 3, 1307. [Google Scholar] [CrossRef]
- Hideyama, T.; Kwak, S. When Does ALS Start? ADAR2-GluA2 Hypothesis for the Etiology of Sporadic ALS. Front. Mol. Neurosci. 2011, 4, 33. [Google Scholar] [CrossRef]
- Tischbein, M.; Baron, D.M.; Lin, Y.C.; Gall, K.V.; Landers, J.E.; Fallini, C.; Bosco, D.A. The RNA-binding protein FUS/TLS undergoes calcium-mediated nuclear egress during excitotoxic stress and is required for GRIA2 mRNA processing. J. Biol. Chem. 2019, 294, 10194–10210. [Google Scholar] [CrossRef]
- Grigoriev, V.V.; Efimova, A.D.; Ustyugov, A.A.; Shevchenko, V.P.; Bachurin, S.O.; Myasoedov, N.F. Glutamate release and uptake processes are altered in a new mouse model of amyotrophic lateral sclerosis. Dokl. Biochem. Biophys. 2016, 468, 165–167. [Google Scholar] [CrossRef]
- Aizawa, H.; Hideyama, T.; Yamashita, T.; Kimura, T.; Suzuki, N.; Aoki, M.; Kwak, S. Deficient RNA-editing enzyme ADAR2 in an amyotrophic lateral sclerosis patient with a FUS(P525L) mutation. J. Clin. Neurosci. 2016, 32, 128–129. [Google Scholar] [CrossRef]
- Moore, S.; Alsop, E.; Lorenzini, I.; Starr, A.; Rabichow, B.E.; Mendez, E.; Levy, J.L.; Burciu, C.; Reiman, R.; Chew, J.; et al. ADAR2 mislocalization and widespread RNA editing aberrations in C9orf72-mediated ALS/FTD. Acta Neuropathol. 2019, 19, 1999. [Google Scholar] [CrossRef]
- Suzuki, H.; Matsuoka, M. Proline-arginine poly-dipeptide encoded by the C9orf72 repeat expansion inhibits adenosine deaminase acting on RNA. J. Neurochem. 2021, 158, 753–765. [Google Scholar] [CrossRef]
- Selvaraj, B.T.; Livesey, M.R.; Zhao, C.; Gregory, J.M.; James, O.T.; Cleary, E.M.; Chouhan, A.K.; Gane, A.B.; Perkins, E.M.; Dando, O.; et al. C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca(2+)-permeable AMPA receptor-mediated excitotoxicity. Nat. Commun. 2018, 9, 347. [Google Scholar] [CrossRef]
- Quinones-Valdez, G.; Tran, S.S.; Jun, H.I.; Bahn, J.H.; Yang, E.W.; Zhan, L.; Brummer, A.; Wei, X.; Van Nostrand, E.L.; Pratt, G.A.; et al. Regulation of RNA editing by RNA-binding proteins in human cells. Commun. Biol. 2019, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Ohgomori, T.; Yamasaki, R.; Takeuchi, H.; Kadomatsu, K.; Kira, J.I.; Jinno, S. Differential involvement of vesicular and glial glutamate transporters around spinal alpha-motoneurons in the pathogenesis of SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Neuroscience 2017, 356, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, T.; Provenzano, F.; Gallia, E.; Ravera, S.; Torazza, C.; Bossi, S.; Ferrando, S.; Puliti, A.; Van Den Bosch, L.; Bonanno, G.; et al. In-vivo genetic ablation of metabotropic glutamate receptor type 5 slows down disease progression in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2019, 129, 79–92. [Google Scholar] [CrossRef]
- Kawahara, Y.; Sun, H.; Ito, K.; Hideyama, T.; Aoki, M.; Sobue, G.; Tsuji, S.; Kwak, S. Underediting of GluR2 mRNA, a neuronal death inducing molecular change in sporadic ALS, does not occur in motor neurons in ALS1 or SBMA. Neurosci. Res. 2006, 54, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, T.; Fujioka, Y.; Tanaka, M.; Honda, D.; Yokoi, S.; Riku, Y.; Ibi, D.; Nagai, T.; Yamada, K.; Watanabe, H.; et al. FUS regulates AMPA receptor function and FTLD/ALS-associated behaviour via GluA1 mRNA stabilization. Nat. Commun. 2015, 6, 7098. [Google Scholar] [CrossRef]
- Capauto, D.; Colantoni, A.; Lu, L.; Santini, T.; Peruzzi, G.; Biscarini, S.; Morlando, M.; Shneider, N.A.; Caffarelli, E.; Laneve, P.; et al. A Regulatory Circuitry Between Gria2, miR-409, and miR-495 Is Affected by ALS FUS Mutation in ESC-Derived Motor Neurons. Mol. Neurobiol. 2018, 55, 7635–7651. [Google Scholar] [CrossRef]
- Dafinca, R.; Barbagallo, P.; Farrimond, L.; Candalija, A.; Scaber, J.; Ababneh, N.A.; Sathyaprakash, C.; Vowles, J.; Cowley, S.A.; Talbot, K. Impairment of Mitochondrial Calcium Buffering Links Mutations in C9ORF72 and TARDBP in iPS-Derived Motor Neurons from Patients with ALS/FTD. Stem Cell Rep. 2020, 14, 892–908. [Google Scholar] [CrossRef]
- Bursch, F.; Kalmbach, N.; Naujock, M.; Staege, S.; Eggenschwiler, R.; Abo-Rady, M.; Japtok, J.; Guo, W.; Hensel, N.; Reinhardt, P.; et al. Altered calcium dynamics and glutamate receptor properties in iPSC-derived motor neurons from ALS patients with C9orf72, FUS, SOD1 or TDP43 mutations. Hum. Mol. Genet. 2019, 28, 2835–2850. [Google Scholar] [CrossRef]
- Xu, L.D.; Ohman, M. ADAR1 Editing and its Role in Cancer. Genes 2018, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Zimmerman, M.B.; Beltz, T.G.; Johnson, A.K. Affect-related behaviors in mice misexpressing the RNA editing enzyme ADAR2. Physiol. Behav. 2009, 97, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Chai, H.L.; Teramoto, S.; Tsuji, S.; Shimazaki, K.; Muramatsu, S.; Kwak, S. Rescue of amyotrophic lateral sclerosis phenotype in a mouse model by intravenous AAV9-ADAR2 delivery to motor neurons. EMBO Mol. Med. 2013, 5, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- High, K.A.; Roncarolo, M.G. Gene Therapy. N. Engl. J. Med. 2019, 381, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Kariyawasam, D.; Alexander, I.E.; Kurian, M.; Farrar, M.A. Great expectations: Virus-mediated gene therapy in neurological disorders. J. Neurol. Neurosurg. Psychiatry 2020, 91, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Qin, S.; Pu, Q.; Wang, Z.; Wu, Q.; Gao, P.; Schettler, J.; Guo, K.; Li, R.; Li, G.; et al. CRISPR-Cas13 Inhibitors Block RNA Editing in Bacteria and Mammalian Cells. Mol. Cell. 2020, 78, 850–861.e5. [Google Scholar] [CrossRef]
- Xu, C.; Zhou, Y.; Xiao, Q.; He, B.; Geng, G.; Wang, Z.; Cao, B.; Dong, X.; Bai, W.; Wang, Y.; et al. Programmable RNA editing with compact CRISPR-Cas13 systems from uncultivated microbes. Nat. Methods 2021, 18, 499–506. [Google Scholar] [CrossRef]
- Van Den Bosch, L.; Vandenberghe, W.; Klaassen, H.; Van Houtte, E.; Robberecht, W. Ca(2+)-permeable AMPA receptors and selective vulnerability of motor neurons. J. Neurol. Sci. 2000, 180, 29–34. [Google Scholar] [CrossRef]
- Van Damme, P.; Leyssen, M.; Callewaert, G.; Robberecht, W.; Van Den Bosch, L. The AMPA receptor antagonist NBQX prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis. Neurosci. Lett. 2003, 343, 81–84. [Google Scholar] [CrossRef]
- Akamatsu, M.; Yamashita, T.; Hirose, N.; Teramoto, S.; Kwak, S. The AMPA receptor antagonist perampanel robustly rescues amyotrophic lateral sclerosis (ALS) pathology in sporadic ALS model mice. Sci. Rep. 2016, 6, 28649. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, B.; Mauricio, E.A.; Shah, J.S.; Li, Z.; Rogawski, M.A. Cortical excitability threshold can be increased by the AMPA blocker Perampanel in amyotrophic lateral sclerosis. Muscle Nerve 2021, 64, 215–219. [Google Scholar] [CrossRef]
- Pascuzzi, R.M.; Shefner, J.; Chappell, A.S.; Bjerke, J.S.; Tamura, R.; Chaudhry, V.; Clawson, L.; Haas, L.; Rothstein, J.D. A phase II trial of talampanel in subjects with amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler. 2010, 11, 266–271. [Google Scholar] [CrossRef]
- Aizawa, H.; Kato, H.; Oba, K.; Kawahara, T.; Okubo, Y.; Saito, T.; Naito, M.; Urushitani, M.; Tamaoka, A.; Nakamagoe, K.; et al. Randomized phase 2 study of perampanel for sporadic amyotrophic lateral sclerosis. J. Neurol. 2021, 7, 21. [Google Scholar] [CrossRef]
- Hosaka, T.; Yamashita, T.; Teramoto, S.; Hirose, N.; Tamaoka, A.; Kwak, S. ADAR2-dependent A-to-I RNA editing in the extracellular linear and circular RNAs. Neurosci. Res. 2018, 11, 5. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Editing Site | Editing Efficiency | ADAR Responsible | Relation to Disease Pathogenesis | Pathogenetic Alteration | Reference |
|---|---|---|---|---|---|---|
| ALS | Q/R site in GluA2 | Decrease | ADAR2 | Exaggerated Ca2+ influx | Lower motor neurons (neuronal death, TDP-43 mislocalization) | [36,71] |
| AD | Q/R site in GluA2 | Decrease | ADAR2 | Not described | Not specified | [73,74] |
| Glioblastoma | Q/R site in GluA2 | Decrease | ADAR2 | Exaggerated Ca2+ influx Activation of Akt pathway | Malignant conversion (promotion of proliferation, migration, and invasion) | [75,76,77] |
| I/M site in GABRA3 | Decrease | ADAR1 ADAR2 | Reduced inhibition of invasion and migration | [78] | ||
| +9 site of miR-336 | Decrease | ADAR2 | Promotion of proliferation, migration, and invasion | [80] | ||
| +6 site of miR-589-3p | Decrease | ADAR2 | Promotion of proliferation, migration, and invasion | [81] | ||
| Ischemia | Q/R site in GluA2 | Decrease | ADAR2 | Exaggerated Ca2+ influx | Not specified | [82,83] |
| Spinal cord injury | R/G site in GluA2 | Decrease | ADAR1 ADAR2 | Reduction of postsynaptic excitatory response to glutamate | Not specified | [84] |
| Site D in 5-HT2c I/V site in Kv1.1 | Decrease | ADAR2 | Not described | [85] | ||
| Epilepsy | R/G site in GluA2 | Increase | ADAR1 ADAR2 | Modulation of neuronal excitability | Not specified | [87,89] |
| Q/R site in GRIK1 Q/R site in GRIK2 | Increase | ADAR1 ADAR2 | Exaggerated Ca2+ influx | [88] | ||
| Depression | Sites B, C, and E in PDE8A | Decrease | Unknown | Not described | Not specified | [100,101] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosaka, T.; Tsuji, H.; Kwak, S. RNA Editing: A New Therapeutic Target in Amyotrophic Lateral Sclerosis and Other Neurological Diseases. Int. J. Mol. Sci. 2021, 22, 10958. https://doi.org/10.3390/ijms222010958
Hosaka T, Tsuji H, Kwak S. RNA Editing: A New Therapeutic Target in Amyotrophic Lateral Sclerosis and Other Neurological Diseases. International Journal of Molecular Sciences. 2021; 22(20):10958. https://doi.org/10.3390/ijms222010958
Chicago/Turabian StyleHosaka, Takashi, Hiroshi Tsuji, and Shin Kwak. 2021. "RNA Editing: A New Therapeutic Target in Amyotrophic Lateral Sclerosis and Other Neurological Diseases" International Journal of Molecular Sciences 22, no. 20: 10958. https://doi.org/10.3390/ijms222010958
APA StyleHosaka, T., Tsuji, H., & Kwak, S. (2021). RNA Editing: A New Therapeutic Target in Amyotrophic Lateral Sclerosis and Other Neurological Diseases. International Journal of Molecular Sciences, 22(20), 10958. https://doi.org/10.3390/ijms222010958