
PD-L1 Inhibitors: Different Classes, Activities, and Mechanisms of Action

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
2.1. The Comparison of In Vitro Activities
2.2. The Mechanisms of PD-1/PD-L1 Blockade
2.3. Specificity Towards the Human and Mouse PD-L1
3. Discussion
4. Materials and Methods
4.1. Homogeneous Time-Resolved Fluorescence Assay
4.2. PD-1/PD-L1 ICB Assay
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

HTRF Limitations
ICB Limitations and Transition of HTRF Potency to Activity in the ICB Assay
References
- Hoos, A. Development of immuno-oncology drugs—From CTLA4 to PD1 to the next generations. Nat. Rev. Drug Discov. 2016, 15, 235–247. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Qu, B.; Yang, H.; Hu, S.; Dong, X. If small molecules immunotherapy comes, can the prime be far behind? Eur. J. Med. Chem. 2021, 218, 113356. [Google Scholar] [CrossRef]
- Chupak, L.S.; Zheng, X. Compounds Useful as Immunomodulators; Bristol-Myers Squibb Co.: New York, NY, USA, 2015. [Google Scholar]
- Chupak, L.S.; Ding, M.; Martin, S.W.; Zheng, X.; Hewawasam, P.; Connoly, T.P.; Xu, N.; Yeung, K.-S.; Zhu, J.; Langley, D.R.; et al. Compounds Useful As Immunomodulators; Bristol-Myers Squibb Co.: New York, NY, USA, 2015. [Google Scholar]
- Skalniak, L.; Zak, K.M.; Guzik, K.; Magiera, K.; Musielak, B.; Pachota, M.; Szelazek, B.; Kocik, J.; Grudnik, P.; Tomala, M.; et al. Small-molecule inhibitors of PD-1/PD-L1 immune checkpoint alleviate the PD-L1-induced exhaustion of T-cells. Oncotarget 2017, 8, 72167–72181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Törner, R.; Skalniak, L.; Dömling, A.; Dubin, G.; Holak, T.A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef] [Green Version]
- Guzik, K.; Tomala, M.; Muszak, D.; Konieczny, M.; Hec, A.; Błaszkiewicz, U.; Pustuła, M.; Butera, R.; Dömling, A.; Holak, T.A. Development of the Inhibitors that Target the PD-1/PD-L1 Interaction-A Brief Look at Progress on Small Molecules, Peptides and Macrocycles. Molecules 2019, 24, 2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, C.; Yang, H.; Lu, Y.; Hu, S.; Wu, Y.; He, Q.; Dong, X. Recent advance of peptide-based molecules and nonpeptidic small-molecules modulating PD-1/PD-L1 protein-protein interaction or targeting PD-L1 protein degradation. Eur. J. Med. Chem. 2021, 213, 113170. [Google Scholar] [CrossRef]
- Miller, M.M.; Mapelli, C.; Allen, M.; Bowsher, M.S.; Boy, K.; Gillis, E.; Langley, D.R.; Mull, E.; Poirier, M.A.; Sanghvt, N.; et al. Macrocyclic Inhibitors of the PD-1/PD-L1 and CD80(B7-1)/PD-L1 Protein/Protein Interactions; Bristol-Myers Squibb Co.: New York, NY, USA, 2014. [Google Scholar]
- Miller, M.M.; Allen, M.P.; Bowsher, M.S.; Boy, K.M.; Gillis, E.P.; Langley, D.R.; Mull, E.; Sun, L.-Q.; Tenney, D.J.; Yeung, K.-S.; et al. Macrocyclic Inhibitors of the PD-1/PD-L1 and CD80(B7-1)/PD-L1 Protein/Protein Interactions; Bristol-Myers Squibb Co.: New York, NY, USA, 2018. [Google Scholar]
- Konieczny, M.; Musielak, B.; Kocik, J.; Skalniak, L.; Sala, D.; Czub, M.; Magiera-Mularz, K.; Rodriguez, I.; Myrcha, M.; Stec, M.; et al. Di-bromo-Based Small-Molecule Inhibitors of the PD-1/PD-L1 Immune Checkpoint. J. Med. Chem. 2020, 63, 11271–11285. [Google Scholar] [CrossRef]
- Liu, L.; Yao, Z.; Wang, S.; Xie, T.; Wu, G.; Zhang, H.; Zhang, P.; Wu, Y.; Yuan, H.; Sun, H. Syntheses, Biological Evaluations, and Mechanistic Studies of Benzo[c][1,2,5]oxadiazole Derivatives as Potent PD-L1 Inhibitors with In Vivo Antitumor Activity. J. Med. Chem. 2021, 64, 8391–8409. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Cao, Q.; Zheng, S.; Tian, Y.; Zhang, H.; Xie, J.; Xie, H.; Liu, Y.; Zhao, Y.; Gong, P. Discovery of [1,2,4]Triazolo [4,3- a]pyridines as Potent Inhibitors Targeting the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction. J. Med. Chem. 2019, 62, 4703–4715. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Wang, W.; Niu, X.; Ren, Y.; Liu, T.; Cao, H.; Wang, S.; Tu, Y.; Chen, J.; Liu, S.; et al. Discovery of Novel and Highly Potent Resorcinol Dibenzyl Ether-Based PD-1/PD-L1 Inhibitors with Improved Drug-like and Pharmacokinetic Properties for Cancer Treatment. J. Med. Chem. 2020, 63, 15946–15959. [Google Scholar] [CrossRef]
- Guo, J.; Luo, L.; Wang, Z.; Hu, N.; Wang, W.; Xie, F.; Liang, E.; Yan, X.; Xiao, J.; Li, S. Design, Synthesis, and Biological Evaluation of Linear Aliphatic Amine-Linked Triaryl Derivatives as Potent Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction with Promising Antitumor Effects In Vivo. J. Med. Chem. 2020, 63, 13825–13850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xia, Y.; Yu, C.; Du, H.; Liu, J.; Li, H.; Huang, S.; Zhu, Q.; Xu, Y.; Zou, Y. Discovery of Novel Small-Molecule Inhibitors of PD-1/PD-L1 Interaction via Structural Simplification Strategy. Molecules 2021, 26, 3347. [Google Scholar] [CrossRef]
- Muszak, D.; Surmiak, E.; Plewka, J.; Magiera-Mularz, K.; Kocik-Krol, J.; Musielak, B.; Sala, D.; Kitel, R.; Stec, M.; Weglarczyk, K.; et al. Terphenyl-Based Small-Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein-Protein Interaction. J. Med. Chem. 2021, 64, 11614–11636. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, K.; Yang, Y.; Huang, X.; Dai, X.; Feng, Z. Design, synthesis, and structure-activity relationship of programmed cell death-1/programmed cell death-ligand 1 interaction inhibitors bearing a benzo[d]isothiazole scaffold. Eur. J. Med. Chem. 2021, 217, 113377. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, K.; Chen, H.; Feng, Z. Design, synthesis, evaluation, and SAR of 4-phenylindoline derivatives, a novel class of small-molecule inhibitors of the programmed cell death-1/programmed cell death-ligand 1 (PD-1/PD-L1) interaction. Eur. J. Med. Chem. 2021, 211, 113001. [Google Scholar] [CrossRef]
- Park, J.-J.; Thi, E.P.; Carpio, V.H.; Bi, Y.; Cole, A.G.; Dorsey, B.D.; Fan, K.; Harasym, T.; Iott, C.L.; Kadhim, S.; et al. Checkpoint inhibition through small molecule-induced internalization of programmed death-ligand 1. Nat. Commun. 2021, 12, 1222. [Google Scholar] [CrossRef]
- Basu, S.; Yang, J.; Xu, B.; Magiera-Mularz, K.; Skalniak, L.; Musielak, B.; Kholodovych, V.; Holak, T.A.; Hu, L. Design, Synthesis, Evaluation, and Structural Studies of C2-Symmetric Small Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein-Protein Interaction. J. Med. Chem. 2019, 62, 7250–7263. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, Y.; Guo, Y.; Pan, Z.; Zhong, S.; Jin, X.; Zhuang, W.; Chen, S.; Gao, J.; Huang, W.; et al. Discovery of phenyl-linked symmetric small molecules as inhibitors of the programmed cell death-1/programmed cell death-ligand 1 interaction. Eur. J. Med. Chem. 2021, 223, 113637. [Google Scholar] [CrossRef] [PubMed]
- Magiera-Mularz, K.; Kuska, K.; Skalniak, L.; Grudnik, P.; Musielak, B.; Plewka, J.; Kocik, J.; Stec, M.; Weglarczyk, K.; Sala, D.; et al. Macrocyclic Peptide Inhibitor of PD-1/PD-L1 Immune Checkpoint. Adv. Ther. 2021, 4, 2000195. [Google Scholar] [CrossRef]
- Magiera-Mularz, K.; Skalniak, L.; Zak, K.M.; Musielak, B.; Rudzinska-Szostak, E.; Berlicki, Ł.; Kocik, J.; Grudnik, P.; Sala, D.; Zarganes-Tzitzikas, T.; et al. Bioactive Macrocyclic Inhibitors of the PD-1/PD-L1 Immune Checkpoint. Angew. Chem. Int. Ed. Engl. 2017, 56, 13732–13735. [Google Scholar] [CrossRef]
- Shi, D.; An, X.; Bai, Q.; Bing, Z.; Zhou, S.; Liu, H.; Yao, X. Computational Insight Into the Small Molecule Intervening PD-L1 Dimerization and the Potential Structure-Activity Relationship. Front. Chem. 2019, 7, 764. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.-F.; Li, Z.; Ma, D.; Yu, Q. Small-molecule PD-L1 inhibitor BMS1166 abrogates the function of PD-L1 by blocking its ER export. Oncoimmunology 2020, 9, 1831153. [Google Scholar] [CrossRef]
- Gou, Q.; Dong, C.; Xu, H.; Khan, B.; Jin, J.; Liu, Q.; Shi, J.; Hou, Y. PD-L1 degradation pathway and immunotherapy for cancer. Cell Death Dis. 2020, 11, 955. [Google Scholar] [CrossRef]
- Banik, S.M.; Pedram, K.; Wisnovsky, S.; Ahn, G.; Riley, N.M.; Bertozzi, C.R. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 2020, 584, 291–297. [Google Scholar] [CrossRef]
- Cotton, A.D.; Nguyen, D.P.; Gramespacher, J.A.; Seiple, I.B.; Wells, J.A. Development of Antibody-Based PROTACs for the Degradation of the Cell-Surface Immune Checkpoint Protein PD-L1. J. Am. Chem. Soc. 2021, 143, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Lazorchak, A.S.; Patterson, T.; Ding, Y.; Sasikumar, P.G.; Sudarshan, N.S.; Gowda, N.M.; Ramachandra, R.K.; Samiulla, D.S.; Giri, S.; Eswarappa, R.; et al. Abstract A36: CA-170, an oral small molecule PD-L1 and VISTA immune checkpoint antagonist, promotes T cell immune activation and inhibits tumor growth in pre-clinical models of cancer. In Proceedings of the Immunomodulation, Special Conference on Tumor Immunology and Immunotherapy, Boston, MA, USA, 20-23 October 2016; 2017; p. A36. [Google Scholar]
- Blevins, D.J.; Hanley, R.; Bolduc, T.; Powell, D.A.; Gignac, M.; Walker, K.; Carr, M.D.; Hof, F.; Wulff, J.E. In Vitro Assessment of Putative PD-1/PD-L1 Inhibitors: Suggestions of an Alternative Mode of Action. ACS Med. Chem. Lett. 2019, 10, 1187–1192. [Google Scholar] [CrossRef]
- Ganesan, A.; Ahmed, M.; Okoye, I.; Arutyunova, E.; Babu, D.; Turnbull, W.L.; Kundu, J.K.; Shields, J.; Agopsowicz, K.C.; Xu, L.; et al. Comprehensive in vitro characterization of PD-L1 small molecule inhibitors. Sci. Rep. 2019, 9, 12392. [Google Scholar] [CrossRef] [PubMed]
- Musielak, B.; Kocik, J.; Skalniak, L.; Magiera-Mularz, K.; Sala, D.; Czub, M.; Stec, M.; Siedlar, M.; Holak, T.A.; Plewka, J. CA-170—A Potent Small-Molecule PD-L1 Inhibitor or Not? Molecules 2019, 24, 2804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasikumar, P.G.; Sudarshan, N.S.; Adurthi, S.; Ramachandra, R.K.; Samiulla, D.S.; Lakshminarasimhan, A.; Ramanathan, A.; Chandrasekhar, T.; Dhudashiya, A.A.; Talapati, S.R.; et al. PD-1 derived CA-170 is an oral immune checkpoint inhibitor that exhibits preclinical anti-tumor efficacy. Commun. Biol. 2021, 4, 699. [Google Scholar] [CrossRef]
- Magiera-Mularz, K.; Kocik, J.; Musielak, B.; Plewka, J.; Sala, D.; Machula, M.; Grudnik, P.; Hajduk, M.; Czepiel, M.; Siedlar, M.; et al. Human and mouse PD-L1: Similar molecular structure, but different druggability profiles. iScience 2021, 24, 101960. [Google Scholar] [CrossRef]
- Vilalta, M.; Punna, S.; Li, S.C.; Malathong, V.; Lange, C.; McMurtrie, D.; Yang, J.; Roth, H.; McMahon, J.; Campbell, J.J.; et al. Abstract B26: Tumor reduction by a small molecule human PD-1/PD-L1 inhibitor in a melanoma/PBMC co-implantation model. In Proceedings of the Poster Presentations—Proffered Abstracts; AACR Special Conference on Tumor Immunology and Immunotherapy, Miami Beach, FL, USA, 27-28 November 2018; 2020; p. B26. [Google Scholar]
- Li, S.C.; Vilalta, M.; Ertl, L.S.; Wang, Y.; Zeng, Y.; Fan, P.; Lange, C.; McMurtrie, D.; Yang, J.; Lui, R.; et al. Abstract 5693: Anti-tumor effect of orally available small molecule PD-L1 inhibitors in a murine model of colon adenocarcinoma. In Proceedings of the Cancer Chemistry; AACR Annual Meeting 2020, Philadelphia, PA, USA, 27–28 April, 22–24 June 2020; 2020; p. 5693. [Google Scholar]
- Wang, T.; Cai, S.; Wang, M.; Zhang, W.; Zhang, K.; Chen, D.; Li, Z.; Jiang, S. Novel Biphenyl Pyridines as Potent Small-Molecule Inhibitors Targeting the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction. J. Med. Chem. 2021, 64, 7390–7403. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Cheng, B.; Liu, T.; Chen, J. Synthesis and pharmacological evaluation of novel resorcinol biphenyl ether analogs as small molecule inhibitors of PD-1/PD-L1 with benign toxicity profiles for cancer treatment. Biochem. Pharmacol. 2021, 188, 114522. [Google Scholar] [CrossRef] [PubMed]
- Qin, M.; Meng, Y.; Yang, H.; Liu, L.; Zhang, H.; Wang, S.; Liu, C.; Wu, X.; Wu, D.; Tian, Y.; et al. Discovery of 4-Arylindolines Containing a Thiazole Moiety as Potential Antitumor Agents Inhibiting the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Interaction. J. Med. Chem. 2021, 64, 5519–5534. [Google Scholar] [CrossRef]
- Scognamiglio, G.; De Chiara, A.; Parafioriti, A.; Armiraglio, E.; Fazioli, F.; Gallo, M.; Aversa, L.; Camerlingo, R.; Cacciatore, F.; Colella, G.; et al. Patient-derived organoids as a potential model to predict response to PD-1/PD-L1 checkpoint inhibitors. Br. J. Cancer 2019, 121, 979–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucherit, N.; Gorvel, L.; Olive, D. 3D Tumor Models and Their Use for the Testing of Immunotherapies. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Grönholm, M.; Feodoroff, M.; Antignani, G.; Martins, B.; Hamdan, F.; Cerullo, V. Patient-Derived Organoids for Precision Cancer Immunotherapy. Cancer Res. 2021, 81, 3149–3155. [Google Scholar] [CrossRef]
- Liu, L.; Yu, L.; Li, Z.; Li, W.; Huang, W. Patient-derived organoid (PDO) platforms to facilitate clinical decision making. J. Transl. Med. 2021, 19, 40. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Name | HTRF IC50 [nM] | ICB Assay | PD-L1 Dimerization | Target Specificity | ||||
|---|---|---|---|---|---|---|---|---|---|
| Other | Ours | EC50 [nM] | %RLUmax | Ref. | hPD-L1 | mPD-L1 | |||
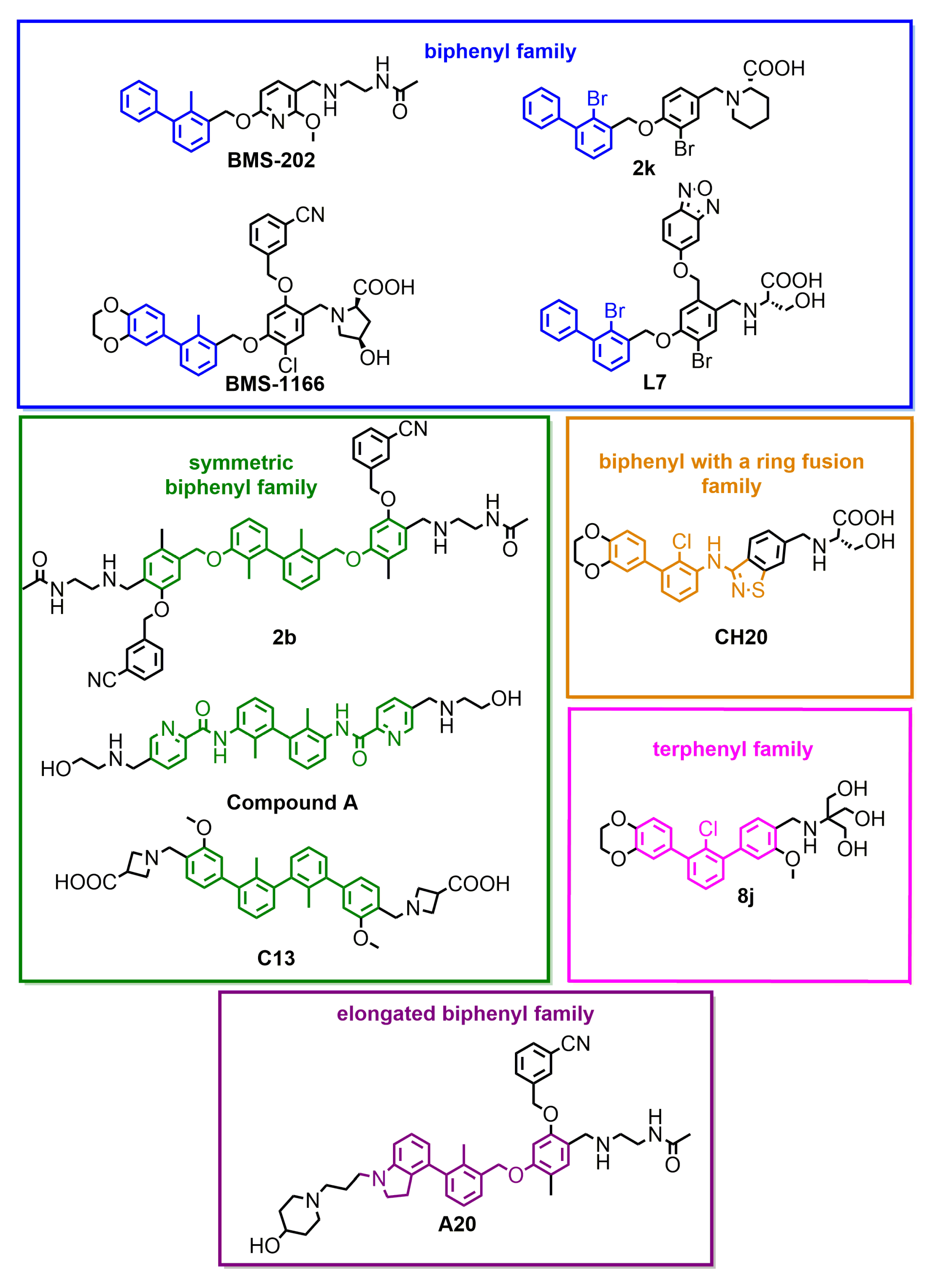
| Small molecules | BMS-202 | 18 [3] | 96 [22] | no act. | √ | √ | No | ||
| BMS-1166 | 1.4 [4] | 3.89 [18] | 1574 | 47 | (Figure A1) | √ | √ | No | |
| 2k | 14.9 [12] | 6632 | 87 | [12] | √ | √ | No | ||
| 8j | <1 [18] | 1026 | 87 | [18] | √ | √ | No | ||
| A20 | 17 [20] | 430 | 55 | [20] | n.d. | √ | n.d. | ||
| CH20 | 8.5 [19] | 5600 | 93 | [19] | n.d. | √ | n.d. | ||
| L7 | 1.8 [13] | 375 | 75 | [13] | n.d. | √ | n.d. | ||
| C13 | 4.23 [23] | 104 | 100 | [23] | n.d. | √ | n.d. | ||
| 2b | 3 [22] | 763 | 93 | [22] | √ | √ | No | ||
| comp. A | 0.4 [21] | 18.9 | 86.3 | [21] | √ | √ | No | ||
| Macrocyclic peptides | p57 | 9 [10] | 566 | 91 | [25] | No | √ | No | |
| p71 | 7 [10] | 293 | 89 | [25] | No | √ | No | ||
| p99 | 153 [10] | 6300 | 83 | [25] | No | √ | No | ||
| p101 | 120 [10] | 27.75 [24] | 7500 | 85 | [24] | No | √ | No | |
| Antibodies | atezolizumab | 0.14 | 100 | [18] | No | √ | √ | ||
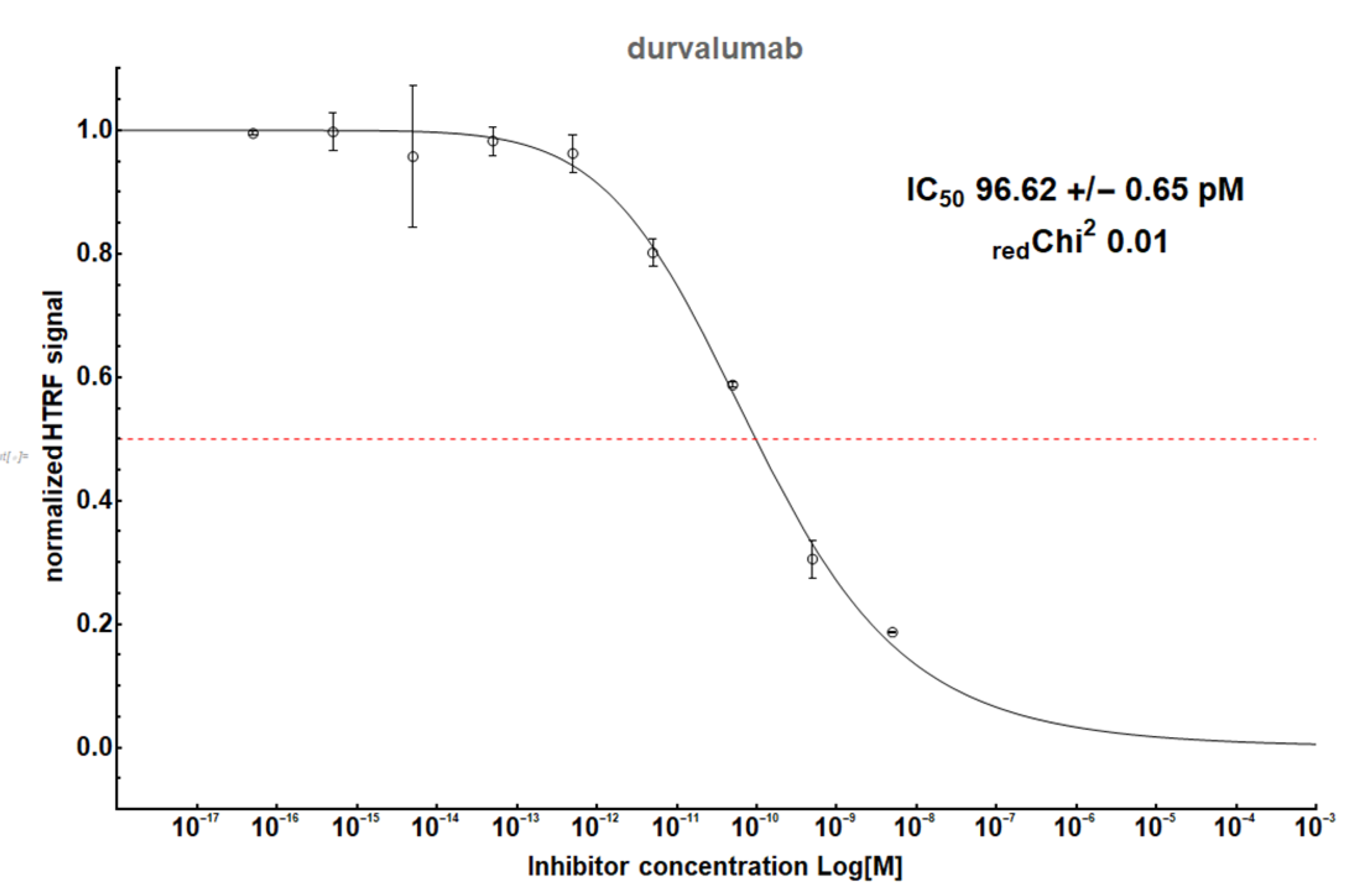
| durvalumab | 0.1 (Figure A2) | 0.23 | 100 | [24] | No | √ | No | ||
| nivolumab | 0.2 [21] | 1.27 | 100 | [25] | - | - | - | ||
| MIH1 | √ | No | |||||||
| MIH5 | No | √ | |||||||
| hPD-1 | √ | √ | |||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Surmiak, E.; Magiera-Mularz, K.; Musielak, B.; Muszak, D.; Kocik-Krol, J.; Kitel, R.; Plewka, J.; Holak, T.A.; Skalniak, L. PD-L1 Inhibitors: Different Classes, Activities, and Mechanisms of Action. Int. J. Mol. Sci. 2021, 22, 11797. https://doi.org/10.3390/ijms222111797
Surmiak E, Magiera-Mularz K, Musielak B, Muszak D, Kocik-Krol J, Kitel R, Plewka J, Holak TA, Skalniak L. PD-L1 Inhibitors: Different Classes, Activities, and Mechanisms of Action. International Journal of Molecular Sciences. 2021; 22(21):11797. https://doi.org/10.3390/ijms222111797
Chicago/Turabian StyleSurmiak, Ewa, Katarzyna Magiera-Mularz, Bogdan Musielak, Damian Muszak, Justyna Kocik-Krol, Radoslaw Kitel, Jacek Plewka, Tad A. Holak, and Lukasz Skalniak. 2021. "PD-L1 Inhibitors: Different Classes, Activities, and Mechanisms of Action" International Journal of Molecular Sciences 22, no. 21: 11797. https://doi.org/10.3390/ijms222111797
APA StyleSurmiak, E., Magiera-Mularz, K., Musielak, B., Muszak, D., Kocik-Krol, J., Kitel, R., Plewka, J., Holak, T. A., & Skalniak, L. (2021). PD-L1 Inhibitors: Different Classes, Activities, and Mechanisms of Action. International Journal of Molecular Sciences, 22(21), 11797. https://doi.org/10.3390/ijms222111797