Abstract
GLOBOCAN 2020 estimated more than 19.3 million new cases, and about 10 million patients were deceased from cancer in 2020. Clinical manifestations showed that several growth factor receptors consisting of transmembrane and cytoplasmic tyrosine kinase (TK) domains play a vital role in cancer progression. Receptor tyrosine kinases (RTKs) are crucial intermediaries of the several cellular pathways and carcinogenesis that directly affect the prognosis and survival of higher tumor grade patients. Tyrosine kinase inhibitors (TKIs) are efficacious drugs for targeted therapy of various cancers. Therefore, RTKs have become a promising therapeutic target to cure cancer. A recent report shows that TKIs are vital mediators of signal transduction and cancer cell proliferation, angiogenesis, and apoptosis. In this review, we discuss the structure and function of RTKs to explore their prime role in cancer therapy. Various TKIs have been developed to date that contribute a lot to treating several types of cancer. These TKI based anticancer drug molecules are also discussed in detail, incorporating their therapeutic efficacy, mechanism of action, and side effects. Additionally, this article focuses on TKIs which are running in the clinical trial and pre-clinical studies. Further, to gain insight into the pathophysiological mechanism of TKIs, we also reviewed the impact of RTK resistance on TKI clinical drugs along with their mechanistic acquired resistance in different cancer types.
1. Introduction
Over the past 250 years, we have seen numerous revolutionary innovations in the war against cancer. However, the causes of cancer are not completely understood to date. Cancer is a result from genetic mutations that lead to uncontrolled propagation of a normal cell [1]. It interferes with the normal cellular program, initiated by the primary tumorigenic site and disseminated to the metastatic site [2]. The current treatment options of cancer are chemotherapy, radiation therapy, combination therapy, laser therapy, and surgery, which often also destroy normal cells and trigger severe side effects in the person with a high burden of the tumor. Therefore, it would be desirable to develop novel therapeutic strategies that can selectively target cancerous cells.
Currently, approximately 90 different tyrosine kinases (TKs) or receptor tyrosine kinases (RTKs) have been identified and further categorized into 20 different subfamilies based on receptors and their ligands [3,4,5,6,7,8]. RTKs are enzymes that play critical roles in most fundamental cell programs in normal cells, including cell proliferation, cell cycle, migration, metabolism, programmed cell death, survival, and differentiation [9]. In contrast, these RTKs catalyze the transfer of gamma-phosphate of ATP to tyrosine hydroxyl groups on target proteins. This posttranslational modification is a crucial phenomenon for cellular communications and homeostasis in normal cells [3,4,5,6,9]. However, RTKs are abnormally implicated in diverse carcinogenic expansion and progression [3,4,5].
Further, RTK associated signaling cascades are able to mediate dysregulated propagation of cells and contribute to sensitivity towards apoptotic stimuli, especially resistance against drugs [6]. In cancer cells, these cascades are epigenetically altered to provide selective advantage. To date, there are more than fifteen different classes of RTKs that have been well explored, with their function and origin shown in Table 1 [9]. EGF receptor/ErbB family, which belongs to RTK class I, is a key mediator in a diverse cellular mechanism including regulation of homeostasis [3,10]. However, the deficit of these ErbBs led to the induction of neurodegenerative diseases and embryonic lethality [10]. On the other hand, elevated levels of ErbB/EGFR are associated with the high grade of tumors [10]. FGFR are also known as an immunoglobulin superfamily, involved in the interaction with various proteins and produced by macrophages [8]. These FGFRs have a unique role in the development of normal cells. Any abnormal levels of FGFs lead to several developmental defects and are linked with angiogenesis, keratinocyte and wound healing processes, and pluripotency via the Wnt signaling pathway [8]. VEGFs are involved in the formation of blood vessels, and permeability and gain or loss results in diabetes, ischemic stroke, and cancers [11]. Another RTK, RET, is also associated with hereditary cancer multiple endocrine neoplasia type 2 [12]. Due to gain or loss of function, deletions, insertions, amplification, point mutation, and elevation in RTKs resulted in dominating oncoprotein translation and malfunctioning of the signaling network that led to onconeogenesis [3,4,5,6].
The breakthrough innovation of RTKs in treating different cancers has become an attractive target, superior to other traditional therapies in selectivity, efficacy, and safety. Drug molecules that inhibit carcinogenesis by binding specifically to their respective targets are the foundation of targeted cancer therapy. To date, a lot of work has been conducted to find synthetic small chemical inhibitors of RTKs to inhibit them selectively [3]. These RTKs play a vital role in regulating cell growth and differentiation, proliferation, motility, adhesion, and apoptosis [4,5,6]. Many TKI drugs are being tested in clinical trials for the treatment of cancers. Clinical results showed that cancer treatment with effective TKIs tends to achieve efficacy, particularly with overcoming TK-resistance (Tables 1 and 2). On the other hand, the non-receptor tyrosine kinases relay intracellular signaling transduction, mediated by dimerization of ligands binding site which collectively induces cytoplasmic domains autophosphorylation of RTKs [5,6,7,8]. Numerous cytoplasmic signaling routes exist, including the Ras/Raf mitogen-activated protein kinase pathway, the Akt pathway/protein kinase -C pathway, the transcription-3 pathway, and the scaffolding proteins mediated signal cascades, which are well reported in several disease models [13,14,15,16].
Chemotherapy is considered a promising option for the treatment of cancers. However, 90% of chemotherapy failures were observed in the invasion and dissemination of malignancies due to drug resistance. A substantial percentage of patient tumor cells turned into resistant cells that create severe problems in patients. [17,18,19]. Apart from this, numerous complications have been shown in antitumor remedies, such as cytotoxic chemotherapy and targeted therapy resistance [20].
To date, there are few promising approaches to control cancer progression based on molecular targets: (1) kinase inhibition at the translational level, (2) immunotherapy to enhance cancer’s rapid immunological defense mechanisms, (3) improvements in treatment choices, (4) delivery of a targeted medicine into proliferating tumor cells, and (5) minimization of the detrimental consequences of chemotherapeutic drugs, among other things [17,21,22]. Chemotherapy resistance is produced by a number of mechanisms, including multidrug resistance (MDR), drug efflux, drug inactivation, cell death prevention (apoptosis suppression), epigenetic, drug metabolism, DNA repair, and gene amplification (Figure 1). We discuss the many anticancer drugs based on tyrosine kinase inhibitors (TKIs) as well as the underlying mechanisms of acquired resistance to RTKI therapy. Non-RTKs, ErbB/HER family members, BCR-ABL, platelet-derived growth factor receptor-alpha/beta (PDGFR-α/β), and vascular endothelial growth factor receptors (VEGFRs) are all modulated by imatinib. Other RTKIs, such as trastuzumab, which targets the HER2/ErbB2 receptor, cetuximab, erlotinib, and gefitinib, which targets the EGFR, also effectively impede cancer growth.
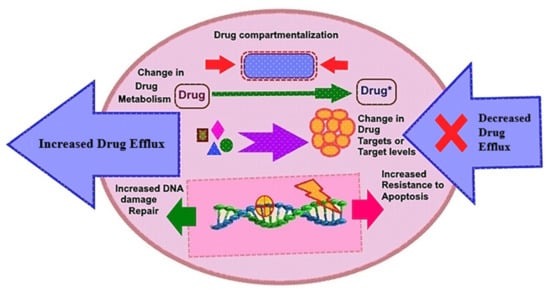
Figure 1.
Depiction of the fundamental drug resistance pathways in cancer cells. Drug efflux mediated cellular programming as drug inactivation, multi-drug resistance, apoptosis suppression, changes in drug metabolism, epigenetic modifications, drug targets, accelerated DNA-repair, and target gene amplification by which cancer cells become resistant to various drugs.
2. Basic Concept on Targeting Receptor Tyrosine Kinases: Effective Strategies to Cure Cancer
The human kinome comprises more than 518 kinases with specific roles based on their subfamilies. Approximately 90 tyrosine kinases and more than 40 tyrosine kinase-like genes control cellular programming and homeostasis [23]. Briefly, tyrosine kinases (TKs) are enzymes involved in transferring phosphate from the high-energy giver ATP to tyrosine residues (serine/threonine) into target amino acids. The use of ATP results in the triggering of intracellular signaling pathways, which closely govern survival, differentiation, and cell development. Certain tyrosine kinase receptors are essential for normal cell regulation and function. However, its dysregulation promotes the growth of tumors and blood vessels via tumor angiogenesis [24,25]. These kinases are generally present as monomers on the cell surface which bind with different growth factors to initiate kinase activities, auto-phosphorylation, and dimerization [26,27]. RTKs such as Met, Axl, Kit, and EGFR are altered in most cancers [28,29]. The proteins PDGFR, VEGFR, and fibroblast growth factor receptor (FGFR) are related to promote metastasis and angiogenesis under tumor microenvironment [30]. Additionally, VEGFRs are expressed on vascular endothelium, and the interactions of VEGF–VEGFR are crucial for the development and propagation of endothelial cells [31,32,33]. It is also well reported that the synergistic effect of fibroblast growth factor with VEGF accelerates vasculature mediated angiogenesis in cancers [34].
RTKs are required for extracellular signal cascades entering the cell, whereas non-RTKs are responsible for internal communication. [34]. The RTK monomer is made up of N-terminus an extracellular ligand-binding domain, C-terminus, and a transmembrane domain containing action of tyrosine kinase (Figure 2). The kinase domain is bi-lobar, with an ATP binding cleft between the N- and C-terminal lobes [9,35]. The ATP-binding site is divided into three sub-regions: sugar binding, adenine binding, and phosphate binding [36]. Kinases’ C-terminal lobes are made up of an activation loop that is defined by a certain amino acid combination at the start of the loop. This combination is known as the ‘DFG motif, composed of the amino acids aspartic acid, phenylalanine, and glycine, denoted as D, F, and G, respectively [35].
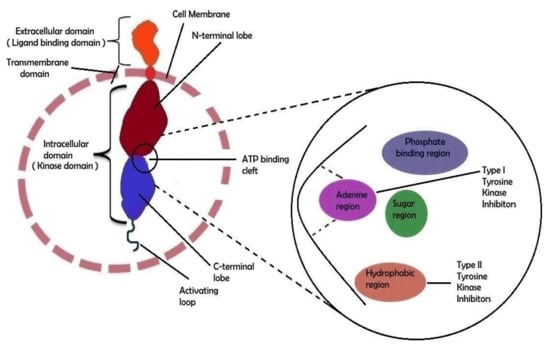
Figure 2.
Molecular structural feature of RTK. A receptor tyrosine kinase’s extracellular domain can bind particular ligands such as growth factors, whereas the intracellular domain is responsible for the kinase’s (auto)phosphorylation. The external and internal domains are separated by the transmembrane region which is fixed in the cell membrane. The ATP-binding cleft is located between the two lobes of the intracellular domain. A schematic depiction of the ATP binding cleft with its numerous regions is shown on the right side of the image. Type I and type II tyrosine kinase inhibitor binding sites have been shown in biochemical general structure model.
The hydrophobic pocket of RTKs is recognized by the subclass of RTK inhibitors that will be addressed in the latter section. Ligand binding to the receptor’s extracellular domain promotes receptor dimerization, which leads to auto-phosphorylation of certain tyrosine residues in the cytoplasmic kinase domain [37]. Additional kinase phosphorylation sites are used to govern a protein–protein interaction for regulating their own kinase activity. When the receptor is activated, it assigns a specific phosphorylation site to the linked proteins [37]. Phosphorylation of respected protein activates numerous signaling pathways, which eventually play a crucial role in biological responses [9].
3. Mutations in RTKs Associated with Diverse Mutational Pathways
RTKs are master key regulators in several diseases, including cancers [4,5,9]. Considerable research has already been undertaken over the last decades to understand the cellular mechanism associated with RTKs. However, mutations in RTKs have attracted significant attention in cancer research [37,38,39]. Figure 3 shows different % mutations of RTKs in several cancers and other oncodriver′s proteins. Here, we summarize a few major pathways that are linked with mutation-induced cancers.
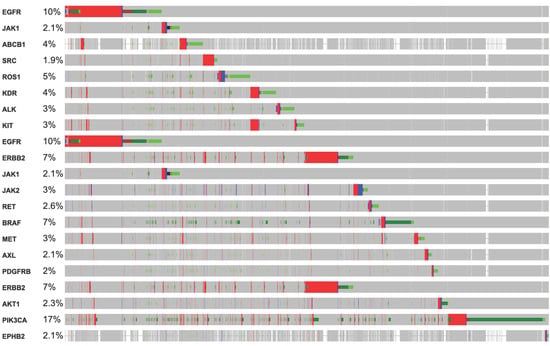
Figure 3.
OncoPrint analysis showing a frequency of mutations in RTKs, and other oncodrivers which suggest a multi-genomic alteration event in various cancers. Among the main RTKs, KRAS, PTEN, BRAF, EGFR, and ERBBs were the most frequently mutated in malignancies.
3.1. Alteration in RTKs Modulate Carcinogenesis via RTK-RAS Mutational Pathway
RTKs and their downstream affecters, such as RAS, appeared as important machinery in the pathogenesis of several cancers [14]. Germline and acquired mutations have been involved in most of the cancer initiation. Constitutive activation of intrinsic receptors by mutation in EGFRs, ERBBs, PDGFRs, and other RTKs has a pivotal role in controlling tumor burden and patients’ survival [15]. Chou et al. demonstrated that 61.1% of mutations were found in EGFR, where the disease control rate and the response rate were 68.5 and 56.8%, respectively [40]. In line with this, the cBioPortal database OncoPrint analysis shows that most of these RTKs have the highest alteration rate, such as EGFR = 10%, ERBB2 = 7%, BRAF = 7%, ROS1 = 5%, and MET = 3%, in 165 (44,752 patients/46,632 samples) cancer cohort studies (Figure 3). Collectively, these mutational changes have exhibited a significant reduction in disease-free and overall survival of patients with cancers. However, negative for EGFR mutations showed better overall survival (OS, p = 0.046) and progression-free survival (PFS, p = 0.011). Additionally, the cBioPortal database RTKs-KRAS-BRAF mutational pathway also revealed an aberration in RTKs which simultaneously induces 16.7% mutation in KRAS, associated with 14.3 and 13.4% mutational changes in BRAF and NF1, respectively. Together these mutational changes alter cancer cell propagation, translation, and survival via the RAS-BRAF mediated signaling pathway (Figure 4A).
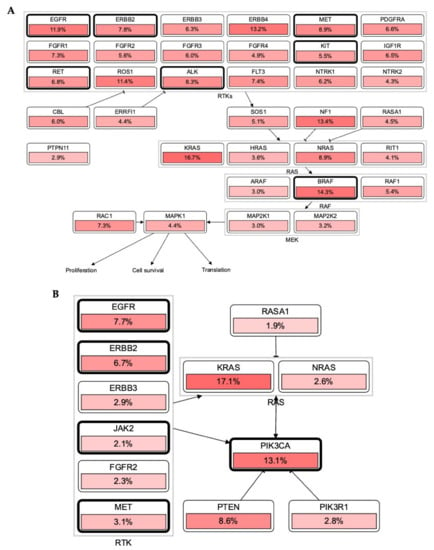
Figure 4.
RTKs involvement in the RAS carcinogenic pathway. (A,B): Most RTKs in different malignancies have undergone mutational alterations (data was retrieved from cBioPortal patient cancer databank). In each pathway, RTKs including other oncodrivers were highly mutated and modulated a number of oncogenic mechanisms which promote tumor initiation, invasiveness, and tumor burden spread in several cancers.
3.2. Association of RTKs via KRAS-PIK3CA Pathway in Cancer
The most altered genes were KRAS with 17.1% in 44,752 patients (46,632 samples in 165 studies) in the KRAS-PIK3CA associated pathway. RAS has been known over the last three decades, and its mutation acts as a driver for cancer progression and maintenance [41]. Jang et al. reported a multigene panel study of mutations where KRAS, NRAS, BRAF, PIK3CA, TP53, and POLE were most mutated in 90 colorectal carcinoma patients [42]. Further, EGFR inhibitors such as erlotinib and gefitinib are beneficial in the management of lung cancer. However, it also revealed limited efficacy in different patients. Patients with KRAS mutations have also been associated with resistance to the EGFR inhibitors. As a result, there is an urgent need to investigate new hybrid small molecules to alleviate tumor burden in patients with KRAS-EGFR mutations. In accordance with this, KYA1797K demonstrated significant effectiveness against KRAS-driven cancer in KrasLA2 mice via inhibiting the Ras-ERK pathway. The destabilization of Ras via RTKs can be a promising strategy to halt a tumor in KRAS-mutated NSCLC [43].
Lewis Cantley discovered the PI3Ks as an unknown phosphoinositide kinase correlated with the polyoma middle T protein [44]. It participates in most of the cellular signaling, including cancers. An elevation or mutation in PIK3CA led to EGFR upregulation and ERK mediated paracrine cascades in breast cancer [45]. In line with this finding, it has been proposed that EGFR inhibitor treatment regulates overall and disease-free survival of NSCLC patients with the EGFR mutation coexisting with the PIK3CA double mutation [46]. In Figure 2, PIK3CA with 13.1% mutation intricately involved in the progression and pathogenesis of cancers via AKT1 (1.8%), mTOR (3.2%), and BRAF (5.7%) mediated proliferation and survival of cancer cells. It also showed that these RTKs directly induced PIK3CA upregulation and mutation, which augmented tumorigenesis (Figure 4B). The significant role of RTKs has been shown in the RTKs-PTEN-KRAS-MTOR and RTKs-PTEN-PIK3CA-AKT1-KRAS mutational pathways and augment translation, proliferation, and survival of tumor cells (Figure 5A,B). Additionally, binding of these RTKs with inhibitors control several cancers via diverse mechanisms (Figure 5C). Collectively, RTKs could be involved in resistance to the drug in cancer cells, and their inhibition may be exploited in target therapies.
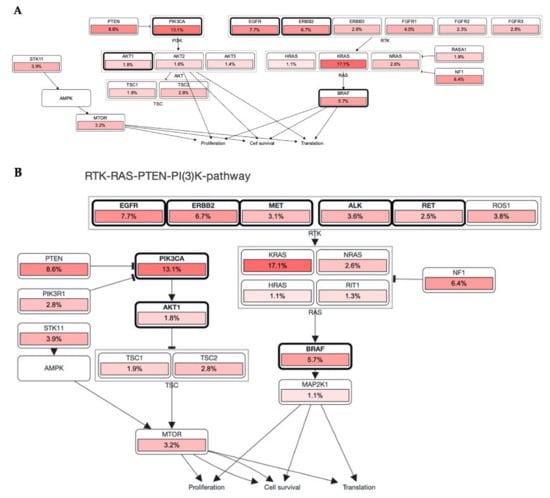
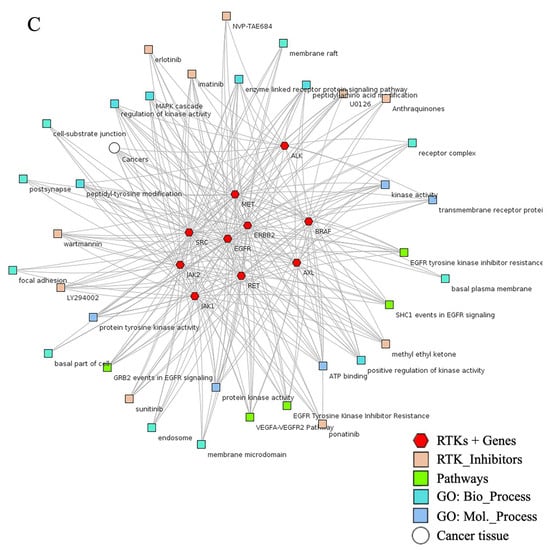
Figure 5.
Mutation of RTKs caused mutation in KRAS, PIK3CA, BRAF, PTEN, and mTOR ablation that induced tumorigenesis. (A,B) Mutational changes occurred in most RTKs, including EGFR, ERBB3, MET, ALK, RET, and FGFRs, which involved diverse mediators that induced cell proliferation. Pathways retrieved from cBioPortal patient cancer databank. (C) Association of RTKs with their inhibitor and major GO were visualized by an abstracted network analysis.
4. Binding Site Architecture of Tyrosine Kinase
In eukaryotic, approximately 2% kinase domains catalyze the phosphate (PO43−) transfer of ATP to a particular substrate of hydroxy-tyrosine by posttranslational modification [47]. Due to the pivotal role of tyrosine phosphorylation via kinase domains, it makes them highly conserved in diverse diseases [3,4,5,6,9,35,48,49]. In EGFR (PDB: 1M14), these domains are made up of a C-lobe (larger) and N-lobe (small). Among them, there are five, the αE, αF, αG, αH, and αI helix, which are found in the large C-lobe. The β1 to β5 five beta sheets are positioned in the N-lobe with one α-helix (αC, amino acid residue 729-744). ATP binding sites are mostly in a deep cleft of both C- and N-lobes and in a highly conserved glycine-rich phosphate-binding loop that bridges β1 with β2 in the N-lobe [50]. The phosphates of ATP significantly interact with a high glycine-rich loop of EGFR [51]. In addition, conserved glutamate (Glu738) in EGFR of the αC helix is formed by actively ion association with a Lysine (Lys721) in β3 of N-lobe, adding α and β phosphates of ATP (Figure 4). In the C-lobe, it creates the ATP-binding cleft with a highly conserved Asp812-Asn818 catalytic loop, among which the interaction of Asn818 by hydrogen bond places Asp812; however, Asp812 binds with the hydroxyl chain of tyrosine substrate [47,51]. Asp831-Val852, together, form an activation loop in the C-lobe that consists of base Asp-Gly-Phe and Asp831-Gly833 DFG motif [51]. The phosphorylation of C-terminal domain containing Tyr974, Tyr992, Tyr1048, Tyr1068, Tyr1086, Tyr1101, and Tyr1173 shows auto-catalytic activities [47]. In contrast, phosphotyrosines residue of the C-terminal is considered for the EGFR-drug or small inhibitors docking site, Src homology, and PTB binding domain. The crystal structure of the EGFR kinase domain, which contains forty amino acids that take a substantial role in the development of EGFRK-specific inhibitors such as erlotinib (OSI-774, CP-358,774, TarcevaTM), is currently in Phase III clinical trials as an anticancer treatment [52]. The phosphorylation of EGFR is linked to a number of downstream pathways, including the RAS-RAF-MEK-ERK and the PI3K-AKT-mTOR circuit (Figure 2 and Figure 3).
Muller and his team reported the crystal structure of VEGF, and they found that it contains a cystine knot connected with two disulfide bonds between C51 and C60. It comprises four β (1,3,5,6) sheets linked by a cystine bond at the terminal end [53]. In β-sheet, β3 and β5 are connected by a hydrogen bond. Another segment of VEGF has α-helix and short β-strand in loop-1; here, loop-1 augments the binding between β1 and β3. Short β-strand, β5, and the beginning of β6 collectively create a three-stranded β-sheet near the cysteine knot. A hydrophobic core was created by combining a three-stranded-sheet, loops L1 and L3 (5–6), and helix 2 with the opposite monomer’s amino terminal -helix. This core is involved in bringing the four-stranded central sheet into equilibrium [53]. Harris et al. released an article detailing the discovery of pazopanib in 2008 [54]. That was a wonderful example of how homology modeling and SBDD were utilized to identify a previously available medication. Because the crystal structure of VEGFR2 was not known at the time, a homology model of the enzyme based on FGFR crystal structures was constructed to predict the binding mechanism of dimethoxyquinazoline analogs. The pyrimidine and quinazoline bound similarly at the ATP binding site, establishing hydrogen bonds with the backbone’s Cys919 residue (Figure 6). The crystallization of these compounds with VEGFR2 was confirmed in silico (PDB: 1Y6A, 1Y6B). In contrast, structural architechture of VEGF also depicting binding sites and its interaction with inhibitor pazopanib in several cancer cells (Figure 7). Finally, pazopanib was discovered when a series of novel analogs were developed, synthesized, and evaluated in vitro [54,55,56].
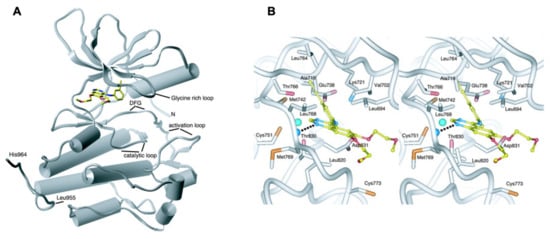
Figure 6.
EGFR-erlotinib binding structure: Erlotinib binds at the gap between the EGFR kinase domain’s amino- and carboxy-terminal lobes (PDB: 1M14). (A,B) A stereo image of the inhibitor binding site and surrounding EGFRK/erlotinib residues. An H-bond from the Met769 amide nitrogen to erlotinib is indicated by a dashed line (It was procured from Stamos et al. [52]).
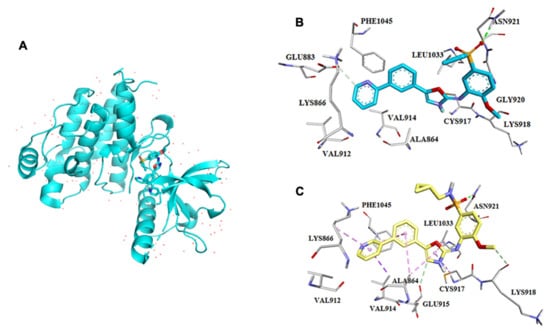
Figure 7.
VEGF structure and interaction with its inhibitor pazopanib. (A) Structure of VEGF, (B) PDB: 1Y6A, (C) PDB: 1Y6B. These docked images were taken from Harris et al., 2005 [54] and 2008 [55].
5. Major Classes in RTKs: Master Regulators of Cell Fate and Homeostasis
Since finding the first RTK, it has been divided into more than twenty subfamilies. The EGF receptor family and ErbB family are under RTK class I [57]. In RTK class II, insulin (IGF) associated receptor families play in diverse pathophysiological cascades that regulate propagation and trans-differentiation of a single cell [57,58]. Dysregulation of IGF-related RTKs has been linked in multiple diseases and tumor initiations [59]. The PDGF receptor family of the third class has a unique contribution in developing connective tissue cells and synthesis of organs [60]. The VEGFR group (class IV) is involved in the growth of endothelial cells and permeability of blood vessels. Dysfunction of VEGFR leads to oncogenesis by angiogenesis and dissemination of secondary metastatic tumors [61]. Apart from that, most of the other RTKs also participate in several cellular mechanism-mediated diseases. To date, numerous small molecule inhibitors were discovered targeting these RTKs with great efficacy in ailments, infection, and malignancies (Table 1).

Table 1.
Major RTKs in human diseases and their effective inhibitors in clinical trial.
6. Selective TKs Inhibitors in the Clinical Trial
RTKs emerged as a key player in most of the cellular processes. Alteration in RTKs resulted in the initiation of cancer growth and other diseases [9,73]. The EGFR, known as HER1, is from the ErbB group and is involved in diverse mechanisms, including invasion and dissemination of tumor cells [15,20,39,43,45,46]. Inhibition and targeting EGFR have undergone extensive studies to date, with the development of four-generation inhibitors, and it has been proved indispensable in the curing of EGFR-mutant patients [74]. First generation inhibitors, such as geftinib, erlotinib and icotinib, have shown significant antineoplastic activities in EGFR-mutant NSCLC patients [75]. Second-generation EGFR inhibitors, such as afatinib and dacomitinib, have been discovered with high affinity binding to EGFR, which are considered pan-HER inhibitors that significantly enhance survival [76]. However, these inhibitors also showed toxicity towards normal adjacent cells, limiting their progress in clinical trials. So, exploration of novel TK inhibitors with minimal toxicity is needed. However, several effective TKs inhibitors have been developed to date, and some of them showed promising anticancer activity in clinical trial studies (Table 2 and Table 3).
Table 2.
Under top 40 selective RTKs inhibitors (drugs) in clinical trial for multiple cancers.
Table 3.
Drug resistance results based on GDSC drug treatment data and the substructure mutations.
7. Recent Updates on Selective Tyrosine Kinase Inhibitors in Pre-Clinical Studies
RTKIs are efficient treatment options for RTKs sensiting-mutant cancers [82]. Abnormal changes in RTKs are responsible for tumor growth and invasion. Elevated levels or mutation of these TKs cause drug resistance and enhance tumor burden and invasion [82,83]. Although many small molecule inhibitors are already developed and in use now, these medicines have significant adverse effects, such as skin rash and gastrointestinal toxicity, thus limiting their therapeutic use. Numerous molecules have been designed and synthesized to discover more effective and safe inhibitors, showing promising anticancer activity in pre-clinical studies.
Li and his team reported pyrimidine derivatives containing cyclopropyl moiety, which exhibited admirable kinase inhibitory activities against EGFR double mutation (L858R/T790M) with an IC50 = 0.26 nM [84]. Similarly, Quinazoline based small molecule comprising benzazepine moiety also reduced the tumor growth with IC50 ranging from 1.06–3.55 μM by targeting EGFR-T790M [85]. Additionally, Pyrazole moiety has been inserted in many FDA-approved drugs such as Pazopanib, which offered more potent inhibitors. These pyrazole derivatives exhibited potent tumoricidal action against EGFR-tyrosine kinase with an IC50 of 395.1, 286.9, and 229.4 nM, respectively, compared to the standard drug, erlotinib [86]. The pyrazoline derivatives consisting of carbothioamide group showed significant EGFR inhibition, showing IC50 values ranging from 1.66 to 1.9 μM, and halted the growth of cancer cells [87]. Our previous studies also suggested that molecules from pyrazolo[3,4-d]pyrimidine, urea, piperazine, and hybrids have shown fabulous anticancer activities via inhibition of various oncoproteins against cancers [87,88,89]. These studies may help to postulate new possibilities for developing novel small molecule drugs with effective anticancer activity.
According to new research, VEGF is a critical activator of both normal and pathological angiogenesis [90]. Inhibition of VEGF mediated signaling by small molecules or antibody has been studied to halt cancer progression. Hennequin et al. identified 6,7-dimethoxyanilinoquinazolines (ZD4190) as a promising VEGF selective inhibitor with KDR/FTK potency ratio ranging from approximately 30-8000-fold [91]. Similarly, Quinazolin-4(3H)-one inhibited VEGFR-2 with an IC50 value of 0.340 ± 0.04 µM, which was higher than that of the reference medication, sorafenib [92]. Biphenylurea/thiourea derivatives tethered with heteroarylsulfonamide motifs were also found to be novel VEGFR2 inhibitors with significant inhibition compared to sorafenib [93]. Novel 2 indolinone-thiazole hybrid molecules effectively bound at the ATP binding site of VEGF-2 and showed a tumoricidal action with an IC50 value of 3.9 ± 0.13 µM, greater than Sunitinib [94]. Apart from that, many peptides-based pan-VEGF inhibitors with druggable binding sites might be crucial for discovering novel and selective VEGF inhibitors to manage the burden of cancer growth [95].
Among tyrosine kinases, the IGF system plays a pivotal role in the maintenance of cell growth and homeostasis [96]. It consists of TKs, insulin receptors (IRs), IGF1R, IGF2R, non-TKs, and some ligands IGFBPs, insulin, IGF1, and IGF2 [96]. However, pharmacological targeting kinases VEGFs may lead to more significant toxicity. Hence, the development of novel multitarget inhibitors that block IGF1Rs is immediately indispensable. One of the most potent IGF-1R inhibitors, phenylpyrazolo[3,4-d]pyrimidine, showed fabulous antitumor activities in the NSCLC induced xenograft and allograft model [97]. Gadekar and colleagues proposed 2,3-dihydroimidazo[2,1-b]thiazoles as dual EGFR and IGF1R inhibitors with reasonable drug likeness properties [98]. Tyrphostin AG 1024, a selective inhibitor of IGF-1R, markedly increases the response of tumor cells to ionizing radiation [99]. Moreover, Pyrazole pyridine-peperidinyl containing propanone also showed highly antitumor potency against cancer cells growth [88,100]. BMS-754807 is a reversible and strong inhibitor of the IGF-1 receptor/insulin receptor family kinases with Ki = 2 nmol/L [101]. It is currently in phase I clinical trial to treat a variety of human cancers [101]. Hence, we have seen the development of several IGFRs inhibitors; however, there is still an urgent need to design and synthesize more potent inhibitors that may be useful for managing cancers.
PDGF group kinases are essential for tumor growth and ablation. It is associated with a higher grade of tumor burden in ER + BRCA [30,65]. Therefore, blockade of PDGF would be one of the most promising strategies to mitigate the invasion of BRCA. In line with this, 3-substituted Quinoline derivatives were found to be effective PDGF inhibitors with IC50 value of 0.340 ± 0.04 µM [102]. Yang et al. found 2-pyrrolidone-fused (2-oxoindolin-3-ylidene) methylpyrrole derivatives that exhibited significant inhibitory activities against PDGFRβ in order to halt angiogenic tumor growth [103]. TK kinase FGFR elevation is also involved in the initiation of the cancerous tumor. Inhibition of FGFR by newly synthesized inhibitor JNJ-42756493 increases survival in patients with advanced or high-grade refractory solid tumors [104]. Similarly, PRN1371 exhibits prolonged target inhibition and highly selective covalent inhibitor against FGFR [105]. Apart from these inhibitors, several active synthesized small molecules efficiently block the translations of other TKs and halt oncogenesis.
Lastly, for the first time, a novel paradigm in which chemo and immunotherapy have been combined to treat cancer seems to be a wonderful antibody–drug conjugate (ADC) technique [106]. In contrast, a particular antibody can bind with an oncoprotein target without harming innocent adjacent cells, improving the efficacy of the drug. To put it another way, ADCs deliver a deactivated drug molecule to cancerous cells. The cytotoxin is produced after internalization of ADC to target cancer cells in which it plays a tumoricidal role. Currently, few ADC based potent drugs are available to cure high burden tumors, especially breast cancers, such as Roche’s Kadcyla (antiHER2 IgG1, trastuzumab, microtubule inhibitory medicine DM1), Gilead Sciences Trodelvy (IgGk–Sacituzumab govitecan-hziy), and Daiichi’s Enhertu (mAb-trastuzumab deruxtecan) [107]. Collectively, RTKs associated ADCs might be efficient and promising drug candidates which can target cancer cells without harming the healthy cells. It has been considered that it is capable to overcome the major clinical barrier of conventional chemotherapy, expanding a novel therapeutic window.
8. Clinical Resistance to Small Molecule Inhibitor Targeting RTKs: Advances and Pitfalls of Targeted Therapy
Today, anticancer therapies including radiotherapy, chemotherapy, combination, surgery, immunotherapy, and laser therapy are well reported; however, there is still dire need to discover a specific target in solid tumors [108,109,110,111]. Although, several new targets such as carbonic anhydrases, BCl2, Bax, TEAD, and HDACs are being explored to overcome drug resistance/drug failure in cancer management [112,113,114]. Additionally, multiple drug resistance due to ablation, mutation, and genomic instabilities is also considered one of the biggest challenges in the clinical model of several cancers [40,41,42,115]. Drug resistance consists of primary and secondary subtypes. Primary resistance is defined as patients who do not reach a stable disease under six months of an initial clinical response. Elsewhere, it shows primary resistance to imatinib is particularly manifest in patients with a KIT exon nine mutation [116]. PDGFRA also depicts primary mutation at kinase domain and lead conformational changes by which it repones primary resistance to imatinib [117]. Similarly, mutation in KRAS is also associated with primary resistance in treatment with EGFR inhibitors [41,42].
Secondary resistance to RTK inhibitors is particularly common in individuals who have been treated with kinase inhibitors [118]. Numerous cellular programs have been modulated due to resistance phenotype changes in tumor cells [118]. Structural changes in the kinase domain of mutated RTKs is the most common cause of secondary resistance. Hence, the catalytic activities of kinase are suppressed due to being unable to bind and inhibit RTKs. KIT and EGFR are always associated with secondary mutations and more than 45% of patients have acquired resistance to imatinib, erlotinib, and gefitinib [119,120]. These mutational changes on RTKs directly affect the clinical response to small molecule inhibitors as well as drugs. Additionally, both primary and secondary resistance significantly change the IC50 value of RTKs inhibitors or drugs. The Genomics of Drug Sensitivity in Cancer (GDSC) is an online database system by which we can obtain big information on drug sensitivity and resistance, especially several cancer cells with their drug responses. Interestingly, we showed that most RTKs inhibitors are efficient in halting cancer progression by targeting a particular kinase with a fabulous IC50 dose (Table 3).
9. Conclusions and Perspectives
Over the last quarter century, we have made significant advances in our knowledge of RTK signaling. Basic research associated with these receptors’ genetics, cellular biology, biochemistry, and structural biology has resulted in a detailed understanding of how this protein family operates. These discoveries have resulted in the creation of a slew of potential medicines, serving as prime instances of laboratory-driven translational research. Similarly, clinical studies of RTKs in several disorders have offered significant mechanistic understanding (i.e., reverse-translational research), allowing for further refinement of treatment methods. The chemical processes behind RTK activation have similar motifs but differ significantly in their details. Only about half of the RTK families are completely known. Exploring other families will undoubtedly yield many new insights regarding RTK activation and signaling in the future. The relevance of direct receptor crosstalk or heterodimerization for signaling specificity remains unknown at the receptor molecule level, as does the precise function performed by intracellular trafficking. Another critical problem for the future is gaining quantitative knowledge of these challenges. RTKs inhibitors have been proven as an effective treatment option for several types of cancer. However, resistance of several RTKs inhibitors towards several cancer cells seems to be a critical obstacle in the successful therapy of cancers. Hence, the development of new effective inhibitors is still required to overcome drug resistance hurdles. Although genetic and pharmacological methods have identified many signaling components in RTK networks, putting these components into mechanistic and quantitative settings is the next significant hurdle. This knowledge might pave the way for new approaches to developing effective therapies for cancer and other illnesses caused by active RTKs.
Author Contributions
Conceptualization, R.K.M., C.B.M., A.K.S. and M.-S.L.; formal analysis, A.P., M.A.-U.-Z. and R.K.M.; funding acquisition, M.-S.L.; investigation, R.K.M., M.A.-U.-Z. and C.B.M.; methodology, R.K.M., C.B.M., A.P., S.J., A.K.S., M.A.-U.-Z. and M.-S.L.; supervision, M.-S.L.; visualization, R.K.M., C.B.M. and M.-S.L.; writing—original draft, R.K.M., M.A.-U.-Z., A.K.S. and C.B.M.; writing—review and editing, R.K.M., C.B.M. and M.-S.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grant from National Research Foundation of Korea (NRF), funded by the Korean Government (MSIP) (Grant nos. NRF-2016R1A5A1011974 and NRF 2020R1A2C1102100).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Michor, F.; Iwasa, Y.; Nowak, M.A. Dynamics of cancer progression. Nat. Rev. Can. 2004, 4, 197–205. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Metibemu, D.S.; Akinloye, O.A.; Akamo, A.J.; Ojo, D.A.; Okeowo, O.T.; Omotuyi, I.O. Exploring receptor tyrosine kinases-inhibitors in cancer treatments. Egypt. J. Med. Hu. Genet. 2019, 20. [Google Scholar] [CrossRef]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef]
- Butti, R.; Das, S.; Gunasekaran, V.P.; Yadav, A.S.; Kumar, D.; Kundu, G.C. Receptor tyrosine kinases (RTKs) in breast cancer: Signaling, therapeutic implications and challenges. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef]
- Receptor Tyrosine Kinases (RTKs). IUPHAR/BPS Guide to PHARMACOLOGY. Available online: http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=304 (accessed on 15 September 2021).
- Alexander, S.P.; Fabbro, D.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; Southan, C.; et al. CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. Br. J. Pharmacol. 2019, 176, S297–S396. [Google Scholar]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target Ther. 2020, 5, 181. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Toledo, S.P.; dos Santos, M.A.; Toledo, R.; Lourenço, D.M., Jr. Impact of RET proto-oncogene analysis on the clinical management of multiple endocrine neoplasia type 2. Clinics 2019, 61, 59–70. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Huang, L.; Fu, L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta. Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Li, R.; Morris, S.W. Development of anaplastic lymphoma kinase (ALK) small-molecule inhibitors for cancer therapy. Med. Res. Rev. 2008, 28, 372–412. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Suzuki, R.; Sakata, S.; Hatano, S.; Asaka, R.; Hamanaka, W.; Ninomiya, H.; Uehara, H.; et al. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012, 18, 378–381. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhou, C.C.; Chen, G.Y.; Cheng, Y.; Huang, C.; Zhang, L.; Xu, C.R.; Li, A.W.; Yan, H.H.; Su, J.; et al. A multicenter phase II study of sorafenib monotherapy in clinically selected patients with advanced lung adenocarcinoma after failure of EGFR-TKI therapy (Chinese Thoracic Oncology Group, CTONG 0805). Lung Cancer 2014, 83, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Tomczak, K.; Li, J.; Ochieng, J.K.; Lee, Y.; Haymaker, C. Next-Generation Immunotherapies to Improve Anticancer Immunity. Front. Pharmacol. 2021, 11, 566401. [Google Scholar] [CrossRef] [PubMed]
- Mongre, R.K.; Sodhi, S.S.; Ghosh, M.; Kim, J.H.; Kim, N.; Park, Y.H.; Kim, S.J.; Heo, Y.J.; Sharma, N.; Jeong, D.K. The novel inhibitor BRM270 downregulates tumorigenesis by suppression of NF-κB signaling cascade in MDR-induced stem like cancer-initiating cells. Int. J. Oncol. 2015, 46, 2573–2585. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duong-Ly, K.C.; Peterson, J.R. The human kinome and kinase inhibition. Curr. Protoc. Pharmacol. 2013, 60, 2–9. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Baier, A.; Szyszka, R. Compounds from Natural Sources as Protein Kinase Inhibitors. Biomolecules 2020, 10, 1546. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef]
- Cybulsky, A.V.; Guillemette, J.; Papillon, J.; Abouelazm, N.T. Regulation of Ste20-like kinase, SLK, activity: Dimerization and activation segment phosphorylation. PLoS ONE 2017, 12, e0177226. [Google Scholar] [CrossRef]
- Falcone, I.; Conciatori, F.; Bazzichetto, C.; Bria, E.; Carbognin, L.; Malaguti, P.; Ferretti, G.; Cognetti, F.; Milella, M.; Ciuffreda, L. AXL Receptor in Breast Cancer: Molecular Involvement and Therapeutic Limitations. Int. J. Mol. Sci. 2020, 21, 8419. [Google Scholar] [CrossRef]
- Myers, S.H.; Brunton, V.G.; Unciti-Broceta, A. AXL Inhibitors in Cancer: A Medicinal Chemistry Perspective. J. Med. Chem. 2015, 59, 3593–3608. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, K.; Yang, Y.; Nakamura, M.; Andersson, P.; Yang, X.; Zhang, Y.; Cao, Y. Dual roles of endothelial FGF-2–FGFR1–PDGF-BB and perivascular FGF-2–FGFR2–PDGFRβ signaling pathways in tumor vascular remodeling. Cell Discov. 2018, 4. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Feng, Y.; Wang, J.; Zhang, X.; Shen, J.; Zou, R.; Yuan, Y. Platelet-derived growth factor-BB promotes proliferation and migration of retinal microvascular pericytes by up-regulating the expression of C-X-C chemokine receptor types 4. Exp. Ther. Med. 2019, 18, 4022–4030. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef]
- Gotink, K.J.; Verheul, H.M. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef]
- Smith, J.A.; Francis, S.H.; Corbin, J.D. Autophosphorylation: A salient feature of protein kinases. Mol. Cell Biochem. 1993, 127–128, 51–70. [Google Scholar] [CrossRef]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.S.; Kim, K.M.; Ting, J.C.; Yu, K.; Fu, J.; Liu, S.; Cristescu, R.; Nebozhyn, M.; Gong, L.; Yue, Y.G.; et al. Genomic landscape and genetic heterogeneity in gastric adenocarcinoma revealed by whole-genome sequencing. Nat. Commun. 2014, 5, 5477. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, E.; Togashi, Y.; Takeuchi, Y.; Shinya, S.; Tada, Y.; Kataoka, K.; Tane, K.; Sato, E.; Ishii, G.; Goto, K.; et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non-small cell lung cancer. Sci. Immunol. 2020, 5, eaav3937. [Google Scholar] [CrossRef]
- Chou, T.Y.; Chiu, C.H.; Li, L.H.; Hsiao, C.Y.; Tzen, C.Y.; Chang, K.T.; Chen, Y.M.; Perng, R.P.; Tsai, S.F.; Tsai, C.M. Mutation in the tyrosine kinase domain of epidermal growth factor receptor is a predictive and prognostic factor for gefitinib treatment in patients with non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 3750–3757. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef]
- Jang, S.; Hong, M.; Shin, M.K.; Kim, B.C.; Shin, H.S.; Yu, E.; Hong, S.M.; Kim, J.; Chun, S.M.; Kim, T.I.; et al. KRAS and PIK3CA mutations in colorectal adenocarcinomas correlate with aggressive histological features and behavior. Hum. Pathol. 2017, 65, 21–30. [Google Scholar] [CrossRef]
- Park, J.; Cho, Y.H.; Shin, W.J.; Lee, S.K.; Lee, J.; Kim, T.; Cha, P.H.; Yang, J.S.; Cho, J.; Min, D.S.; et al. A Ras destabilizer KYA1797K overcomes the resistance of EGFR tyrosine kinase inhibitor in KRAS-mutated non-small cell lung cancer. Sci. Rep. 2019, 9, 648. [Google Scholar] [CrossRef]
- Bradley, D. Biography of Lewis C. Cantley. Proc. Natl. Acad. Sci. USA 2004, 101, 3327–3328. [Google Scholar] [CrossRef] [PubMed]
- Young, C.D.; Zimmerman, L.J.; Hoshino, D.; Formisano, L.; Hanker, A.B.; Gatza, M.L.; Morrison, M.M.; Moore, P.D.; Whitwell, C.A.; Dave, B.; et al. Activating PIK3CA Mutations Induce an Epidermal Growth Factor Receptor (EGFR)/Extracellular Signal-regulated Kinase (ERK) Paracrine Signaling Axis in Basal-like Breast Cancer. Mol. Cell Proteom. 2015, 14, 1959–1976. [Google Scholar] [CrossRef]
- Qiu, X.; Wang, Y.; Liu, F.; Peng, L.; Fang, C.; Qian, X.; Zhang, X.; Wang, Q.; Xiao, Z.; Chen, R.; et al. Survival and prognosis analyses of concurrent PIK3CA mutations in EGFR mutant non-small cell lung cancer treated with EGFR tyrosine kinase inhibitors. Am. J. Cancer Res. 2021, 11, 3189–3200. [Google Scholar] [PubMed]
- Martin-Fernandez, M.L.; Clarke, D.T.; Roberts, S.K.; Zanetti-Domingues, L.C.; Gervasio, F.L. Structure and Dynamics of the EGF Receptor as Revealed by Experiments and Simulations and Its Relevance to Non-Small Cell Lung Cancer. Cells 2019, 8, 316. [Google Scholar] [CrossRef]
- DeBerge, M.; Glinton, K.; Subramanian, M.; Wilsbacher, L.D.; Rothlin, C.V.; Tabas, I.; Thorp, E.B. Macrophage AXL receptor tyrosine kinase inflames the heart after reperfused myocardial infarction. J. Clin. Invest. 2021, 131, e139576. [Google Scholar] [CrossRef] [PubMed]
- Mishra, C.B.; Pandey, P.; Sharma, R.D.; Malik, M.Z.; Mongre, R.K.; Lynn, A.M.; Prasad, R.; Jeon, R.; Prakash, A. Identifying the natural polyphenol catechin as a multi-targeted agent against SARS-CoV-2 for the plausible therapy of COVID-19: An integrated computational approach. Brief. Bioinform. 2021, 22, 1346–1360. [Google Scholar] [CrossRef]
- Ferguson, K.M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Petri, E.T.; Halmos, B.; Boggon, T.J. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J. Clin. Oncol. 2008, 26, 1742–1751. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Muller, Y.A.; Li, B.; Christinger, H.W.; Wells, J.A.; Cunningham, B.C.; de Vos, A.M. Vascular endothelial growth factor: Crystal structure and functional mapping of the kinase domain receptor binding site. Proc. Natl. Acad. Sci. USA 1997, 94, 7192–7197. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Boloor, A.; Cheung, M.; Kumar, R.; Crosby, R.M.; Davis-Ward, R.G.; Epperly, A.H.; Hinkle, K.W.; Hunter, R.N., 3rd; Johnson, J.H.; et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J. Med. Chem. 2008, 51, 4632–4640. [Google Scholar] [CrossRef]
- Harris, P.A.; Cheung, M.; Hunter, R.N., III; Brown, M.L.; Veal, J.M.; Nolte, R.T.; Wang, L.; Liu, W.; Crosby, R.M.; Johnson, J.H.; et al. Discovery and evaluation of 2-anilino-5-aryloxazoles as a novel class of VEGFR-2 kinase inhibitors. J. Med. Chem. 2005, 48, 1610–1619. [Google Scholar] [CrossRef]
- Jia, Y.; Zhang, J.; Feng, J.; Xu, F.; Pan, H.; Xu, W. Design, synthesis and biological evaluation of pazopanib derivatives as antitumor agents. Chem. Biol. Drug. Des. 2014, 83, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Wu, J.; Yu, E. Insulin-like growth factor receptor-1 (IGF-IR) as a target for prostate cancer therapy. Cancer Metastasis Rev. 2014, 33, 607–617. [Google Scholar] [CrossRef]
- Chen, P.H.; Chen, X.; He, X. Platelet-derived growth factors and their receptors: Structural and functional perspectives. Biochim. Biophys. Acta. 2013, 1834, 2176–2186. [Google Scholar] [CrossRef]
- Niu, G.; Chen, X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr. Drug Targets 2010, 11, 1000–1017. [Google Scholar] [CrossRef]
- Ho, J.; Moyes, D.L.; Tavassoli, M.; Naglik, J.R. The Role of ErbB Receptors in Infection. Trends Microbiol. 2017, 25, 942–952. [Google Scholar] [CrossRef]
- Li, Q.; Fu, J.; Xia, Y.; Qi, W.; Ishikado, A.; Park, K.; Yokomizo, H.; Huang, Q.; Cai, W.; Rask-Madsen, C.; et al. Homozygous receptors for insulin and not IGF-1 accelerate intimal hyperplasia in insulin resistance and diabetes. Nat. Commun. 2019, 10, 4427. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. IGF-1R drugs travel from cancer cradle to Graves. Nat. Biotechnol. 2020, 38, 385–388. [Google Scholar] [CrossRef]
- Guérit, E.; Arts, F.; Dachy, G.; Boulouadnine, B.; Demoulin, J.B. PDGF receptor mutations in human diseases. Cell Mol. Life Sci. 2021, 78, 3867–3881. [Google Scholar] [CrossRef]
- Ivy, S.P.; Wick, J.Y.; Kaufman, B.M. An overview of small-molecule inhibitors of VEGFR signaling. Nat. Rev. Clin. Oncol. 2009, 6, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Shen, H.; Buckley, B.; Chen, Y.; Yang, N.; Mussell, A.L.; Chernov, M.; Kobzik, L.; Frangou, C.; Han, S.X.; et al. NTRK1 is a positive regulator of YAP oncogenic function. Oncogene 2019, 38, 2778–2787. [Google Scholar] [CrossRef]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.W.; Bartlett, P.F.; Lackmann, M. Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug. Discov. 2014, 13, 39–62. [Google Scholar] [CrossRef] [PubMed]
- Flem-Karlsen, K.; Nyakas, M.; Farstad, I.N.; McFadden, E.; Wernhoff, P.; Jacobsen, K.D.; Flørenes, V.A.; Mælandsmo, G.M. Soluble AXL as a marker of disease progression and survival in melanoma. PLoS ONE 2020, 15, e0227187. [Google Scholar] [CrossRef]
- Plaza-Menacho, I.; Burzynski, G.M.; de Groot, J.W.; Eggen, B.J.; Hofstra, R.M. Current concepts in RET-related genetics, signaling and therapeutics. Trends Genet. 2006, 22, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Yu, H.A.; Riely, G.J. Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in lung cancers. J. Natl. Compr. Cancer Netw. 2013, 11, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Geller, J.I.; Fox, E.; Turpin, B.K.; Goldstein, S.L.; Liu, X.; Minard, C.G.; Kudgus, R.A.; Reid, J.M.; Berg, S.L.; Weigel, B.J. A study of axitinib, a VEGF receptor tyrosine kinase inhibitor, in children and adolescents with recurrent or refractory solid tumors: A Children’s Oncology Group phase 1 and pilot consortium trial (ADVL1315). Cancer 2018, 124, 4548–4555. [Google Scholar] [CrossRef]
- Baumann, K.H.; du Bois, A.; Meier, W.; Rau, J.; Wimberger, P.; Sehouli, J.; Kurzeder, C.; Hilpert, F.; Hasenburg, A.; Canzler, U.; et al. A phase II trial (AGO 2.11) in platinum-resistant ovarian cancer: A randomized multicenter trial with sunitinib (SU11248) to evaluate dosage, schedule, tolerability, toxicity and effectiveness of a multitargeted receptor tyrosine kinase inhibitor monotherapy. Ann. Oncol. 2012, 23, 2265–2271. [Google Scholar] [PubMed]
- Dragovich, T.; Laheru, D.; Dayyani, F.; Bolejack, V.; Smith, L.; Seng, J.; Burris, H.; Rosen, P.; Hidalgo, M.; Ritch, P.; et al. Phase II trial of vatalanib in patients with advanced or metastatic pancreatic adenocarcinoma after first-line gemcitabine therapy (PCRT O4-001). Cancer Chemother. Pharmacol. 2014, 74, 379–387. [Google Scholar] [CrossRef]
- Huijts, C.M.; Santegoets, S.J.; van den Eertwegh, A.J.; Pijpers, L.S.; Haanen, J.B.; de Gruijl, T.D.; Verheul, H.M.; van der Vliet, H.J. Phase I-II study of everolimus and low-dose oral cyclophosphamide in patients with metastatic renal cell cancer. BMC Cancer 2011, 11, 505. [Google Scholar] [CrossRef]
- Jian, H.; Li, W.; Ma, Z.; Huang, J.; Feng, J.; Song, Y.; Gao, B.; Zhu, H.; Tao, M.; Bai, C.; et al. Intercalating and maintenance gefitinib plus chemotherapy versus chemotherapy alone in selected advanced non-small cell lung cancer with unknown EGFR status. Sci. Rep. 2017, 7, 8483. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.E.; Endicott, J.A.; Johnson, L.N. Protein kinase inhibitors: Insights into drug design from structure. Science 2004, 303, 1800–1805. [Google Scholar] [CrossRef]
- Rosenzweig, S.A. Acquired Resistance to Drugs Targeting Tyrosine Kinases. Adv. Cancer Res. 2018, 138, 71–98. [Google Scholar]
- Li, J.; An, B.; Song, X.; Zhang, Q.; Chen, C.; Wei, S.; Fan, R.; Li, X.; Zou, Y. Design, synthesis and biological evaluation of novel 2,4-diaryl pyrimidine derivatives as selective EGFRL858R/T790M inhibitors. Eur. J. Med. Chem. 2021, 212, 113019. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Wu, B.X.; Zheng, Q.; Hu, C.H.; Tang, X.Z.; Zhang, W.; Rao, G.W. Design, synthesis, biological evaluation and docking study of novel quinazoline derivatives as EGFR-TK inhibitors. Future Med. Chem. 2021, 13, 601–612. [Google Scholar] [CrossRef]
- Raghu, M.S.; Pradeep Kumar, C.B.; Prashanth, M.K.; Yogesh Kumar, K.B.; Prathibha, S.; Kanthimathi, G.; Alissa, S.A.; Alghulikah, H.A.; Osman, S.M. Novel 1,3,5-triazine-based pyrazole derivatives as potential antitumor agents and EFGR kinase inhibitors: Synthesis, cytotoxicity, DNA binding, molecular docking and DFT studies. N. J. Chem. 2021, 45, 13909–13924. [Google Scholar] [CrossRef]
- Mishra, C.B.; Mongre, R.K.; Kumari, S.; Jeong, D.K.; Tiwari, M. Synthesis, in vitro and in vivo anticancer activity of novel 1-(4-imino-1-substituted-1H-pyrazolo[3,4-d]pyrimidin-5(4H)-yl)urea derivatives. RSC Adv. 2016, 6, 24491–24500. [Google Scholar] [CrossRef]
- Mishra, C.B.; Mongre, R.K.; Prakash, A.; Jeon, R.; Supuran, C.T.; Lee, M.S. Anti-breast cancer action of carbonic anhydrase IX inhibitor 4-[4-(4-Benzo[1,3]dioxol-5-ylmethyl-piperazin-1-yl)-benzylidene-hydrazinocarbonyl]-benzenesulfonamide (BSM-0004): In vitro and in vivo studies. J. Enzym. Inhib. Med. Chem. 2021, 36, 954–963. [Google Scholar] [CrossRef]
- Mongre, R.K.; Mishra, C.B.; Prakash, A.; Jung, S.; Lee, B.S.; Kumari, S.; Hong, J.T.; Lee, M.S. Novel Carbazole-Piperazine Hybrid Small Molecule Induces Apoptosis by Targeting BCL-2 and Inhibits Tumor Progression in Lung Adenocarcinoma in Vitro and Xenograft Mice Model. Cancers 2019, 11, 1245. [Google Scholar] [CrossRef]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Hennequin, L.F.; Thomas, A.P.; Johnstone, C.; Stokes, E.S.; Plé, P.A.; Lohmann, J.J.; Ogilvie, D.J.; Dukes, M.; Wedge, S.R.; Curwen, J.O.; et al. Design and structure-activity relationship of a new class of potent VEGF receptor tyrosine kinase inhibitors. J. Med. Chem. 1999, 42, 5369–5389. [Google Scholar] [CrossRef] [PubMed]
- Eissa, I.H.; El-Helby, A.A.; Mahdy, H.A.; Khalifa, M.M.; Elnagar, H.A.; Mehany, A.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; et al. Discovery of new quinazolin-4(3H)-ones as VEGFR-2 inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorg. Chem. 2020, 105, 104380. [Google Scholar] [CrossRef]
- Al-Ansary, G.H.; Nasr, T.; Taha, H.; Fayad, W.; Mahgoub, S. Biphenylurea/thiourea derivatives tagged with heteroarylsulfonamide motifs as novel VEGFR2 inhibitors; Design, synthesis and anti-angiogenic activity. Bioorg. Chem. 2021, 107, 104640. [Google Scholar] [CrossRef]
- Mahmoud, H.K.; Farghaly, T.A.; Abdulwahab, H.G.; Al-Qurashi, N.T.; Shaaban, M.R. Novel 2-indolinone thiazole hybrids as sunitinib analogues: Design, synthesis, and potent VEGFR-2 inhibition with potential anti-renal cancer activity. Eur. J. Med. Chem. 2020, 208, 112752. [Google Scholar] [CrossRef]
- Michaloski, J.S.; Redondo, A.R.; Magalhães, L.S.; Cambui, C.C.; Giordano, R.J. Discovery of pan-VEGF inhibitory peptides directed to the extracellular ligand-binding domains of the VEGF receptors. Sci. Adv. 2016, 2, e1600611. [Google Scholar] [CrossRef]
- Osher, E.; Macaulay, V.M. Therapeutic Targeting of the IGF Axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef]
- Lee, H.J.; Pham, P.C.; Pei, H.; Lim, B.; Hyun, S.Y.; Baek, B.; Kim, B.; Kim, Y.; Kim, M.H.; Kang, N.W.; et al. Development of the phenylpyrazolo[3,4-d]pyrimidine-based, insulin-like growth factor receptor/Src/AXL-targeting small molecule kinase inhibitor. Theranostics 2021, 11, 1918–1936. [Google Scholar] [CrossRef]
- Gadekar, P.K.; Urunkar, G.; Roychowdhury, A.; Sharma, R.; Bose, J.; Khanna, S.; Damre, A.; Sarveswari, S. Design, synthesis and biological evaluation of 2,3-dihydroimidazo[2,1-b]thiazoles as dual EGFR and IGF1R inhibitors. Bioorg. Chem. 2021, 115, 105151. [Google Scholar] [CrossRef]
- Wen, B.; Deutsch, E.; Marangoni, E.; Frascona, V.; Maggiorella, L.; Abdulkarim, B.; Chavaudra, N.; Bourhis, J. Tyrphostin AG 1024 modulates radiosensitivity in human breast cancer cells. Br. J. Cancer 2001, 85, 2017–2021. [Google Scholar] [CrossRef][Green Version]
- Mishra, C.B.; Mongre, R.K.; Kumari, S.; Jeong, D.K.; Tiwari, M. Novel Triazole-Piperazine Hybrid Molecules Induce Apoptosis via Activation of the Mitochondrial Pathway and Exhibit Antitumor Efficacy in Osteosarcoma Xenograft Nude Mice Model. ACS Chem. Biol. 2017, 12, 753–768. [Google Scholar] [CrossRef]
- Carboni, J.M.; Wittman, M.; Yang, Z.; Lee, F.; Greer, A.; Hurlburt, W.; Hillerman, S.; Cao, C.; Cantor, G.H.; Dell-John, J.; et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 2009, 8, 3341–3349. [Google Scholar] [CrossRef]
- Maguire, M.P.; Sheets, K.R.; McVety, K.; Spada, A.P.; Zilberstein, A. A new series of PDGF receptor tyrosine kinase inhibitors: 3-substituted quinoline derivatives. J. Med. Chem. 1994, 37, 2129–2137. [Google Scholar] [CrossRef]
- Yang, T.H.; Lee, C.I.; Huang, W.H.; Lee, A.R. Structural optimization and evaluation of novel 2-pyrrolidone-fused (2-oxoindolin-3-ylidene)methylpyrrole derivatives as potential VEGFR-2/PDGFRβ inhibitors. Chem. Cent. J. 2017, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Bahleda, R.; Italiano, A.; Hierro, C.; Mita, A.; Cervantes, A.; Chan, N.; Awad, M.; Calvo, E.; Moreno, V.; Govindan, R.; et al. Multicenter Phase I Study of Erdafitinib (JNJ-42756493), Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced or Refractory Solid Tumors. Clin. Cancer Res. 2019, 25, 4888–4897. [Google Scholar] [CrossRef]
- Brameld, K.A.; Owens, T.D.; Verner, E.; Venetsanakos, E.; Bradshaw, J.M.; Phan, V.T.; Tam, D.; Leung, K.; Shu, J.; LaStant, J.; et al. Discovery of the Irreversible Covalent FGFR Inhibitor 8-(3-(4-Acryloylpiperazin-1-yl)propyl)-6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)pyrido[2,3-d]pyrimidin-7(8H)-one (PRN1371) for the Treatment of Solid Tumors. J. Med. Chem. 2017, 60, 6516–6527. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Hugo, H.S. Update on antibody-drug conjugates in breast cancer. Clin. Adv. Hematol. Oncol. 2021, 19, 148–151. [Google Scholar]
- Mongre, R.K.; Jung, S.; Mishra, C.B.; Lee, B.S.; Kumari, S.; Lee, M.S. Prognostic and Clinicopathological Significance of SERTAD1 in Various Types of Cancer Risk: A Systematic Review and Retrospective Analysis. Cancers 2019, 11, 337. [Google Scholar] [CrossRef] [PubMed]
- Mongre, R.K.; Mishra, C.B.; Jung, S.; Lee, B.S.; Quynh, N.; Anh, N.H.; Myagmarjav, D.; Jo, T.; Lee, M.S. Exploring the Role of TRIP-Brs in Human Breast Cancer: An Investigation of Expression, Clinicopathological Significance, and Prognosis. Mol. Ther. Oncolytics 2020, 19, 105–126. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, K.C.; Mongre, R.K.; Kim, J.Y.; Kim, Y.R.; Choi, D.Y.; Song, S.; Yun, J.; Han, S.B.; Yoon, D.Y.; et al. IL-32γ suppresses lung cancer stem cell growth via inhibition of ITGAV-mediated STAT5 pathway. Cell Death Dis. 2019, 10, 506. [Google Scholar] [CrossRef] [PubMed]
- Mongre, R.K.; Sodhi, S.S.; Ghosh, M.; Kim, J.H.; Kim, N.; Sharma, N.; Jeong, D.K. A New Paradigm to Mitigate Osteosarcoma by Regulation of MicroRNAs and Suppression of the NF-κB Signaling Cascade. Dev. Reprod. 2014, 18, 197–212. [Google Scholar] [CrossRef]
- Mishra, C.B.; Kumari, S.; Angeli, A.; Bua, S.; Mongre, R.K.; Tiwari, M.; Supuran, C.T. Discovery of Potent Carbonic Anhydrase Inhibitors as effective Anti-convulsant Agents: Drug design, Synthesis, In vitro and In vivo Investigations. J. Med. Chem. 2021, 64, 3100–3114. [Google Scholar] [CrossRef]
- Mishra, C.B.; Tiwari, M.; Supuran, C.T. Progress in the development of human carbonic anhydrases inhibitors and their pharmacological applications: Where are we today? Med. Res. Rev. 2020, 40, 2485–2565. [Google Scholar] [CrossRef] [PubMed]
- Mishra, C.B.; Kumari, S.; Angeli, A.; Bua, S.; Tiwari, M.; Supuran, C.T. Discovery of Benzenesulfonamide Derivatives as Carbonic Anhydrase Inhibitors with Effective Anticonvulsant Action: Design, Synthesis, and Pharmacological Evaluation. J. Med. Chem. 2018, 61, 3151–3165. [Google Scholar] [CrossRef] [PubMed]
- Casals, E.; Gusta, M.F.; Cobaleda-Siles, M.; Garcia-Sanz, A.; Puntes, V.F. Cancer resistance to treatment and antiresistance tools offered by multimodal multifunctional nanoparticles. Cancer Nanotechnol. 2017, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Kee, D.; Zalcberg, J.R. Current and emerging strategies for the management of imatinib-refractory advanced gastrointestinal stromal tumors. Ther. Adv. Med. Oncol. 2012, 4, 255–270. [Google Scholar] [CrossRef]
- Rubin, B.P.; Duensing, A. Mechanisms of resistance to small molecule kinase inhibition in the treatment of solid tumors. Lab. Invest. 2006, 86, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, E.; Nichols, G. Mechanisms of resistance to imatinib in CML patients: A paradigm for the advantages and pitfalls of molecularly targeted therapy. Curr. Cancer Drug Targets 2006, 6, 645–657. [Google Scholar] [CrossRef]
- Tamborini, E.; Bonadiman, L.; Greco, A.; Albertini, V.; Negri, T.; Gronchi, A.; Bertulli, R.; Colecchia, M.; Casali, P.G.; Pierotti, M.A.; et al. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology 2004, 127, 294–299. [Google Scholar] [CrossRef]
- Debiec-Rychter, M.; Cools, J.; Dumez, H.; Sciot, R.; Stul, M.; Mentens, N.; Vranckx, H.; Wasag, B.; Prenen, H.; Roesel, J.; et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology 2005, 128, 270–279. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).