
Hsp90 Inhibition: A Promising Therapeutic Approach for ARSACS

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
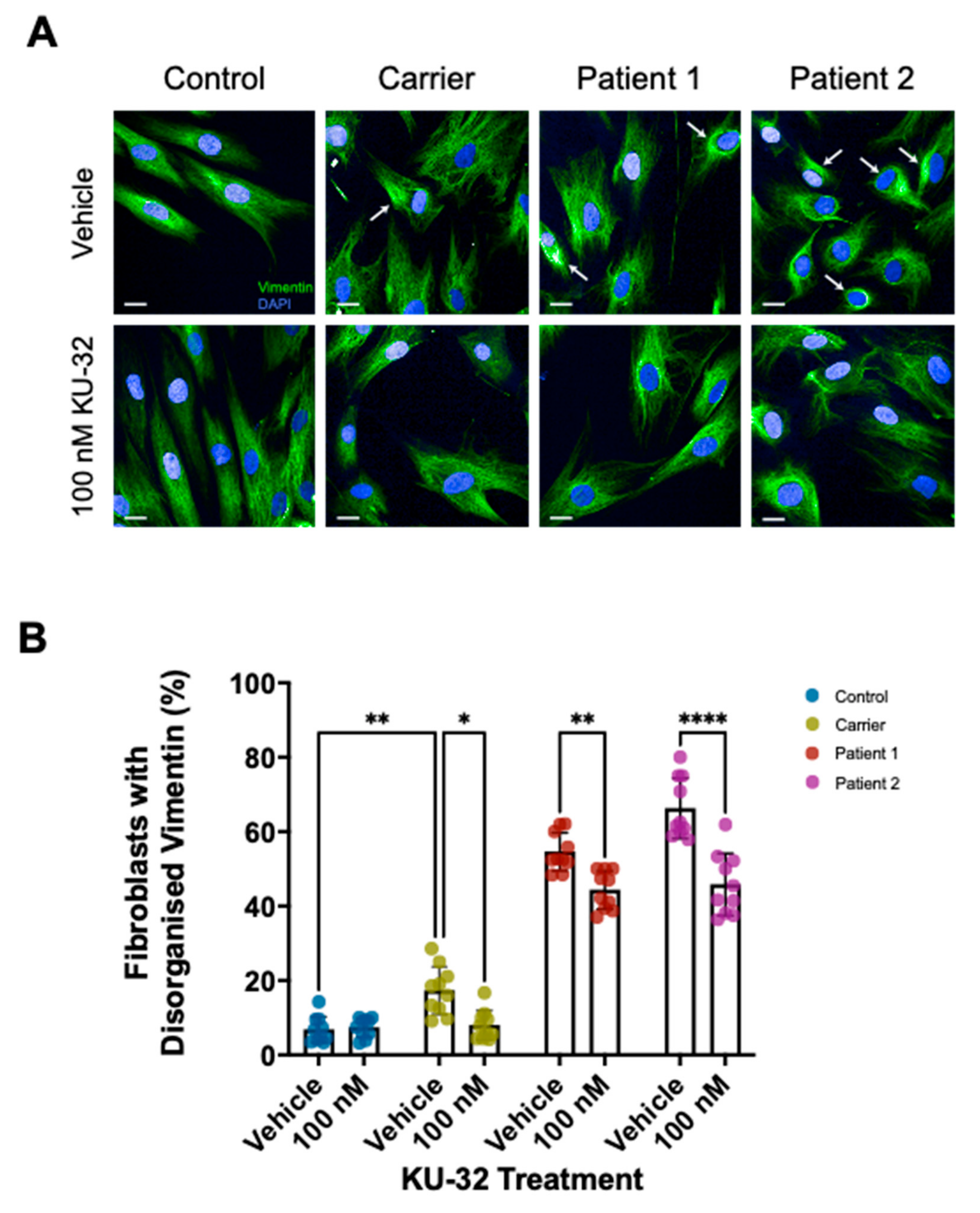
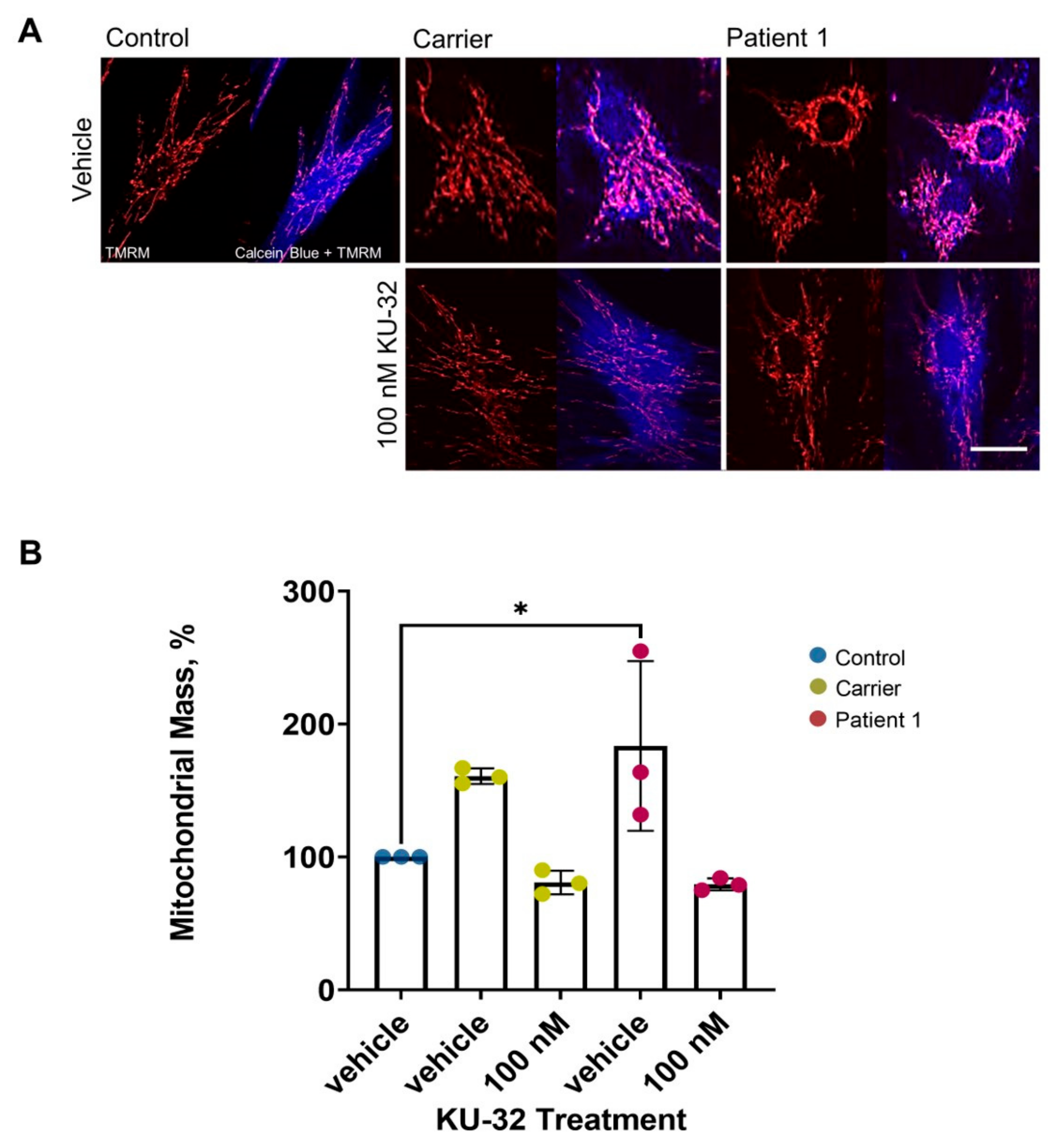
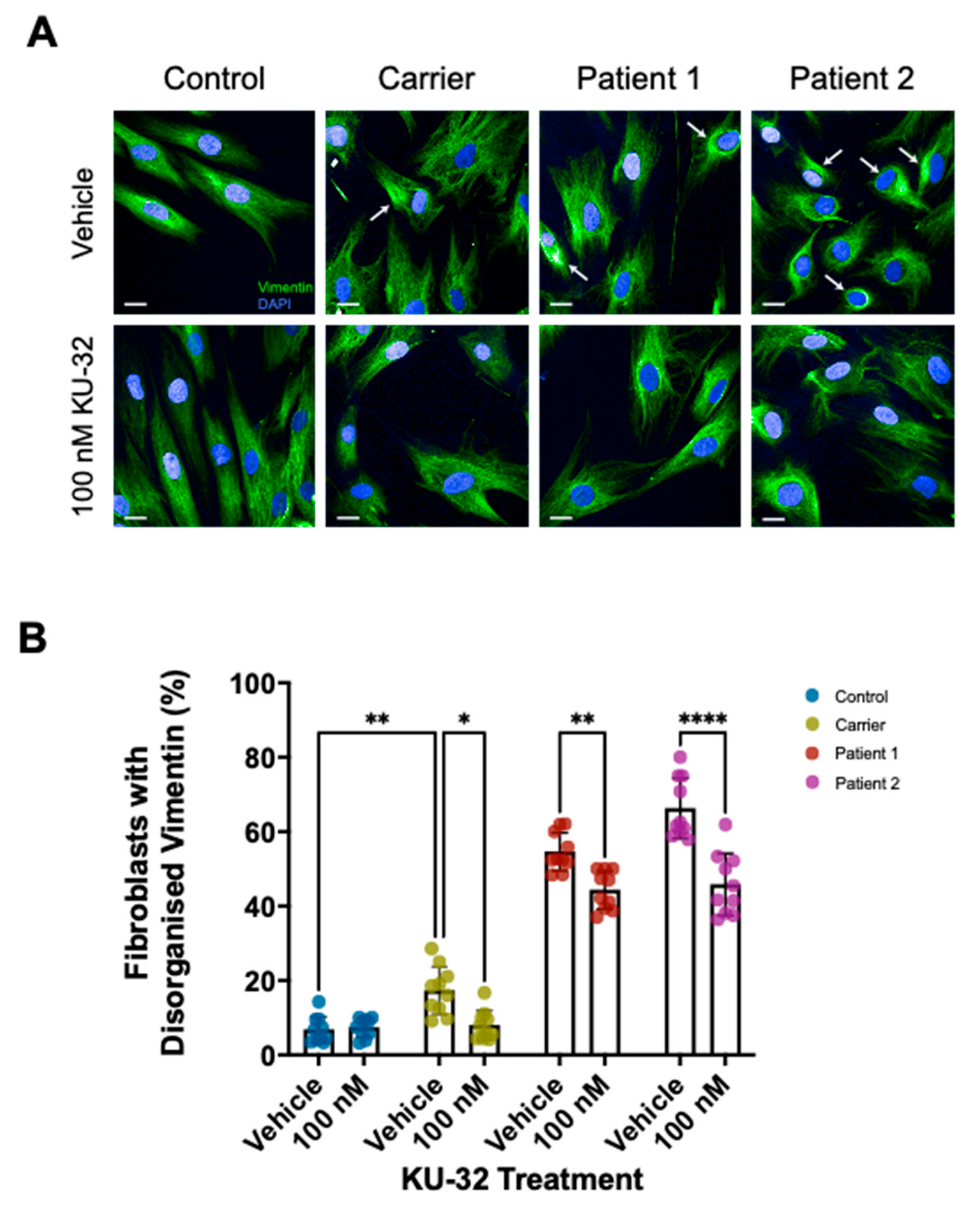
2. Results
Effect of KU-32 Treatment
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Cell Culture
4.3. Immunofluorescence and Imaging
4.4. Mitochondrial Membrane Potential Assay
4.5. Mitochondrial Volume Assay
4.6. Immunoblotting
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bouchard, J.-P.P.; Barbeau, A.; Bouchard, R.W. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Can. J. Neurol. Sci. Le J. Can. Des Sci. Neurol. 1978, 5, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.M.; Prodromou, N.V.; Webb, T.R.; Gallo, J.-M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The Ataxia Protein Sacsin Is a Functional Co-Chaperone That Protects against Polyglutamine-Expanded Ataxin-1. Hum. Mol. Genet. 2009, 18, 1556–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, J.F.; Siller, E.; Barral, J.M. The Sacsin Repeating Region (SRR): A Novel Hsp90-Related Supra-Domain Associated with Neurodegeneration. J. Mol. Biol. 2010, 400, 665–674. [Google Scholar] [CrossRef]
- Anderson, J.F.; Siller, E.; Barral, J.M. The Neurodegenerative-Disease-Related Protein Sacsin Is a Molecular Chaperone. J. Mol. Biol. 2011, 411, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, G.; Denisov, A.Y.; Girard, M.; Dicaire, M.-J.; Hamlin, J.; McPherson, P.S.; Brais, B.; Gehring, K. Structural Basis of Defects in the Sacsin HEPN Domain Responsible for Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS). J. Biol. Chem. 2011, 286, 20407–20412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, A.; Tessa, A.; Barca, A.; Fattori, F.; Fulvia de Leva, M.; Terracciano, A.; Storelli, C.; Maria Santorelli, F.; Verri, T. Comparative Analysis and Functional Mapping of SACS Mutations Reveal Novel Insights into Sacsin Repeated Architecture. Hum. Mutat. 2013, 34, 525–537. [Google Scholar] [CrossRef]
- Larivière, R.; Gaudet, R.; Gentil, B.J.; Girard, M.; Conte, T.C.; Minotti, S.; Leclerc-Desaulniers, K.; Gehring, K.; McKinney, R.A.; Shoubridge, E.A.; et al. Sacs Knockout Mice Present Pathophysiological Defects Underlying Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Hum. Mol. Genet. 2015, 24, 727–739. [Google Scholar] [CrossRef] [Green Version]
- Gentil, B.J.; Lai, G.-T.; Menade, M.; Larivière, R.; Minotti, S.; Gehring, K.; Chapple, J.-P.; Brais, B.; Durham, H.D. Sacsin, Mutated in the Ataxia ARSACS, Regulates Intermediate Filament Assembly and Dynamics. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 2982–2994. [Google Scholar] [CrossRef] [Green Version]
- Duncan, E.J.; Larivière, R.; Bradshaw, T.Y.; Longo, F.; Sgarioto, N.; Hayes, M.J.; Romano, L.E.L.; Nethisinghe, S.; Giunti, P.; Bruntraeger, M.B.; et al. Altered Organization of the Intermediate Filament Cytoskeleton and Relocalization of Proteostasis Modulators in Cells Lacking the Ataxia Protein Sacsin. Hum. Mol. Genet. 2017, 26, 3130–3143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeverenyi, I.; Cassidy, A.J.; Chung, C.W.; Lee, B.T.K.; Common, J.E.A.; Ogg, S.C.; Chen, H.; Sim, S.Y.; Goh, W.L.P.; Ng, K.W.; et al. The Human Intermediate Filament Database: Comprehensive Information on a Gene Family Involved in Many Human Diseases. Hum. Mutat. 2008, 29, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.B.; Blagg, B.S.J. To Fold or Not to Fold: Modulation and Consequences of Hsp90 Inhibition. Future Med. Chem. 2009, 1, 267–283. [Google Scholar] [CrossRef] [Green Version]
- Blagg, B.S.J.; Kerr, T.D. Hsp90 Inhibitors: Small Molecules That Transform the Hsp90 Protein Folding Machinery into a Catalyst for Protein Degradation. Med. Res. Rev. 2006, 26, 310–338. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Dou, F.; Rodina, A.; Chip, S.; Kim, J.; Zhao, Q.; Moulick, K.; Aguirre, J.; Wu, N.; Greengard, P.; et al. Roles of Heat-Shock Protein 90 in Maintaining and Facilitating the Neurodegenerative Phenotype in Tauopathies. Proc. Natl. Acad. Sci. USA 2007, 104, 9511–9516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Rodina, A.; Chiosis, G. Heat Shock Protein 90: Translation from Cancer to Alzheimer’s Disease Treatment? BMC Neurosci. 2008, 9 (Suppl. S2), S7. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Harrell, J.M.; Morishima, Y.; Peng, H.-M.; Pratt, W.B.; Lieberman, A.P. Pharmacologic and Genetic Inhibition of Hsp90-Dependent Trafficking Reduces Aggregation and Promotes Degradation of the Expanded Glutamine Androgen Receptor without Stress Protein Induction. Hum. Mol. Genet. 2006, 15, 1876–1883. [Google Scholar] [CrossRef] [Green Version]
- Waza, M.; Adachi, H.; Katsuno, M.; Minamiyama, M.; Sang, C.; Tanaka, F.; Inukai, A.; Doyu, M.; Sobue, G. 17-AAG, an Hsp90 Inhibitor, Ameliorates Polyglutamine-Mediated Motor Neuron Degeneration. Nat. Med. 2005, 11, 1088–1095. [Google Scholar] [CrossRef]
- Ansar, S.; Burlison, J.A.; Hadden, M.K.; Yu, X.M.; Desino, K.E.; Bean, J.; Neckers, L.; Audus, K.L.; Michaelis, M.L.; Blagg, B.S.J. A Non-Toxic Hsp90 Inhibitor Protects Neurons from Abeta-Induced Toxicity. Bioorganic Med. Chem. Lett. 2007, 17, 1984–1990. [Google Scholar] [CrossRef]
- Lu, Y.; Ansar, S.; Michaelis, M.L.; Blagg, B.S.J. Neuroprotective Activity and Evaluation of Hsp90 Inhibitors in an Immortalized Neuronal Cell Line. Bioorganic Med. Chem. 2009, 17, 1709–1715. [Google Scholar] [CrossRef] [Green Version]
- Urban, M.J.; Li, C.; Yu, C.; Lu, Y.; Krise, J.M.; McIntosh, M.P.; Rajewski, R.A.; Blagg, B.S.J.; Dobrowsky, R.T. Inhibiting Heat-Shock Protein 90 Reverses Sensory Hypoalgesia in Diabetic Mice. ASN Neuro 2010, 2, e00040. [Google Scholar] [CrossRef] [PubMed]
- Nethisinghe, S.; Clayton, L.; Vermeer, S.; Chapple, J.P.; Reilly, M.M.; Bremner, F.; Giunti, P. Retinal Imaging in Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Neuro-Ophthalmology 2011, 35, 197–201. [Google Scholar] [CrossRef]
- Parkinson, M.H.; Bartmann, A.P.; Clayton, L.M.S.; Nethisinghe, S.; Pfundt, R.; Chapple, J.P.; Reilly, M.M.; Manji, H.; Wood, N.J.; Bremner, F.; et al. Optical Coherence Tomography in Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. Brain J. Neurol. 2018, 141, 989–999. [Google Scholar] [CrossRef]
- Rezende Filho, F.M.; Bremner, F.; Pedroso, J.L.; de Andrade, J.B.C.; Marianelli, B.F.; Lourenço, C.M.; Marques-Júnior, W.; França, M.C.; Kok, F.; Sallum, J.M.F.; et al. Retinal Architecture in Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS): Insights into Disease Pathogenesis and Biomarkers. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Deane, C.A.S.; Brown, I.R. Induction of Heat Shock Proteins in Differentiated Human Neuronal Cells Following Co-Application of Celastrol and Arimoclomol. Cell Stress Chaperones 2016, 21, 837–848. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Hamza, A.; Cao, X.; Wang, B.; Yu, S.; Zhan, C.-G.; Sun, D. A Novel Hsp90 Inhibitor to Disrupt Hsp90/Cdc37 Complex against Pancreatic Cancer Cells. Mol. Cancer Ther. 2008, 7, 162–170. [Google Scholar] [CrossRef] [Green Version]
- Sreeramulu, S.; Gande, S.L.; Göbel, M.; Schwalbe, H. Molecular Mechanism of Inhibition of the Human Protein Complex Hsp90-Cdc37, a Kinome Chaperone-Cochaperone, by Triterpene Celastrol. Angew. Chem. (Int. Ed. Engl.) 2009, 48, 5853–5855. [Google Scholar] [CrossRef] [PubMed]
- Girard, M.; Larivière, R.; Parfitt, D.A.; Deane, E.C.; Gaudet, R.; Nossova, N.; Blondeau, F.; Prenosil, G.; Vermeulen, E.G.M.; Duchen, M.R.; et al. Mitochondrial Dysfunction and Purkinje Cell Loss in Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. USA 2012, 109, 1661–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradshaw, T.Y.; Romano, L.E.L.; Duncan, E.J.; Nethisinghe, S.; Abeti, R.; Michael, G.J.; Giunti, P.; Vermeer, S.; Chapple, J.P. A Reduction in Drp1-Mediated Fission Compromises Mitochondrial Health in Autosomal Recessive Spastic Ataxia of Charlevoix Saguenay. Hum. Mol. Genet. 2016, 25, 3232–3244. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, C.; Procaccini, C.; Meschini, M.C.; Cianflone, A.; Carbone, R.; Doccini, S.; Devos, D.; Nesti, C.; Vuillaume, I.; Pellegrino, M.; et al. Powerhouse Failure and Oxidative Damage in Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. J. Neurol. 2015. [Google Scholar] [CrossRef] [Green Version]
- Anesti, V.; Scorrano, L. The Relationship between Mitochondrial Shape and Function and the Cytoskeleton. Biochim. Biophys. Acta 2006, 1757, 692–699. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics-Fusion, Fission, Movement, and Mitophagy-in Neurodegenerative Diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Gentil, B.J.; Minotti, S.; Beange, M.; Baloh, R.H.; Julien, J.; Durham, H.D. Normal Role of the Low-Molecular-Weight Neurofilament Protein in Mitochondrial Dynamics and Disruption in Charcot-Marie-Tooth Disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Mears, J.A.; Lackner, L.L.; Fang, S.; Ingerman, E.; Nunnari, J.; Hinshaw, J.E. Conformational Changes in Dnm1 Support a Contractile Mechanism for Mitochondrial Fission. Nat. Struct. Mol. Biol. 2011, 18, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R.; Surin, A.; Jacobson, J. Imaging Mitochondrial Function in Intact Cells. Methods Enzymol. 2003, 361, 353–389. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nethisinghe, S.; Abeti, R.; Kesavan, M.; Wigley, W.C.; Giunti, P. Hsp90 Inhibition: A Promising Therapeutic Approach for ARSACS. Int. J. Mol. Sci. 2021, 22, 11722. https://doi.org/10.3390/ijms222111722
Nethisinghe S, Abeti R, Kesavan M, Wigley WC, Giunti P. Hsp90 Inhibition: A Promising Therapeutic Approach for ARSACS. International Journal of Molecular Sciences. 2021; 22(21):11722. https://doi.org/10.3390/ijms222111722
Chicago/Turabian StyleNethisinghe, Suran, Rosella Abeti, Maheswaran Kesavan, W. Christian Wigley, and Paola Giunti. 2021. "Hsp90 Inhibition: A Promising Therapeutic Approach for ARSACS" International Journal of Molecular Sciences 22, no. 21: 11722. https://doi.org/10.3390/ijms222111722
APA StyleNethisinghe, S., Abeti, R., Kesavan, M., Wigley, W. C., & Giunti, P. (2021). Hsp90 Inhibition: A Promising Therapeutic Approach for ARSACS. International Journal of Molecular Sciences, 22(21), 11722. https://doi.org/10.3390/ijms222111722