
Emerging Importance of Tyrosine Kinase Inhibitors against Cancer: Quo Vadis to Cure?


 ,
, 
Abstract
1. Introduction
2. Basic Concept on Targeting Receptor Tyrosine Kinases: Effective Strategies to Cure Cancer
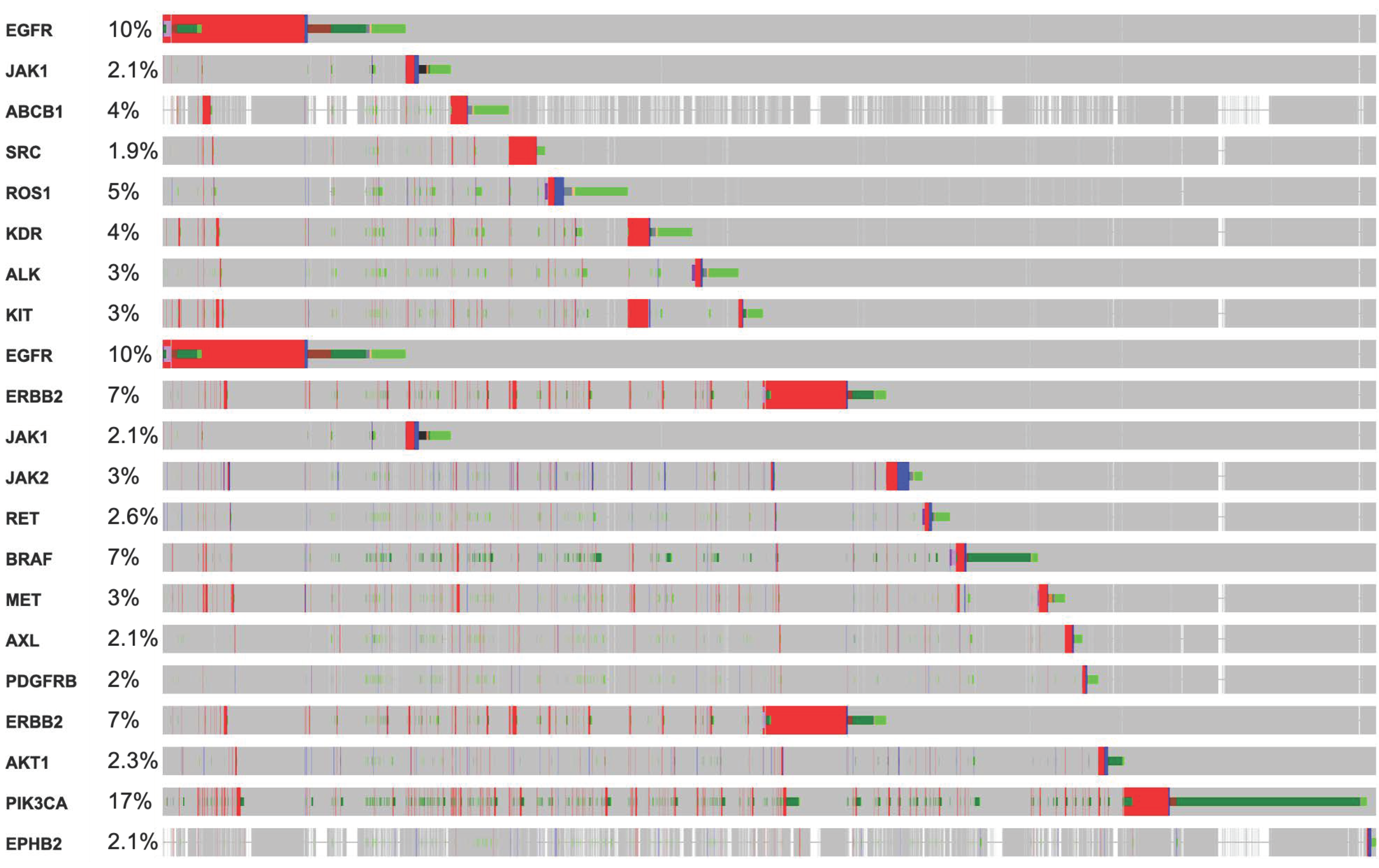
3. Mutations in RTKs Associated with Diverse Mutational Pathways
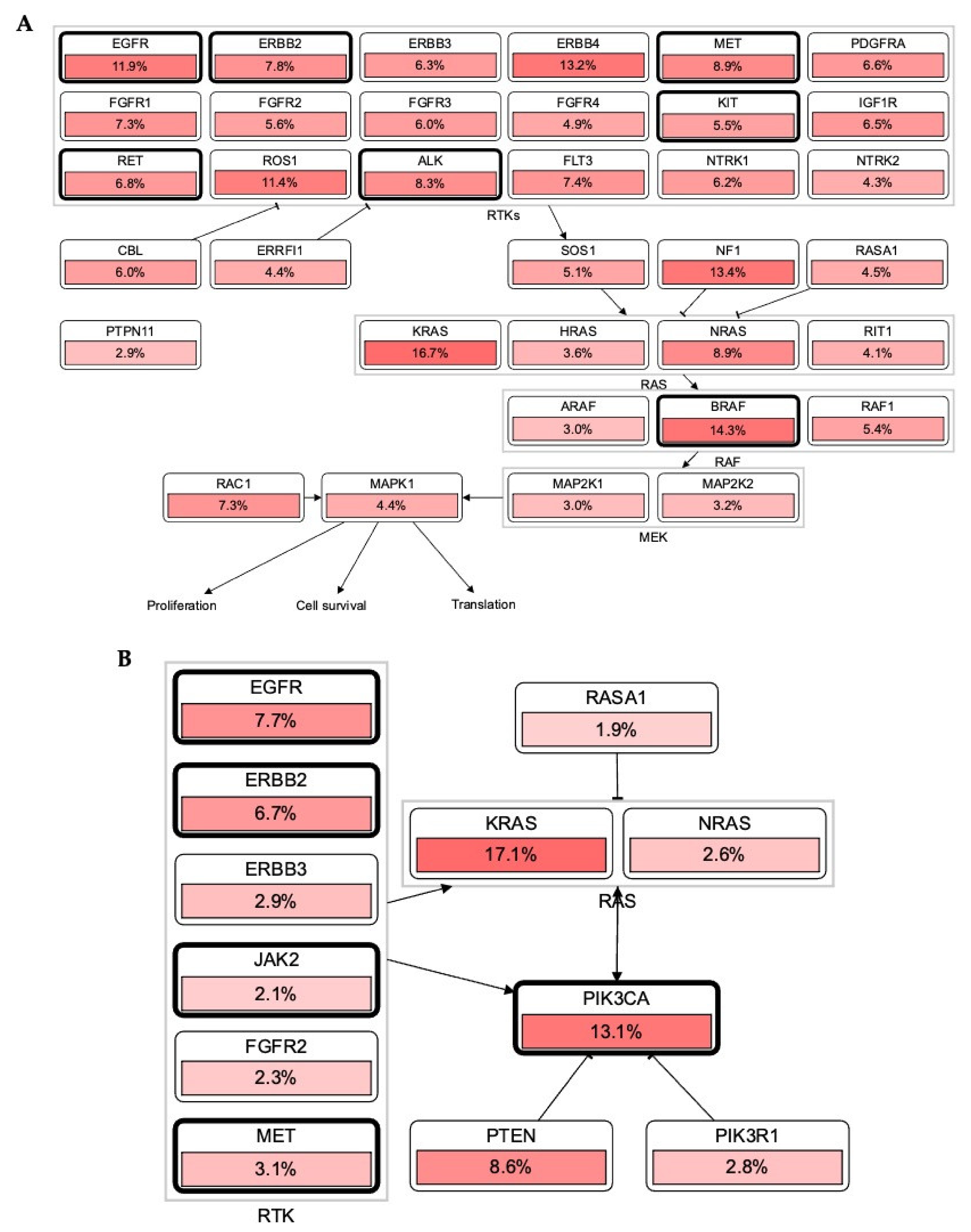
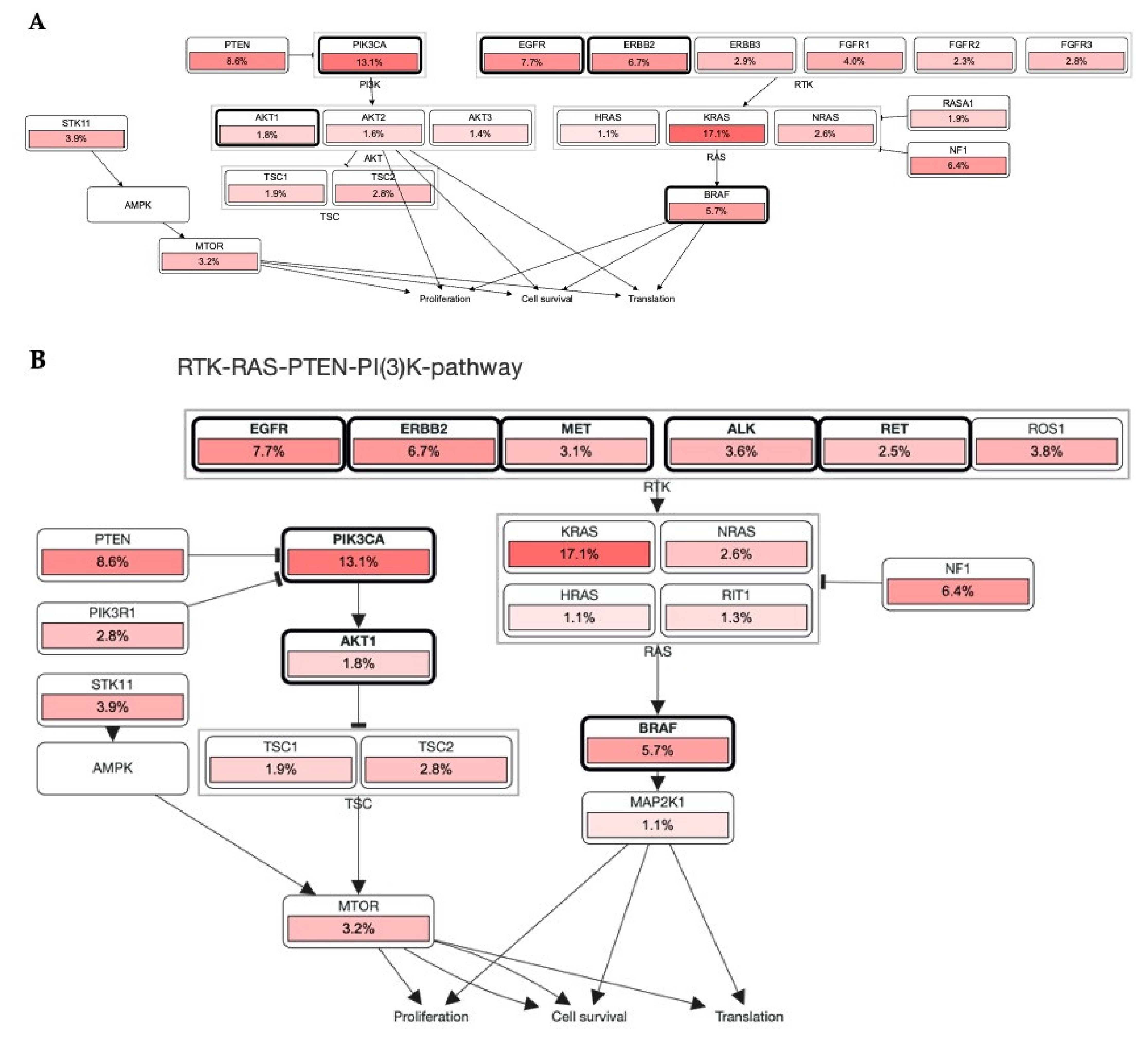
3.1. Alteration in RTKs Modulate Carcinogenesis via RTK-RAS Mutational Pathway
3.2. Association of RTKs via KRAS-PIK3CA Pathway in Cancer
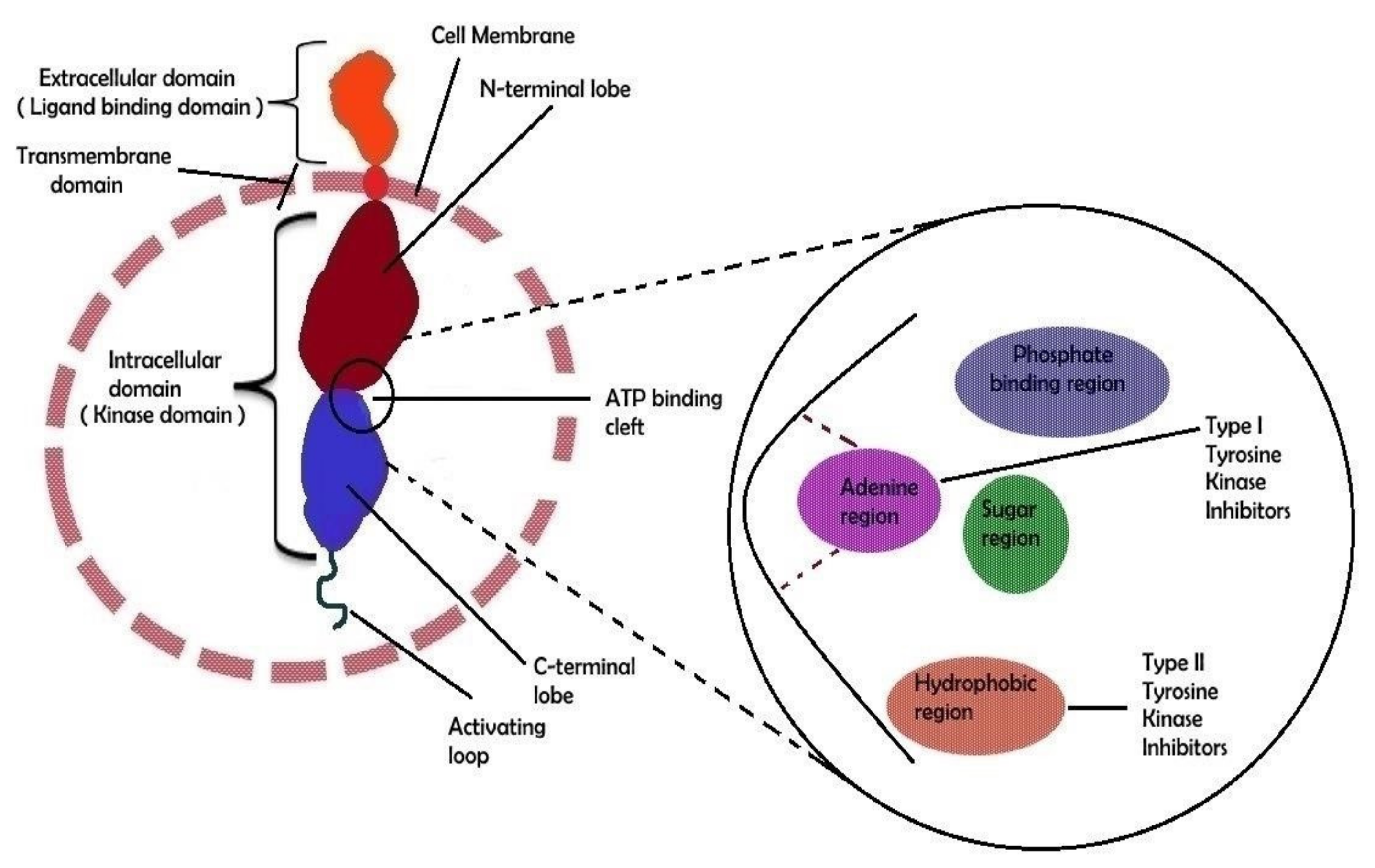
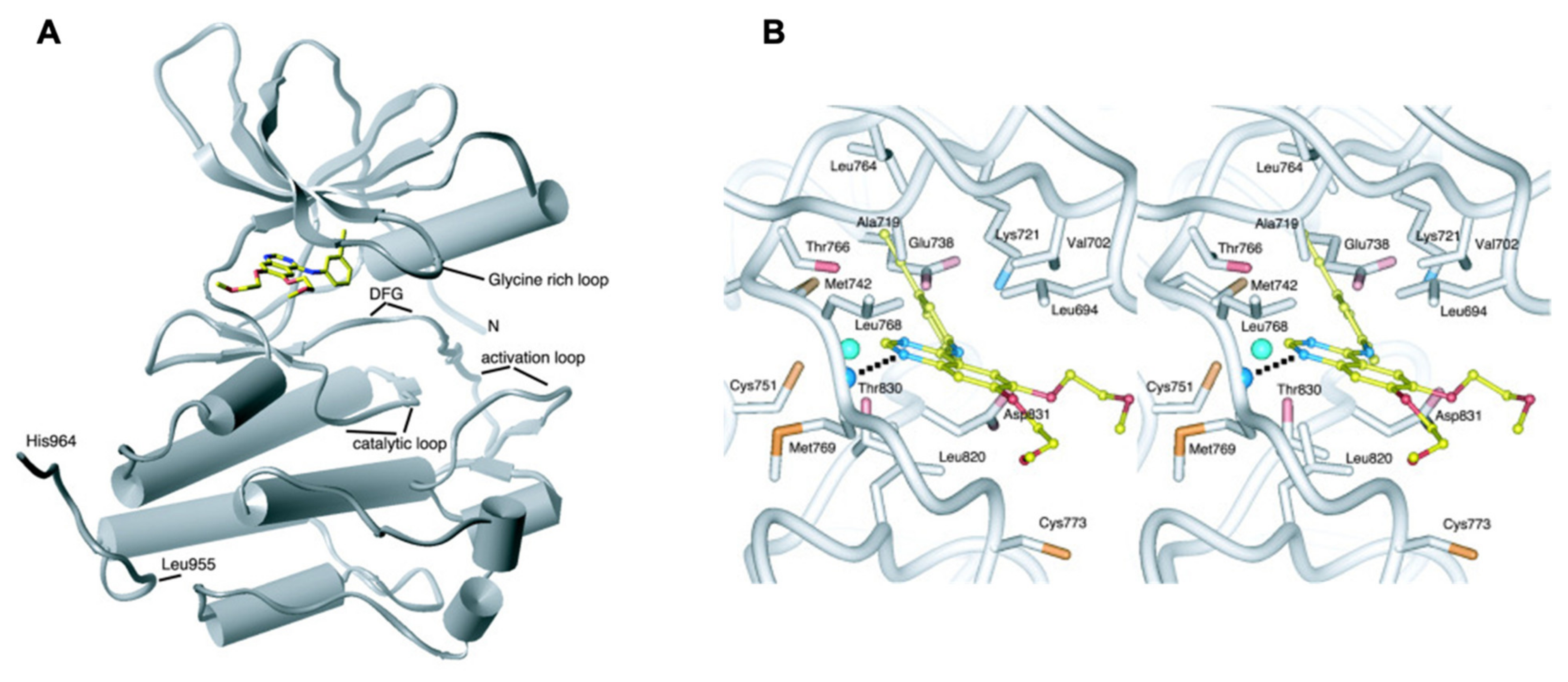
4. Binding Site Architecture of Tyrosine Kinase
5. Major Classes in RTKs: Master Regulators of Cell Fate and Homeostasis
6. Selective TKs Inhibitors in the Clinical Trial
7. Recent Updates on Selective Tyrosine Kinase Inhibitors in Pre-Clinical Studies
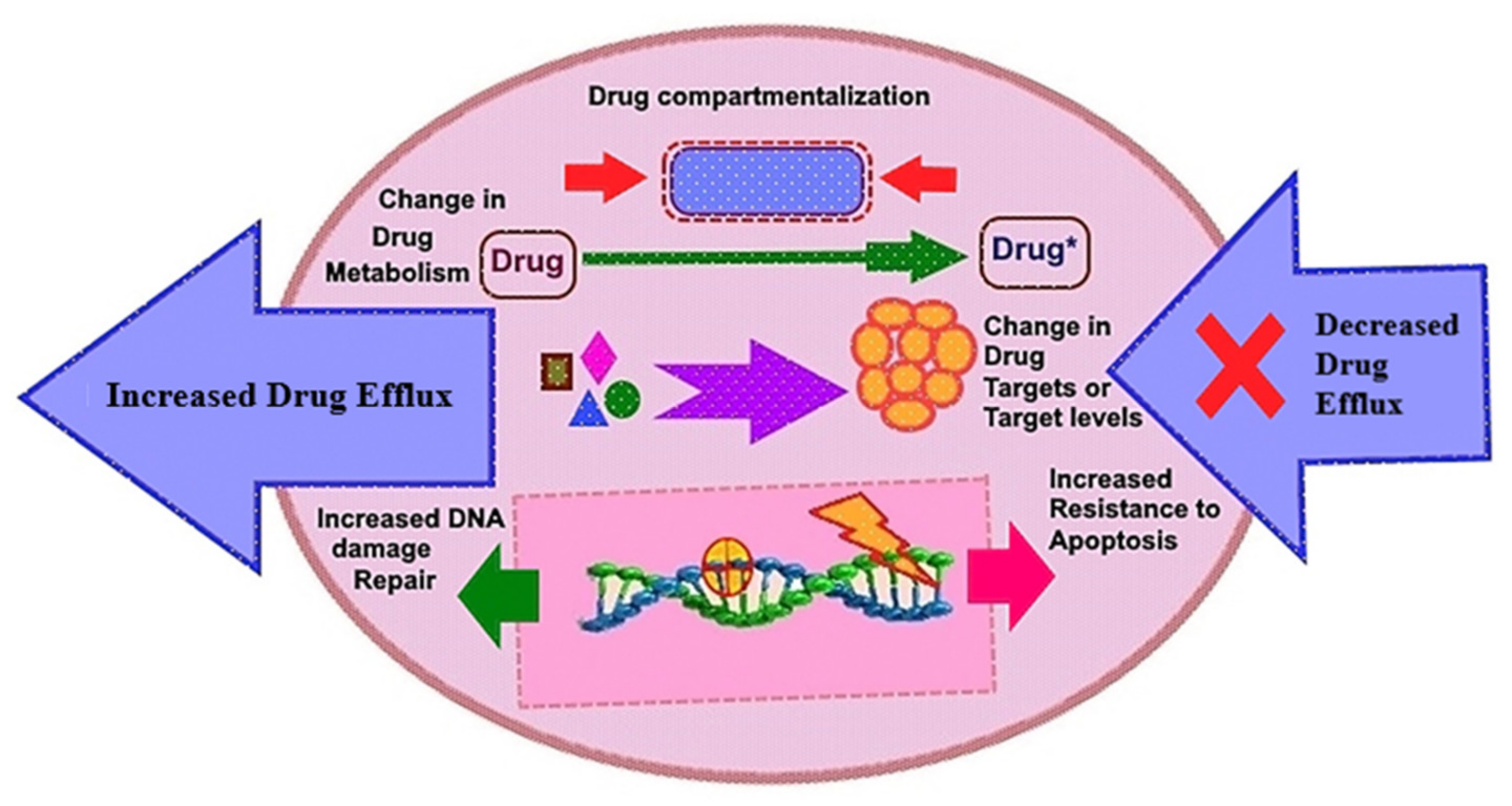
8. Clinical Resistance to Small Molecule Inhibitor Targeting RTKs: Advances and Pitfalls of Targeted Therapy
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Michor, F.; Iwasa, Y.; Nowak, M.A. Dynamics of cancer progression. Nat. Rev. Can. 2004, 4, 197–205. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Metibemu, D.S.; Akinloye, O.A.; Akamo, A.J.; Ojo, D.A.; Okeowo, O.T.; Omotuyi, I.O. Exploring receptor tyrosine kinases-inhibitors in cancer treatments. Egypt. J. Med. Hu. Genet. 2019, 20. [Google Scholar] [CrossRef]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef]
- Butti, R.; Das, S.; Gunasekaran, V.P.; Yadav, A.S.; Kumar, D.; Kundu, G.C. Receptor tyrosine kinases (RTKs) in breast cancer: Signaling, therapeutic implications and challenges. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef]
- Receptor Tyrosine Kinases (RTKs). IUPHAR/BPS Guide to PHARMACOLOGY. Available online: http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=304 (accessed on 15 September 2021).
- Alexander, S.P.; Fabbro, D.; Kelly, E.; Marrion, N.V.; Peters, J.A.; Faccenda, E.; Harding, S.D.; Pawson, A.J.; Sharman, J.L.; Southan, C.; et al. CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. Br. J. Pharmacol. 2019, 176, S297–S396. [Google Scholar]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target Ther. 2020, 5, 181. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Toledo, S.P.; dos Santos, M.A.; Toledo, R.; Lourenço, D.M., Jr. Impact of RET proto-oncogene analysis on the clinical management of multiple endocrine neoplasia type 2. Clinics 2019, 61, 59–70. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Huang, L.; Fu, L. Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta. Pharm. Sin. B 2015, 5, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Dong, X.; Yap, J.; Hu, J. The MAPK and AMPK signalings: Interplay and implication in targeted cancer therapy. J. Hematol. Oncol. 2020, 13, 113. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Li, R.; Morris, S.W. Development of anaplastic lymphoma kinase (ALK) small-molecule inhibitors for cancer therapy. Med. Res. Rev. 2008, 28, 372–412. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Suzuki, R.; Sakata, S.; Hatano, S.; Asaka, R.; Hamanaka, W.; Ninomiya, H.; Uehara, H.; et al. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012, 18, 378–381. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhou, C.C.; Chen, G.Y.; Cheng, Y.; Huang, C.; Zhang, L.; Xu, C.R.; Li, A.W.; Yan, H.H.; Su, J.; et al. A multicenter phase II study of sorafenib monotherapy in clinically selected patients with advanced lung adenocarcinoma after failure of EGFR-TKI therapy (Chinese Thoracic Oncology Group, CTONG 0805). Lung Cancer 2014, 83, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Tomczak, K.; Li, J.; Ochieng, J.K.; Lee, Y.; Haymaker, C. Next-Generation Immunotherapies to Improve Anticancer Immunity. Front. Pharmacol. 2021, 11, 566401. [Google Scholar] [CrossRef] [PubMed]
- Mongre, R.K.; Sodhi, S.S.; Ghosh, M.; Kim, J.H.; Kim, N.; Park, Y.H.; Kim, S.J.; Heo, Y.J.; Sharma, N.; Jeong, D.K. The novel inhibitor BRM270 downregulates tumorigenesis by suppression of NF-κB signaling cascade in MDR-induced stem like cancer-initiating cells. Int. J. Oncol. 2015, 46, 2573–2585. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Duong-Ly, K.C.; Peterson, J.R. The human kinome and kinase inhibition. Curr. Protoc. Pharmacol. 2013, 60, 2–9. [Google Scholar] [CrossRef]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef]
- Baier, A.; Szyszka, R. Compounds from Natural Sources as Protein Kinase Inhibitors. Biomolecules 2020, 10, 1546. [Google Scholar] [CrossRef]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef]
- Cybulsky, A.V.; Guillemette, J.; Papillon, J.; Abouelazm, N.T. Regulation of Ste20-like kinase, SLK, activity: Dimerization and activation segment phosphorylation. PLoS ONE 2017, 12, e0177226. [Google Scholar] [CrossRef]
- Falcone, I.; Conciatori, F.; Bazzichetto, C.; Bria, E.; Carbognin, L.; Malaguti, P.; Ferretti, G.; Cognetti, F.; Milella, M.; Ciuffreda, L. AXL Receptor in Breast Cancer: Molecular Involvement and Therapeutic Limitations. Int. J. Mol. Sci. 2020, 21, 8419. [Google Scholar] [CrossRef]
- Myers, S.H.; Brunton, V.G.; Unciti-Broceta, A. AXL Inhibitors in Cancer: A Medicinal Chemistry Perspective. J. Med. Chem. 2015, 59, 3593–3608. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, K.; Yang, Y.; Nakamura, M.; Andersson, P.; Yang, X.; Zhang, Y.; Cao, Y. Dual roles of endothelial FGF-2–FGFR1–PDGF-BB and perivascular FGF-2–FGFR2–PDGFRβ signaling pathways in tumor vascular remodeling. Cell Discov. 2018, 4. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Xiang, D.; Feng, Y.; Wang, J.; Zhang, X.; Shen, J.; Zou, R.; Yuan, Y. Platelet-derived growth factor-BB promotes proliferation and migration of retinal microvascular pericytes by up-regulating the expression of C-X-C chemokine receptor types 4. Exp. Ther. Med. 2019, 18, 4022–4030. [Google Scholar] [CrossRef]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Yang, H.; Chon, H.J.; Kim, C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp. Mol. Med. 2020, 52, 1475–1485. [Google Scholar] [CrossRef]
- Gotink, K.J.; Verheul, H.M. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef]
- Smith, J.A.; Francis, S.H.; Corbin, J.D. Autophosphorylation: A salient feature of protein kinases. Mol. Cell Biochem. 1993, 127–128, 51–70. [Google Scholar] [CrossRef]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat. Rev. Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.S.; Kim, K.M.; Ting, J.C.; Yu, K.; Fu, J.; Liu, S.; Cristescu, R.; Nebozhyn, M.; Gong, L.; Yue, Y.G.; et al. Genomic landscape and genetic heterogeneity in gastric adenocarcinoma revealed by whole-genome sequencing. Nat. Commun. 2014, 5, 5477. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, E.; Togashi, Y.; Takeuchi, Y.; Shinya, S.; Tada, Y.; Kataoka, K.; Tane, K.; Sato, E.; Ishii, G.; Goto, K.; et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non-small cell lung cancer. Sci. Immunol. 2020, 5, eaav3937. [Google Scholar] [CrossRef]
- Chou, T.Y.; Chiu, C.H.; Li, L.H.; Hsiao, C.Y.; Tzen, C.Y.; Chang, K.T.; Chen, Y.M.; Perng, R.P.; Tsai, S.F.; Tsai, C.M. Mutation in the tyrosine kinase domain of epidermal growth factor receptor is a predictive and prognostic factor for gefitinib treatment in patients with non-small cell lung cancer. Clin. Cancer Res. 2005, 11, 3750–3757. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef]
- Jang, S.; Hong, M.; Shin, M.K.; Kim, B.C.; Shin, H.S.; Yu, E.; Hong, S.M.; Kim, J.; Chun, S.M.; Kim, T.I.; et al. KRAS and PIK3CA mutations in colorectal adenocarcinomas correlate with aggressive histological features and behavior. Hum. Pathol. 2017, 65, 21–30. [Google Scholar] [CrossRef]
- Park, J.; Cho, Y.H.; Shin, W.J.; Lee, S.K.; Lee, J.; Kim, T.; Cha, P.H.; Yang, J.S.; Cho, J.; Min, D.S.; et al. A Ras destabilizer KYA1797K overcomes the resistance of EGFR tyrosine kinase inhibitor in KRAS-mutated non-small cell lung cancer. Sci. Rep. 2019, 9, 648. [Google Scholar] [CrossRef]
- Bradley, D. Biography of Lewis C. Cantley. Proc. Natl. Acad. Sci. USA 2004, 101, 3327–3328. [Google Scholar] [CrossRef] [PubMed]
- Young, C.D.; Zimmerman, L.J.; Hoshino, D.; Formisano, L.; Hanker, A.B.; Gatza, M.L.; Morrison, M.M.; Moore, P.D.; Whitwell, C.A.; Dave, B.; et al. Activating PIK3CA Mutations Induce an Epidermal Growth Factor Receptor (EGFR)/Extracellular Signal-regulated Kinase (ERK) Paracrine Signaling Axis in Basal-like Breast Cancer. Mol. Cell Proteom. 2015, 14, 1959–1976. [Google Scholar] [CrossRef]
- Qiu, X.; Wang, Y.; Liu, F.; Peng, L.; Fang, C.; Qian, X.; Zhang, X.; Wang, Q.; Xiao, Z.; Chen, R.; et al. Survival and prognosis analyses of concurrent PIK3CA mutations in EGFR mutant non-small cell lung cancer treated with EGFR tyrosine kinase inhibitors. Am. J. Cancer Res. 2021, 11, 3189–3200. [Google Scholar] [PubMed]
- Martin-Fernandez, M.L.; Clarke, D.T.; Roberts, S.K.; Zanetti-Domingues, L.C.; Gervasio, F.L. Structure and Dynamics of the EGF Receptor as Revealed by Experiments and Simulations and Its Relevance to Non-Small Cell Lung Cancer. Cells 2019, 8, 316. [Google Scholar] [CrossRef]
- DeBerge, M.; Glinton, K.; Subramanian, M.; Wilsbacher, L.D.; Rothlin, C.V.; Tabas, I.; Thorp, E.B. Macrophage AXL receptor tyrosine kinase inflames the heart after reperfused myocardial infarction. J. Clin. Invest. 2021, 131, e139576. [Google Scholar] [CrossRef] [PubMed]
- Mishra, C.B.; Pandey, P.; Sharma, R.D.; Malik, M.Z.; Mongre, R.K.; Lynn, A.M.; Prasad, R.; Jeon, R.; Prakash, A. Identifying the natural polyphenol catechin as a multi-targeted agent against SARS-CoV-2 for the plausible therapy of COVID-19: An integrated computational approach. Brief. Bioinform. 2021, 22, 1346–1360. [Google Scholar] [CrossRef]
- Ferguson, K.M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Petri, E.T.; Halmos, B.; Boggon, T.J. Structure and clinical relevance of the epidermal growth factor receptor in human cancer. J. Clin. Oncol. 2008, 26, 1742–1751. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Muller, Y.A.; Li, B.; Christinger, H.W.; Wells, J.A.; Cunningham, B.C.; de Vos, A.M. Vascular endothelial growth factor: Crystal structure and functional mapping of the kinase domain receptor binding site. Proc. Natl. Acad. Sci. USA 1997, 94, 7192–7197. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.A.; Boloor, A.; Cheung, M.; Kumar, R.; Crosby, R.M.; Davis-Ward, R.G.; Epperly, A.H.; Hinkle, K.W.; Hunter, R.N., 3rd; Johnson, J.H.; et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J. Med. Chem. 2008, 51, 4632–4640. [Google Scholar] [CrossRef]
- Harris, P.A.; Cheung, M.; Hunter, R.N., III; Brown, M.L.; Veal, J.M.; Nolte, R.T.; Wang, L.; Liu, W.; Crosby, R.M.; Johnson, J.H.; et al. Discovery and evaluation of 2-anilino-5-aryloxazoles as a novel class of VEGFR-2 kinase inhibitors. J. Med. Chem. 2005, 48, 1610–1619. [Google Scholar] [CrossRef]
- Jia, Y.; Zhang, J.; Feng, J.; Xu, F.; Pan, H.; Xu, W. Design, synthesis and biological evaluation of pazopanib derivatives as antitumor agents. Chem. Biol. Drug. Des. 2014, 83, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef]
- Wu, J.; Yu, E. Insulin-like growth factor receptor-1 (IGF-IR) as a target for prostate cancer therapy. Cancer Metastasis Rev. 2014, 33, 607–617. [Google Scholar] [CrossRef]
- Chen, P.H.; Chen, X.; He, X. Platelet-derived growth factors and their receptors: Structural and functional perspectives. Biochim. Biophys. Acta. 2013, 1834, 2176–2186. [Google Scholar] [CrossRef]
- Niu, G.; Chen, X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr. Drug Targets 2010, 11, 1000–1017. [Google Scholar] [CrossRef]
- Ho, J.; Moyes, D.L.; Tavassoli, M.; Naglik, J.R. The Role of ErbB Receptors in Infection. Trends Microbiol. 2017, 25, 942–952. [Google Scholar] [CrossRef]
- Li, Q.; Fu, J.; Xia, Y.; Qi, W.; Ishikado, A.; Park, K.; Yokomizo, H.; Huang, Q.; Cai, W.; Rask-Madsen, C.; et al. Homozygous receptors for insulin and not IGF-1 accelerate intimal hyperplasia in insulin resistance and diabetes. Nat. Commun. 2019, 10, 4427. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E. IGF-1R drugs travel from cancer cradle to Graves. Nat. Biotechnol. 2020, 38, 385–388. [Google Scholar] [CrossRef]
- Guérit, E.; Arts, F.; Dachy, G.; Boulouadnine, B.; Demoulin, J.B. PDGF receptor mutations in human diseases. Cell Mol. Life Sci. 2021, 78, 3867–3881. [Google Scholar] [CrossRef]
- Ivy, S.P.; Wick, J.Y.; Kaufman, B.M. An overview of small-molecule inhibitors of VEGFR signaling. Nat. Rev. Clin. Oncol. 2009, 6, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Shen, H.; Buckley, B.; Chen, Y.; Yang, N.; Mussell, A.L.; Chernov, M.; Kobzik, L.; Frangou, C.; Han, S.X.; et al. NTRK1 is a positive regulator of YAP oncogenic function. Oncogene 2019, 38, 2778–2787. [Google Scholar] [CrossRef]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.W.; Bartlett, P.F.; Lackmann, M. Therapeutic targeting of EPH receptors and their ligands. Nat. Rev. Drug. Discov. 2014, 13, 39–62. [Google Scholar] [CrossRef] [PubMed]
- Flem-Karlsen, K.; Nyakas, M.; Farstad, I.N.; McFadden, E.; Wernhoff, P.; Jacobsen, K.D.; Flørenes, V.A.; Mælandsmo, G.M. Soluble AXL as a marker of disease progression and survival in melanoma. PLoS ONE 2020, 15, e0227187. [Google Scholar] [CrossRef]
- Plaza-Menacho, I.; Burzynski, G.M.; de Groot, J.W.; Eggen, B.J.; Hofstra, R.M. Current concepts in RET-related genetics, signaling and therapeutics. Trends Genet. 2006, 22, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T.; et al. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Yu, H.A.; Riely, G.J. Second-generation epidermal growth factor receptor tyrosine kinase inhibitors in lung cancers. J. Natl. Compr. Cancer Netw. 2013, 11, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Geller, J.I.; Fox, E.; Turpin, B.K.; Goldstein, S.L.; Liu, X.; Minard, C.G.; Kudgus, R.A.; Reid, J.M.; Berg, S.L.; Weigel, B.J. A study of axitinib, a VEGF receptor tyrosine kinase inhibitor, in children and adolescents with recurrent or refractory solid tumors: A Children’s Oncology Group phase 1 and pilot consortium trial (ADVL1315). Cancer 2018, 124, 4548–4555. [Google Scholar] [CrossRef]
- Baumann, K.H.; du Bois, A.; Meier, W.; Rau, J.; Wimberger, P.; Sehouli, J.; Kurzeder, C.; Hilpert, F.; Hasenburg, A.; Canzler, U.; et al. A phase II trial (AGO 2.11) in platinum-resistant ovarian cancer: A randomized multicenter trial with sunitinib (SU11248) to evaluate dosage, schedule, tolerability, toxicity and effectiveness of a multitargeted receptor tyrosine kinase inhibitor monotherapy. Ann. Oncol. 2012, 23, 2265–2271. [Google Scholar] [PubMed]
- Dragovich, T.; Laheru, D.; Dayyani, F.; Bolejack, V.; Smith, L.; Seng, J.; Burris, H.; Rosen, P.; Hidalgo, M.; Ritch, P.; et al. Phase II trial of vatalanib in patients with advanced or metastatic pancreatic adenocarcinoma after first-line gemcitabine therapy (PCRT O4-001). Cancer Chemother. Pharmacol. 2014, 74, 379–387. [Google Scholar] [CrossRef]
- Huijts, C.M.; Santegoets, S.J.; van den Eertwegh, A.J.; Pijpers, L.S.; Haanen, J.B.; de Gruijl, T.D.; Verheul, H.M.; van der Vliet, H.J. Phase I-II study of everolimus and low-dose oral cyclophosphamide in patients with metastatic renal cell cancer. BMC Cancer 2011, 11, 505. [Google Scholar] [CrossRef]
- Jian, H.; Li, W.; Ma, Z.; Huang, J.; Feng, J.; Song, Y.; Gao, B.; Zhu, H.; Tao, M.; Bai, C.; et al. Intercalating and maintenance gefitinib plus chemotherapy versus chemotherapy alone in selected advanced non-small cell lung cancer with unknown EGFR status. Sci. Rep. 2017, 7, 8483. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.E.; Endicott, J.A.; Johnson, L.N. Protein kinase inhibitors: Insights into drug design from structure. Science 2004, 303, 1800–1805. [Google Scholar] [CrossRef]
- Rosenzweig, S.A. Acquired Resistance to Drugs Targeting Tyrosine Kinases. Adv. Cancer Res. 2018, 138, 71–98. [Google Scholar]
- Li, J.; An, B.; Song, X.; Zhang, Q.; Chen, C.; Wei, S.; Fan, R.; Li, X.; Zou, Y. Design, synthesis and biological evaluation of novel 2,4-diaryl pyrimidine derivatives as selective EGFRL858R/T790M inhibitors. Eur. J. Med. Chem. 2021, 212, 113019. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Wu, B.X.; Zheng, Q.; Hu, C.H.; Tang, X.Z.; Zhang, W.; Rao, G.W. Design, synthesis, biological evaluation and docking study of novel quinazoline derivatives as EGFR-TK inhibitors. Future Med. Chem. 2021, 13, 601–612. [Google Scholar] [CrossRef]
- Raghu, M.S.; Pradeep Kumar, C.B.; Prashanth, M.K.; Yogesh Kumar, K.B.; Prathibha, S.; Kanthimathi, G.; Alissa, S.A.; Alghulikah, H.A.; Osman, S.M. Novel 1,3,5-triazine-based pyrazole derivatives as potential antitumor agents and EFGR kinase inhibitors: Synthesis, cytotoxicity, DNA binding, molecular docking and DFT studies. N. J. Chem. 2021, 45, 13909–13924. [Google Scholar] [CrossRef]
- Mishra, C.B.; Mongre, R.K.; Kumari, S.; Jeong, D.K.; Tiwari, M. Synthesis, in vitro and in vivo anticancer activity of novel 1-(4-imino-1-substituted-1H-pyrazolo[3,4-d]pyrimidin-5(4H)-yl)urea derivatives. RSC Adv. 2016, 6, 24491–24500. [Google Scholar] [CrossRef]
- Mishra, C.B.; Mongre, R.K.; Prakash, A.; Jeon, R.; Supuran, C.T.; Lee, M.S. Anti-breast cancer action of carbonic anhydrase IX inhibitor 4-[4-(4-Benzo[1,3]dioxol-5-ylmethyl-piperazin-1-yl)-benzylidene-hydrazinocarbonyl]-benzenesulfonamide (BSM-0004): In vitro and in vivo studies. J. Enzym. Inhib. Med. Chem. 2021, 36, 954–963. [Google Scholar] [CrossRef]
- Mongre, R.K.; Mishra, C.B.; Prakash, A.; Jung, S.; Lee, B.S.; Kumari, S.; Hong, J.T.; Lee, M.S. Novel Carbazole-Piperazine Hybrid Small Molecule Induces Apoptosis by Targeting BCL-2 and Inhibits Tumor Progression in Lung Adenocarcinoma in Vitro and Xenograft Mice Model. Cancers 2019, 11, 1245. [Google Scholar] [CrossRef]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Hennequin, L.F.; Thomas, A.P.; Johnstone, C.; Stokes, E.S.; Plé, P.A.; Lohmann, J.J.; Ogilvie, D.J.; Dukes, M.; Wedge, S.R.; Curwen, J.O.; et al. Design and structure-activity relationship of a new class of potent VEGF receptor tyrosine kinase inhibitors. J. Med. Chem. 1999, 42, 5369–5389. [Google Scholar] [CrossRef] [PubMed]
- Eissa, I.H.; El-Helby, A.A.; Mahdy, H.A.; Khalifa, M.M.; Elnagar, H.A.; Mehany, A.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; et al. Discovery of new quinazolin-4(3H)-ones as VEGFR-2 inhibitors: Design, synthesis, and anti-proliferative evaluation. Bioorg. Chem. 2020, 105, 104380. [Google Scholar] [CrossRef]
- Al-Ansary, G.H.; Nasr, T.; Taha, H.; Fayad, W.; Mahgoub, S. Biphenylurea/thiourea derivatives tagged with heteroarylsulfonamide motifs as novel VEGFR2 inhibitors; Design, synthesis and anti-angiogenic activity. Bioorg. Chem. 2021, 107, 104640. [Google Scholar] [CrossRef]
- Mahmoud, H.K.; Farghaly, T.A.; Abdulwahab, H.G.; Al-Qurashi, N.T.; Shaaban, M.R. Novel 2-indolinone thiazole hybrids as sunitinib analogues: Design, synthesis, and potent VEGFR-2 inhibition with potential anti-renal cancer activity. Eur. J. Med. Chem. 2020, 208, 112752. [Google Scholar] [CrossRef]
- Michaloski, J.S.; Redondo, A.R.; Magalhães, L.S.; Cambui, C.C.; Giordano, R.J. Discovery of pan-VEGF inhibitory peptides directed to the extracellular ligand-binding domains of the VEGF receptors. Sci. Adv. 2016, 2, e1600611. [Google Scholar] [CrossRef]
- Osher, E.; Macaulay, V.M. Therapeutic Targeting of the IGF Axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef]
- Lee, H.J.; Pham, P.C.; Pei, H.; Lim, B.; Hyun, S.Y.; Baek, B.; Kim, B.; Kim, Y.; Kim, M.H.; Kang, N.W.; et al. Development of the phenylpyrazolo[3,4-d]pyrimidine-based, insulin-like growth factor receptor/Src/AXL-targeting small molecule kinase inhibitor. Theranostics 2021, 11, 1918–1936. [Google Scholar] [CrossRef]
- Gadekar, P.K.; Urunkar, G.; Roychowdhury, A.; Sharma, R.; Bose, J.; Khanna, S.; Damre, A.; Sarveswari, S. Design, synthesis and biological evaluation of 2,3-dihydroimidazo[2,1-b]thiazoles as dual EGFR and IGF1R inhibitors. Bioorg. Chem. 2021, 115, 105151. [Google Scholar] [CrossRef]
- Wen, B.; Deutsch, E.; Marangoni, E.; Frascona, V.; Maggiorella, L.; Abdulkarim, B.; Chavaudra, N.; Bourhis, J. Tyrphostin AG 1024 modulates radiosensitivity in human breast cancer cells. Br. J. Cancer 2001, 85, 2017–2021. [Google Scholar] [CrossRef][Green Version]
- Mishra, C.B.; Mongre, R.K.; Kumari, S.; Jeong, D.K.; Tiwari, M. Novel Triazole-Piperazine Hybrid Molecules Induce Apoptosis via Activation of the Mitochondrial Pathway and Exhibit Antitumor Efficacy in Osteosarcoma Xenograft Nude Mice Model. ACS Chem. Biol. 2017, 12, 753–768. [Google Scholar] [CrossRef]
- Carboni, J.M.; Wittman, M.; Yang, Z.; Lee, F.; Greer, A.; Hurlburt, W.; Hillerman, S.; Cao, C.; Cantor, G.H.; Dell-John, J.; et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 2009, 8, 3341–3349. [Google Scholar] [CrossRef]
- Maguire, M.P.; Sheets, K.R.; McVety, K.; Spada, A.P.; Zilberstein, A. A new series of PDGF receptor tyrosine kinase inhibitors: 3-substituted quinoline derivatives. J. Med. Chem. 1994, 37, 2129–2137. [Google Scholar] [CrossRef]
- Yang, T.H.; Lee, C.I.; Huang, W.H.; Lee, A.R. Structural optimization and evaluation of novel 2-pyrrolidone-fused (2-oxoindolin-3-ylidene)methylpyrrole derivatives as potential VEGFR-2/PDGFRβ inhibitors. Chem. Cent. J. 2017, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Bahleda, R.; Italiano, A.; Hierro, C.; Mita, A.; Cervantes, A.; Chan, N.; Awad, M.; Calvo, E.; Moreno, V.; Govindan, R.; et al. Multicenter Phase I Study of Erdafitinib (JNJ-42756493), Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced or Refractory Solid Tumors. Clin. Cancer Res. 2019, 25, 4888–4897. [Google Scholar] [CrossRef]
- Brameld, K.A.; Owens, T.D.; Verner, E.; Venetsanakos, E.; Bradshaw, J.M.; Phan, V.T.; Tam, D.; Leung, K.; Shu, J.; LaStant, J.; et al. Discovery of the Irreversible Covalent FGFR Inhibitor 8-(3-(4-Acryloylpiperazin-1-yl)propyl)-6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)pyrido[2,3-d]pyrimidin-7(8H)-one (PRN1371) for the Treatment of Solid Tumors. J. Med. Chem. 2017, 60, 6516–6527. [Google Scholar] [CrossRef]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Hugo, H.S. Update on antibody-drug conjugates in breast cancer. Clin. Adv. Hematol. Oncol. 2021, 19, 148–151. [Google Scholar]
- Mongre, R.K.; Jung, S.; Mishra, C.B.; Lee, B.S.; Kumari, S.; Lee, M.S. Prognostic and Clinicopathological Significance of SERTAD1 in Various Types of Cancer Risk: A Systematic Review and Retrospective Analysis. Cancers 2019, 11, 337. [Google Scholar] [CrossRef] [PubMed]
- Mongre, R.K.; Mishra, C.B.; Jung, S.; Lee, B.S.; Quynh, N.; Anh, N.H.; Myagmarjav, D.; Jo, T.; Lee, M.S. Exploring the Role of TRIP-Brs in Human Breast Cancer: An Investigation of Expression, Clinicopathological Significance, and Prognosis. Mol. Ther. Oncolytics 2020, 19, 105–126. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, K.C.; Mongre, R.K.; Kim, J.Y.; Kim, Y.R.; Choi, D.Y.; Song, S.; Yun, J.; Han, S.B.; Yoon, D.Y.; et al. IL-32γ suppresses lung cancer stem cell growth via inhibition of ITGAV-mediated STAT5 pathway. Cell Death Dis. 2019, 10, 506. [Google Scholar] [CrossRef] [PubMed]
- Mongre, R.K.; Sodhi, S.S.; Ghosh, M.; Kim, J.H.; Kim, N.; Sharma, N.; Jeong, D.K. A New Paradigm to Mitigate Osteosarcoma by Regulation of MicroRNAs and Suppression of the NF-κB Signaling Cascade. Dev. Reprod. 2014, 18, 197–212. [Google Scholar] [CrossRef]
- Mishra, C.B.; Kumari, S.; Angeli, A.; Bua, S.; Mongre, R.K.; Tiwari, M.; Supuran, C.T. Discovery of Potent Carbonic Anhydrase Inhibitors as effective Anti-convulsant Agents: Drug design, Synthesis, In vitro and In vivo Investigations. J. Med. Chem. 2021, 64, 3100–3114. [Google Scholar] [CrossRef]
- Mishra, C.B.; Tiwari, M.; Supuran, C.T. Progress in the development of human carbonic anhydrases inhibitors and their pharmacological applications: Where are we today? Med. Res. Rev. 2020, 40, 2485–2565. [Google Scholar] [CrossRef] [PubMed]
- Mishra, C.B.; Kumari, S.; Angeli, A.; Bua, S.; Tiwari, M.; Supuran, C.T. Discovery of Benzenesulfonamide Derivatives as Carbonic Anhydrase Inhibitors with Effective Anticonvulsant Action: Design, Synthesis, and Pharmacological Evaluation. J. Med. Chem. 2018, 61, 3151–3165. [Google Scholar] [CrossRef] [PubMed]
- Casals, E.; Gusta, M.F.; Cobaleda-Siles, M.; Garcia-Sanz, A.; Puntes, V.F. Cancer resistance to treatment and antiresistance tools offered by multimodal multifunctional nanoparticles. Cancer Nanotechnol. 2017, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Kee, D.; Zalcberg, J.R. Current and emerging strategies for the management of imatinib-refractory advanced gastrointestinal stromal tumors. Ther. Adv. Med. Oncol. 2012, 4, 255–270. [Google Scholar] [CrossRef]
- Rubin, B.P.; Duensing, A. Mechanisms of resistance to small molecule kinase inhibition in the treatment of solid tumors. Lab. Invest. 2006, 86, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, E.; Nichols, G. Mechanisms of resistance to imatinib in CML patients: A paradigm for the advantages and pitfalls of molecularly targeted therapy. Curr. Cancer Drug Targets 2006, 6, 645–657. [Google Scholar] [CrossRef]
- Tamborini, E.; Bonadiman, L.; Greco, A.; Albertini, V.; Negri, T.; Gronchi, A.; Bertulli, R.; Colecchia, M.; Casali, P.G.; Pierotti, M.A.; et al. A new mutation in the KIT ATP pocket causes acquired resistance to imatinib in a gastrointestinal stromal tumor patient. Gastroenterology 2004, 127, 294–299. [Google Scholar] [CrossRef]
- Debiec-Rychter, M.; Cools, J.; Dumez, H.; Sciot, R.; Stul, M.; Mentens, N.; Vranckx, H.; Wasag, B.; Prenen, H.; Roesel, J.; et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology 2005, 128, 270–279. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RTK Class | Targets | Inhibitors or FDA Approved Drugs | Predicted Localization Based on Proteinatlas.org | Distribution in Tissue or Organs | Diseases | Reference |
|---|---|---|---|---|---|---|
| ErbB/ EGFR | ErbB1, Her2, Her3, Her4 | panitumumab, cetuximab, gefitinib, erlotinib, afatinib, and lapatinib | Cell membrane, cell junctions, secreted | Placenta, most of organs | BRCA, GLIOMA, inflammation, cardiac, etc. | [1,2,62] |
| IGFR | Insulin, IR, IGF-1 | Linsitinib, BMS-754807, XL-228, AXL1717, Masoprocol | Secreted plasma, transmembrane, vesicles | Liver, muscle, pituitary, and hypothelamus cells | Cancer and diabetes | [63,64] |
| PDGFR | PDGFR-α PDGF-β | Nilotinib, Sunitinib, Dasatinib, Pazopanib, Vatalinib, Axitinib | Intracellular membrane, Golgi apparatus, vesicles, nucleoplasm, plasma membrane | Brain, placenta, cervix, kidneys etc. | Cancer, Kosaki syndrome, myofbromatosis, aneurysms, premature aging syndrome, brain calcification | [65] |
| VEGFR | VEGFRs | Bevacizumab, Sorafenib, Sunitinib, Lenvatinib, Pralsetinib | Intracellular, membrane secreted to blood | Lung, Liver, Blood | Cancer, brain diseases, ophthalmic diseases | [66] |
| FGFR | FGFRs, aFGF, FGFs | AZD4547, BGJ398, JNJ42756493, PD173074 | Intracellular, plasma membrane, ER, etc. | Most of all body organs | Cancer, kidney, lungs, skeletal muscle, heart, liver-related diseases | [8,67] |
| NGFR | trkA, trkB, and trkC | Ro 08-2750 | Cytosol and plasma membrane | Lymph node, testis, liver, kidneys, colon | Cancer, wound healing, neuronal diseases | [68] |
| HGFR | c-Met, HGF, MET | K252a SU11274 PHA-665752 ARQ197 SGX523 | Plasma membrane and cytosol; is predicted to be secreted | Epithelial, endothelial, neurons, hepatocytes, hematopoietic, melanocytes And all body organs | Cancer, autism, cardiac dysfunctions | [69] |
| EphR | EPHs, Ephrins | MEDI-547 KB004 XL647 JI-101 | Intracellular and membrane | Brain, lung, endocrine tissue, and other organs | Cancer, brain, and neuronal diseases | [70] |
| AXLR | AXL UFO | Bemcentinib R428 TP-0903 | Plasma membrane, vesicles, and actin filaments | Muscles, lungs, kidney, colon, and other organs | Cancer, encephalomyelitis, cardiac infraction | [48,71] |
| RETR | RETS | Alectinib Cabozantinib Lenvatinib Ruxolitinib | Golgi apparatus, cytosol, and plasma membrane | Endocrine, lungs, bone, skin, colon, and other organs | Cancer | [72] |
| LTKR | LTK ALK TYK1 | Ceritinib, alectinib, RO5424802, AP26113, ASP3026, TSR-011, PF-06463922, RXDX-101, X-396, and CEP-37440 | Intracellular, membrane, vesicles | Enterocytes, mucus-secreting cells, Ito cells, and Kupffer cells | Cancer | [18,19] |
| ROSR | ROS1 | Crizotinib Entrectinib Lorlatinib Ceritinib Cabozantinib | Vesicles, intracellular, membrane | Lung Epididymis Cerebral cortex Olfactory region | Cancer | [19] |
| TKs | Clinical Trial Status | Conditions/Cancer | Interventions | References |
|---|---|---|---|---|
| EGFR | Completed Phase_II | Non-Small Cell Lung Cancer | Drug: sorafenib (Nexavar) | [20] |
| VEGF | Completed Phase-I | Refractory or Recurrent Solid Tumors | Drug: Axitinib | [77] |
| Multiple TKs | Completed Phase 2 | Platinum Refractory Epithelial Ovarian Cancer, Primary Cancer of the Peritoneum, Cancer of the Fallopian Tube | Drug: Sunitinib, Drug: SUNITINIB | [78] |
| VEGF | Completed Phase 1 Phase 2 | Pancreatic Cancer | Drug: Vatalanib, Drug: Gemcitabine | [79] |
| EGFR | Completed Phase 2 | Non-small Cell Lung Cancer | Drug: Aprepitant, Desloratadine, Placebo of aprepitan, Placebo of desloratadine | ClinicalTrials.gov Identifier: NCT02646020 |
| VGFR | Completed | Distal Urethral Cancer, Proximal Urethral Cancer, Recurrent Bladder Cancer, Recurrent Transitional Cell Cancer of the Renal Pelvis and Ureter, Recurrent Urethral Cancer, Stage IV Bladder Cancer Transitional Cell Carcinoma of the Bladder Urethral Cancer Associated With Invasive Bladder Cancer | Drug: pazopanib hydrochloride | ClinicalTrials.gov Identifier: NCT00471536 |
| VGFR | Completed Phase 1 Phase 2 | Metastatic Renal Cell Cancer | Drug: Everolimus | [80] |
| MET and VEGFR2 | Completed | Neoplasms, Head and Neck | Drug: GSK1363089 (foretinib) | ClinicalTrials.gov Identifier: NCT00725764 |
| EGFR | Completed Phase 3 | Non-small Cell Lung Cancer | Drug: Gefitinib, Gemcitabine + Carboplatin | [81] |
| VEGFR, PDGFR, FGFR and c-Kit | Completed Phase 2 | Advanced Malignancy | Drug: Anlotinib | ClinicalTrials.gov Identifier: NCT04216082 |
| VEGFR | Completed Phase 3 | Metastatic Renal Cell Carcinoma | Drug: RAD001, Drug: Placebo | ClinicalTrials.gov Identifier: NCT00410124 |
| Multi TKs | Completed Phase 4 | Colorectal Neoplasms, Gastrointestinal Stromal Tumors | Drug: Esomeprazole 40 mg concomitantly, Esomeprazole 40 mg before, Regorafenib 160 mg or 120 mg | ClinicalTrials.gov Identifier: NCT02800330 |
| Kinase/Target | Drug (Inhibitor) | Cancer Code | Cancer Name | AvgIC50(G1) | AvgIC50(G2) |
|---|---|---|---|---|---|
| ALK | Alectinib | DLBC | B_cell_lymphoma | 37.2207 | 29.0609 |
| ALK | Crizotinib | DLBC | B_cell_lymphoma | 18.5283 | 10.8294 |
| BMX | QL-XII-47 | LAML | acute_myeloid_leukaemia | 5.7441 | 1.6628 |
| BRAF | AZ628 | LUAD | lung_NSCLC_adenocarcinoma | 22.0213 | 9.4282 |
| BRAF | HG6-64-1 | LUAD | lung_NSCLC_adenocarcinoma | 17.6081 | 16.644 |
| DDR1 | QL-XI-92 | MM | myeloma | 17.0624 | 13.4321 |
| DDR1 | QL-XI-92 | UNCLASSIFIED | anaplastic_large_cell_lymphoma | 23.6282 | 3.7261 |
| EGFR | Afatinib | LUAD | lung_NSCLC_adenocarcinoma | 0.9592 | 0.0191 |
| EGFR | AST-1306 | LUAD | lung_NSCLC_adenocarcinoma | 1.1852 | 0.2463 |
| EGFR | AZD8931 | LUAD | lung_NSCLC_adenocarcinoma | 2.622 | 0.0531 |
| EGFR | Cetuximab | LUAD | lung_NSCLC_adenocarcinoma | 69.9293 | 6.8975 |
| EGFR | CI-1033 | LUAD | lung_NSCLC_adenocarcinoma | 2.1347 | 0.2929 |
| EGFR | CUDC-101 | LUAD | lung_NSCLC_adenocarcinoma | 1.3804 | 0.165 |
| EGFR | Erlotinib | UNCLASSIFIED | Burkitt_lymphoma | 20.2694 | 10.3597 |
| EGFR | Foretinib | UNCLASSIFIED | Burkitt_lymphoma | 3.3933 | 1.2532 |
| EGFR | Gefitinib | LUAD | lung_NSCLC_adenocarcinoma | 9.8753 | 0.0121 |
| EGFR | Gefitinib | UNCLASSIFIED | Burkitt_lymphoma | 15.191 | 9.6238 |
| EGFR | Lapatinib | UNCLASSIFIED | Burkitt_lymphoma | 13.9917 | 9.9664 |
| EGFR | Pelitinib | LUAD | lung_NSCLC_adenocarcinoma | 0.0604 | 0.0543 |
| EGFR | PF-00299804 | LUAD | lung_NSCLC_adenocarcinoma | 1.6263 | 0.2417 |
| EGFR | Sapitinib | LUAD | lung_NSCLC_adenocarcinoma | 3.5254 | 0.0979 |
| ERBB4 | AST-1306 | LAML | acute_myeloid_leukaemia | 4.8215 | 4.4793 |
| ERBB4 | AST-1306 | UNCLASSIFIED | lung_NSCLC_carcinoid | 6.282 | 5.0641 |
| ERBB4 | CI-1033 | SKCM | melanoma | 23.8639 | 8.6932 |
| ERBB4 | CI-1033 | UCEC | endometrium | 8.9198 | 6.6994 |
| ERBB4 | PF-00299804 | UCEC | endometrium | 17.4219 | 10.298 |
| FLT1 | Cediranib | UNCLASSIFIED | anaplastic_large_cell_lymphoma | 5.8298 | 4.1008 |
| FLT1 | Linifanib | UNCLASSIFIED | ovary | 11.8566 | 9.8702 |
| FLT1 | Midostaurin | BLCA | bladder | 1.7165 | 1.4931 |
| FLT1 | Tivozanib | BLCA | bladder | 1.8598 | 1.5754 |
| FLT1 | Tivozanib | UNCLASSIFIED | ovary | 2.5587 | 1.3064 |
| FLT3 | Lestaurtinib | LUAD | lung_NSCLC_adenocarcinoma | 6.5216 | 5.5234 |
| FLT3 | Ponatinib | LUAD | lung_NSCLC_adenocarcinoma | 5.3224 | 4.8343 |
| FLT3 | Quizartinib | LUAD | lung_NSCLC_adenocarcinoma | 20.1255 | 14.9548 |
| IGF1R | BMS-754807 | UNCLASSIFIED | fibrosarcoma | 25.2865 | 1.4107 |
| IGF1R | GSK1904529A | UNCLASSIFIED | fibrosarcoma | 70.7571 | 27.3638 |
| JAK1 | AZD1480 | UNCLASSIFIED | endometrium | 3.9166 | 0.4414 |
| JAK1 | JAK1_8709 | UNCLASSIFIED | endometrium | 67.0713 | 33.19 |
| JAK1 | JAK_8517 | GBM | glioma | 173.1292 | 56.1354 |
| JAK1 | JAK_8517 | UNCLASSIFIED | endometrium | 176.2048 | 2.4967 |
| JAK3 | WHI-P97 | UCEC | endometrium | 98.2843 | 80.5835 |
| KDR | Linifanib | COREAD | large_intestine | 18.5488 | 16.919 |
| KDR | Motesanib | PRAD | prostate | 30.6555 | 12.079 |
| KDR | Ponatinib | UNCLASSIFIED | lung_NSCLC_carcinoid | 6.5232 | 5.613 |
| KIT | Dasatinib | LAML | acute_myeloid_leukaemia | 12.8718 | 7.8024 |
| KIT | Imatinib | LAML | acute_myeloid_leukaemia | 23.4979 | 15.9051 |
| KIT | Sorafenib | LAML | acute_myeloid_leukaemia | 40.2854 | 4.4655 |
| KIT | Sunitinib | LAML | acute_myeloid_leukaemia | 35.55 | 6.7039 |
| LCK | JW-7-24-1 | ALL | lymphoblastic_leukemia | 1.4246 | 0.6944 |
| LCK | Staurosporine | ALL | lymphoblastic_leukemia | 0.0532 | 0.0288 |
| LTK | HG-5-113-01 | PRAD | prostate | 7.8398 | 4.7173 |
| NTRK1 | GW441756 | UCEC | endometrium | 16.7441 | 11.9791 |
| NTRK1 | GW441756 | UNCLASSIFIED | B_cell_leukemia | 38.3105 | 24.7002 |
| NTRK3 | AZD1332 | ALL | lymphoblastic_T_cell_leukaemia | 37.467 | 20.9519 |
| NTRK3 | Lestaurtinib | ALL | lymphoblastic_T_cell_leukaemia | 0.4766 | 0.2518 |
| PDGFRA | Ponatinib | COREAD | large_intestine | 13.9216 | 5.3747 |
| PDGFRB | Dasatinib | PRAD | prostate | 52.0138 | 10.5922 |
| PDGFRB | Motesanib | PRAD | prostate | 22.6324 | 13.6836 |
| PDGFRB | Pazopanib | PRAD | prostate | 50.5275 | 40.7679 |
| PDGFRB | Sorafenib | PRAD | prostate | 26.2459 | 26.1382 |
| RET | Cabozantinib | PRAD | prostate | 63.4851 | 12.5383 |
| ROS1 | Crizotinib | GBM | glioma | 560.1637 | 42.8829 |
| SYK | BAY-61-3606 | KIRC | kidney | 319.3293 | 15.0456 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mongre, R.K.; Mishra, C.B.; Shukla, A.K.; Prakash, A.; Jung, S.; Ashraf-Uz-Zaman, M.; Lee, M.-S. Emerging Importance of Tyrosine Kinase Inhibitors against Cancer: Quo Vadis to Cure? Int. J. Mol. Sci. 2021, 22, 11659. https://doi.org/10.3390/ijms222111659
Mongre RK, Mishra CB, Shukla AK, Prakash A, Jung S, Ashraf-Uz-Zaman M, Lee M-S. Emerging Importance of Tyrosine Kinase Inhibitors against Cancer: Quo Vadis to Cure? International Journal of Molecular Sciences. 2021; 22(21):11659. https://doi.org/10.3390/ijms222111659
Chicago/Turabian StyleMongre, Raj Kumar, Chandra Bhushan Mishra, Arvind Kumar Shukla, Amresh Prakash, Samil Jung, Md Ashraf-Uz-Zaman, and Myeong-Sok Lee. 2021. "Emerging Importance of Tyrosine Kinase Inhibitors against Cancer: Quo Vadis to Cure?" International Journal of Molecular Sciences 22, no. 21: 11659. https://doi.org/10.3390/ijms222111659
APA StyleMongre, R. K., Mishra, C. B., Shukla, A. K., Prakash, A., Jung, S., Ashraf-Uz-Zaman, M., & Lee, M.-S. (2021). Emerging Importance of Tyrosine Kinase Inhibitors against Cancer: Quo Vadis to Cure? International Journal of Molecular Sciences, 22(21), 11659. https://doi.org/10.3390/ijms222111659