
Novel Insights into the Role of the Mineralocorticoid Receptor in Human Glioblastoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
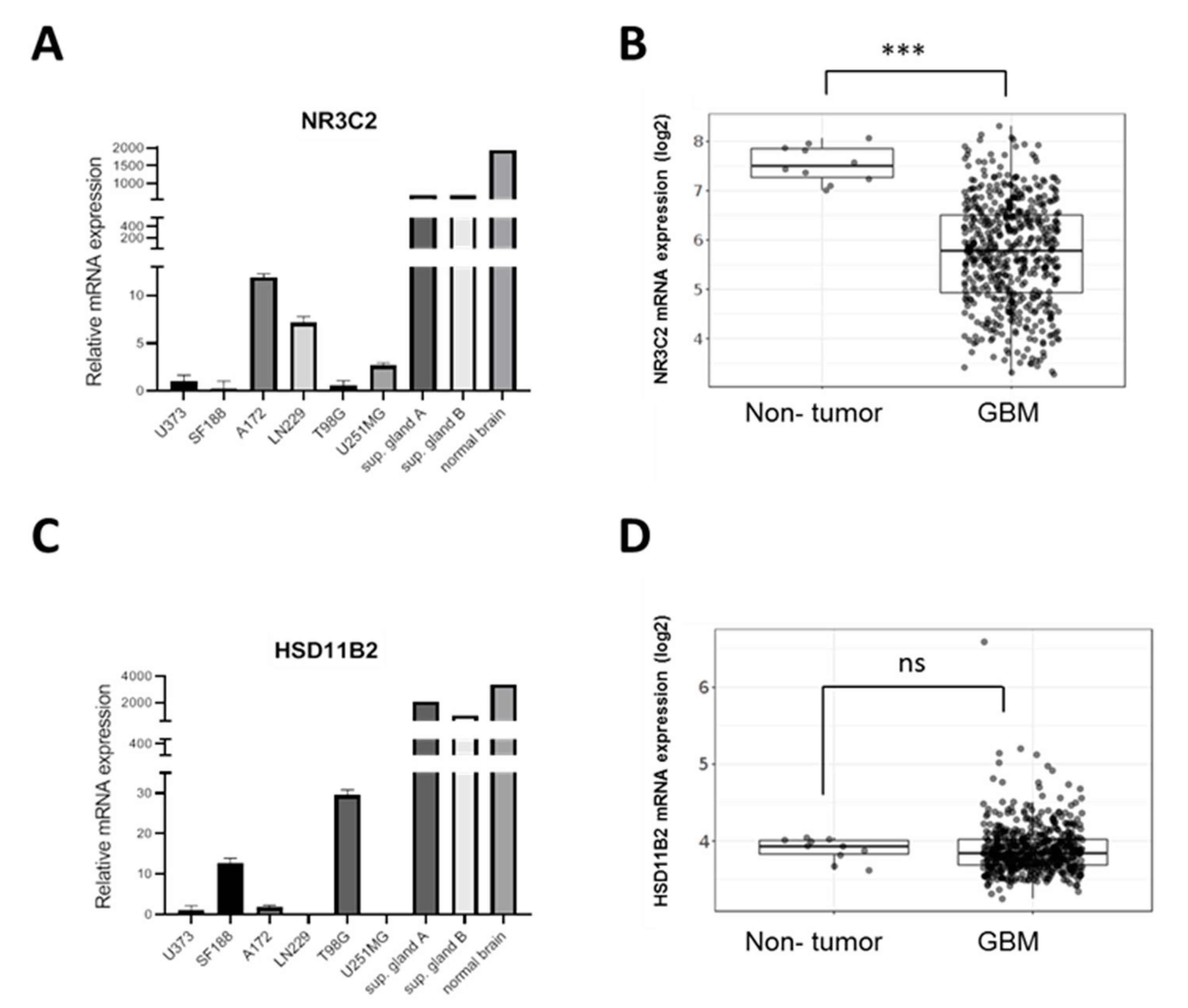
2.1. MR Is Expressed in GBM Cell Lines
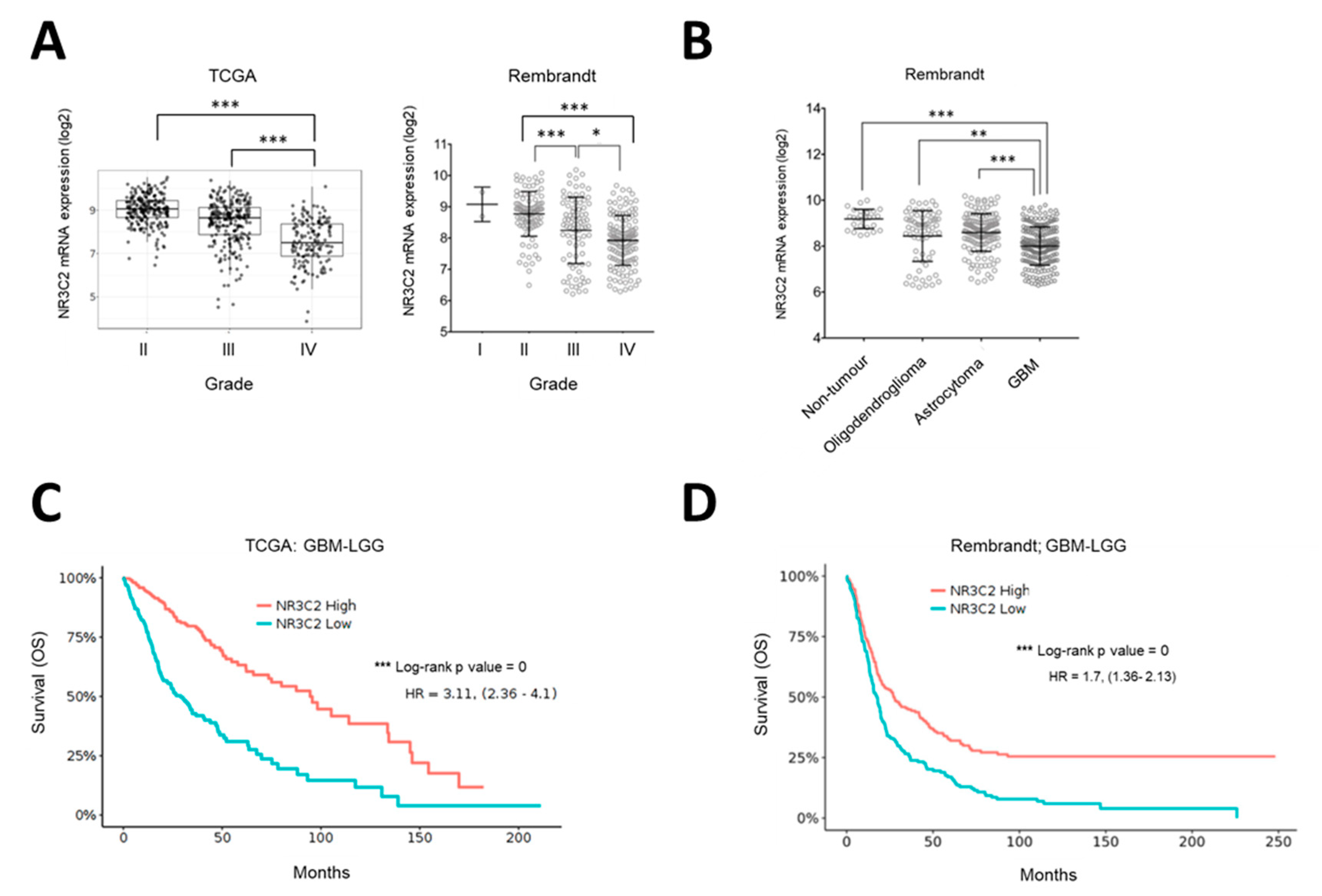
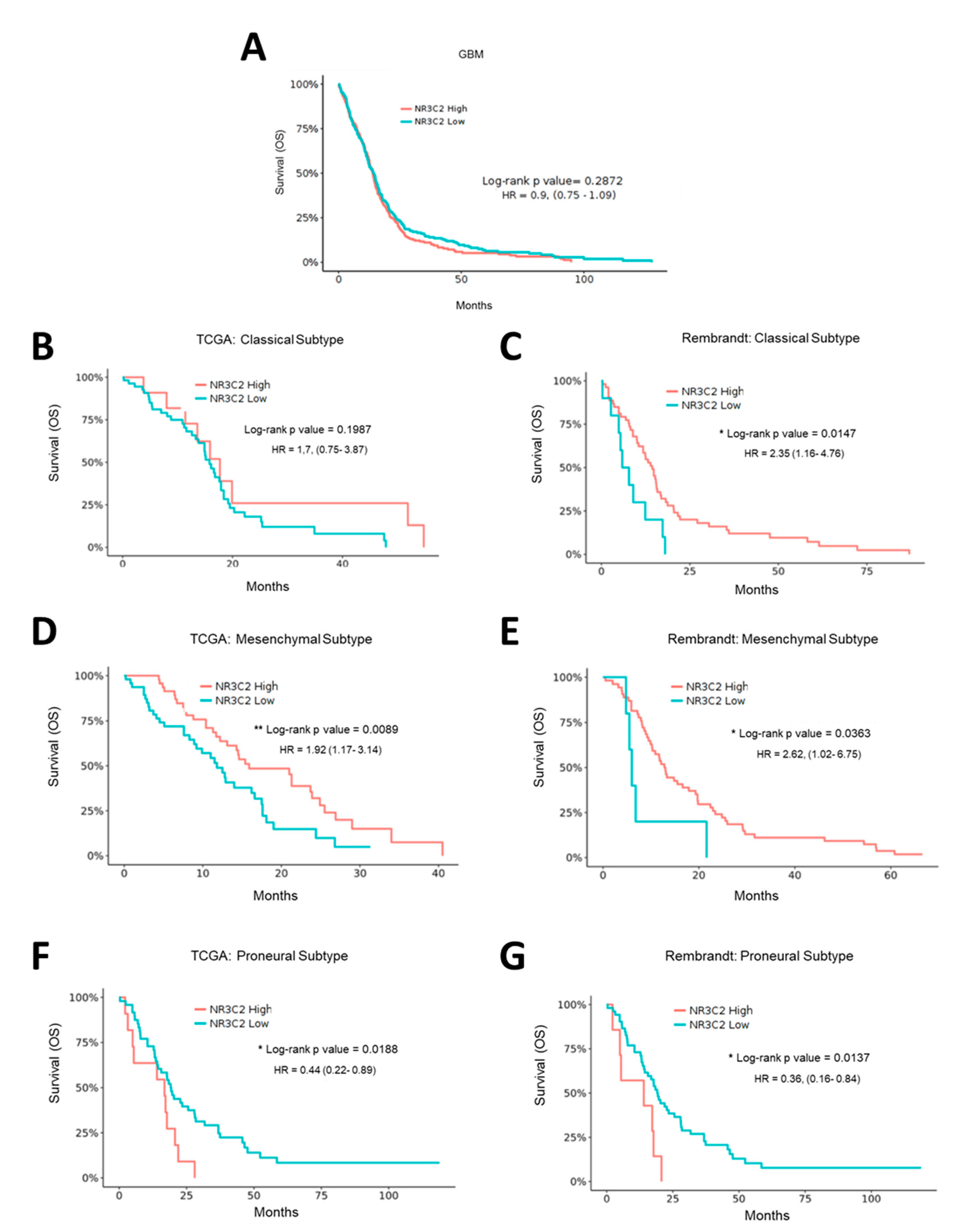
2.2. MR Expression Inversely Correlates with Tumor Progression and Overall Survival
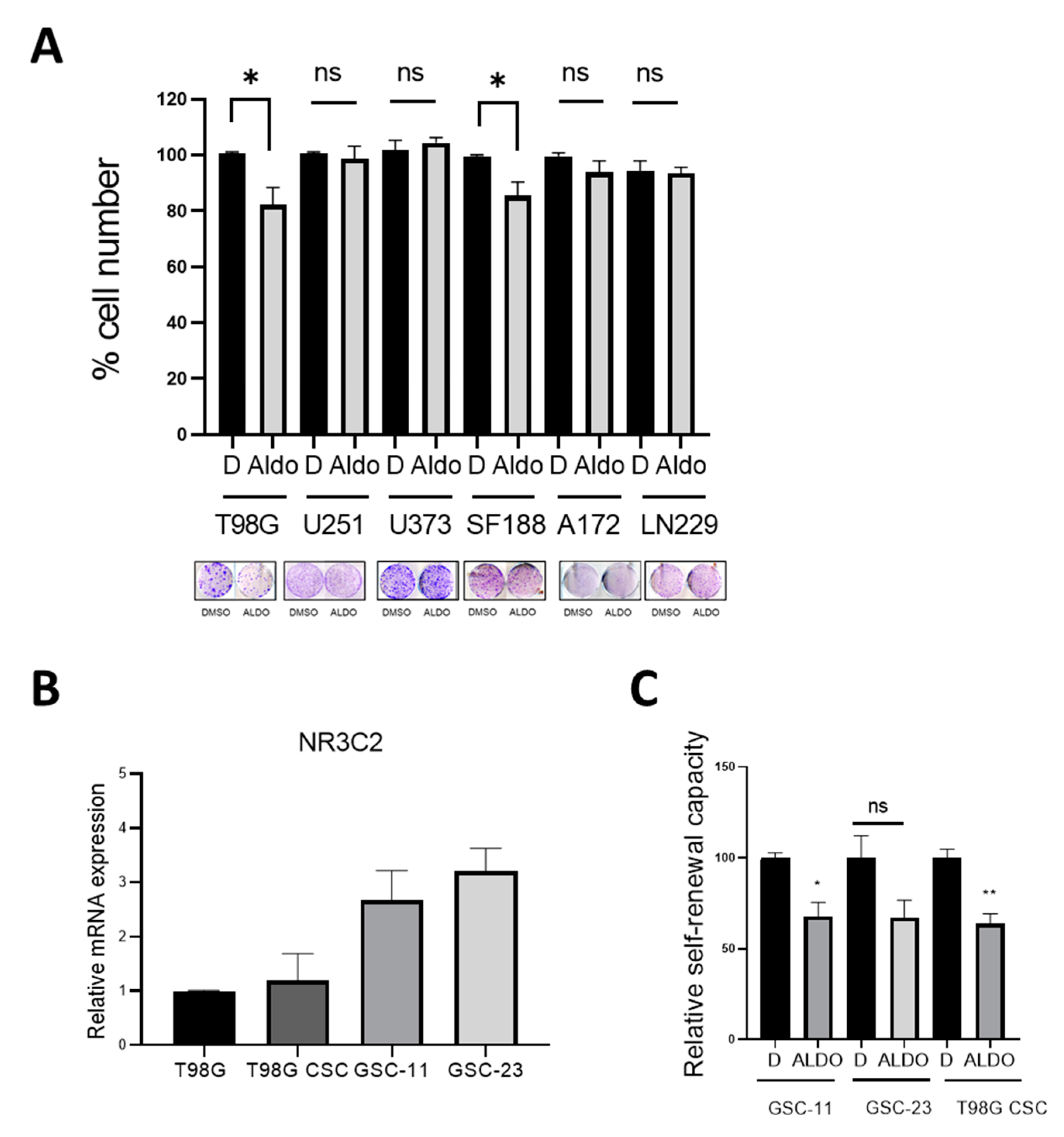
2.3. Effect of Aldosterone on GBM Cells and Glioma Stem Cells
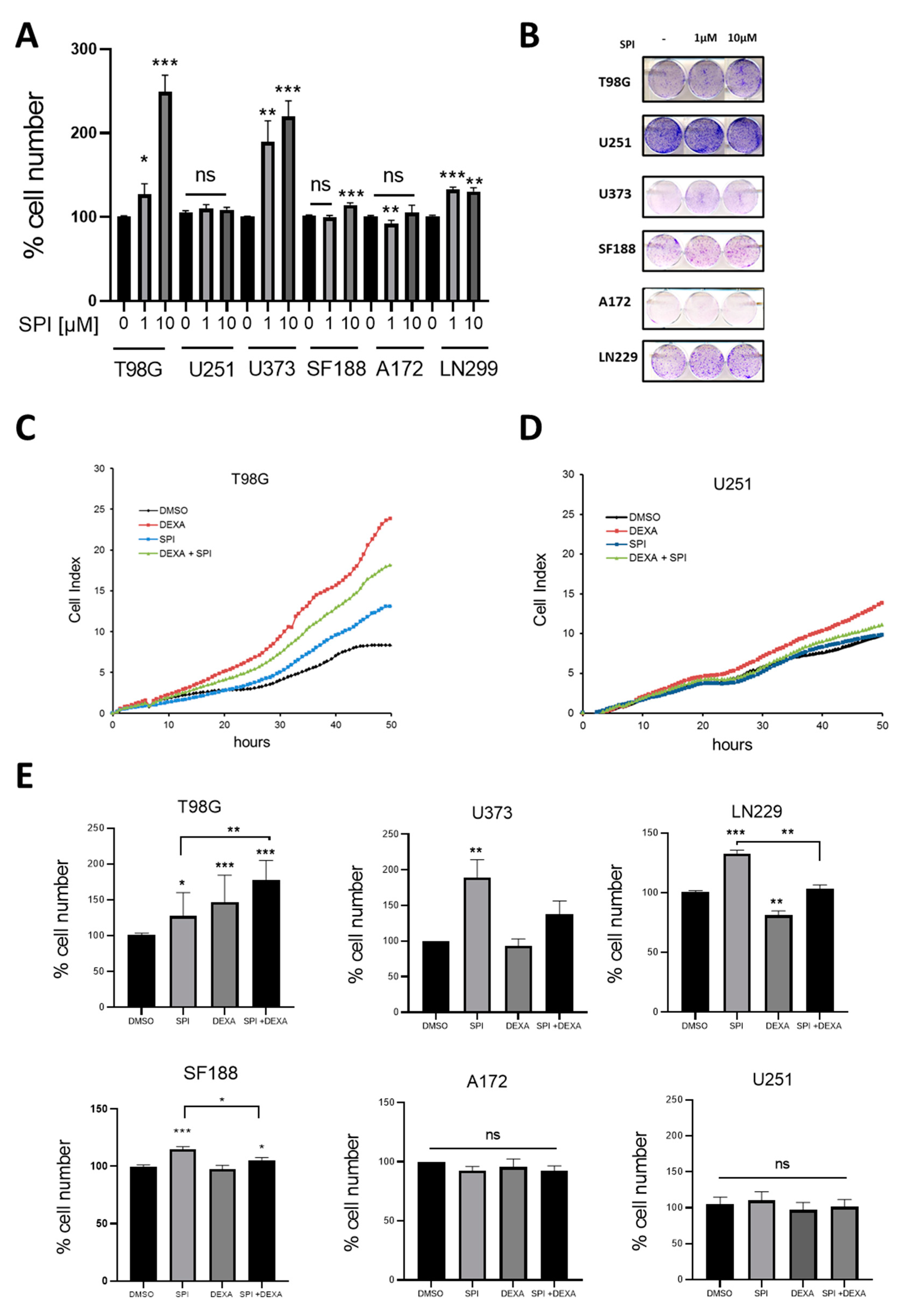
2.4. Effect of the MR Antagonist Spironolactone on GBM Cells
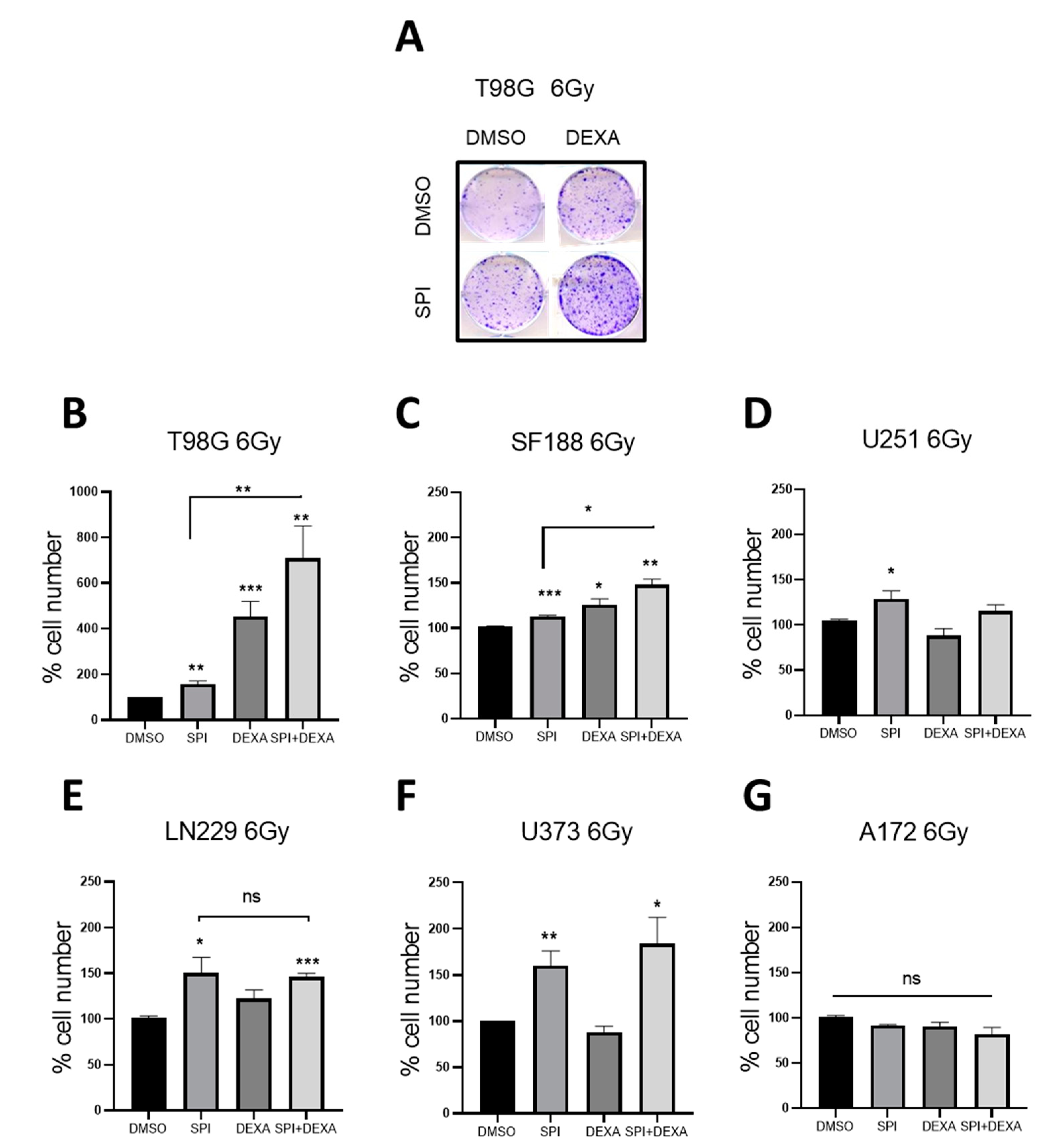
2.5. Spironolactone Protects GBM Cells from Radiation
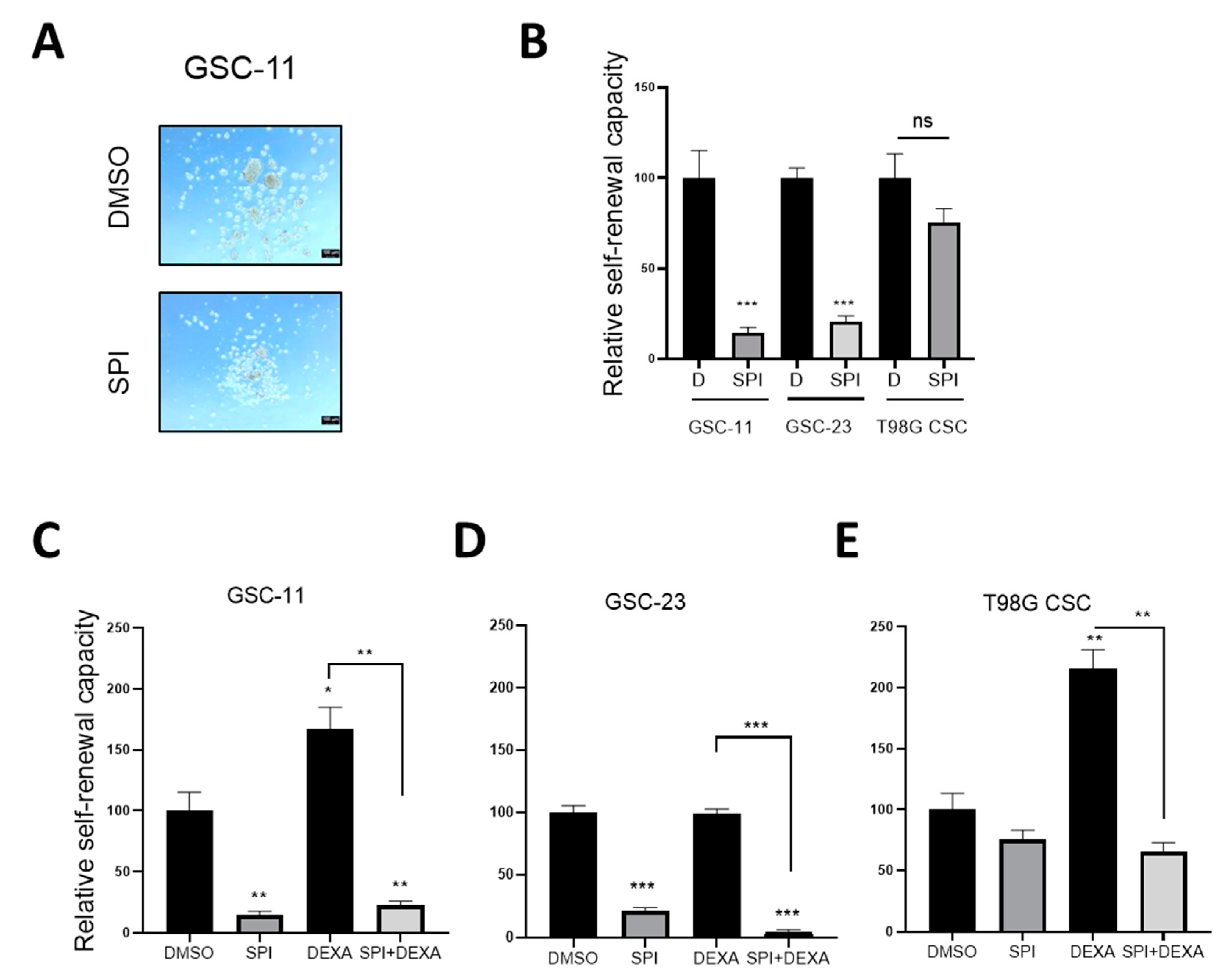
2.6. Spironolactone Impairs GSC Self-Renewal
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Patient Data Analysis
4.3. xCELLigence™ Proliferation Analysis
4.4. Colony Formation Assay
4.5. Neurosphere Formation Assays
4.6. Real-Time Reverse Transcription PCR
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M. The epidemiology of glioma in adults: A state of the science review. Neuro. Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Paul Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta. Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.B.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Dietrich, J.; Rao, K.; Pastorino, S.; Kesari, S. Corticosteroids in brain cancer patients: Benefits and pitfalls. Expert Rev. Clin. Pharmacol. 2011, 4, 233–242. [Google Scholar] [CrossRef]
- Pitter, K.L.; Tamagno, I.; Alikhanyan, K.; Hosni-Ahmed, A.; Pattwell, S.S.; Donnola, S.; Dai, C.; Ozawa, T.; Chang, M.; Chan, T.A. Corticosteroids compromise survival in glioblastoma. Brain 2016, 139, 1458–1471. [Google Scholar] [CrossRef]
- Fan, Z.; Sehm, T.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N.E. Dexamethasone Alleviates Tumor-Associated Brain Damage and Angiogenesis. PLoS ONE 2014, 9, e93264. [Google Scholar] [CrossRef]
- Aldaz, P.; Auzmendi-Iriarte, J.; Durántez, M.; Lasheras-Otero, I.; Carrasco-Garcia, E.; Zelaya, M.; Bragado, L.; Olías-Arjona, A.; Egaña, L.; Samprón, N.; et al. Identification of a Dexamethasone Mediated Radioprotection Mechanism Reveals New Therapeutic Vulnerabilities in Glioblastoma. Cancers 2021, 13, 361. [Google Scholar] [CrossRef]
- Weikum, E.R.; Knuesel, M.; Ortlund, E.; Yamamoto, M.T.K.K.R. Glucocorticoid receptor control of transcription: Precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [CrossRef]
- Ibarrola, J.; Garaikoetxea, M.; Garcia-Peña, A.; Matilla, L.; Jover, E.; Bonnard, B.; Cuesta, M.; Fernández-Celis, A.; Jaisser, F.; López-Andrés, N. Beneficial Effects of Mineralocorticoid Receptor Antagonism on Myocardial Fibrosis in an Experimental Model of the Myxomatous Degeneration of the Mitral Valve. Int. J. Mol. Sci. 2020, 21, 5372. [Google Scholar] [CrossRef]
- Gomez-Sanchez, E.P. Brain mineralocorticoid receptors in cognition and cardiovascular homeostasis. Steroids 2014, 91, 20–31. [Google Scholar] [CrossRef]
- Brilla, C.G. Aldosterone and myocardial fibrosis in heart failure. Herz 2000, 25, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Kober, L.; Ponikowski, P.; Gheorghiade, M.; Filippatos, G.; Krum, H.; Nowack, C.; Kolkhof, P.; Kim, S.-Y.; Zannad, F. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: A randomized, double-blind trial. Eur. Hear. J. 2013, 34, 2453–2463. [Google Scholar] [CrossRef] [PubMed]
- Tiberio, L.; Nascimbeni, R.; Villanacci, V.; Casella, C.; Fra, A.; Vezzoli, V.; Furlan, L.; Meyer, G.; Parrinello, G.; Baroni, M.D.; et al. The Decrease of Mineralcorticoid Receptor Drives Angiogenic Pathways in Colorectal Cancer. PLoS ONE 2013, 8, e59410. [Google Scholar] [CrossRef]
- Jeong, Y.; Xie, Y.; Xiao, G.; Behrens, C.; Girard, L.; Wistuba, I.I.; Minna, J.D.; Mangelsdorf, D. Nuclear Receptor Expression Defines a Set of Prognostic Biomarkers for Lung Cancer. PLoS Med. 2010, 7, e1000378. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Goto, T.; Miyamoto, H. The Role of Mineralocorticoid Receptor Signaling in Genitourinary Cancers. Nucl. Recept. Res. 2019, 6, 101410. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bowman, R.L.; Wang, Q.; Carro, A.; Verhaak, R.G.W.; Squatrito, M. GlioVIs data portal for visualization and analysis of brain tumor expression datasets. Neuro-Oncology 2017, 19, 138–139. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Nandi, S.; Bhattacharjee, S. Combination therapy to checkmate Glioblastoma: Clinical challenges and advances. Clin. Transl. Med. 2018, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.; Mack, S.; Mulkearns-Hubert, E.; Valentim, C.; Rich, J. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Chen, J.; Li, Y.; Yu, T.-S.; McKay, R.M.; Burns, D.K.; Kernie, S.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Osuka, S.; Van Meir, E.G. Overcoming therapeutic resistance in glioblastoma: The way forward. J. Clin. Investig. 2017, 127, 415–426. [Google Scholar] [CrossRef]
- Gold, A.; Eini, L.; Nissim-Rafinia, M.; Viner, R.; Ezer, S.; Erez, K.; Aqaqe, N.; Hanania, R.; Milyavsky, M.; Meshorer, E.; et al. Spironolactone inhibits the growth of cancer stem cells by impairing DNA damage response. Oncogene 2019, 38, 3103–3118. [Google Scholar] [CrossRef] [PubMed]
- Sanomachi, T.; Suzuki, S.; Togashi, K.; Sugai, A.; Seino, S.; Okada, M.; Yoshioka, T.; Kitanaka, C.; Yamamoto, M. Spironolactone, a Classic Potassium-Sparing Diuretic, Reduces Survivin Expression and Chemosensitizes Cancer Cells to Non-DNA-Damaging Anticancer Drugs. Cancers 2019, 11, 1550. [Google Scholar] [CrossRef]
- Shahar, O.D.; Kalousi, A.; Eini, L.; Fisher, B.; Weiss, A.; Darr, J.; Mazina, O.; Bramson, S.; Kupiec, M.; Eden, A.; et al. A high-throughput chemical screen with FDA approved drugs reveals that the antihypertensive drug Spironolactone impairs cancer cell survival by inhibiting homology directed repair. Nucleic Acids Res. 2014, 42, 5689–5701. [Google Scholar] [CrossRef]
- Gabbard, R.; Hooopes, R.; Kemp, M. Spironolactone and XPB: An old drug with a new molecular target. Biomolecules 2020, 10, 756. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Li, J.; Yang, X.; Cao, Q.; Feng, M.; Xue, F.; Wei, L.; Qin, W.; Gu, J.; Xia, Q.; et al. Mineralocorticoid receptor suppresses cancer progression and the Warburg effect by modulating the miR-338-3p-PKLR axis in hepatocellular carcinoma. Hepatology 2015, 62, 1145–1159. [Google Scholar] [CrossRef]
- Shiota, M.; Fujimoto, N.; Kashiwagi, E.; Eto, M. The Role of Nuclear Receptors in Prostate Cancer. Cells 2019, 8, 602. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhang, M.; Duan, X.; Deng, T.; Qiu, H.; Zeng, G. Low NR3C2 levels correlate with aggressive features and poor prognosis in non-distant metastatic clear-cell renal cell carcinoma. J. Cell. Physiol. 2018, 233, 6825–6838. [Google Scholar] [CrossRef]
- Jääskeläinen, A.; Jukkola, A.; Haapasaari, K.-M.; Auvinen, P.; Soini, Y.; Karihtala, P. Cytoplasmic Mineralocorticoid Receptor Expression Predicts Dismal Local Relapse-free Survival in Non-triple-negative Breast Cancer. Anticancer. Res. 2019, 39, 5879–5890. [Google Scholar] [CrossRef]
- Di Fabio, F.; Alvarado, C.; Majdan, A.; Gologan, A.; Voda, L.; Mitmaker, E.; Beitel, L.K.; Gordon, P.H.; Trifiro, M. Underexpression of Mineralocorticoid Receptor in Colorectal Carcinomas and Association with VEGFR-2 Overexpression. J. Gastrointest. Surg. 2007, 11, 1521–1528. [Google Scholar] [CrossRef]
- De La Rocha, A.M.A.; Bertoni, H.L.; Guruceaga, E.; Gonzalez-Huarriz, M.; Martinez-Velez, N.; Xipell, E.; Fueyo, J.; Gomez-Manzano, C.; Alonso, M.M. Analysis of SOX2-Regulated Transcriptome in Glioma Stem Cells. PLoS ONE 2016, 11, e0163155. [Google Scholar] [CrossRef]
- Erice, O.; Smith, M.; White, R.; Goicoechea, I.; Barriuso, J.; Jones, C.; Margison, G.P.; Acosta, J.C.; Wellbrock, C.; Arozarena, I. MGMT Expression Predicts PARP-Mediated Resistance to Temozolomide. Mol. Cancer Ther. 2015, 14, 1236–1246. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldaz, P.; Fernández-Celis, A.; López-Andrés, N.; Arozarena, I. Novel Insights into the Role of the Mineralocorticoid Receptor in Human Glioblastoma. Int. J. Mol. Sci. 2021, 22, 11656. https://doi.org/10.3390/ijms222111656
Aldaz P, Fernández-Celis A, López-Andrés N, Arozarena I. Novel Insights into the Role of the Mineralocorticoid Receptor in Human Glioblastoma. International Journal of Molecular Sciences. 2021; 22(21):11656. https://doi.org/10.3390/ijms222111656
Chicago/Turabian StyleAldaz, Paula, Amaya Fernández-Celis, Natalia López-Andrés, and Imanol Arozarena. 2021. "Novel Insights into the Role of the Mineralocorticoid Receptor in Human Glioblastoma" International Journal of Molecular Sciences 22, no. 21: 11656. https://doi.org/10.3390/ijms222111656
APA StyleAldaz, P., Fernández-Celis, A., López-Andrés, N., & Arozarena, I. (2021). Novel Insights into the Role of the Mineralocorticoid Receptor in Human Glioblastoma. International Journal of Molecular Sciences, 22(21), 11656. https://doi.org/10.3390/ijms222111656