
Current Concepts of Psoriasis Immunopathogenesis


{kind=link}
Abstract
:1. Introduction
2. Genetic Factors in the Development of Psoriasis
3. Environmental Factors in the Development of Psoriasis
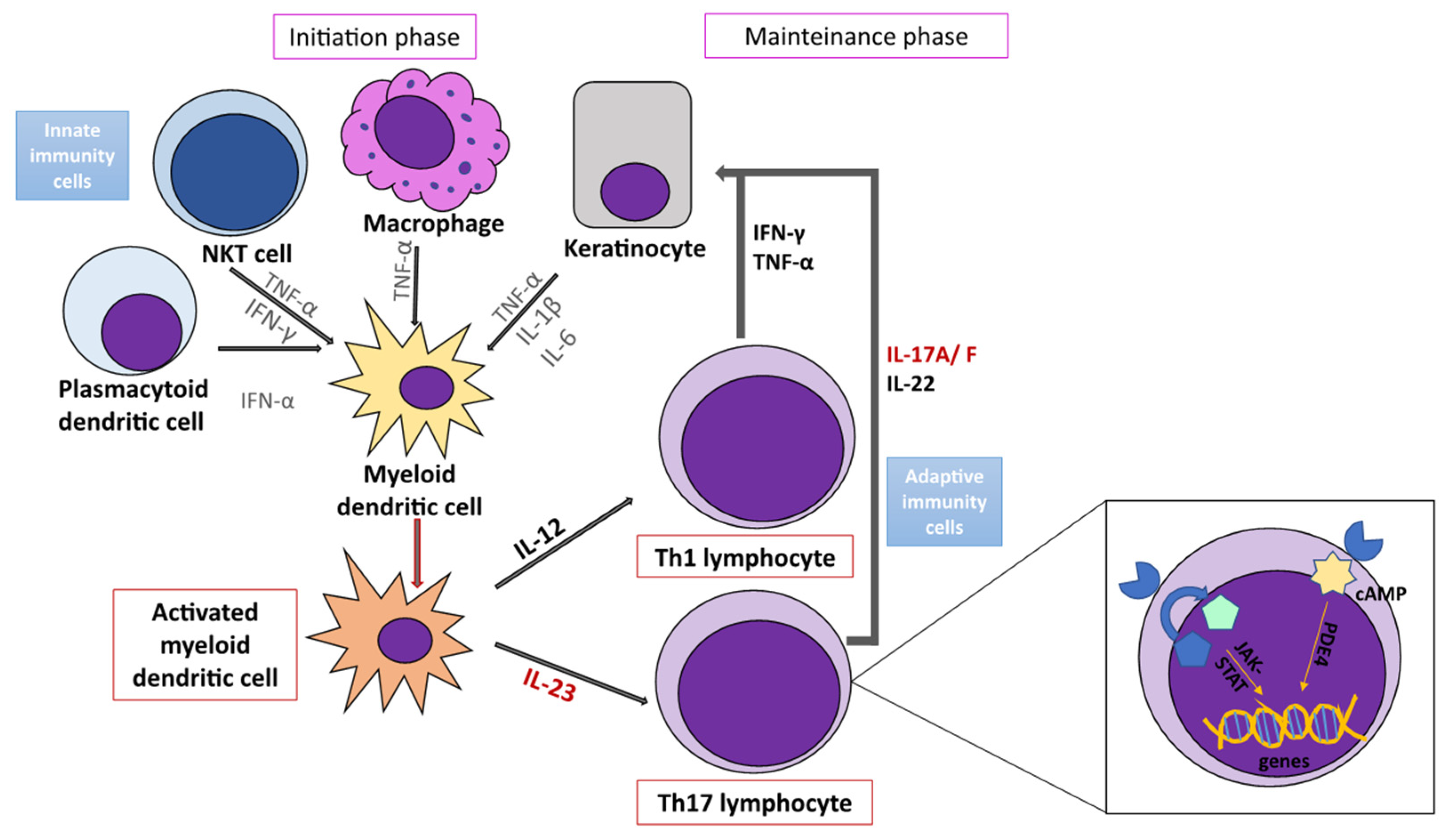
4. Immunopathogenesis of Psoriasis
4.1. Main Cells Involved in Psoriasis Inflammatory Networks
4.1.1. Dendritic Cells
4.1.2. Macrophages
4.1.3. Lymphocytes
4.1.4. NK and NKT Cells
4.1.5. Keratinocytes
4.2. Main Cytokines Involved in Psoriasis Inflammatory Networks
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rendon, A.; Schakel, K. Psoriasis pathogenesis and treatment. Int. J. Mol. Sci. 2019, 20, 1475. [Google Scholar] [CrossRef] [Green Version]
- Pezzolo, E.; Naldi, L. Epidemiology of major chronic inflammatory immune-related skin diseases in 2019. Expert. Rev. Clin. Immunol. 2020, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Ronholt, K.; Iversen, L. Old and new biological therapies for psoriasis. Int. J. Mol. Sci. 2017, 18, 2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalek, I.M.; Loring, B.; John, S.M. A systematic review of worldwide epidemiology of psoriasis. J. Eur. Acad. Dermatol. Veneorol. 2017, 31, 205–212. [Google Scholar] [CrossRef]
- Furue, M.; Kadono, T. “Inflammatory skin march” in atopic dermatitis and psoriasis. Inflamm. Res. 2017, 66, 833–842. [Google Scholar] [CrossRef]
- Machado-Pinto, J.; Diniz Mdos, S.; Bavoso, N.C. Psoriasis: New comorbidities. An. Bras. Dermatol. 2016, 91, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, J.; Grewal, S.; Langan, S.M.; Mehta, N.N.; Ogdie, A.; Van Voorhees, A.S.; Gelfand, J.M. Psoriasis and comorbid diseases: Epidemiology. J. Am. Acad. Dermatol. 2017, 76, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Cottone, M.; Sapienza, C.; Macaluso, F.S.; Cannizzaro, M. Psoriasis and inflammatory bowel disease. Dig. Dis. 2019, 37, 451–457. [Google Scholar] [CrossRef]
- Ferdinando, L.B.; Fukumoto, P.K.; Sanches, S.; Fabricio, L.H.Z.; Skare, T.L. Metabolic syndrome and psoriasis: A study in 97 patients. Rev. Assoc. Med. Bras. 2018, 64, 368–373. [Google Scholar] [CrossRef]
- Ogdie, A.; Grewal, S.K.; Noe, M.H.; Shin, D.B.; Takeshita, J.; Chiesa Fuxench, Z.C.; Carr, R.M.; Gelfand, J.M. Risk of incident liver disease in patients with psoriasis, psoriatic arthritis, and rheumatoid arthritis: A population-based study. J. Investig. Dermatol. 2018, 138, 760–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehncke, W.H. Systemic inflammation and cardiovascular comorbidity in psoriasis patients: Causes and consequences. Front. Immunol. 2018, 9, 579. [Google Scholar] [CrossRef]
- Masson, W.; Lobo, M.; Molinero, G. Psoriasis and Cardiovascular Risk: A Comprehensive Review. Adv. Ther. 2020, 37, 2017–2033. [Google Scholar] [CrossRef] [Green Version]
- Damiani, G.; Radaeli, A.; Olivini, A.; Calvara-Pinton, P.; Malerba, M. Increased airway inflammation in patients with psoriasis. Br. J. Dermatol. 2016, 175, 797–799. [Google Scholar] [CrossRef]
- Malerba, M.; Damiani, G.; Radaeli, A.; Ragnoli, B.; Olivini, A.; Calzavara-Pinton, P.G. Narrowband ultraviolet B phototherapy in psoriasis reduces proinflammatory cytokine levels and improves vitiligo and neutrophilic asthma. Br. J. Dermatol. 2015, 173, 1544–1545. [Google Scholar] [CrossRef]
- Santus, P.; Rizzi, M.; Radovanovic, D.; Airoldi, A.; Cristiano, A.; Conic, R.; Petrou, S.; Pigatto, P.D.M.; Bragazzi, N.; Colombo, D.; et al. Psoriasis and respiratory comorbidities: The added value of fraction of exhaled nitric oxide as a new method to detect, evaluate, and monitor psoriatic systemic involvement and therapeutic efficacy. Biomed. Res. Int. 2018, 2018, 3140682. [Google Scholar] [CrossRef]
- Rousset, L.; Halioua, B. Stress and psoriasis. Int. J. Dermatol. 2018, 57, 1165–1172. [Google Scholar] [CrossRef]
- Springate, D.A.; Parisi, R.; Kontopantelis, E.; Reeves, D.; Griffiths, C.E.; Ashcroft, D.M. Incidence, prevalence and mortality of patients with psoriasis: A U.K. population-based cohort study. Br. J. Dermatol. 2017, 176, 650–658. [Google Scholar] [CrossRef]
- Dhana, A.; Yen, H.; Yen, H.; Cho, E. All-cause and cause-specific mortality in psoriasis: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 2019, 80, 1332–1343. [Google Scholar] [CrossRef]
- Takeshita, J.; Shin, D.B.; Ogdie, A.; Gelfand, J.M. Risk of serious infection, opportunistic infection, and herpes zoster among patients with psoriasis in the United Kingdom. J. Investig. Dermatol. 2018, 138, 1726–1735. [Google Scholar] [CrossRef] [Green Version]
- Capon, F. The genetic basis of psoriasis. Int. J. Mol. Sci. 2017, 18, 2526. [Google Scholar] [CrossRef] [Green Version]
- Nedoszytko, B.; Szczerkowska-Dobosz, A.; Stawczyk-Macieja, M.; Owczarczyk-Saczonek, A.; Reich, A.; Bartosinska, J.; Batycka-Baran, A.; Czajkowski, R.; Dobrucki, I.T.; Dobrucki, L.W.; et al. Pathogenesis of psoriasis in the “omic” era. Part II. Genetic, genomic and epigenetic changes in psoriasis. Postepy. Dermatol. Alergol. 2020, 37, 283–298. [Google Scholar] [CrossRef]
- Dand, N.; Mahil, S.K.; Capon, F.; Smith, C.H.; Simpson, M.A.; Barker, J.N. Psoriasis and Genetics. Acta Derm. Venereol. 2020, 100, adv00030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Tsai, T.F. HLA-Cw6 and psoriasis. Br. J. Dermatol. 2018, 178, 854–862. [Google Scholar] [CrossRef]
- Lee, E.B.; Wu, K.K.; Lee, M.P.; Bhutani, T.; Wu, J.J. Psoriasis risk factors and triggers. Cutis 2018, 102, 18–20. [Google Scholar] [PubMed]
- Kamiya, K.; Kishimoto, M.; Sugai, J.; Komine, M.; Ohtsuki, M. Risk factors for the development of psoriasis. Int. J. Mol. Sci. 2019, 20, 4347. [Google Scholar] [CrossRef] [Green Version]
- Dand, N.; Duckworth, M.; Baudry, D.; Russell, A.; Curtis, C.J.; Lee, S.H.; Evans, I.; Mason, K.J.; Alsharqi, A.; Becher, G.; et al. HLA-C*06:02 genotype is a predictive biomarker of biologic treatment response in psoriasis. J. Allergy Clin. Immunol. 2019, 143, 2120–2130. [Google Scholar] [CrossRef] [Green Version]
- Conrad, C.; Gilliet, M. Psoriasis: From pathogenesis to targeted therapies. Clin. Rev. Allergy Immunol. 2018, 54, 102–113. [Google Scholar] [CrossRef]
- Alexander, H.; Nestle, F.O. Pathogenesis and immunotherapy in cutaneous psoriasis: What can rheumatologists learn? Curr. Opin. Rheumatol. 2017, 29, 71–78. [Google Scholar] [CrossRef]
- De Simone, C.; Caldarola, G.; Moretta, G.; Piscitelli, L.; Ricceri, F.; Prignano, F. Moderate-to-severe psoriasis and pregnancy: Impact on fertility, pregnancy outcome and treatment perspectives. G. Ital. Dermatol. Venereol. 2019, 154, 305–314. [Google Scholar] [CrossRef]
- Passeron, T.; Krutmann, J.; Andersen, M.L.; Katta, R.; Zouboulis, C.C. Clinical and biological impact of the exposome on the skin. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Damiani, G.; Bragazzi, N.L.; McCormick, T.S.; Pigatto, P.D.M.; Leone, S.; Pacifico, A.; Tiodorovic, D.; Di Franco, S.; Alfieri, A.; Fiore, M. Gut microbiota and nutrient interactions with skin in psoriasis: A comprehensive review of animal and human studies. World J. Clin. Cases 2020, 8, 1002–1012. [Google Scholar] [CrossRef]
- Prinz, J.C. Melanocytes: Target cells of an HLA-C*06:02-restricted autoimmune response in psoriasis. J. Investig. Dermatol. 2017, 137, 2053–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgescu, S.R.; Tampa, M.; Caruntu, C.; Sarbu, M.I.; Mitran, C.I.; Mitran, M.I.; Matei, C.; Constantin, C.; Neagu, M. Advances in Understanding the immunological pathways in psoriasis. Int. J. Mol. Sci. 2019, 20, 739. [Google Scholar] [CrossRef] [Green Version]
- Chiricozzi, A.; Romanelli, P.; Volpe, E.; Borsellino, G.; Romanelli, M. Scanning the immunopathogenesis of psoriasis. Int. J. Mol. Sci. 2018, 19, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solimani, F.; Meier, K.; Ghoreschi, K. Emerging topical and systemic JAK inhibitors in dermatology. Front. Immunol. 2019, 10, 2847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alwan, W.; Nestle, F.O. Pathogenesis and treatment of psoriasis: Exploiting pathophysiological pathways for precision medicine. Clin. Exp. Rheumatol. 2015, 33, S2–S6. [Google Scholar]
- Samotij, D.; Nedoszytko, B.; Bartosinska, J.; Batycka-Baran, A.; Czajkowski, R.; Dobrucki, I.T.; Dobrucki, L.W.; Gorecka-Sokolowska, M.; Janaszak-Jasienicka, A.; Krasowska, D.; et al. Pathogenesis of psoriasis in the “omic” era. Part I. Epidemiology, clinical manifestation, immunological and neuroendocrine disturbances. Postepy Dermatol. Alergol. 2020, 37, 135–153. [Google Scholar] [CrossRef]
- Wang, A.; Bai, Y. Dendritic cells: The driver of psoriasis. J. Dermatol. 2020, 47, 104–113. [Google Scholar] [CrossRef]
- Nestle, F.O.; Conrad, C.; Tun-Kyi, A.; Homey, B.; Gombert, M.; Boyman, O.; Burg, G.; Liu, Y.J.; Gilliet, M. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J. Exp. Med. 2005, 202, 135–143. [Google Scholar] [CrossRef]
- Mahil, S.K.; Capon, F.; Barker, J.N. Update on psoriasis immunopathogenesis and targeted immunotherapy. Semin. Immunopathol. 2016, 38, 11–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorthois, I.; Asselineau, D.; Seyler, N.; Pouliot, R. Contribution of in vivo and organotypic 3D models to understanding the role of macrophages and neutrophils in the pathogenesis of psoriasis. Mediat. Inflamm. 2017, 2017, 7215072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Zhu, L.; Tian, H.; Sun, H.X.; Wang, R.; Zhang, L.; Zhao, Y. IL-23-induced macrophage polarization and its pathological roles in mice with imiquimod-induced psoriasis. Protein Cell. 2018, 9, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Edelmayer, R.; Wetter, J.; Salte, K.; Gauvin, D.; Leys, L.; Paulsboe, S.; Su, Z.; Weinberg, I.; Namovic, M.; et al. Monocites/Macrophages play a pathogenic role in IL-23 mediated psoriasis-like skin inflammation. Sci. Rep. 2019, 9, 5310. [Google Scholar] [CrossRef] [Green Version]
- Ayala-Fontanez, N.; Soler, D.C.; McCormick, T.S. Current knowledge on psoriasis and autoimmune diseases. Psoriasis 2016, 6, 7–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, T.; Kabashima, K.; Miyachi, Y. The panoply of αβT cells in the skin. J. Dermatol. Sci. 2014, 76, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Nickoloff, B.J.; Wrone-Smith, T. Injection of pre-psoriatic skin with CD4+ T cells induces psoriasis. Am. J. Pathol. 1999, 155, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Di Meglio, P.; Villanova, F.; Navarini, A.A.; Mylonas, A.; Tosi, I.; Nestle, F.O.; Conrad, C. Targeting CD8(+) T cells prevents psoriasis development. J. Allergy Clin. Immunol. 2016, 138, 274–276.e6. [Google Scholar] [CrossRef] [Green Version]
- Lowes, M.A.; Suarez-Farinas, M.; Krueger, J.G. Immunology of psoriasis. Annu. Rev. Immunol. 2014, 32, 227–255. [Google Scholar] [CrossRef] [Green Version]
- Conrad, C.; Boyman, O.; Tonel, G.; Tun-Kyi, A.; Laggner, U.; de Fougerolles, A.; Kotelianski, V.; Gardner, H.; Nestle, F.O. Alpha1beta1 integrin is crucial for accumulation of epidermal T cells and the development of psoriasis. Nat. Med. 2007, 13, 836–842. [Google Scholar] [CrossRef]
- Karczewski, J.; Dobrowolska, A.; Rychlewska-Hanczewska, A.; Adamski, Z. New insights into the role of T cells in pathogenesis of psoriasis and psoriatic arthritis. Autoimmunity 2016, 49, 435–450. [Google Scholar] [CrossRef]
- Volarić, I.; Vičić, M.; Prpić Massari, L. The role of CD8+ T-cells and their cytokines in the pathogenesis of psoriasis. Acta Dermatovenerol. Croat. 2018, 27, 159–162. [Google Scholar]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lostao, L.; Anel, A.; Pardo, J. How do cytotoxic lymphocytes kill cancer cells? Clin. Cancer. Res. 2015, 21, 5047–5056. [Google Scholar] [CrossRef] [Green Version]
- Prpic Massari, L.; Kastelan, M.; Laskarin, G.; Zamolo, G.; Massari, D.; Rukavina, D. Analysis of perforin expression in peripheral blood and lesions in severe and mild psoriasis. J. Dermatol. Sci. 2007, 47, 29–36. [Google Scholar] [CrossRef]
- Vicic, M.; Kastelan, M.; Sotosek Tokmadzic, V.; Prpic Massari, L. Systemic and local increase of granulysin expression in cytotoxic lymphocytes in severe psoriasis. Acta Derm. Venereol. 2019, 99, 1136–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastelan, M.; Prpic Massari, L.; Gruber, F.; Zamolo, G.; Zauhar, G.; Coklo, M.; Rukavina, D. Perforin expression is upregulated in the epidermis of psoriatic lesions. Br. J. Dermatol. 2004, 151, 831–836. [Google Scholar] [CrossRef]
- Yawalkar, N.; Schmid, S.; Braathen, L.R.; Pichler, W.J. Perforin and granzyme B may contribute to skin inflammation in atopic dermatitis and psoriasis. Br. J. Dermatol. 2001, 144, 1133–1139. [Google Scholar] [CrossRef]
- Vicic, M.; Peternel, S.; Simonic, E.; Sotosek-Tokmadzic, V.; Massari, D.; Brajac, I.; Kastelan, M.; Prpic-Massari, L. Cytotoxic T lymphocytes as a potential brake of keratinocyte proliferation in psoriasis. Med. Hypotheses 2016, 87, 66–68. [Google Scholar] [CrossRef]
- Buhl, T.; Saleh, M.M.; Schon, M.P. More tolerance for dendritic cells in psoriasis. Exp. Dermatol. 2017, 26, 335–337. [Google Scholar] [CrossRef] [Green Version]
- Owczarczyk-Saczonek, A.; Czerwinska, J.; Placek, W. The role of regulatory T cells and anti-inflammatory cytokines in psoriasis. Acta Dermatovenerol. Alp. Pannonica Adriat. 2018, 27, 17–23. [Google Scholar] [CrossRef]
- Schon, M.P. Adaptive and innate immunity in psoriasis and other inflammatory disorders. Front. Immunol. 2019, 10, 1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owczarczyk Saczonek, A.; Krajewska-Wlodarczyk, M.; Kasprowicz-Furmanczyk, M.; Placek, W. Immunological memory of psoriatic lesions. Int. J. Mol. Sci. 2020, 21, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Ogawa, E.; Okuyama, R. Role of innate immune cells in psoriasis. Int. J. Mol. Sci. 2020, 21, 6604. [Google Scholar] [CrossRef] [PubMed]
- Benhadou, F.; Mintoff, D.; Del Marmol, V. Psoriasis: Keratinocytes or immune Cells—Which is the trigger? Dermatology 2019, 235, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Fleming, C.; Yan, J. Dermal γδT cells—A new player in the pathogenesis of psoriasis. Int. Immunopharmacol. 2013, 16, 388–391. [Google Scholar] [CrossRef]
- O’Brien, R.L.; Born, W.K. Dermal γδT cells—What have we learned? Cell. Immunol. 2015, 296, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Laggner, U.; Di Meglio, P.; Perera, G.K.; Hundhausen, C.; Lacy, K.E.; Ali, N.; Smith, C.H.; Hayday, A.C.; Nickoloff, B.J.; Nestle, F.O. Identification of a novel proinflammatory human skin-homing Vγ9Vδ2 T cell subset with a potential role in psoriasis. J. Immunol. 2011, 187, 2783–2793. [Google Scholar] [CrossRef]
- Cai, Y.; Fleming, C.; Yan, J. New insights of T cells in the pathogenesis of psoriasis. Cell. Mol. Immunol. 2012, 9, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Pluzaric, V.; Stefanic, M.; Mihalj, M.; Tolusic Levak, M.; Mursic, I.; Glavas-Obrovac, L.; Petrek, M.; Balogh, P.; Tokic, S. Differential skewing of circulating MR1-Restricted and γδT cells in human psoriasis vulgaris. Front. Immunol. 2020, 11, 572924. [Google Scholar] [CrossRef] [PubMed]
- Polese, B.; Zhang, H.; Thurairajah, B.; King, I.L. Innate lymphocytes in psoriasis. Front. Immunol. 2020, 11, 242. [Google Scholar] [CrossRef] [Green Version]
- Batista, M.D.; Ho, E.L.; Kuebler, P.J.; Milush, J.M.; Lanier, L.L.; Kallas, E.G.; York, V.A.; Chang, D.; Liao, W.; Unemori, P.; et al. Skewed distribution of natural killer cells in psoriasis skin lesions. Exp. Dermatol. 2013, 22, 64–66. [Google Scholar] [CrossRef] [Green Version]
- Ottaviani, C.; Nasorri, F.; Bedini, C.; de Pita, O.; Girolomoni, G.; Cavani, A. CD56brightCD16(-) NK cells accumulate in psoriatic skin in response to CXCL10 and CCL5 and exacerbate skin inflammation. Eur. J. Immunol. 2006, 36, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Mariani, F.; Roncucci, L. Chemerin/chemR23 axis in inflammation onset and resolution. Inflamm. Res. 2015, 64, 85–95. [Google Scholar] [CrossRef]
- Dunphy, S.; Gardiner, C.M. NK cells and psoriasis. J. Biomed. Biotechnol. 2011, 2011, 248317. [Google Scholar] [CrossRef]
- Dunphy, S.E.; Sweeney, C.M.; Kelly, G.; Tobin, A.M.; Kirby, B.; Gardiner, C.M. Natural killer cells from psoriasis vulgaris patients have reduced levels of cytotoxicity associated degranulation and cytokine production. Clin. Immunol. 2017, 177, 43–49. [Google Scholar] [CrossRef] [PubMed]
- PLoSki, R.; Luszczek, W.; Kusnierczyk, P.; Nockowski, P.; Cislo, M.; Krajewski, P.; Malejczyk, J. A role for KIR gene variants other than KIR2DS1 in conferring susceptibility to psoriasis. Hum. Immunol. 2006, 67, 521–526. [Google Scholar] [CrossRef]
- Yip, K.H.; Papadopoulos, M.; Pant, H.; Tumes, D.J. The role of invariant T cells in inflammation of the skin and airways. Semin. Immunopathol. 2019, 41, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Krijgsman, D.; Hokland, M.; Kuppen, P.J.K. The role of natural killer T cells in cancer—A phenotypical and functional approach. Front. Immunol. 2018, 9, 367. [Google Scholar] [CrossRef]
- Bonish, B.; Jullien, D.; Dutronc, Y.; Huang, B.B.; Modlin, R.; Spada, F.M.; Porcelli, S.A.; Nickoloff, B.J. Overexpression of CD1d by keratinocytes in psoriasis and CD1d-dependent IFN-gamma production by NK-T cells. J. Immunol. 2000, 165, 4076–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickoloff, B.J.; Bonish, B.; Huang, B.B.; Porcelli, S.A. Characterization of a T cell line bearing natural killer receptors and capable of creating psoriasis in a SCID mouse model system. J. Dermatol. Sci. 2000, 24, 212–225. [Google Scholar] [CrossRef]
- Curry, L.J.; Qin, J.Z.; Robinson, J.; Nickoloff, B.J. Reactivity of resident immunocytes in normal and prepsoriatic skin using an ex vivo skin-explant model system. Arch. Pathol. Lab. Med. 2003, 127, 289–296. [Google Scholar] [CrossRef]
- Liao, Y.H.; Jee, S.H.; Sheu, B.C.; Huang, Y.L.; Tseng, M.P.; Hsu, S.M.; Tsai, T.F. Increased expression of the natural killer cell inhibitory receptor CD94/NKG2A and CD158b on circulating and lesional T cells in patients with chronic plaque psoriasis. Br. J. Dermatol. 2006, 155, 318–324. [Google Scholar] [CrossRef]
- Furue, M.; Furue, K.; Tsuji, G.; Nakahara, T. Interleukin-17A and keratinocytes in psoriasis. Int. J. Mol. Sci. 2020, 21, 1275. [Google Scholar] [CrossRef] [Green Version]
- Ippagunta, S.K.; Gangwar, R.; Finkelstein, D.; Vogel, P.; Pelletier, S.; Gingras, S.; Redecke, V.; Hacker, H. Keratinocytes contribute intrinsically to psoriasis upon loss of Tnip1 function. Proc. Natl. Acad. Sci. USA 2016, 113, E6162–E6171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Yamasaki, K. Psoriasis and antimicrobial peptides. Int. J. Mol. Sci. 2020, 21, 6791. [Google Scholar] [CrossRef] [PubMed]
- Benhadou, F.; Glitzner, E.; Brisebarre, A.; Swedlund, B.; Song, Y.; Dubois, C.; Rozzi, M.; Paulissen, C.; Del Marmol, V.; Sibilia, M.; et al. Epidermal autonomous VEGFA/Flt1/Nrp1 functions mediate psoriasis-like disease. Sci. Adv. 2020, 6, eaax5849. [Google Scholar] [CrossRef] [Green Version]
- Eberle, F.C.; Bruck, J.; Holstein, J.; Hirahara, K.; Ghoreschi, K. Recent advances in understanding psoriasis. F1000Research 2016, 5, F1000 Faculty Rev-770. [Google Scholar] [CrossRef] [Green Version]
- Lew, W.; Bowcock, A.M.; Krueger, J.G. Psoriasis vulgaris: Cutaneous lymphoid tissue supports T-cell activation and “Type 1” inflammatory gene expression. Trends Immunol. 2004, 25, 295–305. [Google Scholar] [CrossRef]
- Johnson-Huang, L.M.; Suarez-Farinas, M.; Pierson, K.C.; Fuentes-Duculan, J.; Cueto, I.; Lentini, T.; Sullivan-Whalen, M.; Gilleaudeau, P.; Krueger, J.G.; Haider, A.S.; et al. A single intradermal injection of IFN-gamma induces an inflammatory state in both non-lesional psoriatic and healthy skin. J. Investig. Dermatol. 2012, 132, 1177–1187. [Google Scholar] [CrossRef] [Green Version]
- Kryczek, I.; Bruce, A.T.; Gudjonsson, J.E.; Johnston, A.; Aphale, A.; Vatan, L.; Szeliga, W.; Wang, Y.; Liu, Y.; Welling, T.H.; et al. Induction of IL-17+ T cell trafficking and development by IFN-gamma: Mechanism and pathological relevance in psoriasis. J. Immunol. 2008, 181, 4733–4741. [Google Scholar] [CrossRef] [Green Version]
- Harden, J.L.; Johnson-Huang, L.M.; Chamian, M.F.; Lee, E.; Pearce, T.; Leonardi, C.L.; Haider, A.; Lowes, M.A.; Krueger, J.G. Humanized anti-IFN-gamma (HuZAF) in the treatment of psoriasis. J. Allergy Clin. Immunol. 2015, 135, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Downs, A.M.; Dunnill, M.G. Exacerbation of psoriasis by Interferon-alpha therapy for hepatitis C. Clin. Exp. Dermatol. 2000, 25, 351–352. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Papp, K.; Maari, C.; Yao, Y.; Robbie, G.; White, W.I.; Le, C.; White, B. A randomized, double-blind, placebo-controlled, phase I study of MEDI-545, an anti-interferon-alfa monoclonal antibody, in subjects with chronic psoriasis. J. Am. Acad. Dermatol. 2010, 62, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Girolomoni, G.; Strohal, R.; Puig, L.; Bachelez, H.; Barker, J.; Boehncke, W.H.; Prinz, J.C. The role of IL-23 and the IL-23/TH 17 immune axis in the pathogenesis and treatment of psoriasis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 1616–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.R.; Blumenschein, W.; Murphy, E.; Diveu, C.; Wiekowski, M.; Abbondanzo, S.; Lucian, L.; Geissler, R.; Brodie, S.; Kimball, A.B. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J. Exp. Med. 2006, 203, 2577–2587. [Google Scholar] [CrossRef]
- Zheng, Y.; Danilenko, D.M.; Valdez, P.; Kasman, I.; Eastham-Anderson, J.; Wu, J.; Ouyang, W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 2007, 445, 648–651. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.P.; Duffin, K.C.; Helms, C.; Ding, J.; Stuart, P.E.; Goldgar, D.; Gudjonsson, J.E.; Li, Y.; Tejasvi, T.; Feng, B.J.; et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat. Genet. 2009, 41, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Trepicchio, W.L.; Oestreicher, J.L.; Pittman, D.; Wang, F.; Chamian, F.; Dhodapkar, M.; Krueger, J.G. Increased expression of Interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J. Exp. Med. 2004, 199, 125–130. [Google Scholar] [CrossRef]
- Gandhi, M.; Alwawi, E.; Gordon, K.B. Anti-p40 antibodies ustekinumab and briakinumab: Blockade of interleukin-12 and interleukin-23 in the treatment of psoriasis. Semin. Cutan. Med. Surg. 2010, 29, 48–52. [Google Scholar] [CrossRef]
- Lauffer, F.; Eyerich, K.; Boehncke, W.H.; Asadullah, K.; Beissert, S.; Ghoreschi, K.; Schon, M.P. Cytokines of the IL-17 family in psoriasis. J. Dtsch. Dermatol. Ges. 2020, 18, 675–681. [Google Scholar] [CrossRef]
- Blauvelt, A.; Chiricozzi, A. The immunologic role of IL-17 in psoriasis and psoriatic arthritis pathogenesis. Clin. Rev. Allergy Immunol. 2018, 55, 379–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, K.; Kanda, T.; Takaishi, M.; Shiga, T.; Miyoshi, K.; Nakajima, H.; Kamijima, R.; Tarutani, M.; Benson, J.M.; Elloso, M.M.; et al. Distinct roles of IL-23 and IL-17 in the development of psoriasis-like lesions in a mouse model. J. Immunol. 2011, 186, 4481–4489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiricozzi, A.; Nograles, K.E.; Johnson-Huang, L.M.; Fuentes-Duculan, J.; Cardinale, I.; Bonifacio, K.M.; Gulati, N.; Mitsui, H.; Guttman-Yassky, E.; Suarez-Farinas, M.; et al. IL-17 induces an expanded range of downstream genes in reconstituted human epidermis model. PLoS ONE 2014, 9, e90284. [Google Scholar] [CrossRef] [Green Version]
- Arican, O.; Aral, M.; Sasmaz, S.; Ciragil, P. Serum levels of TNF-alpha, IFN-gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediat. Inflamm. 2005, 2005, 273–279. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, C.; Matheson, R.; Zachariae, C.; Cameron, G.; Li, L.; Edson-Heredia, E.; Braun, D.; Banerjee, S. Anti-interleukin-17 monoclonal antibody ixekizumab in chronic plaque psoriasis. N. Engl. J. Med. 2012, 366, 1190–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papp, K.A.; Leonardi, C.; Menter, A.; Ortonne, J.P.; Krueger, J.G.; Kricorian, G.; Aras, G.; Li, J.; Russell, C.B.; Thompson, E.H.Z.; et al. Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N. Engl. J. Med. 2012, 366, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Tohyama, M.; Hanakawa, Y.; Shirakata, Y.; Dai, X.; Yang, L.; Hirakawa, S.; Tokumaru, S.; Okazaki, H.; Sayama, K.; Hashimoto, K. IL-17 and IL-22 mediate IL-20 subfamily cytokine production in cultured keratinocytes via increased IL-22 receptor expression. Eur. J. Immunol. 2009, 39, 2779–2788. [Google Scholar] [CrossRef]
- Wawrzycki, B.; Pietrzak, A.; Grywalska, E.; Krasowska, D.; Chodorowska, G.; Rolinski, J. Interleukin-22 and its correlation with disease activity in plaque psoriasis. Arch. Immunol. Ther. Exp. 2019, 67, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Perera, G.K.; Ainali, C.; Semenova, E.; Hundhausen, C.; Barinaga, G.; Kassen, D.; Williams, A.E.; Mirza, M.M.; Balazs, M.; Wang, X.; et al. Integrative biology approach identifies cytokine targeting strategies for psoriasis. Sci. Transl. Med. 2014, 6, 223. [Google Scholar] [CrossRef]
- Antoniu, S.A. Discontinued drugs 2011 pulmonary, allergy, gastrointestinal and arthritis. Expert. Opin. Investig. Drugs 2012, 21, 1607–1618. [Google Scholar] [CrossRef]
- Ogawa, E.; Sato, Y.; Minagawa, A.; Okuyama, R. Pathogenesis of psoriasis and development of treatment. J. Dermatol. 2018, 45, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furue, K.; Ito, T.; Tsuji, G.; Kadono, T.; Furue, M. Psoriasis and the TNF/IL-23/IL-17 axis. G. Ital. Dermatol. Venereol. 2019, 154, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Damiani, G.; Pacifico, A.; Linder, D.M.; Pigatto, P.D.M.; Conic, R.; Grada, A.; Bragazzi, N.L. Nanodermatology-based solutions for psoriasis: State-of-the art and future prospects. Dermatol. Ther. 2019, 32, e13113. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vičić, M.; Kaštelan, M.; Brajac, I.; Sotošek, V.; Massari, L.P. Current Concepts of Psoriasis Immunopathogenesis. Int. J. Mol. Sci. 2021, 22, 11574. https://doi.org/10.3390/ijms222111574
Vičić M, Kaštelan M, Brajac I, Sotošek V, Massari LP. Current Concepts of Psoriasis Immunopathogenesis. International Journal of Molecular Sciences. 2021; 22(21):11574. https://doi.org/10.3390/ijms222111574
Chicago/Turabian StyleVičić, Marijana, Marija Kaštelan, Ines Brajac, Vlatka Sotošek, and Larisa Prpić Massari. 2021. "Current Concepts of Psoriasis Immunopathogenesis" International Journal of Molecular Sciences 22, no. 21: 11574. https://doi.org/10.3390/ijms222111574
APA StyleVičić, M., Kaštelan, M., Brajac, I., Sotošek, V., & Massari, L. P. (2021). Current Concepts of Psoriasis Immunopathogenesis. International Journal of Molecular Sciences, 22(21), 11574. https://doi.org/10.3390/ijms222111574