
Multi-Omics Analysis of Gut Microbiota in Inflammatory Bowel Diseases: What Benefits for Diagnostic, Prognostic and Therapeutic Tools?


{kind=link}
{kind=link}
Abstract
:1. Introduction
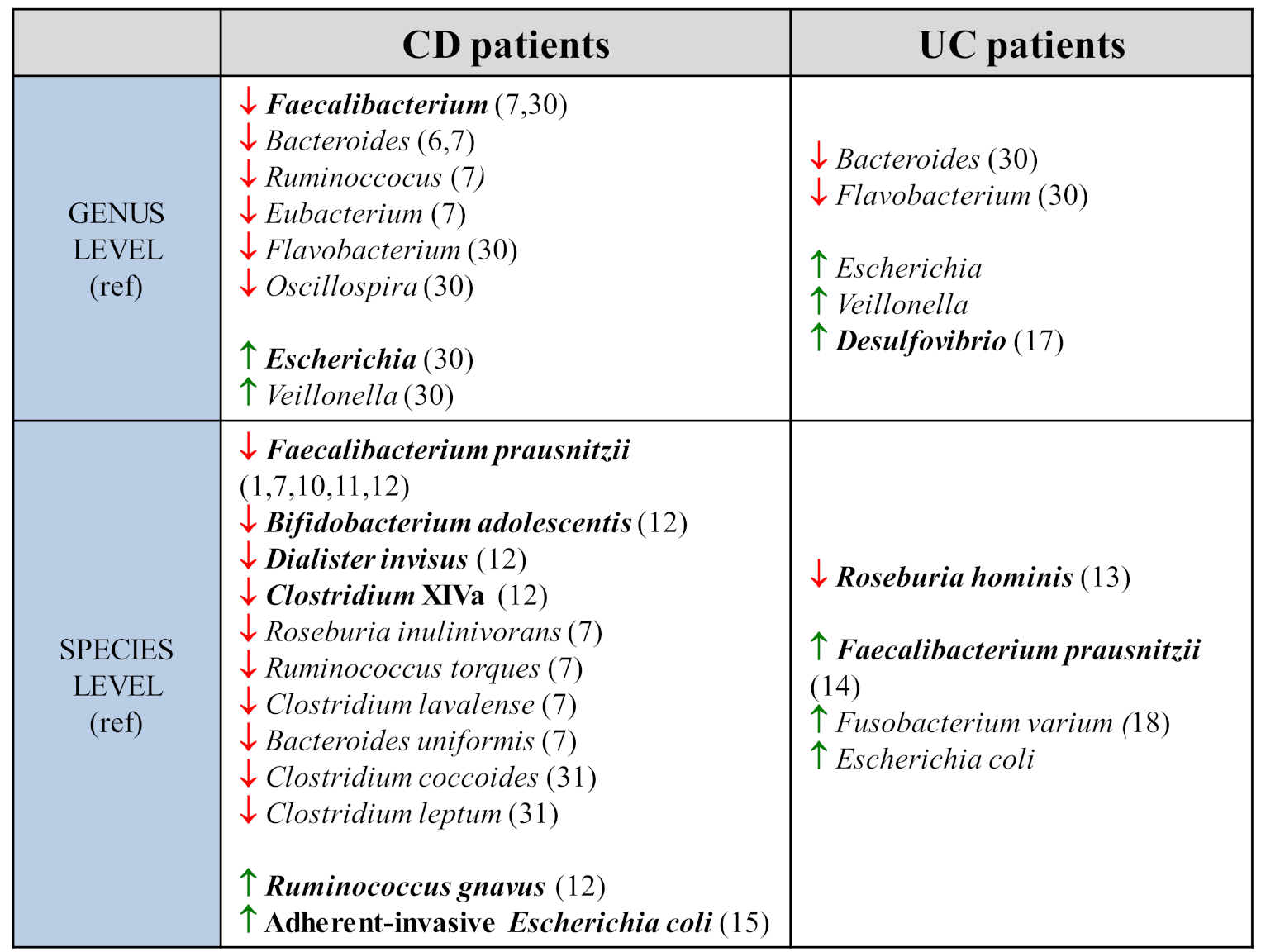
2. Characteristics of Gut Microbiota in IBD
2.1. Metataxonomic and Metagenomic Data
2.2. Limitations of These Techniques in Studying the Dysbiosis Associated with IBD
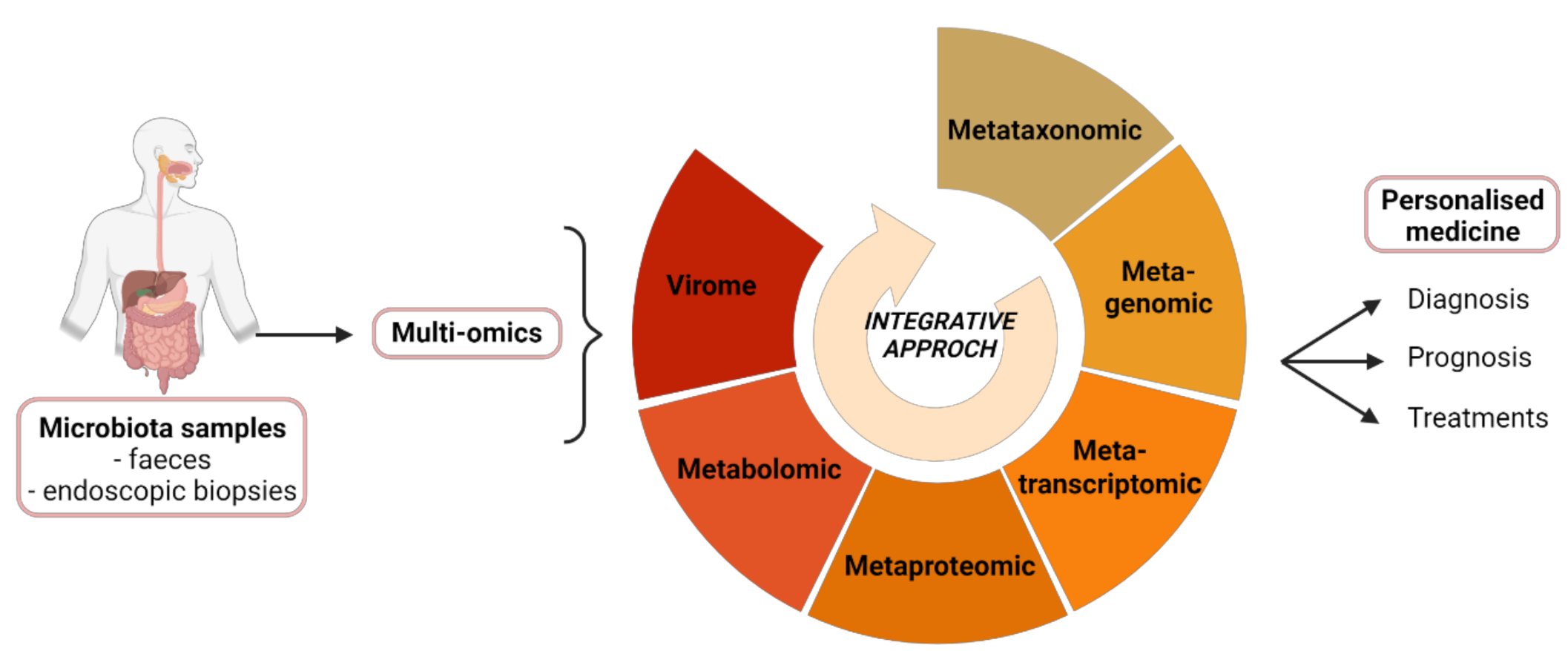
3. Contribution of Multi-Omics Techniques in Studying the Microbiota
3.1. Transcriptomics, Proteomics and Metabolomics
3.2. Limits of Multi-Omics Techniques
3.3. Integrative Mapping of Multi-Omics Data
4. Implications of the Multi-Omics Approach in Studying IBD
4.1. The Microbiota, a New Biomarker for IBD
4.2. Using Omics Techniques to Assess Treatment Response
4.3. Dysbiosis, a Therapeutic Target in the Treatment of IBD
4.4. Perspectives in IBD Management
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-Omics of the Gut Microbial Ecosystem in Inflammatory Bowel Diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Z. Inflammatory Bowel Disease: Pathogenesis. WJG 2014, 20, 91. [Google Scholar] [CrossRef] [PubMed]
- Michielan, A.; D’Incà, R. Intestinal Permeability in Inflammatory Bowel Disease: Pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediators Inflamm. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segal, J.P.; Mullish, B.H.; Quraishi, M.N.; Acharjee, A.; Williams, H.R.T.; Iqbal, T.; Hart, A.L.; Marchesi, J.R. The Application of Omics Techniques to Understand the Role of the Gut Microbiota in Inflammatory Bowel Disease. Therap. Adv. Gastroenterol. 2019, 12, 1756284818822250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, S.J.; Musfeldt, M.; Wenderoth, D.F.; Hampe, J.; Brant, O.; Fölsch, U.R.; Timmis, K.N.; Schreiber, S. Reduction in Diversity of the Colonic Mucosa Associated Bacterial Microflora in Patients with Active Inflammatory Bowel Disease. Gut 2004, 53, 685–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-Phylogenetic Characterization of Microbial Community Imbalances in Human Inflammatory Bowel Diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Nishida, A.; Fujimoto, T.; Fujii, M.; Shioya, M.; Imaeda, H.; Inatomi, O.; Bamba, S.; Sugimoto, M.; Andoh, A. Reduced Abundance of Butyrate-Producing Bacteria Species in the Fecal Microbial Community in Crohn’s Disease. Digestion 2016, 93, 59–65. [Google Scholar] [CrossRef]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vázquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the Human Gut Microbiome in Inflammatory Bowel Disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef] [Green Version]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced Diversity of Faecal Microbiota in Crohn’s Disease Revealed by a Metagenomic Approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low Counts of Faecalibacterium Prausnitzii in Colitis Microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A Decrease of the Butyrate-Producing Species Roseburia Hominis and Faecalibacterium Prausnitzii Defines Dysbiosis in Patients with Ulcerative Colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the Faecal Microbiota in Patients with Crohn’s Disease and Their Unaffected Relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Sankarasubramanian, J.; Ahmad, R.; Avuthu, N.; Singh, A.B.; Guda, C. Gut Microbiota and Metabolic Specificity in Ulcerative Colitis and Crohn’s Disease. Front. Med. (Lausanne) 2020, 7. [Google Scholar] [CrossRef]
- Chassaing, B.; Darfeuille-Michaud, A. The Commensal Microbiota and Enteropathogens in the Pathogenesis of Inflammatory Bowel Diseases. Gastroenterology 2011, 140, 1720–1728. [Google Scholar] [CrossRef]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.-L.; Barnich, N.; Bringer, M.-A.; Swidsinski, A.; Beaugerie, L.; Colombel, J.-F. High Prevalence of Adherent-Invasive Escherichia Coli Associated with Ileal Mucosa in Crohn’s Disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef]
- Rowan, F.; Docherty, N.G.; Murphy, M.; Murphy, B.; Calvin Coffey, J.; O’Connell, P.R. Desulfovibrio Bacterial Species Are Increased in Ulcerative Colitis. Dis. Colon Rectum 2010, 53, 1530–1536. [Google Scholar] [CrossRef]
- Strauss, J.; Kaplan, G.G.; Beck, P.L.; Rioux, K.; Panaccione, R.; Devinney, R.; Lynch, T.; Allen-Vercoe, E. Invasive Potential of Gut Mucosa-Derived Fusobacterium Nucleatum Positively Correlates with IBD Status of the Host. Inflamm. Bowel Dis. 2011, 17, 1971–1978. [Google Scholar] [CrossRef]
- Martinez, C.; Antolin, M.; Santos, J.; Torrejon, A.; Casellas, F.; Borruel, N.; Guarner, F.; Malagelada, J.-R. Unstable Composition of the Fecal Microbiota in Ulcerative Colitis during Clinical Remission. Am. J. Gastroenterol. 2008, 103, 643–648. [Google Scholar] [CrossRef]
- Qian, G.; Ho, J.W.K. Challenges and Emerging Systems Biology Approaches to Discover How the Human Gut Microbiome Impact Host Physiology. Biophys. Rev. 2020, 12, 851–863. [Google Scholar] [CrossRef]
- Jiao, J.-Y.; Liu, L.; Hua, Z.-S.; Fang, B.-Z.; Zhou, E.-M.; Salam, N.; Hedlund, B.P.; Li, W.-J. Microbial Dark Matter Coming to Light: Challenges and Opportunities. Natl. Sci. Rev. 2021, 8, nwaa280. [Google Scholar] [CrossRef]
- Almeida, A.; Mitchell, A.L.; Boland, M.; Forster, S.C.; Gloor, G.B.; Tarkowska, A.; Lawley, T.D.; Finn, R.D. A New Genomic Blueprint of the Human Gut Microbiota. Nature 2019, 568, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Heintz-Buschart, A.; May, P.; Laczny, C.C.; Lebrun, L.A.; Bellora, C.; Krishna, A.; Wampach, L.; Schneider, J.G.; Hogan, A.; de Beaufort, C.; et al. Integrated Multi-Omics of the Human Gut Microbiome in a Case Study of Familial Type 1 Diabetes. Nat. Microbiol. 2016, 2, 1–13. [Google Scholar] [CrossRef]
- Bashiardes, S.; Zilberman-Schapira, G.; Elinav, E. Use of Metatranscriptomics in Microbiome Research. Bioinform. Biol. Insights 2016, 10. [Google Scholar] [CrossRef] [Green Version]
- Schirmer, M.; Franzosa, E.A.; Lloyd-Price, J.; McIver, L.J.; Schwager, R.; Poon, T.W.; Ananthakrishnan, A.N.; Andrews, E.; Barron, G.; Lake, K.; et al. Dynamics of Metatranscription in the Inflammatory Bowel Disease Gut Microbiome. Nat. Microbiol. 2018, 3, 337–346. [Google Scholar] [CrossRef]
- Lehmann, T.; Schallert, K.; Vilchez-Vargas, R.; Benndorf, D.; Püttker, S.; Sydor, S.; Schulz, C.; Bechmann, L.; Canbay, A.; Heidrich, B.; et al. Metaproteomics of Fecal Samples of Crohn’s Disease and Ulcerative Colitis. J. Proteomics 2019, 201, 93–103. [Google Scholar] [CrossRef]
- Erickson, A.R.; Cantarel, B.L.; Lamendella, R.; Darzi, Y.; Mongodin, E.F.; Pan, C.; Shah, M.; Halfvarson, J.; Tysk, C.; Henrissat, B.; et al. Integrated Metagenomics/Metaproteomics Reveals Human Host-Microbiota Signatures of Crohn’s Disease. PLoS ONE 2012, 7, e49138. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Kumita, W.; Ode, T.; Ichinose, S.; Ando, A.; Fujiyama, Y.; Chida, T.; Okamura, N. OmpA Variants Affecting the Adherence of Ulcerative Colitis-Derived Bacteroides Vulgatus. J. Med. Dent. Sci. 2010, 57, 55–64. [Google Scholar]
- Santoru, M.L. Cross Sectional Evaluation of the Gut-Microbiome Metabolome Axis in an Italian Cohort of IBD Patients. Sci. Rep. 2017, 7, 9532. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Holmes, E.; Khan, F.; Kochhar, S.; Scanlan, P.; Shanahan, F.; Wilson, I.D.; Wang, Y. Rapid and Noninvasive Metabonomic Characterization of Inflammatory Bowel Disease. J. Proteome Res. 2007, 6, 546–551. [Google Scholar] [CrossRef]
- Yu, Y.; Yang, W.; Li, Y.; Cong, Y. Enteroendocrine Cells: Sensing Gut Microbiota and Regulating Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2020, 26, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Rajca, S.; Grondin, V.; Louis, E.; Vernier-Massouille, G.; Grimaud, J.-C.; Bouhnik, Y.; Laharie, D.; Dupas, J.-L.; Pillant, H.; Picon, L.; et al. Alterations in the Intestinal Microbiome (Dysbiosis) as a Predictor of Relapse after Infliximab Withdrawal in Crohn’s Disease. Inflamm. Bowel Dis. 2014, 20, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium Prausnitzii Is an Anti-Inflammatory Commensal Bacterium Identified by Gut Microbiota Analysis of Crohn Disease Patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, H.; Brot, L.; Stefanescu, C.; Auzolle, C.; Barnich, N.; Buisson, A.; Fumery, M.; Pariente, B.; Le Bourhis, L.; Treton, X.; et al. Prominence of Ileal Mucosa-Associated Microbiota to Predict Postoperative Endoscopic Recurrence in Crohn’s Disease. Gut 2020, 69, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Kugathasan, S.; Denson, L.A.; Walters, T.D.; Kim, M.-O.; Marigorta, U.M.; Schirmer, M.; Mondal, K.; Liu, C.; Griffiths, A.; Noe, J.D.; et al. Prediction of Complicated Disease Course for Children Newly Diagnosed with Crohn’s Disease: A Multicentre Inception Cohort Study. Lancet 2017, 389, 1710–1718. [Google Scholar] [CrossRef] [Green Version]
- Boumessid, K.; Barreau, F.; Mas, E. How Can a Polymeric Formula Induce Remission in Crohn’s Disease Patients? Int. J. Mol. Sci. 2021, 22, 4025. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N. Microbiome-Based Biomarkers for IBD. Inflamm. Bowel Dis. 2020, 26, 1463–1469. [Google Scholar] [CrossRef]
- Sanchis-Artero, L.; Martínez-Blanch, J.F.; Manresa-Vera, S.; Cortés-Castell, E.; Rodriguez-Morales, J.; Cortés-Rizo, X. Evaluation of Changes in Gut Microbiota in Patients with Crohn’s Disease after Anti-Tnfα Treatment: Prospective Multicenter Observational Study. IJERPH 2020, 17, 5120. [Google Scholar] [CrossRef]
- Fang, H.; Fu, L.; Wang, J. Protocol for Fecal Microbiota Transplantation in Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. Biomed. Res. Int. 2018, 2018, 8941340. [Google Scholar] [CrossRef] [Green Version]
- Paramsothy, S.; Paramsothy, R.; Rubin, D.T.; Kamm, M.A.; Kaakoush, N.O.; Mitchell, H.M.; Castaño-Rodríguez, N. Faecal Microbiota Transplantation for Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. J. Crohns Colitis 2017, 11, 1180–1199. [Google Scholar] [CrossRef] [Green Version]
- Distrutti, E.; Monaldi, L.; Ricci, P.; Fiorucci, S. Gut Microbiota Role in Irritable Bowel Syndrome: New Therapeutic Strategies. WJG 2016, 22, 2219–2241. [Google Scholar] [CrossRef]
- Dietrich, G.; Barreau, F. Le microbiote: Un acteur décisif dans le syndrome de l’intestin irritable? Méd. Thér./Pédiatrie 2018, 11, 203–212. [Google Scholar]
- Al Nabhani, Z.; Lepage, P.; Mauny, P.; Montcuquet, N.; Roy, M.; Le Roux, K.; Dussaillant, M.; Berrebi, D.; Hugot, J.-P.; Barreau, F. Nod2 Deficiency Leads to a Specific and Transmissible Mucosa-Associated Microbial Dysbiosis Which Is Independent of the Mucosal Barrier Defect. J. Crohns Colitis 2016, 10, 1428–1436. [Google Scholar] [CrossRef] [Green Version]
- Barreau, F.; Madre, C.; Meinzer, U.; Berrebi, D.; Dussaillant, M.; Merlin, F.; Eckmann, L.; Karin, M.; Sterkers, G.; Bonacorsi, S.; et al. Nod2 Regulates the Host Response towards Microflora by Modulating T Cell Function and Epithelial Permeability in Mouse Peyer’s Patches. Gut 2010, 59, 207–217. [Google Scholar] [CrossRef]
- Alnabhani, Z.; Hugot, J.-P.; Montcuquet, N.; Le Roux, K.; Dussaillant, M.; Roy, M.; Leclerc, M.; Cerf-Bensussan, N.; Lepage, P.; Barreau, F. Respective Roles of Hematopoietic and Nonhematopoietic Nod2 on the Gut Microbiota and Mucosal Homeostasis. Inflamm. Bowel Dis. 2016, 22, 763–773. [Google Scholar] [CrossRef]
- Couturier-Maillard, A.; Secher, T.; Rehman, A.; Normand, S.; De Arcangelis, A.; Haesler, R.; Huot, L.; Grandjean, T.; Bressenot, A.; Delanoye-Crespin, A.; et al. NOD2-Mediated Dysbiosis Predisposes Mice to Transmissible Colitis and Colorectal Cancer. J. Clin. Investig. 2013, 123, 700–711. [Google Scholar] [CrossRef] [Green Version]
- Barra, M.; Danino, T.; Garrido, D. Engineered Probiotics for Detection and Treatment of Inflammatory Intestinal Diseases. Front. Bioeng. Biotechnol. 2020, 8, 265. [Google Scholar] [CrossRef]
- Mishima, Y.; Sartor, R.B. Manipulating Resident Microbiota to Enhance Regulatory Immune Function to Treat Inflammatory Bowel Diseases. J. Gastroenterol. 2020, 55, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.; Choe, D.; Kim, K.; Cho, B.-K.; Cho, S. Synthetic Biology Approaches in the Development of Engineered Therapeutic Microbes. Int. J. Mol. Sci. 2020, 21, 8744. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lacroix, V.; Cassard, A.; Mas, E.; Barreau, F. Multi-Omics Analysis of Gut Microbiota in Inflammatory Bowel Diseases: What Benefits for Diagnostic, Prognostic and Therapeutic Tools? Int. J. Mol. Sci. 2021, 22, 11255. https://doi.org/10.3390/ijms222011255
Lacroix V, Cassard A, Mas E, Barreau F. Multi-Omics Analysis of Gut Microbiota in Inflammatory Bowel Diseases: What Benefits for Diagnostic, Prognostic and Therapeutic Tools? International Journal of Molecular Sciences. 2021; 22(20):11255. https://doi.org/10.3390/ijms222011255
Chicago/Turabian StyleLacroix, Vickie, Alexis Cassard, Emmanuel Mas, and Frederick Barreau. 2021. "Multi-Omics Analysis of Gut Microbiota in Inflammatory Bowel Diseases: What Benefits for Diagnostic, Prognostic and Therapeutic Tools?" International Journal of Molecular Sciences 22, no. 20: 11255. https://doi.org/10.3390/ijms222011255
APA StyleLacroix, V., Cassard, A., Mas, E., & Barreau, F. (2021). Multi-Omics Analysis of Gut Microbiota in Inflammatory Bowel Diseases: What Benefits for Diagnostic, Prognostic and Therapeutic Tools? International Journal of Molecular Sciences, 22(20), 11255. https://doi.org/10.3390/ijms222011255