
Placental Ischemia Says “NO” to Proper NOS-Mediated Control of Vascular Tone and Blood Pressure in Preeclampsia


Abstract
1. Introduction
2. NOS Isoforms and Biochemistry
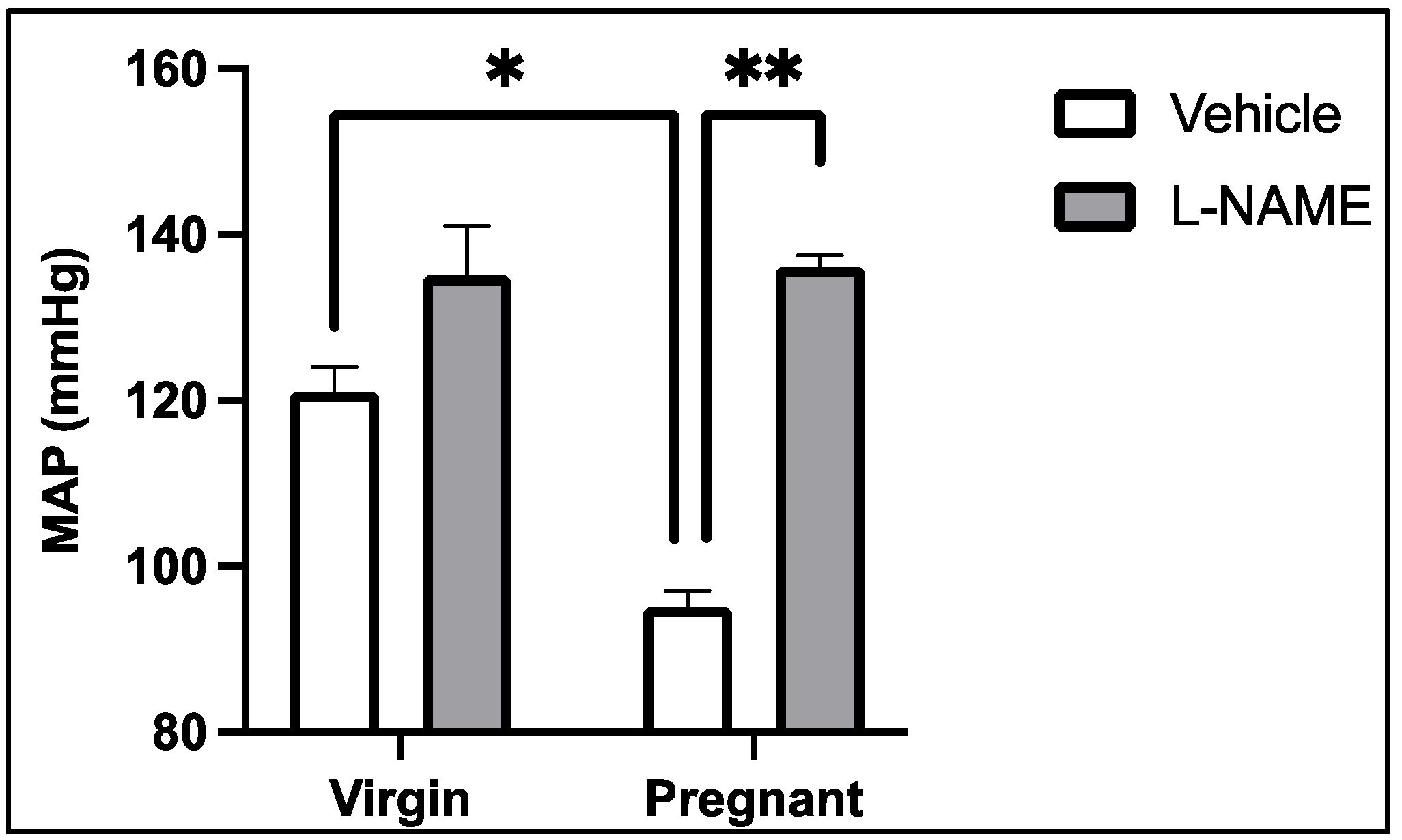
3. NOS-Mediated Control of Renal and Systemic Vascular Function and Blood Pressure Regulation in Normal Pregnancy
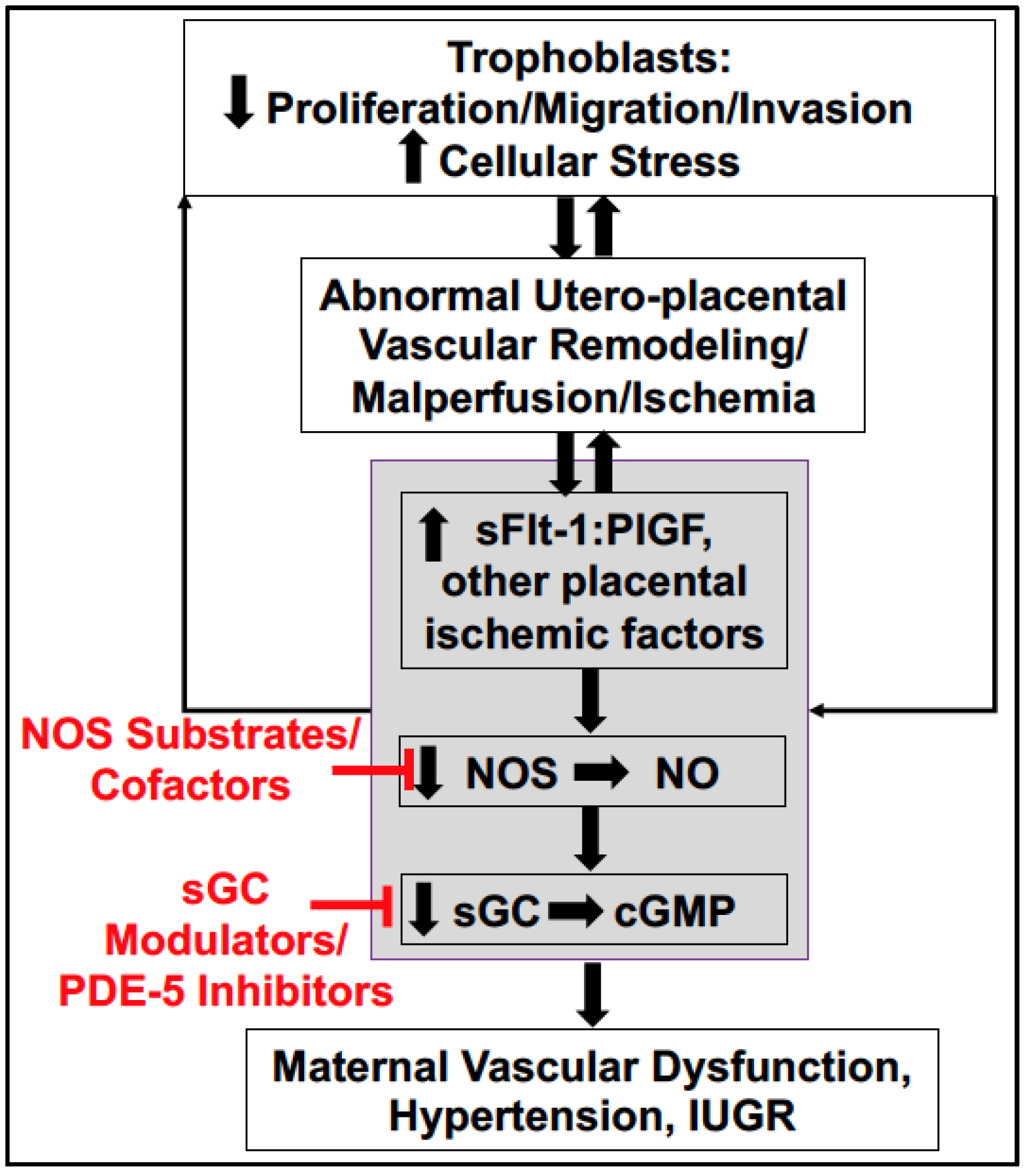
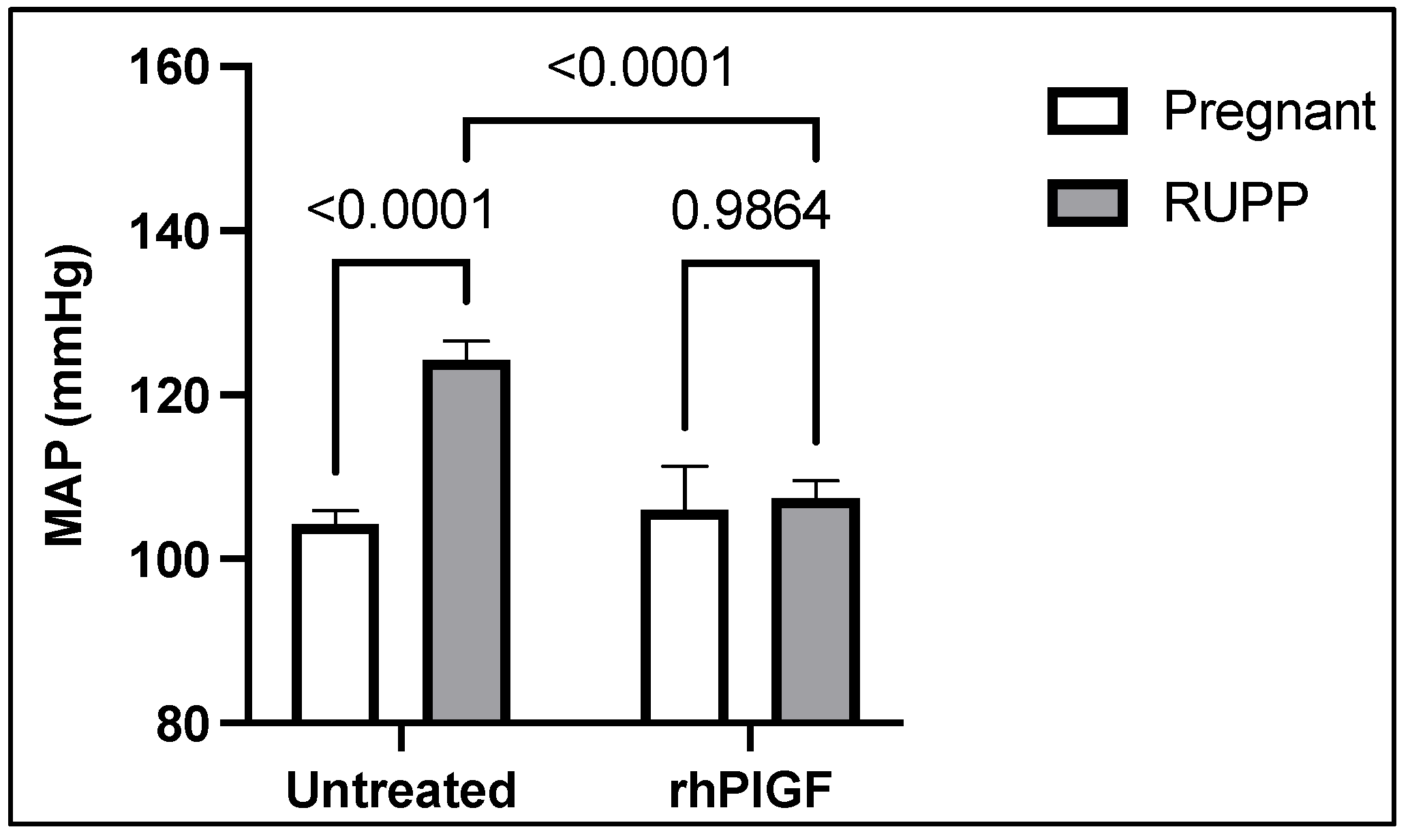
4. Soluble Placental Ischemic Factors and Reduced NO in PE
5. Pro-Inflammatory States as Risk Factors for Placental Ischemic Disease and PE
6. Potential Treatment Strategies Targeting to Increase NO Bioavailability in PE
6.1. L-Arginine Supplementation
6.2. BH4 Supplementation
6.3. L-Citrulline Supplementation
6.4. Downstream Targets: sGC Stimulators, sGC Activators, and PDE-5 Inhibitors
7. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- The American College of Obstetricians and Gynecologists. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists’ Task Force on Hypertension in Pregnancy. Obstet. Gynecol. 2013, 122, 1122–1131. [Google Scholar]
- Ananth, C.V.; Keyes, K.M.; Wapner, R. Pre-eclampsia rates in the United States, 1980–2010: Age-period-cohort analysis. BMJ 2013, 347, f6564. [Google Scholar] [CrossRef] [PubMed]
- Honigberg, M.C.; Riise, H.K.R.; Daltveit, A.K.; Tell, G.S.; Sulo, G.; Igland, J.; Klungsoyr, K.; Scott, N.S.; Wood, M.J.; Natarajan, P.; et al. Heart Failure in Women with Hypertensive Disorders of Pregnancy: Insights from the Cardiovascular Disease in Norway Project. Hypertension 2020, 76, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Andraweera, P.H.; Lassi, Z.S. Cardiovascular Risk Factors in Offspring of Preeclamptic Pregnancies—Systematic Review and Meta-Analysis. J. Pediatr. 2019, 208, 104–113.e6. [Google Scholar] [CrossRef] [PubMed]
- Bakrania, B.A.; Spradley, F.T.; Drummond, H.A.; LaMarca, B.; Ryan, M.J.; Granger, J.P. Preeclampsia: Linking Placental Ischemia with Maternal Endothelial and Vascular Dysfunction. Compr. Physiol. 2020, 11, 1315–1349. [Google Scholar] [CrossRef]
- Redman, C.W.; Staff, A.C.; Roberts, J.M. Syncytiotrophoblast stress in preeclampsia: The convergence point for multiple pathways. Am. J. Obstet. Gynecol. 2021. [Google Scholar] [CrossRef]
- Pereira, M.M.; Torrado, J.; Sosa, C.; Zocalo, Y.; Bia, D. Role of arterial impairment in preeclampsia: Should the paradigm shift? Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2011–H2030. [Google Scholar] [CrossRef]
- Espinoza, J.; Kusanovic, J.P.; Bahado-Singh, R.; Gervasi, M.T.; Romero, R.; Lee, W.; Vaisbuch, E.; Mazaki-Tovi, S.; Mittal, P.; Gotsch, F.; et al. Should Bilateral Uterine Artery Notching Be Used in the Risk Assessment for Preeclampsia, Small-for-Gestational-Age, and Gestational Hypertension? J. Ultrasound Med. 2010, 29, 1103–1115. [Google Scholar] [CrossRef]
- Sharma, S.; Singh, S.; Gujral, U.; Oberoi, U.; Kaur, R. Uterine Artery Notching on Color Doppler Ultrasound and Roll over Test in Prediction of Pregnancy Induced Hypertension. J. Obstet. Gynecol. India 2011, 61, 649–651. [Google Scholar] [CrossRef]
- Lisonkova, S.; Bone, J.N.; Muraca, G.M.; Razaz, N.; Wang, L.Q.; Sabr, Y.; Boutin, A.; Mayer, C.; Joseph, K. Incidence and risk factors for severe preeclampsia, hemolysis, elevated liver enzymes, and low platelet count syndrome, and eclampsia at preterm and term gestation: A population-based study. Am. J. Obstet. Gynecol. 2021. [Google Scholar] [CrossRef]
- Darkwa, E.O.; Djagbletey, R.; Sottie, D.; Owoo, C.; Vanderpuye, N.M.; Essuman, R.; Aryee, G. Serum nitric oxide levels in healthy pregnant women: A case- control study in a tertiary facility in Ghana. Matern. Health Neonatol. Perinatol. 2018, 4, 3. [Google Scholar] [CrossRef]
- Nascimento, R.A.; Possomato-Vieira, J.S.; Bonacio, G.F.; Rizzi, E.; Dias-Junior, C.A. Reductions of Circulating Nitric Oxide are Followed by Hypertension during Pregnancy and Increased Activity of Matrix Metalloproteinases-2 and -9 in Rats. Cells 2019, 8, 1402. [Google Scholar] [CrossRef] [PubMed]
- Cadnapaphornchai, M.A.; Ohara, M.; Morris, K.G.; Knotek, M.; Rogachev, B.; Ladtkow, T.; Carter, E.P.; Schrier, R.W. Chronic NOS inhibition reverses systemic vasodilation and glomerular hyperfiltration in pregnancy. Am. J. Physiol. Physiol. 2001, 280, F592–F598. [Google Scholar] [CrossRef] [PubMed]
- Deng, A.; Engels, K.; Baylis, C. Impact of nitric oxide deficiency on blood pressure and glomerular hemodynamic adaptations to pregnancy in the rat. Kidney Int. 1996, 50, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Bambrana, V.; Dayanand, C.D.; Kotur, P. Relationship between Xanthine Oxidase, Ischemia Modified Albumin, Nitric Oxide with Antioxidants in Non Pregnants, Pre and Post-delivery of Normal Pregnants and Preeclampsia. Indian J. Clin. Biochem. 2016, 32, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.T.; Miller, M.T.; Kassab, S.; Novak, J.; Reckelhoff, J.F.; Kruckeberg, W.C.; Granger, J.P. Differential expression of renal nitric oxide synthase isoforms during pregnancy in rats. Hypertension 1999, 33, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Palei, A.C.; Martin, H.L.; Wilson, B.A.; Anderson, C.D.; Granger, J.P.; Spradley, F.T. Impact of hyperleptinemia during placental ischemia-induced hypertension in pregnant rats. Am. J. Physiol. Circ. Physiol. 2021, 320, H1949–H1958. [Google Scholar] [CrossRef] [PubMed]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357, 593–615. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W. Nitric oxide synthases: Regulation and function. Eur. Hearth J. 2011, 33, 829–837. [Google Scholar] [CrossRef]
- Oliveira-Paula, G.H.; Lacchini, R.; Tanus-Santos, J.E. Endothelial nitric oxide synthase: From biochemistry and gene structure to clinical implications of NOS3 polymorphisms. Gene 2015, 575, 584–599. [Google Scholar] [CrossRef]
- Luizon, M.R.; Palei, A.C.; Cavalli, R.C.; Sandrim, V.C. Pharmacogenetics in the treatment of pre-eclampsia: Current findings, challenges and perspectives. Pharmacogenomics 2017, 18, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Guerby, P.; Tasta, O.; Swiader, A.; Pont, F.; Bujold, E.; Parant, O.; Vayssiere, C.; Salvayre, R.; Negre-Salvayre, A. Role of oxidative stress in the dysfunction of the placental endothelial nitric oxide synthase in preeclampsia. Redox Biol. 2021, 40, 101861. [Google Scholar] [CrossRef] [PubMed]
- Bobin, P.; Belacel-Ouari, M.; Bedioune, I.; Zhang, L.; Leroy, J.; Leblais, V.; Fischmeister, R.; Vandecasteele, G. Cyclic nucleotide phosphodiesterases in heart and vessels: A therapeutic perspective. Arch. Cardiovasc. Dis. 2016, 109, 431–443. [Google Scholar] [CrossRef]
- Vogtmann, R.; Heupel, J.; Herse, F.; Matin, M.; Hagmann, H.; Bendix, I.; Kräker, K.; Dechend, R.; Winterhager, E.; Kimmig, R.; et al. Circulating Maternal sFLT1 (Soluble fms-Like Tyrosine Kinase-1) Is Sufficient to Impair Spiral Arterial Remodeling in a Preeclampsia Mouse Model. Hypertension 2021, 78, 1067–1079. [Google Scholar] [CrossRef]
- Kassab, S.; Miller, M.T.; Hester, R.; Novak, J.; Granger, J.P. Systemic hemodynamics and regional blood flow during chronic nitric oxide synthesis inhibition in pregnant rats. Hypertension 1998, 31, 315–320. [Google Scholar] [CrossRef]
- Baylis, C. Cyclooxygenase products do not contribute to the gestational renal vasodilation in the nitric oxide synthase inhibited pregnant rat. Hypertens. Pregnancy 2002, 21, 109–114. [Google Scholar] [CrossRef]
- Conrad, K.P.; Colpoys, M.C. Evidence against the hypothesis that prostaglandins are the vasodepressor agents of pregnancy. Serial studies in chronically instrumented, conscious rats. J. Clin. Investig. 1986, 77, 236–245. [Google Scholar] [CrossRef]
- Sasser, J.M.; Ni, X.-P.; Humphreys, M.H.; Baylis, C. Increased renal phosphodiesterase-5 activity mediates the blunted natriuretic response to a nitric oxide donor in the pregnant rat. Am. J. Physiol. Physiol. 2010, 299, F810–F814. [Google Scholar] [CrossRef]
- Salas, S.P.; Altermatt, F.; Campos, M.; Giacaman, A.; Rosso, P. Effects of Long-term Nitric Oxide Synthesis Inhibition on Plasma Volume Expansion and Fetal Growth in the Pregnant Rat. Hypertension 1995, 26, 1019–1023. [Google Scholar] [CrossRef]
- Bigonnesse, E.; Sicotte, B.; Brochu, M. Activated NO pathway in uterine arteries during pregnancy in an IUGR rat model. Am. J. Physiol. Circ. Physiol. 2018, 315, H415–H422. [Google Scholar] [CrossRef] [PubMed]
- Tsukimori, K.; Komatsu, H.; Fukushima, K.; Kaku, T.; Nakano, H.; Wake, N. Inhibition of Nitric Oxide Synthetase at Mid-gestation in Rats is Associated with Increases in Arterial Pressure, Serum Tumor Necrosis Factor-, and Placental Apoptosis. Am. J. Hypertens. 2008, 21, 477–481. [Google Scholar] [CrossRef]
- Barron, C.; Mandala, M.; Osol, G. Effects of Pregnancy, Hypertension and Nitric Oxide Inhibition on Rat Uterine Artery Myogenic Reactivity. J. Vasc. Res. 2010, 47, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Kulandavelu, S.; Whiteley, K.J.; Qu, D.; Mu, J.; Bainbridge, S.A.; Adamson, S.L. Endothelial Nitric Oxide Synthase Deficiency Reduces Uterine Blood Flow, Spiral Artery Elongation, and Placental Oxygenation in Pregnant Mice. Hypertension 2012, 60, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Hagaman, J.R.; Kim, H.-S.; Maeda, N.; Jennette, J.C.; Faber, J.E.; Karumanchi, S.A.; Smithies, O.; Takahashi, N. eNOS Deficiency Acts through Endothelin to Aggravate sFlt-1–Induced Pre-Eclampsia–Like Phenotype. J. Am. Soc. Nephrol. 2012, 23, 652–660. [Google Scholar] [CrossRef]
- Shesely, E.G.; Gilbert, C.; Granderson, G.; Carretero, C.; Carretero, O.A.; Beierwaltes, W.H. Nitric oxide synthase gene knockout mice do not become hypertensive during pregnancy. Am. J. Obstet. Gynecol. 2001, 185, 1198–1203. [Google Scholar] [CrossRef]
- Kulandavelu, S.; Qu, D.; Adamson, S.L. Cardiovascular Function in Mice During Normal Pregnancy and in the Absence of Endothelial NO Synthase. Hypertension 2006, 47, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, A.; Neggers, S.J.; Van Der Lely, A.J. Pregnancy and acromegaly. Pituitary 2016, 20, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.L.; Troja, W.; Sumser, E.K.; Maupin, A.; Lampe, K.; Jones, H.N. Insulin-like growth factor 1 signaling in the placenta requires endothelial nitric oxide synthase to support trophoblast function and normal fetal growth. Am. J. Physiol. Integr. Comp. Physiol. 2021, 320, R653–R662. [Google Scholar] [CrossRef]
- Morishita, T.; Tsutsui, M.; Shimokawa, H.; Sabanai, K.; Tasaki, H.; Suda, O.; Nakata, S.; Tanimoto, A.; Wang, K.-Y.; Ueta, Y.; et al. Nephrogenic diabetes insipidus in mice lacking all nitric oxide synthase isoforms. Proc. Natl. Acad. Sci. USA 2005, 102, 10616–10621. [Google Scholar] [CrossRef]
- Hyndman, K.A.; Mironova, E.V.; Giani, J.F.; Dugas, C.; Collins, J.; McDonough, A.A.; Stockand, J.D.; Pollock, J.S. Collecting Duct Nitric Oxide Synthase 1ß Activation Maintains Sodium Homeostasis during High Sodium Intake Through Suppression of Aldosterone and Renal Angiotensin II Pathways. J. Am. Hearth Assoc. 2017, 6, e006896. [Google Scholar] [CrossRef]
- Hyndman, K.; Boesen, E.; Elmarakby, A.A.; Brands, M.W.; Huang, P.; Kohan, D.E.; Pollock, D.M.; Pollock, J.S. Renal Collecting Duct NOS1 Maintains Fluid–Electrolyte Homeostasis and Blood Pressure. Hypertension 2013, 62, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Zhang, J.; Jiang, S.; Xu, L.; Qu, L.; Pang, B.; Jiang, K.; Wang, L.; Intapad, S.; Buggs, J.; et al. Macula Densa NOS1beta Modulates Renal Hemodynamics and Blood Pressure during Pregnancy: Role in Gestational Hypertension. J. Am. Soc. Nephrol. 2021, 32, 2485–2500. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.C.; Blantz, R.C. Ions and signal transduction in the macula densa. J. Clin. Investig. 2000, 106, 633–635. [Google Scholar] [CrossRef] [PubMed]
- Baylis, C.; Blantz, R.C. Tubuloglomerular feedback activity in virgin and 12-day-pregnant rats. Am. J. Physiol. Content 1985, 249, F169–F173. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wei, J.; Stec, D.E.; Roman, R.J.; Ge, Y.; Cheng, L.; Liu, E.Y.; Zhang, J.; Hansen, P.B.; Fan, F.; et al. Macula Densa Nitric Oxide Synthase 1beta Protects against Salt-Sensitive Hypertension. J. Am. Soc. Nephrol. 2016, 27, 2346–2356. [Google Scholar] [CrossRef]
- Sasser, J.M.; Molnar, M.; Baylis, C. Relaxin ameliorates hypertension and increases nitric oxide metabolite excretion in angiotensin II but not N(omega)-nitro-L-arginine methyl ester hypertensive rats. Hypertension 2011, 58, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Uiterweer, E.D.P.; Koster, M.P.; Jeyabalan, A.; Kuc, S.; Siljee, J.E.; Stewart, D.R.; Conrad, K.P.; Franx, A. Circulating pregnancy hormone relaxin as a first trimester biomarker for preeclampsia. Pregnancy Hypertens. 2020, 22, 47–53. [Google Scholar] [CrossRef]
- Bakrania, B.A.; George, E.M.; Granger, J.P. Animal models of preeclampsia: Investigating pathophysiology and therapeutic targets. Am. J. Obstet. Gynecol. 2021. [Google Scholar] [CrossRef]
- Santiago-Font, J.A.; Amaral, L.M.; Faulkner, J.; Ibrahim, T.; Vaka, V.R.; Cunningham, M.W.; LaMarca, B. Serelaxin improves the pathophysiology of placental ischemia in the reduced uterine perfusion pressure rat model of preeclampsia. Am. J. Physiol. Integr. Comp. Physiol. 2016, 311, R1158–R1163. [Google Scholar] [CrossRef]
- Conrad, K.P.; Kerchner, L.J.; Mosher, M.D. Plasma and 24-h NO(x) and cGMP during normal pregnancy and preeclampsia in women on a reduced NO(x) diet. Am. J. Physiol. 1999, 277, F48–F57. [Google Scholar] [CrossRef]
- Baksu, B.; Davas, I.; Baksu, A.; Akyol, A.; Gulbaba, G. Plasma nitric oxide, endothelin-1 and urinary nitric oxide and cyclic guanosine monophosphate levels in hypertensive pregnant women. Int. J. Gynecol. Obstet. 2005, 90, 112–117. [Google Scholar] [CrossRef]
- Tropea, T.; Wareing, M.; Greenwood, S.L.; Feelisch, M.; Sibley, C.P.; Cottrell, E.C. Nitrite mediated vasorelaxation in human chorionic plate vessels is enhanced by hypoxia and dependent on the NO-sGC-cGMP pathway. Nitric Oxide 2018, 80, 82–88. [Google Scholar] [CrossRef]
- Mannaerts, D.; Faes, E.; Cornette, J.; Gyselaers, W.; Spaanderman, M.; Goovaerts, I.; Stoop, T.; Roelant, E.; Jacquemyn, Y.; Van Craenenbroeck, E.M. Low-flow mediated constriction as a marker of endothelial function in healthy pregnancy and preeclampsia: A pilot study. Pregnancy Hypertens. 2019, 17, 75–81. [Google Scholar] [CrossRef]
- Noori, M.; Donald, A.E.; Angelakopoulou, A.; Hingorani, A.; Williams, D. Prospective Study of Placental Angiogenic Factors and Maternal Vascular Function before and after Preeclampsia and Gestational Hypertension. Circulation 2010, 122, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Li, F.-F.; He, M.-Z.; Xie, Y.; Wu, Y.-Y.; Yang, M.-T.; Fan, Y.; Qiao, F.-Y.; Deng, D.-R. Involvement of dysregulated IKCa and SKCa channels in preeclampsia. Placenta 2017, 58, 9–16. [Google Scholar] [CrossRef]
- Osol, G.; Celia, G.; Gokina, N.; Barron, C.; Chien, E.; Mandala, M.; Luksha, L.; Kublickiene, K. Placental growth factor is a potent vasodilator of rat and human resistance arteries. Am. J. Physiol. Circ. Physiol. 2008, 294, H1381–H1387. [Google Scholar] [CrossRef] [PubMed]
- Perdigao, J.L.; Chinthala, S.; Mueller, A.; Minhas, R.; Ramadan, H.; Nasim, R.; Naseem, H.; Young, D.; Shahul, S.; Chan, S.L.; et al. Angiogenic Factor Estimation as a Warning Sign of Preeclampsia-Related Peripartum Morbidity among Hospitalized Patients. Hypertension 2019, 73, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, G.; Jahan, S.; Bibi, N.; Ullah, A.; Faryal, R.; Almajwal, A.; Afsar, T.; Al-Disi, D.; Abulmeaty, M.; Al Khuraif, A.A.; et al. Additional file 1 of Association of endothelial nitric oxide synthase gene variants with preeclampsia. Reprod. Health 2021, 18, 163. [Google Scholar] [CrossRef] [PubMed]
- Possomato-Vieira, J.S.; Palei, A.C.; Pinto-Souza, C.C.; Cavalli, R.; Dias-Junior, C.A.; Sandrim, V. Circulating levels of hydrogen sulphide negatively correlate to nitrite levels in gestational hypertensive and preeclamptic pregnant women. Clin. Exp. Pharmacol. Physiol. 2021, 48, 1224–1230. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.A.; Sandrim, V.C.; Palei, A.C.; Amaral, L.M.; Belo, V.A.; Lacchini, R.; Cavalli, R.C.; Tanus-Santos, J.E.; Luizon, M.R. NAMPT single-nucleotide polymorphism rs1319501 and visfatin/NAMPT affect nitric oxide formation, sFlt-1 and antihypertensive therapy response in preeclampsia. Pharmacogenomics 2021, 22, 451–464. [Google Scholar] [CrossRef]
- Haworth, S.M.M.; Zhuge, Z.; Nihlén, C.; Von Rosen, M.F.; Weitzberg, E.; Lundberg, J.O.; Krmar, R.T.; Nasiell, J.; Carlström, M. Red blood cells from patients with pre-eclampsia induce endothelial dysfunction. J. Hypertens. 2021, 39, 1628–1641. [Google Scholar] [CrossRef] [PubMed]
- Tashie, W.; Fondjo, L.A.; Owiredu, W.K.B.A.; Ephraim, R.K.D.; Asare, L.; Adu-Gyamfi, E.A.; Seidu, L. Altered Bioavailability of Nitric Oxide and L-Arginine Is a Key Determinant of Endothelial Dysfunction in Preeclampsia. BioMed Res. Int. 2020, 2020, 3251956. [Google Scholar] [CrossRef]
- Kim, S.; Park, M.; Kim, J.-Y.; Kim, T.; Hwang, J.; Ha, K.-S.; Won, M.-H.; Ryoo, S.; Kwon, Y.-G.; Kim, Y.-M. Circulating miRNAs Associated with Dysregulated Vascular and Trophoblast Function as Target-Based Diagnostic Biomarkers for Preeclampsia. Cells 2020, 9, 2003. [Google Scholar] [CrossRef] [PubMed]
- Mazloomi, S.; Khodadadi, I.; Alimohammadi, S.; Shafiee, G. Correlation of thioredoxin reductase (TrxR) and nitric oxide synthase (NOS) activities with serum trace elements in preeclampsia. Clin. Exp. Hypertens. 2020, 43, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.; Nie, L.; Zeng, X.; Xin, S.; Wu, M.; Yang, B.; Luo, Y.; Liu, B.; Zheng, J.; Liu, H. Enhancement of heat shock protein 70 attenuates inducible nitric oxide synthase in preeclampsia complicated with fetal growth restriction. J. Matern.-Neonatal Med. 2020, 1–9. [Google Scholar] [CrossRef]
- Ajadi, I.; Maduray, K.; Eche, S.; Gathiram, P.; Mackraj, I. Serum levels of vasoactive factors in HIV-infected pre-eclamptic women on HAART. J. Obstet. Gynaecol. 2020, 41, 546–551. [Google Scholar] [CrossRef]
- Serrano-Berrones, M.; Barragán-Padilla, S.B. Study on the association of hypertriglyceridemia with hypertensive states of pregnancy. Gac. Med. Mex. 2019, 155, S17–S21. [Google Scholar] [CrossRef]
- Deniz, R.; Baykus, Y.; Ustebay, S.; Ugur, K.; Yavuzkir, S.; Aydin, S. Evaluation of elabela, apelin and nitric oxide findings in maternal blood of normal pregnant women, pregnant women with pre-eclampsia, severe pre-eclampsia and umbilical arteries and venules of newborns. J. Obstet. Gynaecol. 2019, 39, 907–912. [Google Scholar] [CrossRef]
- Bos, M.; Schoots, M.H.; Fernandez, B.O.; Mikus-Lelinska, M.; Lau, L.C.; Eikmans, M.; Van Goor, H.; Gordijn, S.J.; Pasch, A.; Feelisch, M.; et al. Reactive Species Interactome Alterations in Oocyte Donation Pregnancies in the Absence and Presence of Pre-Eclampsia. Int. J. Mol. Sci. 2019, 20, 1150. [Google Scholar] [CrossRef]
- ElMonier, A.A.; El-Boghdady, N.A.; Abdelaziz, M.A.; Shaheen, A.A. Association between endoglin/transforming growth factor beta receptors 1, 2 gene polymorphisms and the level of soluble endoglin with preeclampsia in Egyptian women. Arch. Biochem. Biophys. 2018, 662, 7–14. [Google Scholar] [CrossRef]
- Hodžić, J.; Izetbegović, S.; Muračević, B.; Iriškić, R.; Jović, H. Nitric oxide biosynthesis during normal pregnancy and pregnancy complicated by preeclampsia. Med. Glas. 2017, 211–217. [Google Scholar] [CrossRef]
- Rocha-Penha, L.; Caldeira-Dias, M.; Tanus-Santos, J.E.; Cavalli, R.D.C.; Sandrim, V.C. Myeloperoxidase in Hypertensive Disorders of Pregnancy and Its Relation with Nitric Oxide. Hypertension 2017, 69, 1173–1180. [Google Scholar] [CrossRef]
- Lorca, R.A.; Lane, S.L.; Bales, E.S.; Nsier, H.; Yi, H.; Donnelly, M.A.; Euser, A.G.; Julian, C.G.; Moore, L.G. High Altitude Reduces NO-Dependent Myometrial Artery Vasodilator Response During Pregnancy. Hypertension 2019, 73, 1319–1326. [Google Scholar] [CrossRef]
- Chen, J.; Gao, Q.; Jiang, L.; Feng, X.; Zhu, X.; Fan, X.; Mao, C.; Xu, Z. The NOX2-derived reactive oxygen species damaged endothelial nitric oxide system via suppressed BKCa/SKCa in preeclampsia. Hypertens. Res. 2017, 40, 457–464. [Google Scholar] [CrossRef]
- Salsoso, R.; Mate, A.; Toledo, F.; Vázquez, C.M.; Sobrevia, L. Insulin requires A2B adenosine receptors to modulate the L-arginine/nitric oxide signalling in the human fetoplacental vascular endothelium from late-onset preeclampsia. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1867, 165993. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.S.; Kumar, S. Activation of angiotensin type 2 receptor attenuates testosterone-induced hypertension and uterine vascular resistance in pregnant ratsdagger. Biol. Reprod. 2021, 105, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.-Y.; Uprety, L.P.; Jang, Y.-J.; Yang, J.I. Pro-inflammatory mediators and signaling proteins in the decidua of pre-eclampsia. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 12016–12024. [Google Scholar] [PubMed]
- Mukosera, G.T.; Clark, T.C.; Ngo, L.; Liu, T.; Schroeder, H.; Power, G.G.; Yellon, S.M.; Parast, M.M.; Blood, A.B. Nitric oxide metabolism in the human placenta during aberrant maternal inflammation. J. Physiol. 2020, 598, 2223–2241. [Google Scholar] [CrossRef]
- Shaheen, G.; Jahan, S.; Ain, Q.U.; Ullah, A.; Afsar, T.; Almajwal, A.; Alam, I.; Razak, S. Placental endothelial nitric oxide synthase expression and role of oxidative stress in susceptibility to preeclampsia in Pakistani women. Mol. Genet. Genom. Med. 2020, 8, e1019. [Google Scholar]
- Guerby, P.; Swiader, A.; Tasta, O.; Pont, F.; Rodriguez, F.; Parant, O.; Vayssière, C.; Shibata, T.; Uchida, K.; Salvayre, R.; et al. Modification of endothelial nitric oxide synthase by 4-oxo-2(E)-nonenal(ONE) in preeclamptic placentas. Free Radic. Biol. Med. 2019, 141, 416–425. [Google Scholar] [CrossRef]
- Hitzerd, E.; Broekhuizen, M.; Colafella, K.M.M.; Glisic, M.; de Vries, R.; Koch, B.C.; de Raaf, M.A.; Merkus, D.; Schoenmakers, S.; Reiss, I.K.; et al. Placental effects and transfer of sildenafil in healthy and preeclamptic conditions. EBioMedicine 2019, 45, 447–455. [Google Scholar] [CrossRef]
- Guerby, P.; Swiader, A.; Augé, N.; Parant, O.; Vayssière, C.; Uchida, K.; Salvayre, R.; Negre-Salvayre, A. High glutathionylation of placental endothelial nitric oxide synthase in preeclampsia. Redox Biol. 2019, 22, 101126. [Google Scholar] [CrossRef] [PubMed]
- Motta-Mejia, C.; Kandzija, N.; Zhang, W.; Mhlomi, V.; Cerdeira, A.S.; Burdujan, A.; Tannetta, D.; Dragovic, R.; Sargent, I.L.; Redman, C.W.; et al. Placental Vesicles Carry Active Endothelial Nitric Oxide Synthase and Their Activity is Reduced in Preeclampsia. Hypertension 2017, 70, 372–381. [Google Scholar] [CrossRef]
- Du, L.; He, F.; Kuang, L.; Tang, W.; Li, Y.; Chen, D. eNOS/iNOS and endoplasmic reticulum stress-induced apoptosis in the placentas of patients with preeclampsia. J. Hum. Hypertens. 2016, 31, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, E.D.; Babischkin, J.S.; Aberdeen, G.W.; Burch, M.G.; Pepe, G.J. Maternal systemic vascular dysfunction in a primate model of defective uterine spiral artery remodeling. Am. J. Physiol. Circ. Physiol. 2021, 320, H1712–H1723. [Google Scholar] [CrossRef]
- Travis, O.K.; Tardo, G.A.; Giachelli, C.; Siddiq, S.; Nguyen, H.T.; Crosby, M.T.; Johnson, T.; Brown, A.K.; Williams, J.M.; Cornelius, D.C. Tumor Necrosis Factor-Alpha Blockade Improves Uterine Artery Resistance, Maternal Blood Pressure, and Fetal Growth in Placental Ischemic Rats. Pregnancy Hypertens. 2021, 25, 39–47. [Google Scholar] [CrossRef]
- Cottrell, J.N.; Witcher, A.C.; Comley, K.M.; Cunningham, M.W., Jr.; Ibrahim, T.; Cornelius, D.C.; LaMarca, B.D.; Amaral, L.M. Progesterone-induced blocking factor improves blood pressure, inflammation, and pup weight in response to reduced uterine perfusion pressure (RUPP). Am. J. Physiol. Integr. Comp. Physiol. 2021, 320, R719–R727. [Google Scholar] [CrossRef]
- El-Saka, M.H.; Madi, N.M.; Ibrahim, R.R.; Alghazaly, G.M.; Elshwaikh, S.; El-Bermawy, M. The ameliorative effect of angiotensin 1-7 on experimentally induced-preeclampsia in rats: Targeting the role of peroxisome proliferator-activated receptors gamma expression & asymmetric dimethylarginine. Arch. Biochem. Biophys. 2019, 671, 123–129. [Google Scholar] [CrossRef]
- Wang, C.; Liu, X.; Kong, D.; Qin, X.; Li, Y.; Teng, X.; Huang, X. Apelin as a novel drug for treating preeclampsia. Exp. Ther. Med. 2017, 14, 5917–5923. [Google Scholar] [CrossRef]
- Amaral, L.M.; Faulkner, J.L.; Elfarra, J.; Cornelius, D.C.; Cunningham, M.W.; Ibrahim, T.; Vaka, V.R.; McKenzie, J.; LaMarca, B. Continued Investigation Into 17-OHPC: Results from the Preclinical RUPP Rat Model of Preeclampsia. Hypertension 2017, 70, 1250–1255. [Google Scholar] [CrossRef] [PubMed]
- Jammalamadaga, V.S.; Abraham, P. Spectrum of Factors Triggering Endothelial Dysfunction in PIH. J. Clin. Diagn. Res. 2016, 10, BC14–BC17. [Google Scholar] [CrossRef] [PubMed]
- Younes, S.T.; Maeda, K.J.; Sasser, J.; Ryan, M.J. The glucagon-like peptide 1 receptor agonist liraglutide attenuates placental ischemia-induced hypertension. Am. J. Physiol. Circ. Physiol. 2020, 318, H72–H77. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.-L.; Tian, X.-Y.; Wang, Y.-Q.; Zhang, H.-F.; Zhang, L. Vitamin D Supplementation Prevents Placental Ischemia Induced Endothelial Dysfunction by Downregulating Placental Soluble FMS-Like Tyrosine Kinase-1. DNA Cell Biol. 2017, 36, 1134–1141. [Google Scholar] [CrossRef]
- Zhu, M.; Ren, Z.; Possomato-Vieira, J.S.; Khalil, R.A. Restoring placental growth factor-soluble fms-like tyrosine kinase-1 balance reverses vascular hyper-reactivity and hypertension in pregnancy. Am. J. Physiol. Integr. Comp. Physiol. 2016, 311, R505–R521. [Google Scholar] [CrossRef]
- Zhang, T.; Guo, D.; Zheng, W.; Dai, Q. Effects of S1PR2 antagonist on blood pressure and angiogenesis imbalance in preeclampsia rats. Mol. Med. Rep. 2021, 23, 456. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.-J.; Yang, Z.; Huai, J.; Xiang, Q.-Q. Pravastatin alleviates oxidative stress and decreases placental trophoblastic cell apoptosis through IL-6/STAT3 signaling pathway in preeclampsia rats. Mol. Med. Rep. 2020, 24, 12955–12962. [Google Scholar]
- Chimini, J.S.; Possomato-Vieira, J.S.; da Silva, M.L.S.; Dias-Junior, C.A. Placental nitric oxide formation and endothelium-dependent vasodilation underlie pravastatin effects against angiogenic imbalance, hypertension in pregnancy and intrauterine growth restriction. Basic Clin. Pharmacol. Toxicol. 2019, 124, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Ou, M.; Zhang, Q.; Zhao, H.; Shu, C. Polyunsaturated Fatty Acid Diet and Upregulation of Lipoxin A4 Reduce the Inflammatory Response of Preeclampsia. J. Proteome Res. 2020, 20, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhang, J.; Zhu, B. Protective effect of metformin on a rat model of lipopolysaccharide-induced preeclampsia. Fundam. Clin. Pharmacol. 2019, 33, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Lefkou, E.; Varoudi, K.; Pombo, J.; Jurisic, A.; Jurisic, Z.; Contento, G.; Girardi, G. Triple therapy with pravastatin, low molecular weight heparin and low dose aspirin improves placental haemodynamics and pregnancy outcomes in obstetric antiphospholipid syndrome in mice and women through a nitric oxide-dependent mechanism. Biochem. Pharmacol. 2020, 182, 114217. [Google Scholar] [CrossRef]
- Purnamayanti, N.M.D.; Windu, S.C.; Poeranto, S. Effect of Nigella sativa Ethanol Extract on the Nitric Oxide Content and Renal Arteriole Diameter of a Pre-eclampsia Mouse Model. Eurasian J. Med. 2018, 50, 148–151. [Google Scholar] [CrossRef]
- Chang, A.S.; Grant, R.; Tomita, H.; Kim, H.-S.; Smithies, O.; Kakoki, M. Prolactin alters blood pressure by modulating the activity of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2016, 113, 12538–12543. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Chen, X.; Ding, Y.; Chen, C.; Liu, T.; Zhang, H. Abnormal angiogenesis of placenta in progranulindeficient mice. Mol. Med. Rep. 2020, 22, 3482–3492. [Google Scholar]
- Lane, S.L.; Doyle, A.S.; Bales, E.S.; Lorca, R.A.; Julian, C.G.; Moore, L.G. Increased uterine artery blood flow in hypoxic murine pregnancy is not sufficient to prevent fetal growth restrictiondagger. Biol. Reprod. 2020, 102, 660–670. [Google Scholar] [CrossRef]
- Zhang, S.; Zou, C.; Zhang, Q. Deletion of GIT1 Impacts eNOS Activity to Aggravate sFlt-1–Induced Preeclampsia Phenotype in Mice. G3 Genes Genomes Genet. 2018, 8, 3377–3382. [Google Scholar] [CrossRef] [PubMed]
- Makris, A.; Yeung, K.R.; Lim, S.M.; Sunderland, N.; Heffernan, S.; Thompson, J.; Iliopoulos, J.; Killingsworth, M.C.; Yong, J.; Xu, B.; et al. Placental Growth Factor Reduces Blood Pressure in a Uteroplacental Ischemia Model of Preeclampsia in Nonhuman Primates. Hypertension 2016, 67, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.K.; English, F.A.; Johns, E.J.; Kenny, L.C. Plasma-mediated vascular dysfunction in the reduced uterine perfusion pressure model of preeclampsia: A microvascular characterization. Hypertension 2009, 54, 345–351. [Google Scholar] [CrossRef]
- Turanov, A.A.; Lo, A.; Hassler, M.R.; Makris, A.; Ashar-Patel, A.; Alterman, J.F.; Coles, A.H.; Haraszti, R.A.; Roux, L.; Godinho, B.M.D.C.; et al. RNAi modulation of placental sFLT1 for the treatment of preeclampsia. Nat. Biotechnol. 2018, 36, 1164–1173. [Google Scholar] [CrossRef]
- Murphy, S.R.; LaMarca, B.; Cockrell, K.; Arany, M.; Granger, J.P. L-arginine supplementation abolishes the blood pressure and endothelin response to chronic increases in plasma sFlt-1 in pregnant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R259–R263. [Google Scholar] [CrossRef] [PubMed]
- Amraoui, F.; Spijkers, L.; Lahsinoui, H.H.; Vogt, L.; Van Der Post, J.; Peters, S.; Afink, G.; Ris-Stalpers, C.; Born, B.-J.V.D. SFlt-1 Elevates Blood Pressure by Augmenting Endothelin-1-Mediated Vasoconstriction in Mice. PLoS ONE 2014, 9, e91897. [Google Scholar] [CrossRef]
- Davenport, A.P.; Hyndman, K.A.; Dhaun, N.; Southan, C.; Kohan, D.E.; Pollock, J.S.; Pollock, D.M.; Webb, D.J.; Maguire, J.J. Endothelin. Pharmacol. Rev. 2016, 68, 357–418. [Google Scholar] [CrossRef]
- Spradley, F.T. Abstract 037: Exaggerated Placental Ischemia-induced Hypertension in Endothelin Receptor Type B (ETB)-deficient Pregnant Rats s Independent of Increased sFlt-1 or ROS Levels. Hypertension 2017, 70, A037. [Google Scholar] [CrossRef]
- Morton, J.S.; Levasseur, J.; Ganguly, E.; Quon, A.; Kirschenman, R.; Dyck, J.R.B.; Fraser, G.M.; Davidge, S.T. Characterisation of the Selective Reduced Uteroplacental Perfusion (sRUPP) Model of Preeclampsia. Sci. Rep. 2019, 9, 9565. [Google Scholar] [CrossRef] [PubMed]
- Brennan, L.; Morton, J.S.; Quon, A.; Davidge, S.T. Postpartum Vascular Dysfunction in the Reduced Uteroplacental Perfusion Model of Preeclampsia. PLoS ONE 2016, 11, e0162487. [Google Scholar] [CrossRef]
- Oe, Y.; Ko, M.; Fushima, T.; Sato, E.; Karumanchi, S.A.; Sato, H.; Sugawara, J.; Ito, S.; Takahashi, N. Hepatic dysfunction and thrombocytopenia induced by excess sFlt1 in mice lacking endothelial nitric oxide synthase. Sci. Rep. 2018, 8, 102. [Google Scholar] [CrossRef]
- Spradley, F.T.; Tan, A.Y.; Joo, W.S.; Daniels, G.; Kussie, P.; Karumanchi, S.A.; Granger, J.P. Placental Growth Factor Administration Abolishes Placental Ischemia-Induced Hypertension. Hypertension 2016, 67, 740–747. [Google Scholar] [CrossRef]
- Mbah, A.K.; Kornosky, J.L.; Kristensen, S.; August, E.; Alio, A.P.; Marty, P.J.; Belogolovkin, V.; Bruder, K.; Salihu, H.M. Super-obesity and risk for early and late pre-eclampsia. BJOG Int. J. Obstet. Gynaecol. 2010, 117, 997–1004. [Google Scholar] [CrossRef]
- Battineni, G.; Sagaro, G.; Chintalapudi, N.; Amenta, F.; Tomassoni, D.; Tayebati, S. Impact of Obesity-Induced Inflammation on Cardiovascular Diseases (CVD). Int. J. Mol. Sci. 2021, 22, 4798. [Google Scholar] [CrossRef]
- Aksin, S.; Andan, C. Protein-9 (CTRP9) levels associated with C1q tumor necrosis factor in obese preeclamptic, non-obese preeclamptic, obese and normal pregnant women. J. Matern.-Neonatal Med. 2020, 34, 2540–2547. [Google Scholar] [CrossRef]
- Lindsay, K.L.; Buss, C.; Wadhwa, P.D.; Entringer, S. Maternal Stress Potentiates the Effect of an Inflammatory Diet in Pregnancy on Maternal Concentrations of Tumor Necrosis Factor Alpha. Nutrients 2018, 10, 1252. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.W.; Young, S.L.; Grattan, D.; Jasoni, C.L. Obesity during Pregnancy Disrupts Placental Morphology, Cell Proliferation, and Inflammation in a Sex-Specific Manner across Gestation in the Mouse. Biol. Reprod. 2014, 90, 130. [Google Scholar] [CrossRef]
- Mahany, E.B.; Han, X.; Borges, B.C.; Cruz-Machado, S.D.S.; Allen, S.J.; Galiano, D.G.; Hoenerhoff, M.J.; Bellefontaine, N.H.; Elias, C.F. Obesity and High-Fat Diet Induce Distinct Changes in Placental Gene Expression and Pregnancy Outcome. Endocrinology 2018, 159, 1718–1733. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, Y.; Wang, C.; Shi, R.; Zhou, X.; Li, Z.; Sun, W.; Zhao, L.; Yuan, L. Maternal obesity increases the risk of fetal cardiac dysfunction via visceral adipose tissue derived exosomes. Placenta 2021, 105, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Salvolini, E.; Vignini, A.; Sabbatinelli, J.; Lucarini, G.; Pompei, V.; Sartini, D.; Cester, A.M.; Ciavattini, A.; Mazzanti, L.; Emanuelli, M. Nitric oxide synthase and VEGF expression in full-term placentas of obese women. Histochem. Cell Biol. 2019, 152, 415–422. [Google Scholar] [CrossRef]
- Santos-Rosendo, C.; Bugatto, F.; González-Domínguez, A.; Lechuga-Sancho, A.M.; Mateos, R.M.; Visiedo, F. Placental Adaptive Changes to Protect Function and Decrease Oxidative Damage in Metabolically Healthy Maternal Obesity. Antioxidants 2020, 9, 794. [Google Scholar] [CrossRef]
- Amaral, L.M.; Pinheiro, L.C.; Guimaraes, D.A.; Palei, A.C.; Sertório, J.T.; Portella, R.L.; Tanus-Santos, J.E. Antihypertensive effects of inducible nitric oxide synthase inhibition in experimental pre-eclampsia. J. Cell. Mol. Med. 2013, 17, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, C.; Bulla, R.; Tripodo, C.; Gismondi, A.; Stabile, H.; Bossi, F.; Guarnotta, C.; Garlanda, C.; De Seta, F.; Spessotto, P.; et al. An Alternative Role of C1q in Cell Migration and Tissue Remodeling: Contribution to Trophoblast Invasion and Placental Development. J. Immunol. 2010, 185, 4420–4429. [Google Scholar] [CrossRef] [PubMed]
- Sutton, E.F.; Gemmel, M.; Brands, J.; Gallaher, M.J.; Powers, R.W. Paternal deficiency of complement component C1q leads to a preeclampsia-like pregnancy in wild-type female mice and vascular adaptations postpartum. Am. J. Physiol. Integr. Comp. Physiol. 2020, 318, R1047–R1057. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Ahmed, A.; Girardi, G. Role of Complement Component C1q in the Onset of Preeclampsia in Mice. Hypertension 2011, 58, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Jia, K.; Ma, L.; Wu, S.; Yang, W. Serum Levels of Complement Factors C1q, Bb, and H in Normal Pregnancy and Severe Pre-Eclampsia. Med. Sci. Monit. 2019, 25, 7087–7093. [Google Scholar] [CrossRef] [PubMed]
- Lillegard, K.E.; Johnson, A.C.; Lojovich, S.J.; Bauer, A.J.; Marsh, H.C.; Gilbert, J.S.; Regal, J.F. Complement activation is critical for placental ischemia-induced hypertension in the rat. Mol. Immunol. 2013, 56, 91–97. [Google Scholar] [CrossRef]
- Kouser, L.; Madhukaran, S.P.; Shastri, A.; Saraon, A.; Ferluga, J.; Al-Mozaini, M.; Kishore, U. Emerging and Novel Functions of Complement Protein C1q. Front. Immunol. 2015, 6, 317. [Google Scholar] [CrossRef]
- De Jesus, G.R.; Mendoza-Pinto, C.; De Jesus, N.R.; Dos Santos, F.C.; Klumb, E.M.; Carrasco, M.G.; Levy, R.A. Understanding and Managing Pregnancy in Patients with Lupus. Autoimmune Dis. 2015, 2015, 943490. [Google Scholar] [CrossRef]
- Kim, M.Y.; Buyon, J.P.; Guerra, M.M.; Rana, S.; Zhang, D.; Laskin, C.A.; Petri, M.; Lockshin, M.D.; Sammaritano, L.R.; Branch, D.W.; et al. Angiogenic factor imbalance early in pregnancy predicts adverse outcomes in patients with lupus and antiphospholipid antibodies: Results of the PROMISSE study. Am. J. Obstet. Gynecol. 2016, 214, 108.e1–108.e14. [Google Scholar] [CrossRef] [PubMed]
- Villar, J.; Ariff, S.; Gunier, R.B.; Thiruvengadam, R.; Rauch, S.; Kholin, A.; Roggero, P.; Prefumo, F.; do Vale, M.S.; Cardona-Perez, J.A.; et al. Maternal and Neonatal Morbidity and Mortality among Pregnant Women with and without COVID-19 Infection: The INTERCOVID Multinational Cohort Study. Jama Pediatr. 2021, 175, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Papageorghiou, A.T.; Deruelle, P.; Gunier, R.B.; Rauch, S.; García-May, P.K.; Mhatre, M.; Usman, M.A.; Abd-Elsalam, S.; Etuk, S.; Simmons, L.E.; et al. Preeclampsia and COVID-19: Results from the INTERCOVID prospective longitudinal study. Am. J. Obstet. Gynecol. 2021, 225, 289.e1–289.e17. [Google Scholar] [CrossRef]
- Coronado-Arroyo, J.C.; Concepción-Zavaleta, M.J.; Zavaleta-Gutiérrez, F.E.; Concepción-Urteaga, L.A. Is COVID-19 a risk factor for severe preeclampsia? Hospital experience in a developing country. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 256, 502–503. [Google Scholar] [CrossRef] [PubMed]
- Conde-Agudelo, A.; Romero, R. SARS-COV-2 infection during pregnancy and risk of preeclampsia: A systematic review and meta-analysis. Am. J. Obstet. Gynecol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.P.; Khong, T.Y.; Tan, G.C. The Effects of COVID-19 on Placenta and Pregnancy: What Do We Know So Far? Diagnostics 2021, 11, 94. [Google Scholar] [CrossRef]
- Lesnic, A.; Haj Hamoud, B.; Poenaru, M.O.; Moldovan, V.T.; Chicea, R.; Sima, R.M.; Popescu, M.; Ples, L. Can SARS-CoV-2 Induce Uterine Vascular Anomalies and Poor Contractile Response? A Case Report. Medicina 2021, 57, 670. [Google Scholar] [CrossRef]
- Murphy, S.R.; Lamarca, B.B.D.; Parrish, M.; Cockrell, K.; Granger, J.P. Control of soluble fms-like tyrosine-1 (sFlt-1) production response to placental ischemia/hypoxia: Role of tumor necrosis factor-α. Am. J. Physiol. Integr. Comp. Physiol. 2013, 304, R130–R135. [Google Scholar] [CrossRef]
- Johal, T.; Lees, C.C.; Everett, T.R.; Wilkinson, I.B. The nitric oxide pathway and possible therapeutic options in pre-eclampsia. Br. J. Clin. Pharmacol. 2013, 78, 244–257. [Google Scholar] [CrossRef]
- Kalidindi, M.; Velauthar, L.; Khan, K.; Aquilina, J. The role of nitrates in the prevention of preeclampsia: An update. Curr. Opin. Obstet. Gynecol. 2012, 24, 361–367. [Google Scholar] [CrossRef]
- D’Aniello, G.; Tolino, A.; Fisher, G. Plasma L-arginine is markedly reduced in pregnant women affected by preeclampsia. J. Chromatogr. B Biomed. Sci. Appl. 2001, 753, 427–431. [Google Scholar] [CrossRef]
- Kim, Y.; Park, H.; Lee, H.; Ha, E.; Suh, S.; Oh, S.; Yoo, H.-S. Reduced l-arginine Level and Decreased Placental eNOS Activity in Preeclampsia. Placenta 2006, 27, 438–444. [Google Scholar] [CrossRef]
- Noris, M.; Todeschini, M.; Cassis, P.; Pasta, F.; Cappellini, A.; Bonazzola, S.; Macconi, D.; Maucci, R.; Porrati, F.; Benigni, A.; et al. l-Arginine Depletion in Preeclampsia Orients Nitric Oxide Synthase Toward Oxidant Species. Hypertension 2004, 43, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Tamás, P.; Bódis, J.; Sulyok, E.; Kovács, G.L.; Hantosi, E.; Molnar, G.; Martens-Lobenhoffer, J.; Bode-Böger, S.M. L-arginine metabolism in early-onset and late-onset pre-eclamptic pregnancies. Scand. J. Clin. Lab. Investig. 2013, 73, 436–443. [Google Scholar] [CrossRef]
- Khalil, A.A.; Tsikas, D.; Akolekar, R.; Jordan, J.; Nicolaides, K.H. Asymmetric dimethylarginine, arginine and homoarginine at 11–13 weeks’ gestation and preeclampsia: A case-control study. J. Hum. Hypertens. 2013, 27, 38–43. [Google Scholar] [CrossRef]
- Facchinetti, F.; Longo, M.; Piccinini, F.; Neri, I.; Volpe, A. L-arginine infusion reduces blood pressure in preeclamptic women through nitric oxide release. J. Soc. Gynecol. Investig. 1999, 6, 202–207. [Google Scholar]
- Facchinetti, F.; Saade, G.R.; Neri, I.; Pizzi, C.; Longo, M.; Volpe, A. L-Arginine Supplementation in Patients with Gestational Hypertension: A Pilot Study. Hypertens. Pregnancy 2007, 26, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Neri, I.; Jasonni, V.M.; Gori, G.F.; Blasi, I.; Facchinetti, F. Effect of L-arginine on blood pressure in pregnancy-induced hypertension: A randomized placebo-controlled trial. J. Matern.-Fetal Neonatal Med. 2006, 19, 277–281. [Google Scholar] [CrossRef]
- Rytlewski, K.; Olszanecki, R.; Korbut, R.; Zdebski, Z. Effects of prolonged oral supplementation with l-arginine on blood pressure and nitric oxide synthesis in preeclampsia. Eur. J. Clin. Investig. 2005, 35, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Pulido, E.E.C.; Benavides, L.G.; Barón, J.G.P.; Gonzalez, S.P.; Saray, A.J.M.; Padilla, F.E.G.; Sutto, S.E.T. Efficacy of L-arginine for preventing preeclampsia in high-risk pregnancies: A double-blind, randomized, clinical trial. Hypertens. Pregnancy 2016, 35, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Vadillo-Ortega, F.; Perichart-Perera, O.; Espino, S.; Vergara, M.A.A.; Ibarra-Gonzalez, I.; Ahued, R.; Godines, M.; Parry, S.; Macones, G.; Strauss, J.F. Effect of supplementation during pregnancy with L-arginine and antioxidant vitamins in medical food on pre-eclampsia in high risk population: Randomised controlled trial. BMJ 2011, 342, d2901. [Google Scholar] [CrossRef] [PubMed]
- Hladunewich, M.A.; Derby, G.C.; Lafayette, R.A.; Blouch, K.L.; Druzin, M.L.; Myers, B.D. Effect of L-arginine therapy on the glomerular injury of preeclampsia: A randomized controlled trial. Obstet. Gynecol. 2006, 107, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C.; Berge, L.; Haugen, G.; Lorentzen, B.; Mikkelsen, B.; Henriksen, T. Dietary supplementation with l -arginine or placebo in women with pre-eclampsia. Acta Obstet. Gynecol. Scand. 2003, 83, 103–107. [Google Scholar] [CrossRef]
- Dorniak-Wall, T.; Grivell, R.M.; Dekker, G.A.; Hague, W.; Dodd, J. The role of L-arginine in the prevention and treatment of pre-eclampsia: A systematic review of randomised trials. J. Hum. Hypertens. 2013, 28, 230–235. [Google Scholar] [CrossRef]
- Gui, S.; Jia, J.; Niu, X.; Bai, Y.; Zou, H.; Deng, J.; Zhou, R. Arginine supplementation for improving maternal and neonatal outcomes in hypertensive disorder of pregnancy: A systematic review. J. Renin-Angiotensin-Aldosterone Syst. 2013, 15, 88–96. [Google Scholar] [CrossRef]
- Alexander, B.T.; Llinas, M.T.; Kruckeberg, W.C.; Granger, J.P. L-arginine Attenuates Hypertension in Pregnant Rats with Reduced Uterine Perfusion Pressure. Hypertension 2004, 43, 832–836. [Google Scholar] [CrossRef]
- Arikawe, A.; Udenze, I.; Olusanya, A.; Akinnibosun, O.A.; Dike, I.; Duru, B. L-arginine supplementation lowers blood pressure, protein excretion and plasma lipid profile in experimental salt-induced hypertension in pregnancy: Relevance to preeclampsia. Pathophysiology 2019, 26, 191–197. [Google Scholar] [CrossRef]
- Oludare, G.; Jinadu, H.; Aro, O. L-arginine attenuates blood pressure and reverses the suppression of angiogenic risk factors in a rat model of preeclampsia. Pathophysiology 2018, 25, 389–395. [Google Scholar] [CrossRef]
- Kukor, Z.; Valent, S.; Tóth, M. Regulation of Nitric Oxide Synthase Activity by Tetrahydrobiopterin in Human Placentae from Normal and Pre-eclamptic Pregnancies. Placenta 2000, 21, 763–772. [Google Scholar] [CrossRef]
- Mitchell, B.M.; Cook, L.G.; Danchuk, S.; Puschett, J.B. Uncoupled Endothelial Nitric Oxide Synthase and Oxidative Stress in a Rat Model of Pregnancy-Induced Hypertension. Am. J. Hypertens. 2007, 20, 1297–1304. [Google Scholar] [CrossRef]
- Powers, R.W.; Gandley, R.E.; Lykins, D.L.; Roberts, J.M. Moderate Hyperhomocysteinemia Decreases Endothelial-Dependent Vasorelaxation in Pregnant but Not Nonpregnant Mice. Hypertension 2004, 44, 327–333. [Google Scholar] [CrossRef]
- Gaiday, A.; Tussupkaliyev, A.B.; Bermagambetova, S.K.; Zhumagulova, S.S.; Sarsembayeva, L.K.; Dossimbetova, M.B.; Daribay, Z.Z. Effect of homocysteine on pregnancy: A systematic review. Chem. Interact. 2018, 293, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Mignini, L.E.; Latthe, P.M.; Villar, J.; Kilby, M.D.; Carroli, G.; Khan, K.S. Mapping the Theories of Preeclampsia: The Role of Homocysteine. Obstet. Gynecol. 2005, 105, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Visser, S.; Hermes, W.; Ket, J.C.; Otten, R.H.; van Pampus, M.G.; Bloemenkamp, K.W.; Franx, A.; Mol, B.W.; de Groot, C.J. Systematic review and metaanalysis on nonclassic cardiovascular biomarkers after hypertensive pregnancy disorders. Am. J. Obstet. Gynecol. 2014, 211, 373.e1–373.e9. [Google Scholar] [CrossRef] [PubMed]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Fukuda, Y.; Matsuura, H.; Oshima, T.; Chayama, K. Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am. J. Hypertens. 2002, 15, 326–332. [Google Scholar] [CrossRef]
- Porkert, M.; Sher, S.A.; Reddy, U.; Cheema, F.A.; Niessner, C.; Kolm, P.; Jones, D.P.; Hooper, C.C.; Taylor, W.R.; Harrison, D.G.; et al. Tetrahydrobiopterin: A novel antihypertensive therapy. J. Hum. Hypertens. 2008, 22, 401–407. [Google Scholar] [CrossRef]
- Fortepiani, L.A.; Reckelhoff, J.F. Treatment with tetrahydrobiopterin reduces blood pressure in male SHR by reducing testosterone synthesis. Am. J. Physiol. Integr. Comp. Physiol. 2005, 288, R733–R736. [Google Scholar] [CrossRef]
- Hong, H.-J.; Hsiao, G.; Cheng, T.-H.; Yen, M.-H. Supplemention with Tetrahydrobiopterin Suppresses the Development of Hypertension in Spontaneously Hypertensive Rats. Hypertension 2001, 38, 1044–1048. [Google Scholar] [CrossRef]
- Kang, K.-T.; Sullivan, J.C.; Spradley, F.T.; D’Uscio, L.V.; Katušić, Z.S.; Pollock, J.S. Antihypertensive therapy increases tetrahydrobiopterin levels and NO/cGMP signaling in small arteries of angiotensin II-infused hypertensive rats. Am. J. Physiol. Circ. Physiol. 2011, 300, H718–H724. [Google Scholar] [CrossRef] [PubMed]
- Kase, H.; Hashikabe, Y.; Uchida, K.; Nakanishi, N.; Hattori, Y. Supplementation with tetrahydrobiopterin prevents the cardiovascular effects of angiotensin II-induced oxidative and nitrosative stress. J. Hypertens. 2005, 23, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Podjarny, E.; Benchetrit, S.; Rathaus, M.; Pomeranz, A.; Rashid, G.; Shapira, J.; Bernheim, J. Effect of tetrahydrobiopterin on blood pressure in rats after subtotal nephrectomy. Nephron 2003, 94, p6–p9. [Google Scholar] [CrossRef] [PubMed]
- Podjarny, E.; Hasdan, G.; Bernheim, J.; Rashid, G.; Green, J.; Korzets, Z.; Bernheim, J. Effect of chronic tetrahydrobiopterin supplementation on blood pressure and proteinuria in 5/6 nephrectomized rats. Nephrol. Dial. Transplant. 2004, 19, 2223–2227. [Google Scholar] [CrossRef] [PubMed]
- Alacam, H.; Dikmen, Z.G.; Yaman, H.; Cakir, E.; Deren, O.; Akgul, E.O.; Aydin, I.; Kurt, Y.G.; Keskin, U.; Akalin, S.; et al. The Role of Asymmetric Dimethyl Arginine and Oxidant/Antioxidant System in Preeclampsia. Fetal Pediatr. Pathol. 2011, 30, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Rijvers, C.; Marzano, S.; Winkens, B.; Bakker, J.; Kroon, A.; Spaanderman, M.; Peeters, L. Early-pregnancy asymmetric dimethylarginine (ADMA) levels in women prone to develop recurrent hypertension. Pregnancy Hypertens. 2013, 3, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, C.; Marozio, L.; Neri, I.; Giarola, M.; Volpe, A.; Facchinetti, F. Increased L-Citrulline/ L-Arginine Plasma Ratio in Severe Preeclampsia. Obstet. Gynecol. 2000, 96, 395–399. [Google Scholar] [CrossRef]
- Khalaf, D.; Krüger, M.; Wehland, M.; Infanger, M.; Grimm, D. The Effects of Oral l-Arginine and l-Citrulline Supplementation on Blood Pressure. Nutrients 2019, 11, 1679. [Google Scholar] [CrossRef]
- Weckman, A.M.; McDonald, C.R.; Baxter, J.-A.B.; Fawzi, W.W.; Conroy, A.L.; Kain, K.C. Perspective: L-arginine and L-citrulline Supplementation in Pregnancy: A Potential Strategy to Improve Birth Outcomes in Low-Resource Settings. Adv. Nutr. 2019, 10, 765–777. [Google Scholar] [CrossRef]
- Gemmel, M.; Sutton, E.F.; Brands, J.; Burnette, L.; Gallaher, M.J.; Powers, R.W. L-Citrulline supplementation during pregnancy improves perinatal and postpartum maternal vascular function in a mouse model of preeclampsia. Am. J. Physiol. Integr. Comp. Physiol. 2021. [Google Scholar] [CrossRef]
- Bourdon, A.; Parnet, P.; Nowak, C.; Tran, N.-T.; Winer, N.; Darmaun, D. L-Citrulline Supplementation Enhances Fetal Growth and Protein Synthesis in Rats with Intrauterine Growth Restriction. J. Nutr. 2016, 146, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.-T.; Amarger, V.; Bourdon, A.; Misbert, E.; Grit, I.; Winer, N.; Darmaun, D. Maternal citrulline supplementation enhances placental function and fetal growth in a rat model of IUGR: Involvement of insulin-like growth factor 2 and angiogenic factors. J. Matern.-Fetal Neonatal Med. 2017, 30, 1906–1911. [Google Scholar] [CrossRef]
- Barkhidarian, B.; Khorshidi, M.; Shab-Bidar, S.; Hashemi, B. Effects of L-citrulline supplementation on blood pressure: A systematic review and meta-analysis. Avicenna J. Phytomed. 2019, 9, 10–20. [Google Scholar] [PubMed]
- Abbasi, H.; Dastgheib, S.A.; Hadadan, A.; Karimi-Zarchi, M.; Javaheri, A.; Meibodi, B.; Zanbagh, L.; Tabatabaei, R.S.; Neamatzadeh, H. Association of Endothelial Nitric Oxide Synthase 894G > T Polymorphism with Preeclampsia Risk: A Systematic Review and Meta-Analysis based on 35 Studies. Fetal Pediatr. Pathol. 2020, 40, 455–470. [Google Scholar] [CrossRef]
- Zeng, F.; Zhu, S.; Wong, M.; Yang, Z.; Tang, J.; Li, K.; Su, X. Associations between nitric oxide synthase 3 gene polymorphisms and preeclampsia risk: A meta-analysis. Sci. Rep. 2016, 6, 23407. [Google Scholar] [CrossRef]
- Joshi, M.S.; Mineo, C.; Shaul, P.W.; Bauer, J.A. Biochemical consequences of the NOS3 Glu298Asp variation in human endothelium: Altered caveolar localization and impaired response to shear. FASEB J. 2007, 21, 2655–2663. [Google Scholar] [CrossRef] [PubMed]
- Tesauro, M.; Thompson, W.C.; Rogliani, P.; Qi, L.; Chaudhary, P.P.; Moss, J. Intracellular processing of endothelial nitric oxide synthase isoforms associated with differences in severity of cardiopulmonary diseases: Cleavage of proteins with aspartate vs. glutamate at position. Proc. Natl. Acad. Sci. USA 2000, 97, 2832–2835. [Google Scholar] [CrossRef] [PubMed]
- Sakar, M.N.; Atay, A.E.; Demir, S.; Bakır, V.L.; Demir, B.; Balsak, D.; Akay, E.; Ulusoy, A.I.; Verit, F.F. Association of endothelial nitric oxide synthase gene G894T polymorphism and serum nitric oxide levels in patients with preeclampsia and gestational hypertension. J. Matern.-Neonatal Med. 2014, 28, 1907–1911. [Google Scholar] [CrossRef]
- Sharma, D.; Hussain, S.; Akhter, N.; Singh, A.; Trivedi, S.; Bhatttacharjee, J. Endothelial nitric oxide synthase (eNOS) gene Glu298Asp polymorphism and expression in North Indian preeclamptic women. Pregnancy Hypertens. 2014, 4, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Muniz, L.; Luizon, M.R.; Palei, A.C.; Lacchini, R.; Duarte, G.; Cavalli, R.C.; Tanus-Santos, J.E.; Sandrim, V.C. eNOS tag SNP haplotypes in hypertensive disorders of pregnancy. DNA Cell Biol. 2012, 31, 1665–1670. [Google Scholar] [CrossRef]
- Sandrim, V.C.; Palei, A.C.T.; Sertorio, J.T.; Cavalli, R.C.; Duarte, G.; Tanus-Santos, J.E. Effects of eNOS polymorphisms on nitric oxide formation in healthy pregnancy and in pre-eclampsia. Mol. Hum. Reprod. 2010, 16, 506–510. [Google Scholar] [CrossRef]
- Lauria, M.R.; Standley, C.A.; Sorokin, Y.; Todt, J.C.; Bottoms, S.F.; Yelian, F.D.; Cotton, D.B. Brain natriuretic peptide and cyclic guanosine-3′,5′ monophosphate in pre-eclampsia. J. Matern.-Fetal Med. 1996, 5, 128–131. [Google Scholar] [CrossRef]
- Schiessl, B.; Strasburger, C.; Bidlingmaier, M.; Mylonas, I.; Jeschke, U.; Kainer, F.; Friese, K. Plasma- and urine concentrations of nitrite/nitrate and cyclic Guanosinemonophosphate in intrauterine growth restricted and preeclamptic pregnancies. Arch. Gynecol. Obstet. 2006, 274, 150–154. [Google Scholar] [CrossRef] [PubMed]
- Boccardo, P.; Soregaroli, M.; Aiello, S.; Noris, M.; Donadelli, R.; Lojacono, A.; Benigni, A. Systemic and fetal-maternal nitric oxide synthesis in normal pregnancy and pre-eclampsia. BJOG Int. J. Obstet. Gynaecol. 1996, 103, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, C.; Nisell, H.; Carlstrom, K.; Kublickas, M.; Randmaa, I.; Nylund, L. Acute volume expansion in normal pregnancy and preeclampsia: Effects on plasma atrial natriuretic peptide (ANP) and cyclic guanosine monophosphate (cGMP) concentrations and feto-maternal circulation. Acta Obstet. Gynecol. Scand. 1994, 73, 294–299. [Google Scholar] [CrossRef]
- Itoh, H.; Sagawa, N.; Nanno, H.; Mori, T.; Mukoyama, M.; Itoh, H.; Nakao, K. Impaired Guanosine 3′,5′-Cyclic Phosphate Production in Severe Pregnancy-Induced Hypertension with High Plasma Levels of Atrial and Brain Natriuretic Peptides. Endocr. J. 1997, 44, 389–393. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sandrim, V.C.; Palei, A.C.; Sertório, J.T.; Amaral, L.M.; Cavalli, R.C.; Tanus-Santos, J.E. Alterations in cyclic GMP levels in preeclampsia may reflect increased B-type natriuretic peptide levels and not impaired nitric oxide activity. Clin. Biochem. 2011, 44, 1012–1014. [Google Scholar] [CrossRef]
- Schneider, F.; Lutun, P.; Baldauf, J.-J.; Quirin, L.; Dreyfus, M.; Ritter, J.; Tempé, J.-D. Plasma cyclic GMP concentrations and their relationship with changes of blood pressure levels in pre-eclampsia. Acta Obstet. Gynecol. Scand. 1996, 75, 40–44. [Google Scholar] [CrossRef]
- Dusse, L.M.; Alpoim, P.N.; Lwaleed, B.A.; de Sousa, L.P.; Carvalho, M.; Gomes, K.B. Is there a link between endothelial dysfunction, coagulation activation and nitric oxide synthesis in preeclampsia? Clin. Chim. Acta 2013, 415, 226–229. [Google Scholar] [CrossRef]
- Lopez-Jaramillo, P.; Narvaez, M.; Calle, A.; Rivera, J.; Jacome, P.; Ruano, C.; Nava, E. Cyclic guanosine 3′,5′ monophosphate concentrations in pre-eclampsia: Effects of hydralazine. Br. J. Obstet. Gynaecol. 1996, 103, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Manninen, A.; Vuorinen, P.; Laippala, P.; Tuimala, R.; Vapaatalo, H. Atrial Natriuretic Peptide and Cyclic Guanosine-3′5′-monophosphate in Hypertensive Pregnancy and during Nifedipine Treatment. Pharmacol. Toxicol. 1994, 74, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Okuno, S.; Hamada, H.; Yasuoka, M.; Watanabe, H.; Fujiki, Y.; Yamada, N.; Sohda, S.; Kubo, T. Brain Natriuretic Peptide (BNP) and Cyclic Guanosine Monophosphate (cGMP) Levels in Normal Pregnancy and Preeclampsia. J. Obstet. Gynaecol. Res. 1999, 25, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ren, W.; Lin, L.; Zeng, S.; Huang, L.; Tang, J.; Bi, S.; Pan, J.; Chen, D.; Du, L. Abnormal cGMP-dependent protein kinase I-mediated decidualization in preeclampsia. Hypertens. Res. 2021, 44, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Tang, J.; Li, N.; Zhou, X.; Zhu, X.; Li, W.; Liu, B.; Feng, X.; Tao, J.; Han, B.; et al. New conception for the development of hypertension in preeclampsia. Oncotarget 2016, 7, 78387–78395. [Google Scholar] [CrossRef]
- Coats, L.E.; Bamrick-Fernandez, D.R.; Ariatti, A.M.; Bakrania, B.A.; Rawls, A.Z.; Ojeda, N.B.; Alexander, B.T. Stimulation of soluble guanylate cyclase diminishes intrauterine growth restriction in a rat model of placental ischemia. Am. J. Physiol. Integr. Comp. Physiol. 2021, 320, R149–R161. [Google Scholar] [CrossRef]
- Takushima, S.; Nishi, Y.; Nonoshita, A.; Mifune, H.; Hirata, R.; Tanaka, E.; Doi, R.; Hori, D.; Kamura, T.; Ushijima, K. Changes in the nitric oxide-soluble guanylate cyclase system and natriuretic peptide receptor system in placentas of pregnant Dahl salt-sensitive rats. J. Obstet. Gynaecol. Res. 2014, 41, 540–550. [Google Scholar] [CrossRef]
- Turgut, N.H.; Temiz, T.K.; Turgut, B.; Karadas, B.; Parlak, M.; Bagcivan, I. Investigation of the role of the NO-cGMP pathway on YC-1 and DEA/NO effects on thoracic aorta smooth muscle responses in a rat preeclampsia model. Can. J. Physiol. Pharmacol. 2013, 91, 797–803. [Google Scholar] [CrossRef]
- Sandner, P.; Zimmer, D.P.; Milne, G.T.; Follmann, M.; Hobbs, A.; Stasch, J.-P. Soluble Guanylate Cyclase Stimulators and Activators. Handb. Exp. Pharmacol. 2018, 355–394. [Google Scholar] [CrossRef]
- Bakrania, B.A.; Spradley, F.T.; Patel, B.R.; Travis, A.B.; Sandner, P.; Granger, J.P. Soluble Guanylate Cyclase Activators Increase cGMP Expression and Improve Vascular Function and Placental Ischemia-Induced Hypertension. FASEB J. 2019, 33, 865.13. [Google Scholar] [CrossRef]
- Coats, L.E.; Bakrania, B.A.; Bamrick-Fernandez, D.R.; Ariatti, A.M.; Rawls, A.Z.; Ojeda, N.B.; Alexander, B.T. Soluble guanylate cyclase stimulation in late gestation does not mitigate asymmetric intrauterine growth restriction or cardiovascular risk induced by placental ischemia in the rat. Am. J. Physiol. Circ. Physiol. 2021, 320, H1923–H1934. [Google Scholar] [CrossRef]
- Da Costa, B.P.; Scocco, C.; De Figueiredo, C.P.; Guimaraes, J.A.; Da Costa, B.E.P.; De Figueiredo, C.E.P. Maternal medicine: Increased serum phosphodiesterase activity in women with pre-eclampsia. BJOG Int. J. Obstet. Gynaecol. 2006, 113, 577–579. [Google Scholar] [CrossRef] [PubMed]
- George, E.; Palei, A.C.; Dent, E.A.; Granger, J.P. Sildenafil attenuates placental ischemia-induced hypertension. Am. J. Physiol. Integr. Comp. Physiol. 2013, 305, R397–R403. [Google Scholar] [CrossRef] [PubMed]
- Samangaya, R.A.; Mires, G.; Shennan, A.; Skillern, L.; Howe, D.; McLeod, A.; Baker, P.N. A Randomised, Double-Blinded, Placebo-Controlled Study of the Phosphodiesterase Type 5 Inhibitor Sildenafil for the Treatment of Preeclampsia. Hypertens. Pregnancy 2009, 28, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Trapani, A.J.; Gonçalves, L.F.; Trapani, T.F.; Franco, M.J.; Galluzzo, R.N.; Pires, M.M.S. Comparison between transdermal nitroglycerin and sildenafil citrate in intrauterine growth restriction: Effects on uterine, umbilical and fetal middle cerebral artery pulsatility indices. Ultrasound Obstet. Gynecol. 2015, 48, 61–65. [Google Scholar] [CrossRef]
- Trapani, A., Jr.; Goncalves, L.F.; Trapani, T.F.; Vieira, S.; Pires, M.; Pires, M.M.S. Perinatal and Hemodynamic Evaluation of Sildenafil Citrate for Preeclampsia Treatment: A Randomized Controlled Trial. Obstet. Gynecol. 2016, 128, 253–259. [Google Scholar] [CrossRef]
- Groom, K.M.; McCowan, L.M.; Mackay, L.K.; Lee, A.C.; Gardener, G.; Unterscheider, J.; Sekar, R.; Dickinson, J.E.; Muller, P.; Reid, R.A.; et al. STRIDER NZAus: A multicentre randomised controlled trial of sildenafil therapy in early-onset fetal growth restriction. BJOG Int. J. Obstet. Gynaecol. 2019, 126, 997–1006. [Google Scholar] [CrossRef]
- Pels, A.; Derks, J.; Elvan-Taspinar, A.; van Drongelen, J.; de Boer, M.; Duvekot, H.; van Laar, J.; van Eyck, J.; Al-Nasiry, S.; Sueters, M.; et al. Maternal Sildenafil vs Placebo in Pregnant Women with Severe Early-Onset Fetal Growth Restriction: A Randomized Clinical Trial. JAMA Netw. Open 2020, 3, e205323. [Google Scholar] [CrossRef]
- Ferreira, R.D.D.S.; Negrini, R.; Bernardo, W.M.; Simões, R.; Piato, S. The effects of sildenafil in maternal and fetal outcomes in pregnancy: A systematic review and meta-analysis. PLoS ONE 2019, 14, e0219732. [Google Scholar] [CrossRef]
- Furuhashi, F.; Tanaka, H.; Maki, S.; Tsuji, M.; Magawa, S.; Kaneda, M.K.; Nii, M.; Tanaka, K.; Ogura, T.; Nishimura, Y.; et al. Tadalafil treatment for preeclampsia (medication in preeclampsia; MIE): A multicenter phase II clinical trial. J. Matern.-Neonatal Med. 2019, 34, 3709–3715. [Google Scholar] [CrossRef]
- Furuhashi, F.H.; Tanaka, H.; Kaneda, M.K.; Maki, S.; Nii, M.; Umekawa, T.; Osato, K.; Kamimoto, Y.; Ikeda, T. Safety trial of tadalafil administered for the treatment of preeclampsia. J. Matern.-Neonatal Med. 2018, 33, 167–170. [Google Scholar] [CrossRef]
- Herraiz, S.; Pellicer, B.; Serra, V.; Cauli, O.; Cortijo, J.; Felipo, V.; Pellicer, A. Sildenafil citrate improves perinatal outcome in fetuses from pre-eclamptic rats. BJOG Int. J. Obstet. Gynaecol. 2012, 119, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, N.; Wang, B.; Niu, X.; Cai, W.; Li, Y.; Li, Y.; Chen, S. Effect and mechanism of prophylactic use of tadalafil during pregnancy on l-NAME-induced preeclampsia-like rats. Placenta 2020, 99, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Motta, C.; Grosso, C.; Zanuzzi, C.; Molinero, D.; Picco, N.; Bellingeri, R.; Alustiza, F.; Barbeito, C.; Vivas, A.; Romanini, M. Effect of Sildenafil on Pre-Eclampsia-Like Mouse Model Induced By L-Name. Reprod. Domest. Anim. 2015, 50, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Ramesar, S.; Mackraj, I.; Gathiram, P.; Moodley, J. Sildenafil citrate improves fetal outcomes in pregnant, l-NAME treated, Sprague–Dawley rats. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 149, 22–26. [Google Scholar] [CrossRef]
- Soobryan, N.; Murugesan, S.; Phoswa, W.; Gathiram, P.; Moodley, J.; Mackraj, I. The effects of sildenafil citrate on uterine angiogenic status and serum inflammatory markers in an L-NAME rat model of pre-eclampsia. Eur. J. Pharmacol. 2016, 795, 101–107. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Umekawa, T.; Maki, S.; Kubo, M.; Nii, M.; Tanaka, K.; Tanaka, H.; Osato, K.; Kamimoto, Y.; Kondo, E.; et al. Tadalafil Improves L-NG-Nitroarginine Methyl Ester-Induced Preeclampsia with Fetal Growth Restriction-Like Symptoms in Pregnant Mice. Am. J. Hypertens. 2017, 31, 89–96. [Google Scholar] [CrossRef]
- Gillis, E.E.; Mooney, J.N.; Garrett, M.R.; Granger, J.P.; Sasser, J.M. Sildenafil Treatment Ameliorates the Maternal Syndrome of Preeclampsia and Rescues Fetal Growth in the Dahl Salt–Sensitive Rat. Hypertension 2016, 67, 647–653. [Google Scholar] [CrossRef]
- Stanley, J.L.; Andersson, I.J.; Poudel, R.; Rueda-Clausen, C.F.; Sibley, C.P.; Davidge, S.T.; Baker, P.N. Sildenafil Citrate Rescues Fetal Growth in the Catechol-O-Methyl Transferase Knockout Mouse Model. Hypertension 2012, 59, 1021–1028. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Species/Experimental Model | Circulating | Tissue |
|---|---|---|
| Human | Shaheen G. et al. [58] ↓ NOx Possomato-Vieira J.S. et al. [59] ↓ NOx Pereira D.A. et al. [60] ↓ NOx McCann Haworth S.M. et al. [61] ↓ NOx Tashie W. et al. [62] ↓ NOx Kim S. et al. [63] ↑ NOx Mazloomi S. et al. [64] ↓ NOS Lai H. et al. [65] ↓ NOx Ajadi I. et al. [66] ↓ NOx Serrano-Berrones et al. [67] ↓ NOx Deniz R. et al. [68] ↓ NOx Bos M. et al. [69] ↓ NOx ElMonier A.A. et al. [70] ↓ NOx Hodzic J. et al. [71] ↓ NOx Rocha-Penha L. et al. [72] ↓ NOx Bambrana V. et al. [15] ↓ NOx Lai H. et al. [65] ↓ NOx | Blood vessels: Lorca R.A. et al. [73] ↓ NOS function Primary HUVECs: Chen J. et al. [74] ↓ NOS3 Salsoso R. et al. [75] ↓ NOS activity Placenta: Mishra J.S. et al. [76] ↓ NOS3 K.-Y. Jung et al. [77] ↑ NOS2 Kim S. et al. [63] ↑ NOS2, ↓ NOS3 Mukosera G.T. et al. [78] ↑ NOx Shaheen G. et al. [79] ↓ NOS3 Guerby P. et al. [80] ↓ NOS3 |
| Hitzerd E. et al. [81] ↑ maternal placenta NOS3, ↓ maternal placental NOS2, = fetal placenta NOS2, ↑ fetal placenta NOS2 Guerby P. et al. [82] ↓ NOS3 Li F.F. et al. [55] ↓ NOS2, NOS3 Motta-Mejia C. et al. [83] ↓ NOS3 Du L. et al. [84] ↑ NOS2, ↓ NOS3 | ||
| Non-human primate/EarlyPregnancy Excess of Estradiol | Albrecht E.D. et al. [85] ↓ NOx | Blood vessels: Albrecht E.D. et al. [85] ↓ NOS3 |
| Rat/RUPP | Travis O.K. et al. [86] ↓ NOx Palei A.C. et al. [17] ↓ NOx Cottrell J.N. et al. [87] ↓ NOx El-Saka M.H. et al. [88] ↓ NOx Wang C. et al. [89] ↓ NOx Amaral L.M. et al. [90] ↓ NOx Jammalamadaga V.S. et al. [91] ↓ NOx Santiago-Font J.A. et al. [49] ↓ NOx | Blood vessels: Younes S.T. et al. [92] = NOS3 Ma S.L. et al. [93] ↓ NOS3, NOx Zhu M. et al. [94] ↓ NOS3 Placenta: Tengfei Z. et al. [95] ↓ NOS3, ↑ NOS2 Wang C. et al. [89] ↓ NOS3 |
| Rat/DOCA-salt | Wang G.-J. et al. [96] ↓ NOx | Placenta: Chimini J.S. et al. [97] ↓ NOx Tyurenkov I.N. et al. [74] ↓ NOS3, ↑ NOS2 |
| Rat/Elevated Testosterone | Mishra J.S. et al. [76] ↓ NOx | Blood vessels: Mishra JS et al. [76] ↓ NOS3 |
| Rat/Lipopolysaccharide (LPS) | Ou M. et al. [98] ↓ NOx Hu J. et al. [99] ↑ NOx | - |
| Mouse/AntiphospholipidSyndrome | Lefkou E. et al. [100] ↓ NOx | - |
| Mouse/Human PE Serum Injection | Purnamayanti N.M.D. et al. [101] ↓ NOx | - |
| Mouse/Prolactin Overexpression | - | Kidney: Chang A.S. et al. [102] ↓ NOx, ↑ NOS2 |
| Mouse/Progranulin Deficiency | - | Placenta: Xu B. et al. [103] ↓ NOS3 |
| Mouse/Hypoxia Chamber | - | Blood vessels: Lane S.L. et al. [104] ↓ NOS function |
| Mouse/sFlt-1 Adenovirus | - | Blood vessels: Zhang S. et al. [105] ↓ NOS3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palei, A.C.; Granger, J.P.; Spradley, F.T. Placental Ischemia Says “NO” to Proper NOS-Mediated Control of Vascular Tone and Blood Pressure in Preeclampsia. Int. J. Mol. Sci. 2021, 22, 11261. https://doi.org/10.3390/ijms222011261
Palei AC, Granger JP, Spradley FT. Placental Ischemia Says “NO” to Proper NOS-Mediated Control of Vascular Tone and Blood Pressure in Preeclampsia. International Journal of Molecular Sciences. 2021; 22(20):11261. https://doi.org/10.3390/ijms222011261
Chicago/Turabian StylePalei, Ana C., Joey P. Granger, and Frank T. Spradley. 2021. "Placental Ischemia Says “NO” to Proper NOS-Mediated Control of Vascular Tone and Blood Pressure in Preeclampsia" International Journal of Molecular Sciences 22, no. 20: 11261. https://doi.org/10.3390/ijms222011261
APA StylePalei, A. C., Granger, J. P., & Spradley, F. T. (2021). Placental Ischemia Says “NO” to Proper NOS-Mediated Control of Vascular Tone and Blood Pressure in Preeclampsia. International Journal of Molecular Sciences, 22(20), 11261. https://doi.org/10.3390/ijms222011261