
BET Proteins as Attractive Targets for Cancer Therapeutics


Abstract
1. Introduction
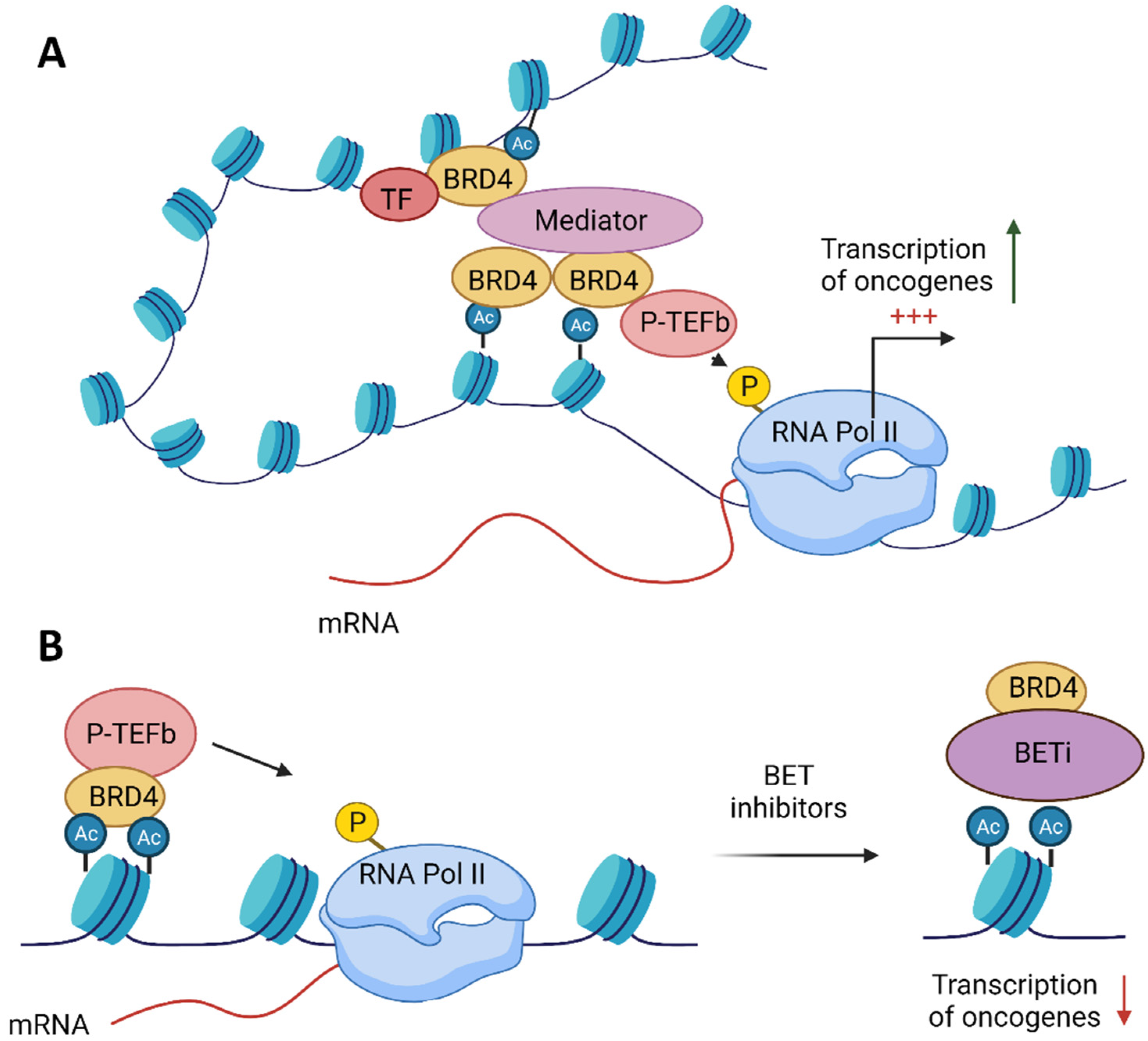
2. BET Proteins’ Function
3. BET Proteins in Cancer
4. BET Inhibitors Target MYC
5. BET Inhibitors in Clinical Trials
6. Resistance to BET Inhibitors
7. BET Inhibitors in Combination Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classes of Compounds | Compounds | Preclinical Study | Clinical Trial | |||
|---|---|---|---|---|---|---|
| Cancer | References | Phase/Status | Cancer | References | ||
| Small molecular weight inhibitors | ALK inhibitors | Lymphoma | [57] | |||
| BTK inhibitors | Lymphoma | [57,89,100,102,103,104,105,106,107] | ||||
| CDK inhibitors | Lymphoma | [105,106] | ||||
| Osteosarcoma | [108] | |||||
| BCL2/MCL1 inhibitors | ALL | [109] | I/not yet recruiting | MF | NCT04480086 | |
| AML | [62,64,110] | I, completed | Lymphoma | NCT03255096 | ||
| LC | [111] | I, completed | Advanced solid tumours and haematological malignancies | NCT02391480 | ||
| Lymphoma | [105,106,112,113,114] | I, recruiting | MF | NCT04454658 | ||
| EGFR/ERBB2 inhibitors | BC | [115] | ||||
| FLT3/ERBB2 inhibitors | AML | [116] | ||||
| Hedgehog inhibitors | Lymphoma | [117] | ||||
| JAK inhibitors | AML | [118] | I/II, recruiting | MF | NCT02158858 | |
| MF | [66,67] | I, not yet recruiting | MF | NCT04480086 | ||
| I, recruiting | MF | NCT04454658 | ||||
| I/II, terminated | Solid tumours | NCT02711137 | ||||
| MEK/ERK inhibitors | AML | [119] | I/II, withdrawn | Solid tumours | NCT03266159 | |
| BC | [119] | |||||
| CRC | [119,120] | |||||
| Lymphoma | [89] | |||||
| MM | [119] | |||||
| Neuroblastoma | [121] | |||||
| NSCLC | [110] | |||||
| OC | [81] | |||||
| PrC | [119] | |||||
| Thyroid cancer | [122] | |||||
| mTOR inhibitors | BC | [123] | ||||
| Glioblastoma | [124] | |||||
| Lymphoma | [89,102,103,104,107] | |||||
| OS | [125] | |||||
| PARP inhibitors | BC | [99,100,126] | I/II, terminated | Solid tumours | NCT02711137 | |
| Bladder cancer | [100] | |||||
| Endometrial cancer | [100] | |||||
| LC | [127] | |||||
| OC | [35,100,101] | |||||
| PC | [100] | |||||
| PI3K inhibitors | BC | [81,126] | ||||
| CRC | [126] | |||||
| Lymphoma | [102,104,128] | |||||
| Glioblastoma | [126] | |||||
| OC | [80,126] | |||||
| PIKK inhibitors | Lymphoma | [129] | ||||
| Proteasome inhibitors | MM | [62,130] | ||||
| Antibodies | Anti-CD20 monoclonal antibodies | Lymphoma | [104,107,131] | |||
| Immune modulators | Immunomodulatory drugs (IMiDs) | Lymphoma | [89,104,132,133] | |||
| MM | [134] | |||||
| Anti-PD-1 monoclonal antibodies | Lymphoma | [135] | I/II, not yet recruiting | Solid tumours and haematological malignancies | NCT02419417 | |
| II, recruiting | Metastatic CRPC | NCT04471974 | ||||
| I, not yet recruiting | Solid tumours | NCT04840589 | ||||
| I/II, active, not recruiting, | Advanced tumours | NCT02419417 | ||||
| Anti-4-1BB monoclonal antibodies | Lymphoma | [135] | ||||
| Chimeric antigen receptor (CAR) T-cells | ALL | [136] | ||||
| Epigenetic drugs | EZH2 inhibitors | Lymphoma | [103,137] | |||
| HDAC inhibitors | AML | [116] | I/II withdrawn | Advanced and refractory solid tumours and lymphomas | NCT03925428 | |
| Bladder cancer | [138,139] | |||||
| BC | [140] | |||||
| Chondrosarcoma | [93] | |||||
| Glioblastoma | [141] | |||||
| LC | [142] | |||||
| Lymphoma | [57,104,106,107,113,143,144,145] | |||||
| Melanoma | [146] | |||||
| Neuroblastoma | [147] | |||||
| PC | [148,149,150] | |||||
| Sarcoma | [151] | |||||
| Azacytidine | AML | [62] | I/II, withdrawn | AML | NCT02303782 | |
| I/II completed | Haematological malignancies | NCT02543879 | ||||
| I/II, terminated | Solid tumours | NCT02711137 | ||||
| Decitabine | Lymphoma | [104] | ||||
| Chemotherapy | Gemcitabine | I/II, terminated | Solid tumours | NCT02711137 | ||
| Paclitaxel | I/II, terminated | Solid tumours | NCT02711137 | |||
| Temozolomide | Glioblastoma [124] | |||||
| Hormone therapy | Antiandrogen | PrC [152] | I/II, active, completed | PrC | NCT02711956 | |
| II, recruiting | PrC | NCT04471974 | ||||
| I/II, terminated | PrC | NCT02607228 | ||||
| I/II, terminated | Solid tumours | NCT02711137 | ||||
| Estrogen receptor degrader | BC [153] | I, completed | BC | NCT02392611 | ||
| I/II, terminated | BC | NCT02983604 | ||||
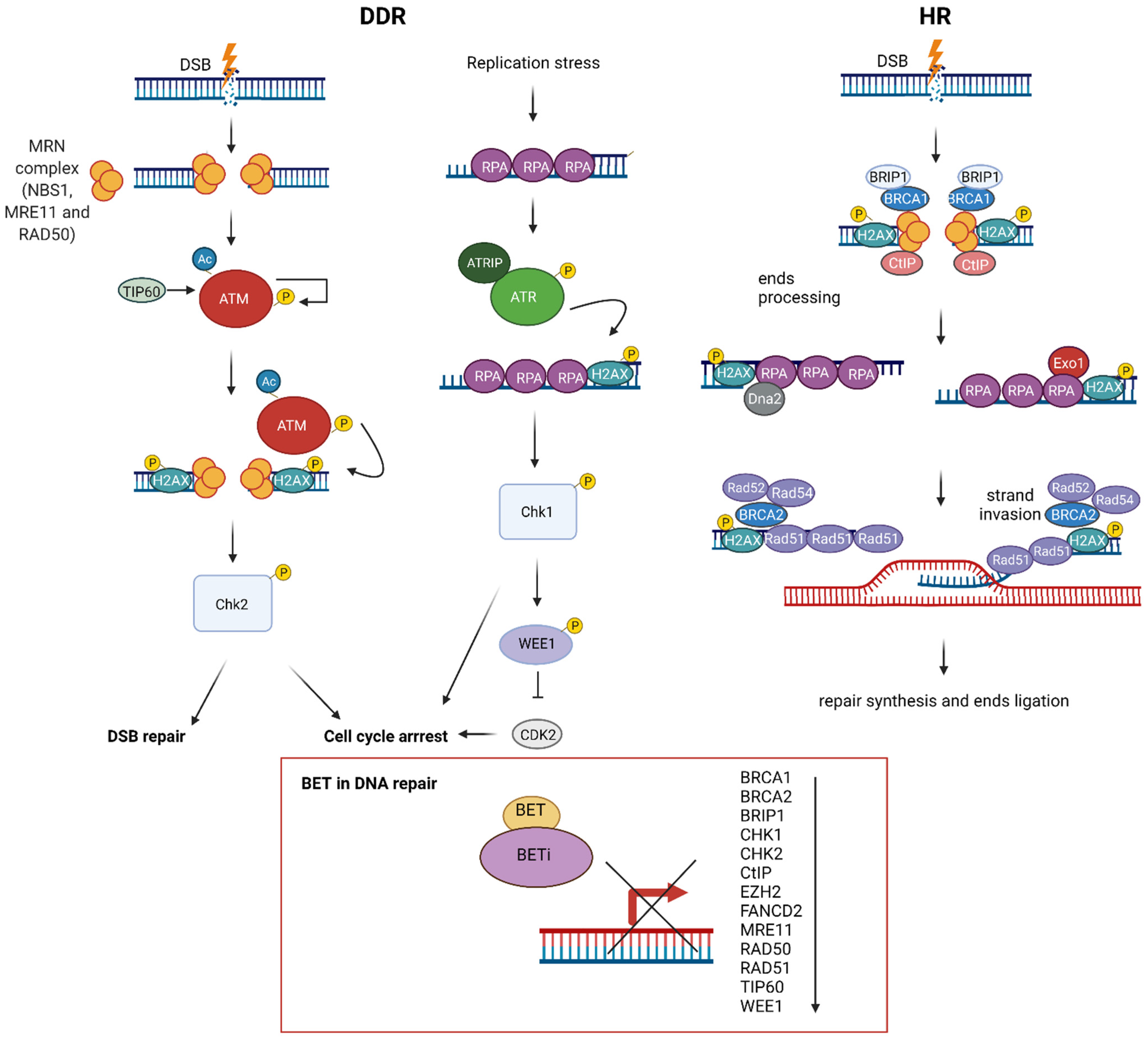
8. BET in DNA Repair
9. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hnisz, D.; Abraham, B.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-Y.; Chiang, C.-M. The Double Bromodomain-containing Chromatin Adaptor Brd4 and Transcriptional Regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef] [PubMed]
- Florence, B. You bet-cha: A novel family of transcriptional regulators. Front. Biosci. 2001, 6, D1008–D1018. [Google Scholar] [CrossRef] [PubMed]
- Belkina, A.; Denis, G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Thota, A.; Rao, M.R.S. Insights into Role of Bromodomain, Testis-specific (Brdt) in Acetylated Histone H4-dependent Chromatin Remodeling in Mammalian Spermiogenesis. J. Biol. Chem. 2012, 287, 6387–6405. [Google Scholar] [CrossRef]
- Pivot-Pajot, C.; Caron, C.; Govin, J.; Vion, A.; Rousseaux, S.; Khochbin, S. Acetylation-Dependent Chromatin Reorganization by BRDT, a Testis-Specific Bromodomain-Containing Protein. Mol. Cell. Biol. 2003, 23, 5354–5365. [Google Scholar] [CrossRef]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 Extraterminal Domain Confers Transcription Activation Independent of pTEFb by Recruiting Multiple Proteins, Including NSD. Mol. Cell. Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef]
- Shen, C.; Ipsaro, J.; Shi, J.; Milazzo, J.P.; Wang, E.; Roe, J.-S.; Suzuki, Y.; Pappin, D.J.; Joshua-Tor, L.; Vakoc, C.R. NSD3-Short Is an Adaptor Protein that Couples BRD4 to the CHD8 Chromatin Remodeler. Mol. Cell 2015, 60, 847–859. [Google Scholar] [CrossRef]
- Zhang, Q.; Zeng, L.; Shen, C.; Ju, Y.; Konuma, T.; Zhao, C.; Vakoc, C.R.; Zhou, M.-M. Structural Mechanism of Transcriptional Regulator NSD3 Recognition by the ET Domain of BRD. Structure 2016, 24, 1201–1208. [Google Scholar] [CrossRef]
- Zaware, N.; Zhou, M.-M. Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol. 2019, 26, 870–879. [Google Scholar] [CrossRef]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.-S.; Brady, J.N.; Ozato, K. The Bromodomain Protein Brd4 Is a Positive Regulatory Component of P-TEFb and Stimulates RNA Polymerase II-Dependent Transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Yang, Z.; Yik, J.H.N.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for Stimulation of Transcriptional Elongation by the Bromodomain Protein Brd. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Jiang, Y.W.; Veschambre, P.; Erdjument-Bromage, H.; Tempst, P.; Conaway, J.W.; Conaway, R.C.; Kornberg, R.D. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc. Natl. Acad. Sci. USA 1998, 95, 8538–8543. [Google Scholar] [CrossRef]
- Denis, G.V.; McComb, M.E.; Faller, D.V.; Sinha, A.; Romesser, P.B.; Costello, C.E. Identification of Transcription Complexes that Contain the Double Bromodomain Protein Brd2 and Chromatin Remodeling Machines. J. Proteome Res. 2006, 5, 502–511. [Google Scholar] [CrossRef]
- LeRoy, G.; Rickards, B.; Flint, S. The Double Bromodomain Proteins Brd2 and Brd3 Couple Histone Acetylation to Transcription. Mol. Cell 2008, 30, 51–60. [Google Scholar] [CrossRef]
- Sinha, A.; Faller, D.V.; Denis, G.V. Bromodomain analysis of Brd2-dependent transcriptional activation of cyclin A. Biochem. J. 2005, 387, 257–269. [Google Scholar] [CrossRef]
- Denis, G.V.; Vaziri, C.; Guo, N.; Faller, U.V. RING3 kinase transactivates promoters of cell cycle regulatory genes through E2F. Cell Growth Differ. Mol. Boil. J. Am. Assoc. Cancer Res. 2000, 11, 417–424. [Google Scholar]
- Dai, J.; Zhou, S.; Ge, Q.; Qin, J.; Li, J.; Ju, H.; Cao, Y.; Zheng, M.; Li, C.; Gao, X.; et al. Recruitment of Brd3 and Brd4 to acetylated chromatin is essential for proinflammatory cytokine-induced matrix-degrading enzyme expression. J. Orthop. Surg. Res. 2019, 14, 59. [Google Scholar] [CrossRef] [PubMed]
- Lamonica, J.M.; Deng, W.; Kadauke, S.; Campbell, A.E.; Gamsjaeger, R.; Wang, H.; Cheng, Y.; Billin, A.; Hardison, R.; Mackay, J.; et al. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc. Natl. Acad. Sci. USA 2011, 108, E159–E168. [Google Scholar] [CrossRef] [PubMed]
- Stonestrom, A.; Hsu, S.C.; Jahn, K.S.; Huang, P.; Keller, C.A.; Giardine, B.M.; Kadauke, S.; Campbell, A.E.; Evans, P.; Hardison, R.; et al. Functions of BET proteins in erythroid gene expression. Blood 2015, 125, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, H.; Blanton, W.P.; Belkina, A.; LeBrasseur, N.; Denis, G.V. Brd2 disruption in mice causes severe obesity without Type 2 diabetes. Biochem. J. 2009, 425, 71–85. [Google Scholar] [CrossRef]
- Berkovits, B.D.; Wang, L.; Guarnieri, P.; Wolgemuth, D.J. The testis-specific double bromodomain-containing protein BRDT forms a complex with multiple spliceosome components and is required for mRNA splicing and 3′-UTR truncation in round spermatids. Nucleic Acids Res. 2012, 40, 7162–7175. [Google Scholar] [CrossRef]
- Floyd, S.; Pacold, M.E.; Huang, Q.; Clarke, S.M.; Lam, F.C.; Cannell, I.; Bryson, B.D.; Rameseder, J.; Lee, M.; Blake, E.J.; et al. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nat. Cell Biol. 2013, 498, 246–250. [Google Scholar] [CrossRef]
- Phelps, M.A.; Lin, T.S.; Johnson, A.J.; Hurh, E.; Rozewski, D.M.; Farley, K.L.; Wu, D.; Blum, K.A.; Fischer, B.; Mitchell, S.M.; et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood 2009, 113, 2637–2645. [Google Scholar] [CrossRef]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. c-Myc Regulates Transcriptional Pause Release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef]
- Peng, J.; Zhu, Y.; Milton, J.T.; Price, D. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998, 12, 755–762. [Google Scholar] [CrossRef]
- Dey, A.; Nishiyama, A.; Karpova, T.; McNally, J.; Ozato, K. Brd4 Marks Select Genes on Mitotic Chromatin and Directs Postmitotic Transcription. Mol. Biol. Cell 2009, 20, 4899–4909. [Google Scholar] [CrossRef]
- French, C.A.; Miyoshi, I.; Aster, J.C.; Kubonishi, I.; Kroll, T.G.; Cin, P.D.; Vargas, S.O.; Perez-Atayde, A.R.; Fletcher, J.A. BRD4 Bromodomain Gene Rearrangement in Aggressive Carcinoma with Translocation t(15;19). Am. J. Pathol. 2001, 159, 1987–1992. [Google Scholar] [CrossRef]
- French, C.A.; Miyoshi, I.; Kubonishi, I.; Grier, H.E.; Perez-Atayde, A.R.; Fletcher, J.A. BRD4-NUT fusion oncogene: A novel mechanism in aggressive carcinoma. Cancer Res. 2003, 63, 304–307. [Google Scholar] [PubMed]
- French, C.A.; Ramirez, C.; Kolmakova, J.; Hickman, T.; Cameron, M.J.; Thyne, M.E.; Kutok, J.L.; Toretsky, J.A.; Tadavarthy, A.K.; Kees, U.R.; et al. BRD–NUT oncoproteins: A family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 2007, 27, 2237–2242. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-K.; Louzada, S.; An, Y.; Kim, S.Y.; Youk, J.; Park, S.; Koo, S.H.; Keam, B.; Jeon, Y.K.; Ku, J.-L.; et al. Complex chromosomal rearrangements by single catastrophic pathogenesis in NUT midline carcinoma. Ann. Oncol. 2017, 28, 890–897. [Google Scholar] [CrossRef]
- Grayson, A.R.; Walsh, E.M.; Cameron, M.J.; Godec, J.; Ashworth, T.; Ambrose, J.M.; Aserlind, A.B.; Wang, H.; Evan, G.I.; Kluk, M.J.; et al. MYC, a downstream target of BRD-NUT, is necessary and sufficient for the blockade of differentiation in NUT midline carcinoma. Oncogene 2014, 33, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination–proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.A.; Conery, A.R.; Bryant, B.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nat. Cell Biol. 2011, 478, 524–528. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Fujisawa, T.; Filippakopoulos, T.F.P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-S.; Kim, K.-H.; Jeon, Y.-K.; Kim, S.-H.; Jang, S.-G.; Ku, J.-L.; Park, J.-G. Promoter hypermethylation of the ADAM23 gene in colorectal cancer cell lines and cancer tissues. Int. J. Cancer 2009, 124, 1258–1262. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.-W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nat. Cell Biol. 2011, 478, 529–533. [Google Scholar] [CrossRef]
- Puissant, A.; Frumm, S.M.; Alexe, G.; Bassil, C.F.; Qi, J.; Chanthery, Y.H.; Nekritz, E.A.; Zeid, R.; Gustafson, W.C.; Greninger, P.; et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013, 3, 308–323. [Google Scholar] [CrossRef]
- Coudé, M.-M.; Braun, T.; Berrou, J.; Dupont, M.; Bertrand, S.; Masse, A.; Raffoux, E.; Itzykson, R.; Delord, M.; Riveiro, M.E.; et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 2015, 6, 17698–17712. [Google Scholar] [CrossRef] [PubMed]
- Wyce, A.; Degenhardt, Y.; Bai, Y.; Le, B.; Korenchuk, S.; Crouthamel, M.-C.; McHugh, C.F.; Vessella, R.; Creasy, C.L.; Tummino, P.J.; et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget 2013, 4, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Eder, J.P.; LoRusso, P.M. BET inhibitors: A novel epigenetic approach. Ann. Oncol. 2017, 28, 1776–1787. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Bertoni, F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2017, 8, 24–36. [Google Scholar] [CrossRef]
- Stathis, A.; Zucca, E.; Bekradda, M.; Gomez-Roca, C.; Delord, J.-P.; de La Motte Rouge, T.; Uro-Coste, E.; De Braud, F.; Pelosi, G.; French, C.A. Clinical Response of Carcinomas Harboring the BRD4–NUT Oncoprotein to the Targeted Bromodomain Inhibitor OTX015/MK-8628. Cancer Discov. 2016, 6, 492–500. [Google Scholar] [CrossRef]
- Bauer, D.E.; Mitchell, C.M.; Strait, K.M.; Lathan, C.S.; Stelow, E.B.; Lüer, S.C.; Muhammed, S.; Evans, A.G.; Sholl, L.M.; Rosai, J.; et al. Clinicopathologic Features and Long-term Outcomes of NUT Midline Carcinoma. Clin. Cancer Res. 2012, 18, 5773–5779. [Google Scholar] [CrossRef]
- Lewin, J.; Soria, J.-C.; Stathis, A.; Delord, J.-P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib Trial With Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef]
- Piha-Paul, S.A.; Hann, C.L.; French, C.A.; Cousin, S.; Braña, I.; Cassier, P.A.; Moreno, V.; De Bono, J.S.; Harward, S.D.; Ferron-Brady, G.; et al. Phase 1 Study of Molibresib (GSK525762), a Bromodomain and Extra-Terminal Domain Protein Inhibitor, in NUT Carcinoma and Other Solid Tumors. JNCI Cancer Spectr. 2019, 4, pkz093. [Google Scholar] [CrossRef]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, e186–e195. [Google Scholar] [CrossRef]
- Bradbury, R.H.; Callis, R.; Carr, G.R.; Chen, H.; Clark, E.; Feron, L.; Glossop, S.; Graham, M.A.; Hattersley, M.; Jones, C.; et al. Optimization of a Series of Bivalent Triazolopyridazine Based Bromodomain and Extraterminal Inhibitors: The Discovery of (3R)-4-[2-[4-[1-(3-Methoxy-[1,2,4]triazolo[4,3-b]pyridazin-6-yl)-4-piperidyl]phenoxy]ethyl]-1,3-dimethyl-piperazin-2-one (AZD5153). J. Med. Chem. 2016, 59, 7801–7817. [Google Scholar] [CrossRef] [PubMed]
- Rhyasen, G.W.; Hattersley, M.M.; Yao, Y.; Dulak, A.; Wang, W.; Petteruti, P.; Dale, I.L.; Boiko, S.; Cheung, T.; Zhang, J.; et al. AZD5153: A Novel Bivalent BET Bromodomain Inhibitor Highly Active against Hematologic Malignancies. Mol. Cancer Ther. 2016, 15, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.; Chen, J.; Zhou, Y.; Wang, Z.; Ma, Z.; Xu, C.; Jiang, M. AZD5153 Inhibits Prostate Cancer Cell Growth In Vitro and In Vivo. Cell. Physiol. Biochem. 2018, 50, 798–809. [Google Scholar] [CrossRef]
- Xu, K.; Chen, D.; Qian, D.; Zhang, S.; Zhang, Y.; Guo, S.; Ma, Z.; Wang, S. AZD5153, a novel BRD4 inhibitor, suppresses human thyroid carcinoma cell growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2018, 499, 531–537. [Google Scholar] [CrossRef]
- Wang, J.S.-Z.; De Vita, S.; Karlix, J.L.; Cook, C.; Littlewood, G.M.; Hattersley, M.M.; Moorthy, G.; Edlund, H.; Fabbri, G.; Sachsenmeier, K.F.; et al. First-in-human study of AZD5153, a small molecule inhibitor of bromodomain protein 4 (BRD4), in patients (pts) with relapsed/refractory (RR) malignant solid tumor and lymphoma: Preliminary data. J. Clin. Oncol. 2019, 37, 3085. [Google Scholar] [CrossRef]
- Postel-Vinay, S.; Herbschleb, K.; Massard, C.; Woodcock, V.; Soria, J.-C.; Walter, A.O.; Ewerton, F.; Poelman, M.; Benson, N.; Ocker, M.; et al. First-in-human phase I study of the bromodomain and extraterminal motif inhibitor BAY 1238097: Emerging pharmacokinetic/pharmacodynamic relationship and early termination due to unexpected toxicity. Eur. J. Cancer 2019, 109, 103–110. [Google Scholar] [CrossRef]
- Bui, M.H.; Lin, X.; Albert, D.H.; Li, L.; Lam, L.T.; Faivre, E.J.; Warder, S.E.; Huang, X.; Wilcox, D.; Donawho, C.K.; et al. Preclinical Characterization of BET Family Bromodomain Inhibitor ABBV-075 Suggests Combination Therapeutic Strategies. Cancer Res. 2017, 77, 2976–2989. [Google Scholar] [CrossRef]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.-W.; Bamborough, P.; Petretich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef]
- Borthakur, G.; Odenike, O.; Aldoss, I.; Rizzieri, D.A.; Prebet, T.; Chen, C.; Popovic, R.; Modi, D.A.; Joshi, R.H.; Wolff, J.E.; et al. A phase 1 study of the pan-bromodomain and extraterminal inhibitor mivebresib (ABBV-075) alone or in combination with venetoclax in patients with relapsed/refractory acute myeloid leukemia. Cancer 2021, 127, 2943–2953. [Google Scholar] [CrossRef]
- Blum, K.; Abramson, J.; Maris, M.; Flinn, I.; Goy, A.; Mertz, J.; Sims, R.; Garner, F.; Senderowicz, A.; Younes, A. A phase I study of CPI-0610, a bromodomain and extra terminal protein (BET) inhibitor in patients with relapsed or refractory lymphoma. Ann. Oncol. 2018, 29, iii7–iii9. [Google Scholar] [CrossRef]
- Mascarenhas, J.O.; Rampal, R.K.; Kosiorek, H.E.; Bhave, R.; Hexner, E.; Wang, E.S.; Gerds, A.; Abboud, C.N.; Kremyanskaya, M.; Berenzon, D.; et al. Phase 2 study of ruxolitinib and decitabine in patients with myeloproliferative neoplasm in accelerated and blast phase. Blood Adv. 2020, 4, 5246–5256. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mascarenhas, J.; Kremyanskaya, M.; Hoffman, R.; Rampal, R.K.; Gupta, V.; Talpaz, M.; Granacher, N.; Leber, B.; Kiladjian, J.J.; et al. CPI-0610, Bromodomain and Extraterminal Domain Protein (BET) Inhibitor, as “Add-on” to Ruxolitinib, in Advanced Myelofibrosis Patients with Suboptimal Response: Update of MANIFEST Phase 2 Study. Available online: https://ash.confex.com/ash/2020/webprogram/Paper140891.html (accessed on 1 September 2021).
- Parikh, S.A.; French, C.A.; Costello, B.A.; Marks, R.S.; Dronca, R.S.; Nerby, C.L.; Roden, A.C.; Peddareddigari, V.G.; Hilton, J.; Shapiro, G.I.; et al. NUT Midline Carcinoma: An Aggressive Intrathoracic Neoplasm. J. Thorac. Oncol. 2013, 8, 1335–1338. [Google Scholar] [CrossRef] [PubMed]
- Falchook, G.; Rosen, S.; Lorusso, P.; Watts, J.; Gupta, S.; Coombs, C.C.; Talpaz, M.; Kurzrock, R.; Mita, M.; Cassaday, R.; et al. Development of 2 Bromodomain and Extraterminal Inhibitors With Distinct Pharmacokinetic and Pharmacodynamic Profiles for the Treatment of Advanced Malignancies. Clin. Cancer Res. 2020, 26, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Ameratunga, M.; Braña, I.; Bono, P.; Postel-Vinay, S.; Plummer, R.; Aspegren, J.; Korjamo, T.; Snapir, A.; de Bono, J.S. First-in-human Phase 1 open label study of the BET inhibitor ODM-207 in patients with selected solid tumours. Br. J. Cancer 2020, 123, 1730–1736. [Google Scholar] [CrossRef]
- Roboz, G.J.; Desai, P.; Lee, S.; Ritchie, E.K.; Winer, E.S.; DeMario, M.; Brennan, B.; Nüesch, E.; Chesne, E.; Brennan, L.; et al. A dose escalation study of RO6870810/TEN-10 in patients with acute myeloid leukemia and myelodysplastic syndrome. Leuk. Lymphoma 2021, 62, 1740–1748. [Google Scholar] [CrossRef]
- Shapiro, G.I.; LoRusso, P.; Dowlati, A.; Do, K.T.; Jacobson, C.A.; Vaishampayan, U.; Weise, A.; Caimi, P.F.; Eder, J.P.; French, C.A.; et al. A Phase 1 study of RO6870810, a novel bromodomain and extra-terminal protein inhibitor, in patients with NUT carcinoma, other solid tumours, or diffuse large B-cell lymphoma. Br. J. Cancer 2021, 124, 744–753. [Google Scholar] [CrossRef]
- Settleman, J. Bet on drug resistance. Nat. Cell Biol. 2016, 529, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.N.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.-S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nat. Cell Biol. 2015, 525, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.E.; Eom, J.-I.; Jeung, H.-K.; Cheong, J.-W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. AMPK–ULK1-Mediated Autophagy Confers Resistance to BET Inhibitor JQ1 in Acute Myeloid Leukemia Stem Cells. Clin. Cancer Res. 2016, 23, 2781–2794. [Google Scholar] [CrossRef]
- Jang, J.E.; Eom, J.-I.; Jeung, H.-K.; Cheong, J.-W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. Targeting AMPK-ULK1-mediated autophagy for combating BET inhibitor resistance in acute myeloid leukemia stem cells. Autophagy 2017, 13, 761–762. [Google Scholar] [CrossRef]
- Wu, T.; Wang, G.; Chen, W.; Zhu, Z.; Liu, Y.; Huang, Z.; Huang, Y.; Du, P.; Yang, Y.; Liu, C.-Y.; et al. Co-inhibition of BET proteins and NF-κB as a potential therapy for colorectal cancer through synergistic inhibiting MYC and FOXM1 expressions. Cell Death Dis. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nat. Cell Biol. 2016, 529, 413–417. [Google Scholar] [CrossRef]
- Kumar, K.; Raza, S.S.; Knab, L.M.; Chow, C.R.; Kwok, B.; Bentrem, D.J.; Popovic, R.; Ebine, K.; Licht, J.D.; Munshi, H.G. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci. Rep. 2015, 5, 9489. [Google Scholar] [CrossRef]
- Kurimchak, A.M.; Shelton, C.; Duncan, K.E.; Johnson, K.J.; Brown, J.; O’Brien, S.; Gabbasov, R.; Fink, L.S.; Li, Y.; Lounsbury, N.; et al. Resistance to BET Bromodomain Inhibitors Is Mediated by Kinome Reprogramming in Ovarian Cancer. Cell Rep. 2016, 16, 1273–1286. [Google Scholar] [CrossRef]
- Calder, J.; Nagelberg, A.; Luu, J.; Lu, D.; Lockwood, W.W. Resistance to BET inhibitors in lung adenocarcinoma is mediated by casein kinase phosphorylation of BRD4. Oncogenesis 2021, 10, 27. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.; Gollavilli, P.N.; Wang, S.; Asangani, I.A. Resistance to BET Inhibitor Leads to Alternative Therapeutic Vulnerabilities in Castration-Resistant Prostate Cancer. Cell Rep. 2018, 22, 2236–2245. [Google Scholar] [CrossRef]
- Shu, S.; Wu, H.-J.; Ge, J.Y.; Zeid, R.; Harris, I.S.; Jovanović, B.; Murphy, K.; Wang, B.; Qiu, X.; Endress, J.E.; et al. Synthetic Lethal and Resistance Interactions with BET Bromodomain Inhibitors in Triple-Negative Breast Cancer. Mol. Cell 2020, 78, 1096–1113.e8. [Google Scholar] [CrossRef]
- Dawson, M.S.; Stein, E.M.; Huntly, B.J.P.; Karadimitris, A.; Kamdar, M.; Fernandez de Larrea, C.; Dickinson, M.J.; Yeh, P.S.H.; Daver, N.; Chaidos, A.; et al. A Phase I Study of GSK525762, a Selective Bromodomain (BRD) and Extra Terminal Pro-tein (BET) Inhibitor: Results from Part 1 of Phase I/II Open Label Single Agent Study in Patients with Acute Myeloid Leu-kemia (AML). Blood 2017, 130 (Suppl. S1), 1377. [Google Scholar] [CrossRef]
- Iniguez, A.B.; Alexe, G.; Wang, E.J.; Roti, G.; Patel, S.; Chen, L.; Kitara, S.; Conway, A.; Robichaud, A.L.; Stolte, B.; et al. Resistance to Epigenetic-Targeted Therapy Engenders Tumor Cell Vulnerabilities Associated with Enhancer Remodeling. Cancer Cell 2018, 34, 922–938.e7. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Tarantelli, C.; Bernasconi, E.; Gaudio, E.; Cascione, L.; Restelli, V.; Arribas, A.J.; Spriano, F.; Rinaldi, A.; Mensah, A.A.; Kwee, I.; et al. BET bromodomain inhibitor birabresib in mantle cell lymphoma: In vivo activity and identification of novel combinations to overcome adaptive resistance. ESMO Open 2018, 3, e000387. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Cheng, Y.-C.; Lin, H.; Huang, M.-J.; Chow, J.-M.; Lin, S.; Liu, H.E. Downregulation of c-Myc is critical for valproic acid-induced growth arrest and myeloid differentiation of acute myeloid leukemia. Leuk. Res. 2007, 31, 1403–1411. [Google Scholar] [CrossRef]
- Slaughter, M.J.; Shanle, E.K.; Khan, A.; Chua, K.F.; Hong, T.; Boxer, L.D.; Allis, C.D.; Josefowicz, S.Z.; Garcia, B.A.; Rothbart, S.B.; et al. HDAC inhibition results in widespread alteration of the histone acetylation landscape and BRD4 targeting to gene bodies. Cell Rep. 2021, 34, 108638. [Google Scholar] [CrossRef]
- Huan, S.; Gui, T.; Xu, Q.; Zhuang, S.; Li, Z.; Shi, Y.; Lin, J.; Gong, B.; Miao, G.; Tam, M.; et al. Combination BET Family Protein and HDAC Inhibition Synergistically Elicits Chondrosarcoma Cell Apoptosis Through RAD51-Related DNA Damage Repair. Cancer Manag. Res. 2020, 12, 4429–4439. [Google Scholar] [CrossRef]
- Ren, Q.; Gao, W. Current status in the discovery of dual BET/HDAC inhibitors. Bioorg. Med. Chem. Lett. 2021, 31, 127671. [Google Scholar] [CrossRef]
- Carlino, L.; Rastelli, G. Dual Kinase-Bromodomain Inhibitors in Anticancer Drug Discovery: A Structural and Pharmacological Perspective. J. Med. Chem. 2016, 59, 9305–9320. [Google Scholar] [CrossRef]
- Burgoyne, A.M.; Vann, K.R.; Joshi, S.; Morales, G.A.; Vega, F.M.; Singh, A.; Pal, D.; Merati, A.B.; Kutateladze, T.G.; Durden, D.L. A triple action CDK4/6-PI3K-BET inhibitor with augmented cancer cell cytotoxicity. Cell Discov. 2020, 6, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib Monotherapy in Patients With Advanced Cancer and a Germline BRCA1/2 Mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef]
- Mio, C.; Gerratana, L.; Bolis, M.; Caponnetto, F.; Zanello, A.; Barbina, M.; Di Loreto, C.; Garattini, E.; Damante, G.; Puglisi, F. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int. J. Cancer 2019, 144, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Yin, J.; Fang, Y.; Chen, J.; Jeong, K.J.; Chen, X.; Vellano, C.P.; Ju, Z.; Zhao, W.; Zhang, D.; et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 2018, 33, 401–416.e8. [Google Scholar] [CrossRef]
- Wilson, A.J.; Stubbs, M.; Liu, P.; Ruggeri, B.; Khabele, D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 2018, 149, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Ceribelli, M.; Kelly, P.N.; Shaffer, A.L.; Wright, G.W.; Xiao, W.; Yang, Y.; Griner, L.A.M.; Guha, R.; Shinn, P.; Keller, J.M.; et al. Blockade of oncogenic I B kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, 11365–11370. [Google Scholar] [CrossRef] [PubMed]
- Bernasconi, E.; Gaudio, E.; Lejeune, P.; Tarantelli, C.; Cascione, L.; Kwee, I.; Spriano, F.; Rinaldi, A.; Mensah, A.A.; Chung, E.; et al. Preclinical evaluation of the BET bromodomain inhibitor BAY 1238097 for the treatment of lymphoma. Br. J. Haematol. 2017, 178, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Boi, M.; Gaudio, E.; Bonetti, P.; Kwee, I.; Bernasconi, E.; Tarantelli, C.; Rinaldi, A.; Testoni, M.; Cascione, L.; Ponzoni, M.; et al. The BET Bromodomain Inhibitor OTX015 Affects Pathogenetic Pathways in Preclinical B-cell Tumor Models and Synergizes with Targeted Drugs. Clin. Cancer Res. 2015, 21, 1628–1638. [Google Scholar] [CrossRef]
- Sun, B.; Fiskus, W.; Qian, Y.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Saenz, D.T.; Mill, C.P.; et al. BET protein proteolysis targeting chimera (PROTAC) exerts potent lethal activity against mantle cell lymphoma cells. Leukemia 2017, 32, 343–352. [Google Scholar] [CrossRef]
- Sun, B.; Shah, B.A.; Fiskus, W.; Qi, J.; Rajapakshe, K.; Coarfa, C.; Li, L.; Devaraj, S.G.T.; Sharma, S.K.; Zhang, L.; et al. Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood 2015, 126, 1565–1574. [Google Scholar] [CrossRef]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef]
- Baker, E.K.; Taylor, S.; Gupte, A.; Sharp, P.; Walia, M.; Walsh, N.C.; Zannettino, A.; Chalk, A.; Burns, C.; Walkley, C.R. BET inhibitors induce apoptosis through a MYC independent mechanism and synergise with CDK inhibitors to kill osteosarcoma cells. Sci. Rep. 2015, 5, 10120. [Google Scholar] [CrossRef]
- Peirs, S.; Frismantas, V.; Matthijssens, F.; Van Loocke, W.; Pieters, T.; Vandamme, N.; Lintermans, B.; Dobay, M.P.; Berx, G.; Poppe, B.; et al. Targeting BET proteins improves the therapeutic efficacy of BCL-2 inhibition in T-cell acute lymphoblastic leukemia. Leukemia 2017, 31, 2037–2047. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, Q.; Acharya, P.; Stengel, K.; Sheng, Q.; Zhou, X.; Kwak, H.; Fischer, M.A.; Bradner, J.E.; Strickland, S.A.; et al. High-Resolution Mapping of RNA Polymerases Identifies Mechanisms of Sensitivity and Resistance to BET Inhibitors in t(8;21) AML. Cell Rep. 2016, 16, 2003–2016. [Google Scholar] [CrossRef]
- Lam, L.T.; Lin, X.; Faivre, E.J.; Yang, Z.; Huang, X.; Wilcox, D.M.; Bellin, R.; Jin, S.; Tahir, S.K.; Mitten, M.; et al. Vulnerability of Small-Cell Lung Cancer to Apoptosis Induced by the Combination of BET Bromodomain Proteins and BCL2 Inhibitors. Mol. Cancer Ther. 2017, 16, 1511–1520. [Google Scholar] [CrossRef]
- Lasorsa, E.; Smonksey, M.; Kirk, J.S.; Rosario, S.; Hernandez-Ilizaliturri, F.J.; Ellis, L. Mitochondrial protection impairs BET bromodomain inhibitor-mediated cell death and provides rationale for combination therapeutic strategies. Cell Death Dis. 2015, 6, e2014. [Google Scholar] [CrossRef][Green Version]
- Kim, S.R.; Lewis, J.M.; Cyrenne, B.M.; Monico, P.F.; Mirza, F.N.; Carlson, K.R.; Foss, F.M.; Girardi, M. BET inhibition in advanced cutaneous T cell lymphoma is synergistically potentiated by BCL2 inhibition or HDAC inhibition. Oncotarget 2018, 9, 29193–29207. [Google Scholar] [CrossRef]
- Cummin, T.E.C.; Cox, K.L.; Murray, T.D.; Turaj, A.H.; Dunning, L.; English, V.L.; Fell, R.; Packham, G.; Ma, Y.; Powell, B.; et al. BET inhibitors synergize with venetoclax to induce apoptosis in MYC-driven lymphomas with high BCL-2 expression. Blood Adv. 2020, 4, 3316–3328. [Google Scholar] [CrossRef]
- Stuhlmiller, T.J.; Miller, S.M.; Zawistowski, J.S.; Nakamura, K.; Beltran, A.S.; Duncan, J.S.; Angus, S.; Collins, K.A.; Granger, D.A.; Reuther, R.A.; et al. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Rep. 2015, 11, 390–404. [Google Scholar] [CrossRef]
- Fiskus, W.; Sharma, S.; Qi, J.; Shah, B.; Devaraj, S.G.T.; Leveque, C.; Portier, B.P.; Iyer, S.P.; Bradner, J.E.; Bhalla, K.N. BET Protein Antagonist JQ1 Is Synergistically Lethal with FLT3 Tyrosine Kinase Inhibitor (TKI) and Overcomes Resistance to FLT3-TKI in AML Cells Expressing FLT-ITD. Mol. Cancer Ther. 2014, 13, 2315–2327. [Google Scholar] [CrossRef]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef]
- Saenz, D.T.; Fiskus, W.; Qian, Y.; Manshouri, T.; Rajapakshe, K.; Raina, K.; Coleman, K.G.; Crew, A.P.; Shen, A.; Mill, C.P.; et al. Novel BET protein proteolysis-targeting chimera exerts superior lethal activity than bromodomain inhibitor (BETi) against post-myeloproliferative neoplasm secondary (s) AML cells. Leukemia 2017, 31, 1951–1961. [Google Scholar] [CrossRef]
- Wyce, A.; Matteo, J.J.; Foley, S.W.; Felitsky, D.J.; Rajapurkar, S.R.; Zhang, X.-P.; Musso, M.C.; Korenchuk, S.; Karpinich, N.O.; Keenan, K.M.; et al. MEK inhibitors overcome resistance to BET inhibition across a number of solid and hematologic cancers. Oncogenesis 2018, 7, 35. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, L.; Neitzel, L.R.; Loganathan, S.N.; Tang, N.; Qin, L.; Crispi, E.E.; Guo, Y.; Knapp, S.; Beauchamp, R.D.; et al. The MAPK Pathway Regulates Intrinsic Resistance to BET Inhibitors in Colorectal Cancer. Clin. Cancer Res. 2017, 23, 2027–2037. [Google Scholar] [CrossRef]
- Healy, J.R.; Hart, L.S.; Shazad, A.L.; Gagliardi, M.E.; Tsang, M.; Elias, J.; Ruden, J.; Farrel, A.; Rokita, J.L.; Li, Y.; et al. Limited antitumor activity of combined BET and MEK inhibition in neuroblastoma. Pediatr. Blood Cancer 2020, 67, e28267. [Google Scholar] [CrossRef]
- Zhu, X.; Holmsen, E.; Park, S.; Willingham, M.C.; Qi, J.; Cheng, S.-Y. Synergistic effects of BET and MEK inhibitors promote regression of anaplastic thyroid tumors. Oncotarget 2018, 9, 35408–35421. [Google Scholar] [CrossRef][Green Version]
- Vázquez, R.; Riveiro, M.E.; Astorgues-Xerri, L.; Odore, E.; Rezai, K.; Erba, E.; Panini, N.; Rinaldi, A.; Kwee, I.; Beltrame, L.; et al. The bromodomain inhibitor OTX015 (MK-8628) exerts anti-tumor activity in triple-negative breast cancer models as single agent and in combination with everolimus. Oncotarget 2017, 8, 7598–7613. [Google Scholar] [CrossRef]
- Berenguer-Daizé, C.; Astorgues-Xerri, L.; Odore, E.; Cayol, M.; Cvitkovic, E.; Noel, K.; Bekradda, M.; MacKenzie, S.; Rezai, K.; Lokiec, F.; et al. OTX015 (MK-8628), a novel BET inhibitor, displaysin vitroandin vivoantitumor effects alone and in combination with conventional therapies in glioblastoma models. Int. J. Cancer 2016, 139, 2047–2055. [Google Scholar] [CrossRef]
- Lee, D.H.; Qi, J.; Bradner, J.E.; Said, J.W.; Doan, N.B.; Forscher, C.; Yang, H.; Koeffler, H.P. Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma. Int. J. Cancer 2015, 136, 2055–2064. [Google Scholar] [CrossRef] [PubMed]
- Stratikopoulos, E.E.; Dendy, M.; Szabolcs, M.; Khaykin, A.J.; Lefebvre, C.; Zhou, M.-M.; Parsons, R. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell 2015, 27, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.P.; Marchesi, I.; Schröder, C.; Schmidt, R.; Yokota, J.; Bagella, L. BET-inhibitor I-BET762 and PARP-Inhibitor Talazoparib Synergy in Small Cell Lung Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9595. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, S.; Meja, K.; Shepherd, C.; Khwaja, A. Synergistic induction of cell death in haematological malignancies by combined phosphoinositide-3-kinase and BET bromodomain inhibition. Br. J. Haematol. 2015, 170, 275–278. [Google Scholar] [CrossRef]
- Muralidharan, S.V.; Bhadury, J.; Nilsson, L.M.; Green, L.C.; McLure, K.G.; Nilsson, J.A. BET bromodomain inhibitors synergize with ATR inhibitors to induce DNA damage, apoptosis, senescence-associated secretory pathway and ER stress in Myc-induced lymphoma cells. Oncogene 2016, 35, 4689–4697. [Google Scholar] [CrossRef]
- Siegel, M.B.; Liu, S.Q.; Davare, M.; Spurgeon, S.E.; Loriaux, M.M.; Druker, B.; Scott, E.C.; Tyner, J.W. Small molecule inhibitor screen identifies synergistic activity of the bromodomain inhibitor CPI203 and bortezomib in drug resistant myeloma. Oncotarget 2015, 6, 18921–18932. [Google Scholar] [CrossRef]
- Emadali, A.; Rousseaux, S.; Bruder-Costa, J.; Rome, C.; Duley, S.; Hamaidia, S.; Betton, P.; Debernardi, A.; Leroux, D.; Bernay, B.; et al. Identification of a novel BET bromodomain inhibitor-sensitive, gene regulatory circuit that controls Rituximab response and tumour growth in aggressive lymphoid cancers. EMBO Mol. Med. 2013, 5, 1180–1195. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, R.; Matta, H.; Tolani, B.; Triche, T., Jr.; Chaudhary, P.M. Immunomodulatory drugs target IKZF1-IRF4-MYC axis in primary effusion lymphoma in a cereblon-dependent manner and display synergistic cytotoxicity with BRD4 inhibitors. Oncogene 2016, 35, 1797–1810. [Google Scholar] [CrossRef]
- Moros, A.; Rodríguez, V.; Saborit-Villarroya, I.; Montraveta, A.; Balsas, P.; Sandy, P.; Martínez, A.; Wiestner, A.; Normant, E.; Campo, E.; et al. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia 2014, 28, 2049–2059. [Google Scholar] [CrossRef]
- Siu, K.T.; Ramachandran, J.; Yee, A.J.; Eda, H.; Santo, L.; Panaroni, C.; Mertz, J.A.; Iii, R.J.S.; Cooper, M.R.; Raje, N. Preclinical activity of CPI-0610, a novel small-molecule bromodomain and extra-terminal protein inhibitor in the therapy of multiple myeloma. Leukemia 2017, 31, 1760–1769. [Google Scholar] [CrossRef]
- Hogg, S.; Vervoort, S.J.; Deswal, S.; Ott, C.J.; Li, J.; Cluse, L.A.; Beavis, P.; Darcy, P.; Martin, B.P.; Spencer, A.; et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 2017, 18, 2162–2174. [Google Scholar] [CrossRef]
- Tanaka, M.; Roberts, J.M.; Seo, H.S.; Souza, A.; Paulk, J.; Scott, T.G.; DeAngelo, S.L.; Dhe-Paganon, S.; Bradner, J.E. Design and characterization of bivalent BET inhibitors. Nat. Chem. Biol. 2016, 12, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Lwin, T.; Zhang, X.; Huang, A.; Wang, J.; Marquez, V.E.; Chen-Kiang, S.; Dalton, W.S.; Sotomayor, E.; Tao, J. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia 2013, 27, 2341–2350. [Google Scholar] [CrossRef]
- Liu, S.; Li, F.; Pan, L.; Yang, Z.; Shu, Y.; Lv, W.; Dong, P.; Gong, W. BRD4 inhibitor and histone deacetylase inhibitor synergistically inhibit the proliferation of gallbladder cancer in vitro and in vivo. Cancer Sci. 2019, 110, 2493–2506. [Google Scholar] [CrossRef]
- Hölscher, A.S.; Schulz, W.A.; Pinkerneil, M.; Niegisch, G.; Hoffmann, M.J. Combined inhibition of BET proteins and class I HDACs synergistically induces apoptosis in urothelial carcinoma cell lines. Clin. Epigenetics 2018, 10, 1–14. [Google Scholar] [CrossRef]
- Borbely, G.; Haldosen, L.-A.; Dahlman-Wright, K.; Zhao, C. Induction of USP17 by combining BET and HDAC inhibitors in breast cancer cells. Oncotarget 2015, 6, 33623–33635. [Google Scholar] [CrossRef]
- Meng, W.; Wang, B.; Mao, W.; Wang, J.; Zhao, Y.; Li, Q.; Zhang, C.; Tang, Y.; Ma, J. Enhanced efficacy of histone deacetylase inhibitor combined with bromodomain inhibitor in glioblastoma. J. Exp. Clin. Cancer Res. 2018, 37, 241. [Google Scholar] [CrossRef]
- Adeegbe, D.; Liu, Y.; Lizotte, P.; Kamihara, Y.; Aref, A.R.; Almonte, C.; Dries, R.; Li, Y.; Liu, S.; Wang, X.; et al. Synergistic Immunostimulatory Effects and Therapeutic Benefit of Combined Histone Deacetylase and Bromodomain Inhibition in Non–Small Cell Lung Cancer. Cancer Discov. 2017, 7, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Bhadury, J.; Nilsson, L.M.; Muralidharan, S.V.; Green, L.C.; Li, Z.; Gesner, E.M.; Hansen, H.C.; Keller, U.B.; McLure, K.G.; Nilsson, J.A. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E2721–E2730. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Okhovat, J.-P.; Hong, E.K.; Kim, Y.H.; Wood, G.S. Preclinical Studies Support Combined Inhibition of BET Family Proteins and Histone Deacetylases as Epigenetic Therapy for Cutaneous T-Cell Lymphoma. Neoplasia 2019, 21, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Cortiguera, M.G.; García-Gaipo, L.; Wagner, S.D.; León, J.; Batlle-López, A.; Delgado, M.D. Suppression of BCL6 function by HDAC inhibitor mediated acetylation and chromatin modification enhances BET inhibitor effects in B-cell lymphoma cells. Sci. Rep. 2019, 9, 16495. [Google Scholar] [CrossRef]
- Heinemann, A.; Cullinane, C.; De Paoli-Iseppi, R.; Wilmott, J.; Gunatilake, D.; Madore, J.; Strbenac, D.; Yang, J.; Gowrishankar, K.; Tiffen, J.C.; et al. Combining BET and HDAC inhibitors synergistically induces apoptosis of melanoma and suppresses AKT and YAP signaling. Oncotarget 2015, 6, 21507–21521. [Google Scholar] [CrossRef]
- Shahbazi, J.; Liu, P.Y.; Atmadibrata, B.; Bradner, J.E.; Marshall, G.M.; Lock, R.; Liu, T. The Bromodomain Inhibitor JQ1 and the Histone Deacetylase Inhibitor Panobinostat Synergistically Reduce N-Myc Expression and Induce Anticancer Effects. Clin. Cancer Res. 2016, 22, 2534–2544. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.K.; Herner, A.; Mello, S.S.; Wirth, M.; Hausmann, S.; Sánchez-Rivera, F.J.; Lofgren, S.M.; Kuschma, T.; Hahn, S.; Vangala, D.; et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat. Med. 2015, 21, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zegar, T.; Weiser, T.; Hamdan, F.; Berger, B.; Lucas, R.; Balourdas, D.-I.; Ladigan, S.; Cheung, P.F.; Liffers, S.; et al. Characterization of a dual BET/HDAC inhibitor for treatment of pancreatic ductal adenocarcinoma. Int. J. Cancer 2020, 147, 2847–2861. [Google Scholar] [CrossRef]
- He, S.; Dong, G.; Li, Y.; Wu, S.; Wang, W.; Sheng, C. Potent Dual BET/HDAC Inhibitors for Efficient Treatment of Pancreatic Cancer. Angew. Chem. Int. Ed. 2020, 59, 3028–3032. [Google Scholar] [CrossRef] [PubMed]
- Enßle, J.C.; Boedicker, C.; Wanior, M.; Vogler, M.; Knapp, S.; Fulda, S. Co-targeting of BET proteins and HDACs as a novel approach to trigger apoptosis in rhabdomyosarcoma cells. Cancer Lett. 2018, 428, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.; Wilder-Romans, K.; Dommeti, V.L.; Krishnamurthy, P.M.; Apel, I.J.; Escara-Wilke, J.; Plymate, S.R.; Navone, N.M.; Wang, S.; Feng, F.Y.; et al. BET Bromodomain Inhibitors Enhance Efficacy and Disrupt Resistance to AR Antagonists in the Treatment of Prostate Cancer. Mol. Cancer Res. 2016, 14, 324–331. [Google Scholar] [CrossRef]
- Feng, Q.; Zhang, Z.; Shea, M.J.; Creighton, C.J.; Coarfa, C.; Hilsenbeck, S.G.; Lanz, R.; He, B.; Wang, L.; Fu, X.; et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014, 24, 809–819. [Google Scholar] [CrossRef]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.-E.; Malki, M.I. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef]
- Scully, R.; Chen, J.; Plug, A.; Xiao, Y.; Weaver, D.; Feunteun, J.; Ashley, T.; Livingston, D.M. Association of BRCA1 with Rad51 in Mitotic and Meiotic Cells. Cell 1997, 88, 265–275. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.-D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Steinfeld, J.B.; Liang, F.; Chen, X.; Maranon, D.G.; Ma, C.J.; Kwon, Y.; Rao, T.; Wang, W.; Sheng, C.; et al. BRCA1–BARD1 promotes RAD51-mediated homologous DNA pairing. Nat. Cell Biol. 2017, 550, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Litman, R.; Peng, M.; Jin, Z.; Zhang, F.; Zhang, J.; Powell, S.; Andreassen, P.R.; Cantor, S.B. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 2005, 8, 255–265. [Google Scholar] [CrossRef]
- Cantor, S.B.; Bell, D.W.; Ganesan, S.; Kass, E.M.; Drapkin, R.; Grossman, S.; Wahrer, D.C.; Sgroi, D.C.; Lane, W.S.; Haber, D.A.; et al. BACH1, a Novel Helicase-like Protein, Interacts Directly with BRCA1 and Contributes to Its DNA Repair Function. Cell 2001, 105, 149–160. [Google Scholar] [CrossRef]
- Taniguchi, T.; Garcia-Higuera, I.; Andreassen, P.R.; Gregory, R.C.; Grompe, M.; D’Andrea, A.D. S-phase–specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood 2002, 100, 2414–2420. [Google Scholar] [CrossRef]
- Hussain, S.; Wilson, J.B.; Medhurst, A.L.; Hejna, J.; Witt, E.; Ananth, S.; Davies, A.; Masson, J.-Y.; Moses, R.; West, S.C.; et al. Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways. Hum. Mol. Genet. 2004, 13, 1241–1248. [Google Scholar] [CrossRef]
- Alcón, P.; Shakeel, S.; Chen, Z.A.; Rappsilber, J.; Patel, K.J.; Passmore, L.A. FANCD2–FANCI is a clamp stabilized on DNA by monoubiquitination of FANCD2 during DNA repair. Nat. Struct. Mol. Biol. 2020, 27, 240–248. [Google Scholar] [CrossRef]
- Neizer-Ashun, F.; Bhattacharya, R. Reality CHEK: Understanding the biology and clinical potential of CHK1. Cancer Lett. 2021, 497, 202–211. [Google Scholar] [CrossRef]
- Stracker, T.H.; Petrini, J.H.J. The MRE11 complex: Starting from the ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef]
- Bassi, C.; Li, Y.-T.; Khu, K.; Mateo, F.; Baniasadi, P.S.; Elia, A.; Mason, J.; Stambolic, V.; Pujana, M.A.; Mak, T.W.; et al. The acetyltransferase Tip60 contributes to mammary tumorigenesis by modulating DNA repair. Cell Death Differ. 2016, 23, 1198–1208. [Google Scholar] [CrossRef]
- Russell, P.; Nurse, P. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell 1987, 49, 559–567. [Google Scholar] [CrossRef]
- Zeidler, M.; Varambally, S.; Cao, Q.; Chinnaiyan, A.M.; Ferguson, D.O.; Merajver, S.D.; Kleer, C.G. The Polycomb Group Protein EZH2 Impairs DNA Repair in Breast Epithelial Cells. Neoplasia 2005, 7, 1011–1019. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, S.Y.; Gong, F.; Battenhouse, A.M.; Boutz, D.R.; Bashyal, A.; Refvik, S.T.; Chiang, C.-M.; Xhemalce, B.; Paull, T.T.; et al. Systematic bromodomain protein screens identify homologous recombination and R-loop suppression pathways involved in genome integrity. Genes Dev. 2019, 33, 1751–1774. [Google Scholar] [CrossRef] [PubMed]
- Stanlie, A.; Yousif, A.; Akiyama, H.; Honjo, T.; Begum, N.A. Chromatin Reader Brd4 Functions in Ig Class Switching as a Repair Complex Adaptor of Nonhomologous End-Joining. Mol. Cell 2014, 55, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Jauset, T.; Massó-Vallés, D.; Martínez-Martín, S.; Beaulieu, M.-E.; Foradada, L.; Fiorentino, F.P.; Yokota, J.; Haendler, B.; Siegel, S.; Whitfield, J.R.; et al. BET inhibition is an effective approach against KRAS-driven PDAC and NSCLC. Oncotarget 2018, 9, 18734–18746. [Google Scholar] [CrossRef]
- Gainor, J.F.; Longo, D.L.; Chabner, B.A. Pharmacodynamic Biomarkers: Falling Short of the Mark? Clin. Cancer Res. 2014, 20, 2587–2594. [Google Scholar] [CrossRef][Green Version]
- Mita, M.M.; Mita, A.C. Bromodomain inhibitors a decade later: A promise unfulfilled? Br. J. Cancer 2020, 123, 1713–1714. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, Z.; Zhao, L.; Li, L.; Zuo, W.; Han, W.Z.A.L. High expression of RAD51 promotes DNA damage repair and survival in KRAS-mutant lung cancer cells. BMB Rep. 2019, 52, 151–156. [Google Scholar] [CrossRef]
- Luoto, K.R.; Meng, A.X.; Wasylishen, A.; Zhao, H.; Coackley, C.L.; Penn, L.; Bristow, R. Tumor Cell Kill by c-MYC Depletion: Role of MYC-Regulated Genes that Control DNA Double-Strand Break Repair. Cancer Res. 2010, 70, 8748–8759. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, J.; Borowicz, S.; Collins, C.; Huo, D.; Olopade, O.I. c-Myc activates BRCA1 gene expression through distal promoter elements in breast cancer cells. BMC Cancer 2011, 11, 246. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Liu, C.; Tao, Z.; Wang, M.; Jia, Y.; Sang, X.; Shen, L.; Xue, Y.; Jiang, K.; Luo, F.; et al. MYC status as a determinant of synergistic response to Olaparib and Palbociclib in ovarian cancer. EBioMedicine 2019, 43, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Carey, J.P.; Karakas, C.; Bui, T.; Chen, X.; Vijayaraghavan, S.; Zhao, Y.; Wang, J.; Mikule, K.; Litton, J.K.; Hunt, K.K.; et al. Synthetic Lethality of PARP Inhibitors in Combination with MYC Blockade Is Independent of BRCA Status in Triple-Negative Breast Cancer. Cancer Res. 2017, 78, 742–757. [Google Scholar] [CrossRef]





| Compound | Target | Combination | Tumour Type | Results | Phase/Status | Reference |
|---|---|---|---|---|---|---|
| ABBV-075 | BRD2/4, BRDT | N/A | Solid tumours: BC, CRC, PR, PrC, uveal melanoma, head and neck | SD: 26 n = 71 | I, completed | NCT02391480 [53] |
| Venetoclax | AML | CR: 3 PR: 2 MLFS: 2 n = 44 | [64] | |||
| Navitocla xRuxolitinib | MF | Not posted | I, not yet recruiting | NCT04480086 | ||
| ABBV-744 | BRD2/3/4, BRDT | N/A | AML | Not posted | I, terminated | NCT03360006 |
| Navitocla xRuxolitinib | MF | Not posted | I, recruiting | NCT04454658 | ||
| BAY1238097 | BRD4 | N/A | Solid tumours, myeloma, lymphoma | NR: 8 SD: 2 n = 11 | I, terminated | NCT02369029 [61] |
| BI 894999 | BRD2/3/4, BRDT | N/A | Solid tumours and lymphoma | Not posted | I, not yet recruiting | NCT02516553 |
| BMS-986158 | Undisclosed | N/A | Solid tumours and lymphoma in children | Not posted | I, recruiting | NCT03936465 |
| Nivolumab | Advanced solid tumours and haematological malignancies | Not posted | I/II, not yet recruiting | NCT02419417 | ||
| CC-90010 | Undisclosed | N/A | Solid tumours and NHL | Not posted | I, recruiting | NCT03220347 |
| CPI-0610 | BRD4 | N/A | Lymphoma | CR: 2 PR: 3 SD: 5 n = 64 | I, completed | NCT01949883 [65] |
| N/A | MM | Not posted | I, completed | NCT02157636 | ||
| N/A | Peripheral nerve tumours | Not posted | II, withdrawn | NCT02986919 | ||
| Ruxolitinib | MF | Not posted | I/II, recruiting | NCT02158858 [66,67] | ||
| FT-1101 | BRD2/3/4, BRDT | Azacitidine | AML, MDS, NHL | Not posted | I, completed | NCT02543879 |
| GS-5829 | N/A | Solid tumours, lymphoma | Not posted | I, completed | NCT02392611 | |
| Exemestane Fulvestrant | ER-positive BC | Not posted | ||||
| Exemestane Fulvestrant | ER-positive and HER-negative BC | Not posted | I/II, terminated | NCT02983604 | ||
| Enzalutamide | Castrate-resistant PrC | Not posted | I/II, terminated | NCT02607228 | ||
| I-BET151 (GSK2820151) | BRD2/3/4 | N/A | Solid tumours | Not posted | I, terminated | NCT02630251 |
| I-BET762 (GSK525762) | BRD2/3/4, BRDT | N/A | Hematologic malignancies | Not posted | I/II, completed | NCT01943851 |
| N/A | NMC | PR: 2 SD: 7 n = 19 | I, completed | NCT01587703 [53,68] | ||
| Trametinib | Solid tumours | Not posted | I/II, withdrawn | NCT03266159 | ||
| Entinostat | Solid tumours and haematological malignancies | Not posted | I, withdrawn | NCT03925428 | ||
| INCB054329 | BRD2/3/4, BRDT | N/A | Solid tumours and haematological malignancies | Not posted | I/II, withdrawn | NCT02431260 |
| INCB057643 | BRD2/3/4 | Abiraterone, Azacitidine, Gemcitabine, Paclitaxel, Rucaparib, Ruxolitinib | Solid tumours | CR: 2 PR: 4 n = 134 | I/II, terminated | NCT02711137 [69] |
| ODM-207 | Undisclosed | N/A | Solid tumours | SD: 6 n = 27s | I/II, completed | NCT03035591 [70] |
| OTX015 (MK-8628) | BRD2/3/4 | N/A | AML, DLBCL | Not posted | I, active, not recruiting | NCT02698189 |
| N/A | CRPC, NMC, NSCLC, TNBC | Not posted | I, terminated | NCT02698176 | ||
| N/A | Glioblastoma multiforme | Not posted | II, terminated | NCT02296476 | ||
| N/A | CRPC, NMC, NSCLC, PC, TNBC | PR: 3 (NMC) SD: 25 (3 NMC) n = 46 | I, completed | NCT02259114 [52] [50] | ||
| N/A | AML, acute lymphoblastic leukaemia, DLBCL, MM | CR:2 (DLBCL) PR:1 (DLBCL) n = 33 CR: 2 (AL) PR: 3 (AL) n = 41 | I, completed | NCT01713582 [47] [55] | ||
| Azacitidine | AML | Not posted | I/II, withdrawn | NCT02303782 | ||
| PLX51107 | BRD2/3/4, BRDT | N/A | Solid tumours and haematological malignancies | Not posted | I/II, terminated | NCT02683395 |
| RO6870810 (TEN-010) | Undisclosed | N/A | AML, MDS | CR: 1 SD: 13 n = 32 | I, completed | NCT02308761 [71] |
| N/A | Solid tumours | PR: 1 (solid tumours) SD: 24 (solid tumours) n = 47 PR: 2 (NMC) SD: 5 (NMC) n = 8 PR: 2 (DLBCL) SD: 4 (DLBCL) n = 19 | I, completed | NCT01987362 [72] | ||
| Venetocla xRituximab | DLBCL, B-cell lymphoma | Not posted | I, completed | NCT03255096 | ||
| ZEN-3694 | Undisclosed | N/A | Metastatic CRPC | Not posted | I, completed | NCT02705469 |
| Enzalutamide | Metastatic CRPC | Not posted | I/II, completed | NCT02711956 | ||
| Enzalutamide Pembrolizumab | Metastatic CRPC | Not posted | II, recruiting | NCT04471974 | ||
| Ipilimumab Nivolumab | Solid tumours | Not posted | I, not yet recruiting | NCT04840589 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarnik, J.; Popławski, T.; Tokarz, P. BET Proteins as Attractive Targets for Cancer Therapeutics. Int. J. Mol. Sci. 2021, 22, 11102. https://doi.org/10.3390/ijms222011102
Sarnik J, Popławski T, Tokarz P. BET Proteins as Attractive Targets for Cancer Therapeutics. International Journal of Molecular Sciences. 2021; 22(20):11102. https://doi.org/10.3390/ijms222011102
Chicago/Turabian StyleSarnik, Joanna, Tomasz Popławski, and Paulina Tokarz. 2021. "BET Proteins as Attractive Targets for Cancer Therapeutics" International Journal of Molecular Sciences 22, no. 20: 11102. https://doi.org/10.3390/ijms222011102
APA StyleSarnik, J., Popławski, T., & Tokarz, P. (2021). BET Proteins as Attractive Targets for Cancer Therapeutics. International Journal of Molecular Sciences, 22(20), 11102. https://doi.org/10.3390/ijms222011102