
The Trace Element Selenium Is Important for Redox Signaling in Phorbol Ester-Differentiated THP-1 Macrophages

 , , , ,
, , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Se Does Not Affect Differentiation of THP-1 Cells into Macrophages
2.2. PMA-Induced Differentiation of Monocytes to Macrophages Alters the Selenoprotein Expression Profile
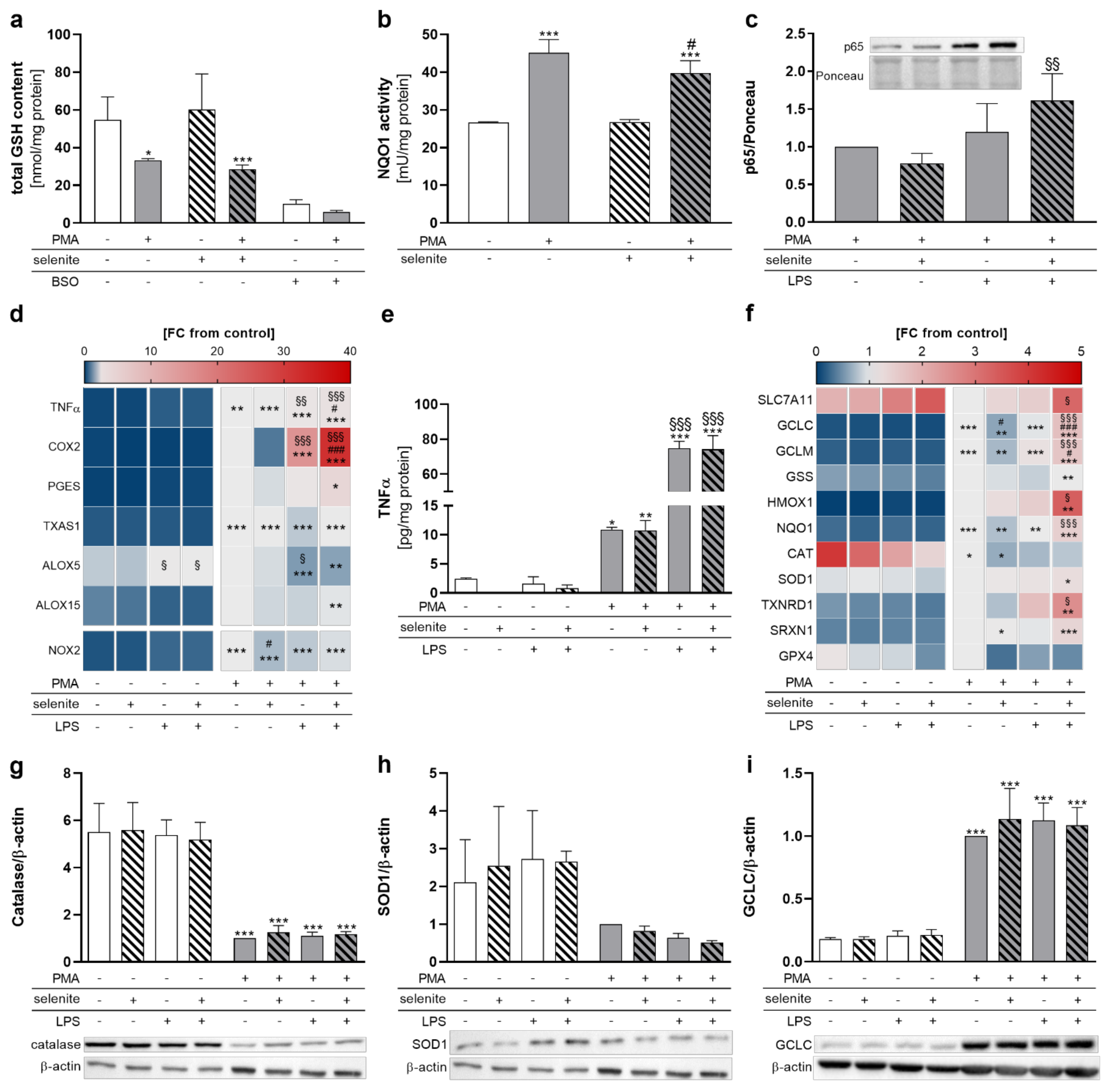
2.3. PMA and Se Treatment Modulate the Cellular Redox Status and the Activity of the Redox-Sensitive Transcription Factors NRF2 and NF-κB
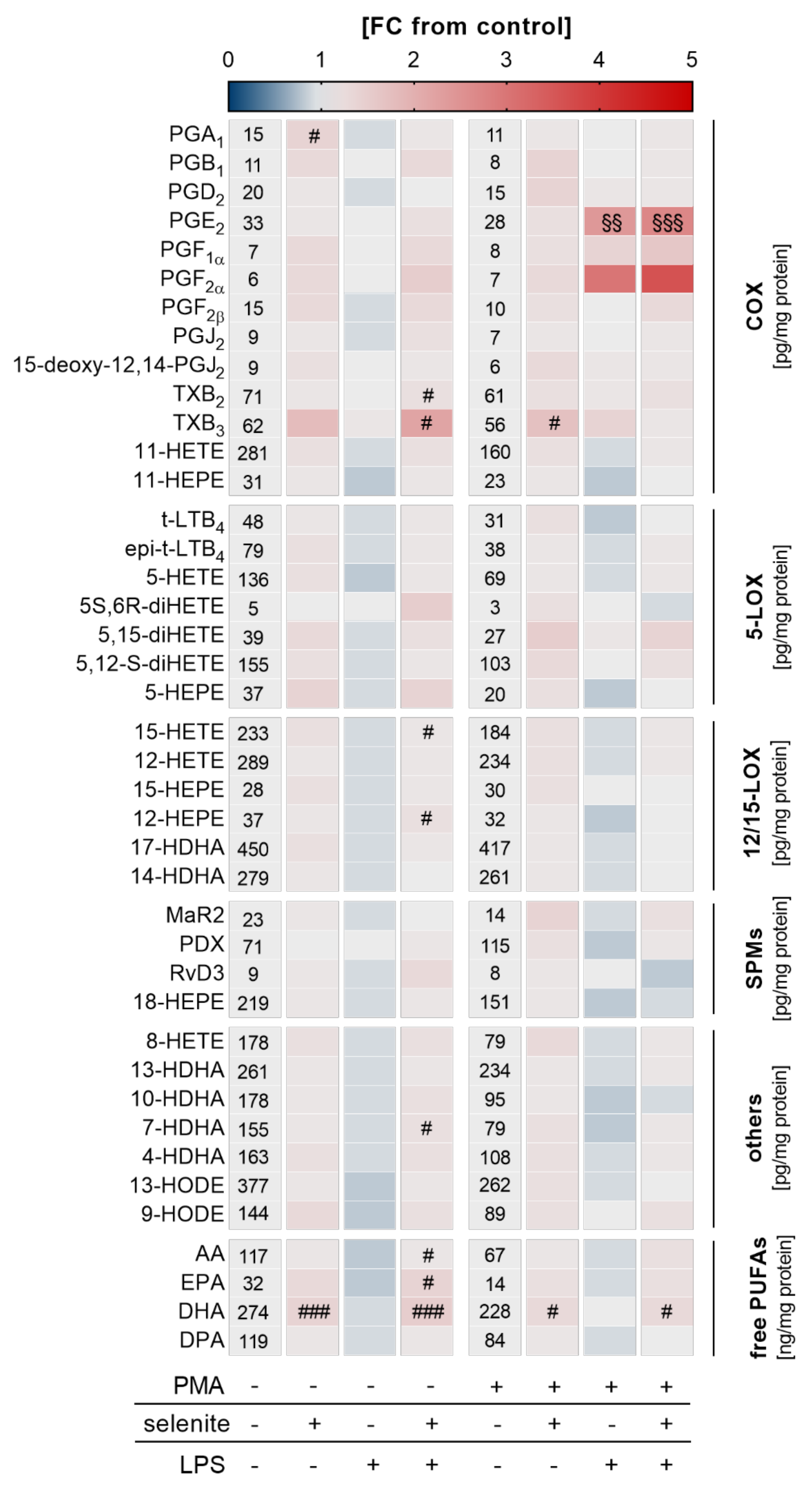
2.4. Effects of LPS Treatment on LM profiles in THP-1 Monocytes and Macrophages
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Determination of Adherence, Morphology and Cell Number
4.4. Flow Cytometry
4.5. Nuclear Isolation
4.6. Western Blot
4.7. Quantitative Real-Time PCR
4.8. ELISA
4.9. Trace Element Analysis by Total Reflection X-ray Fluorescence (TXRF) Spectroscopy
4.10. GSH Assay
4.11. Enzyme Activities
4.12. Solid Phase Extraction of LMs
4.13. LM Analysis by UPLC-MS/MS
4.14. Statistics
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Labunskyy, V.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular Pathways and Physiological Roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; Rose, A.H.; Hoffmann, P.R. The Role of Selenium in Inflammation and Immunity: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2012, 16, 705–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avery, J.C.; Hoffmann, P.R. Selenium, Selenoproteins, and Immunity. Nutrients 2018, 10, 1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinbrenner, H.; Speckmann, B.; Klotz, L.-O. Selenoproteins: Antioxidant selenoenzymes and beyond. Arch. Biochem. Biophys. 2016, 595, 113–119. [Google Scholar] [CrossRef]
- Bertz, M.; Kühn, K.; Koeberle, S.C.; Müller, M.F.; Hoelzer, D.; Thies, K.; Deubel, S.; Thierbach, R.; Kipp, A.P. Selenoprotein H controls cell cycle progression and proliferation of human colorectal cancer cells. Free. Radic. Biol. Med. 2018, 127, 98–107. [Google Scholar] [CrossRef]
- Peng, X.; Giménez-Cassina, A.; Petrus, P.; Conrad, M.; Rydén, M.; Arnér, E.S.J. Thioredoxin reductase 1 suppresses adipocyte differentiation and insulin responsiveness. Sci. Rep. 2016, 6, 28080. [Google Scholar] [CrossRef] [Green Version]
- Rajalin, A.-M.; Micoogullari, M.; Sies, H.; Steinbrenner, H. Upregulation of the thioredoxin-dependent redox system during differentiation of 3T3-L1 cells to adipocytes. Biol. Chem. 2014, 395, 667–677. [Google Scholar] [CrossRef]
- Wang, C.; Li, R.; Huang, Y.; Wang, M.; Yang, F.; Huang, D.; Wu, C.; Li, Y.; Tang, Y.; Zhang, R.; et al. Selenoprotein K modulate intracellular free Ca2+ by regulating expression of calcium homoeostasis endoplasmic reticulum protein. Biochem. Biophys. Res. Commun. 2017, 484, 734–739. [Google Scholar] [CrossRef]
- Yan, J.; Fei, Y.; Han, Y.; Lu, S. Selenoprotein O deficiencies suppress chondrogenic differentiation of ATDC5 cells. Cell Biol. Int. 2016, 40, 1033–1040. [Google Scholar] [CrossRef]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of Monocytes, Macrophages, and Dendritic Cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Dall’Asta, M.; Derlindati, E.; Ardigò, D.; Zavaroni, I.; Brighenti, F.; Del Rio, D. Macrophage polarization: The answer to the diet/inflammation conundrum? Nutr. Metab. Cardiovasc. Dis. 2012, 22, 387–392. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Dorrington, M.G.; Fraser, I.D.C.; Dorrington, M.G.; Fraser, I.D.C. NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Michalke, B. Selenium; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar] [CrossRef]
- Schomburg, L.; Schweizer, U. Hierarchical regulation of selenoprotein expression and sex-specific effects of selenium. Biochim. Biophys. Acta 2009, 1790, 1453–1462. [Google Scholar] [CrossRef]
- Papp, L.V.; Lu, J.; Holmgren, A.; Khanna, K.K. From Selenium to Selenoproteins: Synthesis, Identity, and Their Role in Human Health. Antioxid. Redox Signal. 2007, 9, 775–806. [Google Scholar] [CrossRef]
- Reeves, M.A.; Hoffmann, P.R. The human selenoproteome: Recent insights into functions and regulation. Cell. Mol. Life Sci. 2009, 66, 2457–2478. [Google Scholar] [CrossRef] [Green Version]
- Auwerx, J. The human leukemia cell line, THP-1: A multifacetted model for the study of monocyte-macrophage differentiation. Cell. Mol. Life Sci. 1991, 47, 22–31. [Google Scholar] [CrossRef]
- Daigneault, M.; Preston, J.; Marriott, H.; Whyte, M.K.B.; Dockrell, D.H. The Identification of Markers of Macrophage Differentiation in PMA-Stimulated THP-1 Cells and Monocyte-Derived Macrophages. PLoS ONE 2010, 5, e8668. [Google Scholar] [CrossRef]
- Lund, M.E.; To, J.; O’Brien, B.A.; Donnelly, S. The choice of phorbol 12-myristate 13-acetate differentiation protocol influences the response of THP-1 macrophages to a pro-inflammatory stimulus. J. Immunol. Methods 2016, 430, 64–70. [Google Scholar] [CrossRef]
- Maeß, M.B.; Wittig, B.; Cignarella, A.; Lorkowski, S. Reduced PMA enhances the responsiveness of transfected THP-1 macrophages to polarizing stimuli. J. Immunol. Methods 2014, 402, 76–81. [Google Scholar] [CrossRef]
- Li, T.; Yang, W.; Li, M.; Byun, D.-S.; Tong, C.; Nasser, S.; Zhuang, M.; Arango, D.; Mariadason, J.M.; Augenlicht, L.H. Expression of selenium-binding protein 1 characterizes intestinal cell maturation and predicts survival for patients with colorectal cancer. Mol. Nutr. Food Res. 2008, 52, 1289–1299. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Micoogullari, M.; Hoang, N.A.; Bergheim, I.; Klotz, L.-O.; Sies, H. Selenium-binding protein 1 (SELENBP1) is a marker of mature adipocytes. Redox Biol. 2018, 20, 489–495. [Google Scholar] [CrossRef]
- Chen, G.; Wang, H.; Miller, C.T.; Thomas, D.G.; Gharib, T.G.; E Misek, D.; Giordano, T.J.; Orringer, M.B.; Hanash, S.M.; Beer, D.G. Reduced selenium-binding protein 1 expression is associated with poor outcome in lung adenocarcinomas. J. Pathol. 2004, 202, 321–329. [Google Scholar] [CrossRef]
- Wang, N.; Chen, Y.; Yang, X.; Jiang, Y. Selenium-binding protein 1 is associated with the degree of colorectal cancer differentiation and is regulated by histone modification. Oncol. Rep. 2014, 31, 2506–2514. [Google Scholar] [CrossRef]
- Sivandzade, F.; Prasad, S.; Bhalerao, A.; Cucullo, L. NRF2 and NF-қB interplay in cerebrovascular and neurodegenerative disorders: Molecular mechanisms and possible therapeutic approaches. Redox Biol. 2018, 21, 101059. [Google Scholar] [CrossRef]
- Koeberle, S.C.; Gollowitzer, A.; Laoukili, J.; Kranenburg, O.; Werz, O.; Koeberle, A.; Kipp, A.P. Distinct and overlapping functions of glutathione peroxidases 1 and 2 in limiting NF-κB-driven inflammation through redox-active mechanisms. Redox Biol. 2019, 28, 101388. [Google Scholar] [CrossRef]
- Vunta, H.; Davis, F.; Palempalli, U.D.; Bhat, D.; Arner, R.J.; Thompson, J.T.; Peterson, D.G.; Reddy, C.C.; Prabhu, K.S. The Anti-inflammatory Effects of Selenium Are Mediated through 15-Deoxy-Δ12,14-prostaglandin J2 in Macrophages. J. Biol. Chem. 2007, 282, 17964–17973. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Bi, C.; Wang, Y.; Sun, J.; Meng, X.; Li, J. Selenium ameliorates Staphylococcus aureus-induced inflammation in bovine mammary epithelial cells by inhibiting activation of TLR2, NF-κB and MAPK signaling pathways. BMC Vet. Res. 2018, 14, 197. [Google Scholar] [CrossRef] [Green Version]
- Hiller, F.; Oldorff, L.; Besselt, K.; Kipp, A.P. Differential Acute Effects of Selenomethionine and Sodium Selenite on the Severity of Colitis. Nutrients 2015, 7, 2687–2706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Ding, T.; Fang, L.; Cui, L.; Li, J.; Meng, X.; Zhu, G.; Qian, C.; Wang, H.; Li, J. Organic Selenium Ameliorates Staphylococcus aureus-Induced Mastitis in Rats by Inhibiting the Activation of NF-κB and MAPK Signaling Pathways. Front. Vet. Sci. 2020, 7, 443. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.W.; Reddy, V.K.; Short, S.; Motley, A.K.; Lintel, M.K.; Bradley, A.M.; Freeman, T.; Vallance, J.; Ning, W.; Parang, B.; et al. Selenoprotein P influences colitis-induced tumorigenesis by mediating stemness and oxidative damage. J. Clin. Investig. 2015, 125, 2646–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Carlson, B.; Yoo, M.-H.; Sano, Y.; Sengupta, A.; Kim, J.Y.; Irons, R.; Gladyshev, V.N.; Hatfield, D.L.; Park, J.M. Selenoproteins regulate macrophage invasiveness and extracellular matrix-related gene expression. BMC Immunol. 2009, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Nelson, S.M.; Lei, X.; Prabhu, K.S. Selenium Levels Affect the IL-4–Induced Expression of Alternative Activation Markers in Murine Macrophages. J. Nutr. 2011, 141, 1754–1761. [Google Scholar] [CrossRef] [Green Version]
- Norton, R.L.; Fredericks, G.J.; Huang, Z.; Fay, J.D.; Hoffmann, F.W.; Hoffmann, P.R. Selenoprotein K regulation of palmitoylation and calpain cleavage of ASAP2 is required for efficient FcγR-mediated phagocytosis. J. Leukoc. Biol. 2016, 101, 439–448. [Google Scholar] [CrossRef]
- Verma, S.; Hoffmann, F.W.; Kumar, M.; Huang, Z.; Roe, K.; Nguyen-Wu, E.; Hashimoto, A.S.; Hoffmann, P.R. Selenoprotein K Knockout Mice Exhibit Deficient Calcium Flux in Immune Cells and Impaired Immune Responses. J. Immunol. 2011, 186, 2127–2137. [Google Scholar] [CrossRef] [Green Version]
- Brigelius-Flohé, R.; Friedrichs, B.; Maurer, S.; Schultz, M.; Streicher, R. Interleukin-1-induced nuclear factor κB activation is inhibited by overexpression of phospholipid hydroperoxide glutathione peroxidase in a human endothelial cell line. Biochem. J. 1997, 328, 199–203. [Google Scholar] [CrossRef] [Green Version]
- Bi, C.-L.; Wang, H.; Wang, Y.-J.; Sun, J.; Dong, J.-S.; Meng, X.; Li, J.-J. Selenium inhibits Staphylococcus aureus-induced inflammation by suppressing the activation of the NF-κB and MAPK signalling pathways in RAW264.7 macrophages. Eur. J. Pharmacol. 2016, 780, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; Zhao, Y.; Li, H.; Lei, L. Ferroptosis as an emerging target in inflammatory diseases. Prog. Biophys. Mol. Biol. 2020, 155, 20–28. [Google Scholar] [CrossRef]
- Mattmiller, S.A.; Carlson, B.A.; Gandy, J.C.; Sordillo, L.M. Reduced macrophage selenoprotein expression alters oxidized lipid metabolite biosynthesis from arachidonic and linoleic acid. J. Nutr. Biochem. 2014, 25, 647–654. [Google Scholar] [CrossRef]
- Zamamiri-Davis, F.; Lu, Y.; Thompson, J.T.; Prabhu, K.; Reddy, P.V.; Sordillo, L.M.; Reddy, C. Nuclear factor-κB mediates over-expression of cyclooxygenase-2 during activation of RAW 264.7 macrophages in selenium deficiency. Free. Radic. Biol. Med. 2002, 32, 890–897. [Google Scholar] [CrossRef]
- Wan, J.M.-F.; Lee, C.-Y.J. Immunoregulatory and Antioxidant Performance of α-Tocopherol and Selenium on Human Lymphocytes. Biol. Trace Elem. Res. 2002, 86, 123–136. [Google Scholar] [CrossRef]
- Spallholz, J.E. Free radical generation by selenium compounds and their prooxidant toxicity. Biomed. Environ. Sci. 1997, 10, 260–270. [Google Scholar]
- Stewart, M.S.; E Spallholz, J.; Neldner, K.H.; Pence, B.C. Selenium compounds have disparate abilities to impose oxidative stress and induce apoptosis. Free. Radic. Biol. Med. 1998, 26, 42–48. [Google Scholar] [CrossRef]
- Karwaciak, I.; Gorzkiewicz, M.; Bartosz, G.; Pulaski, L. TLR2 activation induces antioxidant defence in human monocyte-macrophage cell line models. Oncotarget 2017, 8, 54243–54264. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.; Banning, A.; Brigelius-Flohé, R.; Kipp, A. Nrf2 target genes are induced under marginal selenium-deficiency. Genes Nutr. 2010, 5, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.-C.; Chiang, S.-K.; Chen, S.-E.; Yu, Y.-L.; Chou, R.-H.; Chang, W.-C. Heme oxygenase-1 mediates BAY 11–7085 induced ferroptosis. Cancer Lett. 2018, 416, 124–137. [Google Scholar] [CrossRef]
- Iida, Y.; Okamoto-Κatsuyama, M.; Maruoka, S.; Mizumura, K.; Shimizu, T.; Shikano, S.; Hikichi, M.; Takahashi, M.; Tsuya, K.; Okamoto, S.; et al. Effective ferroptotic small-cell lung cancer cell death from SLC7A11 inhibition by sulforaphane. Oncol. Lett. 2020, 21, 71. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Al-Quraishy, S.; Dkhil, M.; Wunderlich, F.; Sies, H. Dietary Selenium in Adjuvant Therapy of Viral and Bacterial Infections. Adv. Nutr. 2015, 6, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Saad, R.; Taylor, E.W.; Rayman, M.P. Selenium and selenoproteins in viral infection with potential relevance to COVID-19. Redox Biol. 2020, 37, 101715. [Google Scholar] [CrossRef]
- Diwakar, B.T.; Korwar, A.M.; Paulson, R.F.; Prabhu, K.S. The Regulation of Pathways of Inflammation and Resolution in Immune Cells and Cancer Stem Cells by Selenium. Adv. Cancer Res. 2017, 136, 153–172. [Google Scholar] [CrossRef]
- Kaur, S.; Harjai, K.; Chhibber, S. In Vivo Assessment of Phage and Linezolid Based Implant Coatings for Treatment of Methicillin Resistant S. aureus (MRSA) Mediated Orthopaedic Device Related Infections. PLoS ONE 2016, 11, e0157626. [Google Scholar] [CrossRef] [Green Version]
- Nettleford, S.K.; Zhao, L.; Qian, F.; Herold, M.; Arner, B.; Desai, D.; Amin, S.; Xiong, N.; Singh, V.; Carlson, B.A.; et al. The Essential Role of Selenoproteins in the Resolution of Citrobacter rodentium-Induced Intestinal Inflammation. Front. Nutr. 2020, 7, 96. [Google Scholar] [CrossRef]
- Schwarz, M.; Lossow, K.; Schirl, K.; Hackler, J.; Renko, K.; Kopp, J.F.; Schwerdtle, T.; Schomburg, L.; Kipp, A.P. Copper interferes with selenoprotein synthesis and activity. Redox Biol. 2020, 37, 101746. [Google Scholar] [CrossRef]
- Winther, J.R.; Thorpe, C. Quantification of thiols and disulfides. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2013, 1840, 838–846. [Google Scholar] [CrossRef] [Green Version]
- Prochaska, H.J.; Santamaria, A.B. Direct measurement of NAD(P)H:quinone reductase from cells cultured in microtiter wells: A screening assay for anticarcinogenic enzyme inducers. Anal. Biochem. 1988, 169, 328–336. [Google Scholar] [CrossRef]
- Werz, O.; Gerstmeier, J.; Libreros, S.; De La Rosa, X.; Werner, M.; Norris, P.; Chiang, N.; Serhan, C.N. Human macrophages differentially produce specific resolvin or leukotriene signals that depend on bacterial pathogenicity. Nat. Commun. 2018, 9, 59. [Google Scholar] [CrossRef]
- Schädel, P.; Troisi, F.; Czapka, A.; Gebert, N.; Pace, S.; Ori, A.; Werz, O. Aging drives organ-specific alterations of the inflammatory microenvironment guided by immunomodulatory mediators in mice. FASEB J. 2021, 35, e21558. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | RefSeq-ID | Sequence (5′→3′) |
|---|---|---|
| ALOX15 (arachidonate 15-lipoxygenase) | NM_001140.3 | TGGAGCCTTCCTAACCTACAG TCCACATACCGATAGATGATTTCC |
| ALOX5 (arachidonate 5-lipoxygenase) | NM_000698.3 | GCTGCAACCCTGTGTTGATCC AAATGTTCCCTTGCTGGACCTC |
| CAT (catalase) | NM_001752.4 | CCTATCCTGACACTCACCGCCA GAGCACCACCCTGATTGTCCTG |
| COX2 (cyclooxygenase 2; prostaglandin-endoperoxide synthase 2 (PTGS2)) | NM_000963.2 | CCCAGCACTTCACGCATCAG CTGTCTAGCCAGAGTTTCACCGT |
| EEFSEC (eukaryotic elongation factor, selenocysteine-tRNA-specific) | NM_021937.3 | CCCTAGAGAACACCAAGTTCCGAG TCAATGAGCTCTGGAATGCCCT |
| EIF4A3 (eukaryotic translation initiation factor 4A3) | NM_014740.3 | AAAGAAAGGTGGACTGGCTGACGG ACTCCTTCATGATGGACTCCCGCT |
| GCLC (glutamate-cysteine ligase catalytic subunit) | NM_001498.3 | TGCTGTCTCCAGGTGACATTCCA GGAGATGCAGCACTCAAAGCCA |
| GCLM (glutamate-cysteine ligase modifier subunit) | NM_002061.3 | GTTGACATGGCCTGTTCAGTCCT CCCAGTAAGGCTGTAAATGCTCCA |
| GPX1 (glutathione peroxidase 1) | NM_000581.2 | TACTTATCGAGAATGTGGCGTCCC TTGGCGTTCTCCTGATGCCC |
| GPX3 (glutathione peroxidase 3) | NM_002084.5 | GTCGAAGATGGACTGCCATGGT AGCTGGCCACGTTGACAAAGAG |
| GPX4 (glutathione peroxidase 4) | NM_002085.3 | AGGCAAGACCGAAGTAAACTACAC TCTCTTCGTTACTCCCTGGCT |
| GSS (glutathione synthesis) | NM_000178.2 | CCAAGTGCCCAGACATTGCCA CCTTCTTCACCCACATCCAGTGAG |
| HMOX1 (heme oxygenase 1) | NM_002133.2 | CAACAAAGTGCAAGATTCTGCCC CTACAGCAACTGTCGCCACC |
| NOX2 (NADPH oxidase 2) | NM_000397.3 | TCACCAAGGTGGTCACTCACCC TGCCACTCCAGCTTGGACAC |
| NQO1 (NAD(P)H quinone oxidoreductase 1) | NM_001025434.1 | CATCACAGGTAAACTGAAGGACCC CTCTGGAATATCACAAGGTCTGCG |
| PGES (prostaglandin E synthase) | NM_004878.3 | ACGCTGCTGGTCATCAAGATG TGGCAAAGGCCTTCTTCCGC |
| PSTK (phosphoseryl-tRNA kinase) | NM_153336 | TTTGAGGCCCAGTCTTGCTACC GCCCAACGAATATTTCCGAGCC |
| RPL13a (ribosomal protein L13a) | NM_012423.4 | GAGGTTGGCTGGAAGTACCAGG TGTTTCCGTAGCCTCATGAGCTG |
| RPS9 (ribosomal protein S9) | NM_001013.4 | CCATATCAGGGTCCGCAAGCA GGCCCTTCTTGGCATTCTTCCT |
| SECISBP2 (SECIS-binding protein 2) | NM_024077.4 | TGAAGAGCCACCAGGCACAG GCATCTGGCTGCAGTAATCCCT |
| SELENBP1 (selenium-binding protein 1) | NM_009150.3 | TTCCCTTGGAGATCCGCTTCCT ACTGACCATGTACCTCCCTCGT |
| SELENOF (selenoprotein F) | NM_203341.1 | TGATCTTCTCGGACAGTTCAACCT CACGGACATACTTGGACTTGAGGG |
| SELENOH (C11orf31, chromosome 11 open reading frame 31; selenoprotein H) | NM_170746.2 | GCTTCCAGTAAAGGTGAACCCGA TCAGGGAATTTGAGTTTGCGTGG |
| SELENOI (selenoprotein I) | NM_033505.4 | ATGCCTCAGCACCAGGTCAC GTTCTGCGAGCTTGCTTTCCGT |
| SELENOK (selenoprotein K) | NM_021237.3 | GATGATGGAAGAGGGCCACCAG CGCATGTCCGGTTGTCTGCT |
| SELENOM (selenoprotein M) | NM_080430.2 | TGAAGGCTTTCGTCACGCAG AGTGGGATGCGCTCTAGTTCCT |
| SELENON (selenoprotein N) | NM_206926.2 | TGTGATGTTCCGGATCCATGCC TGTGGTTGGGCACGAAGAGC |
| SELENOO (selenoprotein O) | NM_031454.1 | TGACGCCGAGTTCCAAAGGCA TTGTGAAGTCGGCACCGGTCAG |
| SELENOP (selenoprotein P) | NM_005410 | GAAACTCCATCGCCTCATTACCAT CTGCCTATGCTGACCCTTGTG |
| SELENOS (selenoprotein S, VIMP) | NM_203472.1 | GCTGCATCCTTCTCTACGTGGTC CAACAACATCAGGTTCCACAGCA |
| SELENOT (selenoprotein T) | NM_016275.3 | CGATCATAGCACCACCTATCAGCA GAGCCTGCCAAGAAAGCATCTG |
| SEPHS2 (selenophosphate synthetase 2) | NM_012248.4 | GACGGTTTGGGCTTCTTCAAGG TCCACAATGCCAACGATCCAC |
| SEPSECS (sep (o-phosphoserine) tRNA:sec (selenocysteine) tRNA synthase) | NM_016955.3 | CTAGTGCTCCCGCTTATTCGCC CTGGACACTTGCCCTTCTCCAG |
| SLC7A11 (solute carrier family 7 member 11) | NM_014331.3 | TGCTCTTCTCTGGAGACCTCGAC ACAGTGGCACCTTGAAAGGACG |
| SOD1 (superoxide dismutase 1) | NM_000454.4 | TACAGCAGGCTGTACCAGTGCA TCGGCCACACCATCTTTGTCAG |
| SRXN1 (sulfiredoxin 1) | NM_080725.1 | CTCAGTGCTCGTTACTTCATGGTC GTTTGGCCCTTCCTCTTCCTCC |
| TNFA (tumor necrosis factor α) | NM_000594.2 | AGCCCATGTTGTAGCAAACCCT GGAGTAGATGAGGTACAGGCCC |
| TXAS1 (thromboxane A synthase 1) | NM_001061.4 | CCTGAAAGGTTCACGGCTGAG CAACTTGACCTCAAGCAGCCCT |
| TXNRD1 (thioredoxin reductase 1) | NM_182742.1 | GTGTTGTGGGCTTTCACGTACTG TGTTGTGAATACCTCTGCACAGAC |
| TXNRD2 (thioredoxin reductase 2) | NM_006440.5 | GTTCCCACGACCGTCTTCAC GTGATAGACCTCAACATGCTCCTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wolfram, T.; Weidenbach, L.M.; Adolf, J.; Schwarz, M.; Schädel, P.; Gollowitzer, A.; Werz, O.; Koeberle, A.; Kipp, A.P.; Koeberle, S.C. The Trace Element Selenium Is Important for Redox Signaling in Phorbol Ester-Differentiated THP-1 Macrophages. Int. J. Mol. Sci. 2021, 22, 11060. https://doi.org/10.3390/ijms222011060
Wolfram T, Weidenbach LM, Adolf J, Schwarz M, Schädel P, Gollowitzer A, Werz O, Koeberle A, Kipp AP, Koeberle SC. The Trace Element Selenium Is Important for Redox Signaling in Phorbol Ester-Differentiated THP-1 Macrophages. International Journal of Molecular Sciences. 2021; 22(20):11060. https://doi.org/10.3390/ijms222011060
Chicago/Turabian StyleWolfram, Theresa, Leonie M. Weidenbach, Johanna Adolf, Maria Schwarz, Patrick Schädel, André Gollowitzer, Oliver Werz, Andreas Koeberle, Anna P. Kipp, and Solveigh C. Koeberle. 2021. "The Trace Element Selenium Is Important for Redox Signaling in Phorbol Ester-Differentiated THP-1 Macrophages" International Journal of Molecular Sciences 22, no. 20: 11060. https://doi.org/10.3390/ijms222011060
APA StyleWolfram, T., Weidenbach, L. M., Adolf, J., Schwarz, M., Schädel, P., Gollowitzer, A., Werz, O., Koeberle, A., Kipp, A. P., & Koeberle, S. C. (2021). The Trace Element Selenium Is Important for Redox Signaling in Phorbol Ester-Differentiated THP-1 Macrophages. International Journal of Molecular Sciences, 22(20), 11060. https://doi.org/10.3390/ijms222011060