
The Kynurenine Pathway as a Potential Target for Neuropathic Pain Therapy Design: From Basic Research to Clinical Perspectives


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
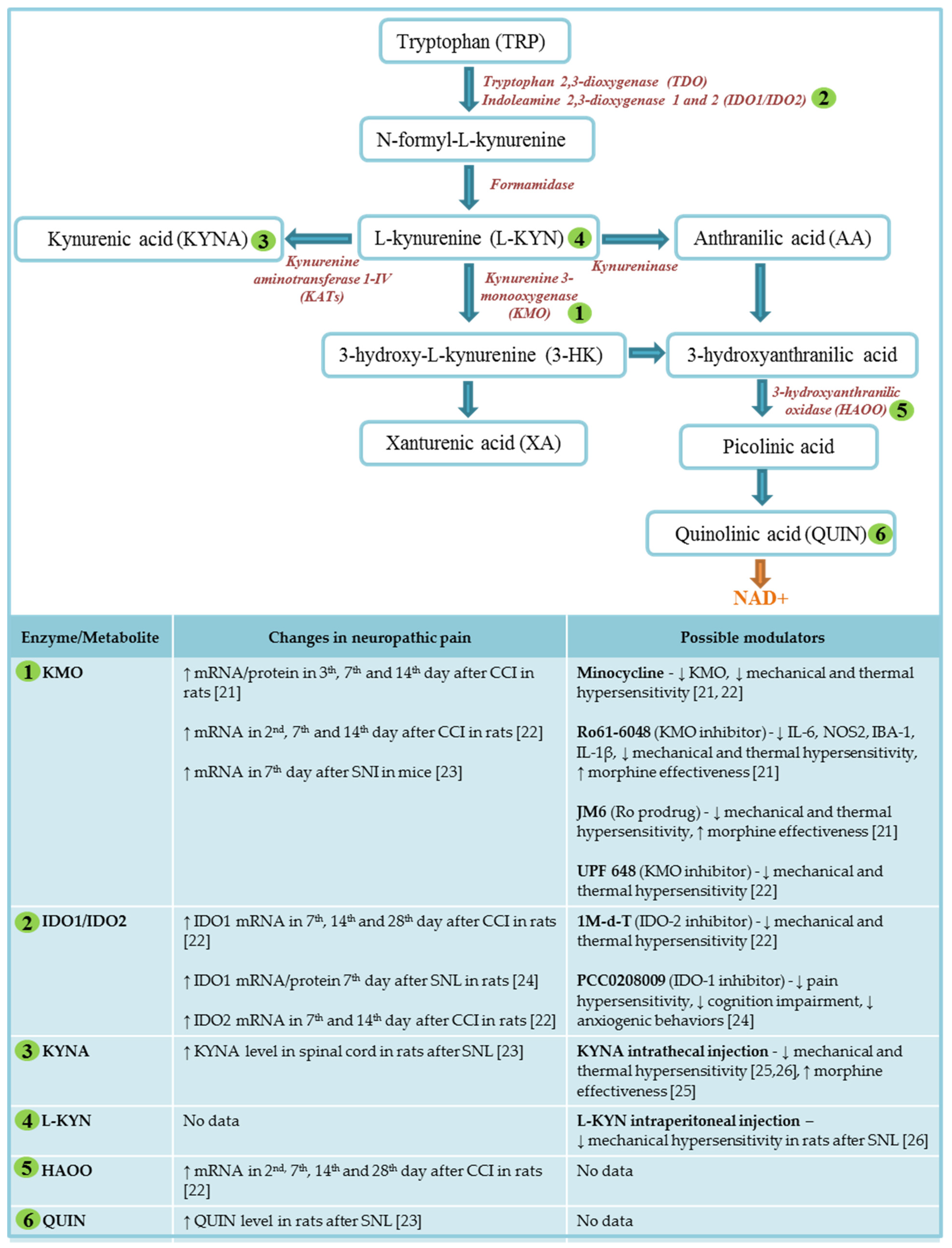
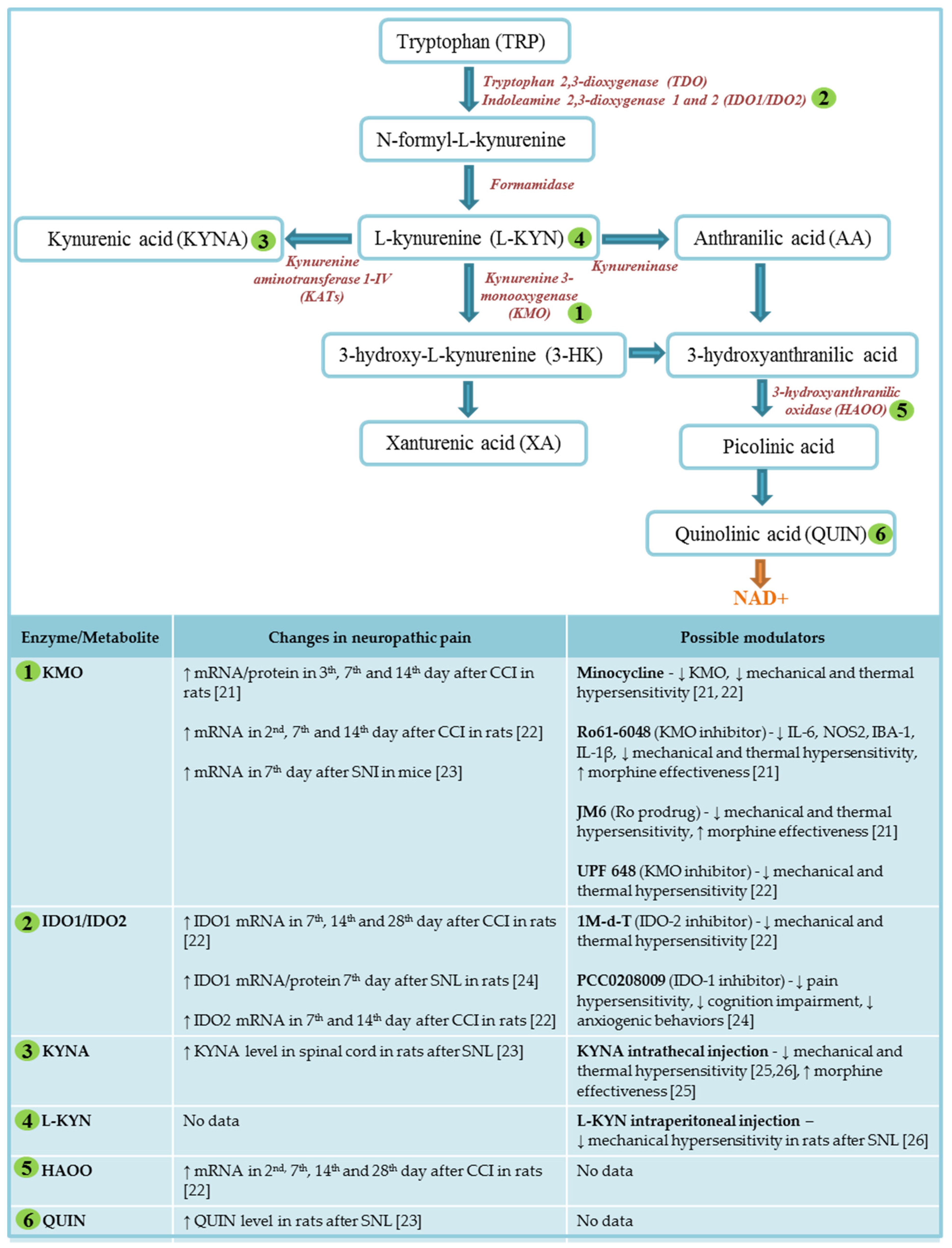
2. Kynurenine Pathway of Tryptophan Metabolism
3. Neuroactive Metabolites of Kynurenine Pathway
3.1. L-kynurenine
3.2. Kynurenic Acid
3.3. 3-Hydroxy-L-kynurenine
3.4. Quinolinic Acid
4. Major Enzymes of Kynurenine Pathway
4.1. Indolamine, 2,3-dioxygenase-1 and 2
4.2. Kynurenine-3-monooxygenase
4.3. Kynurenine aminotransferases
4.4. Kynureninase
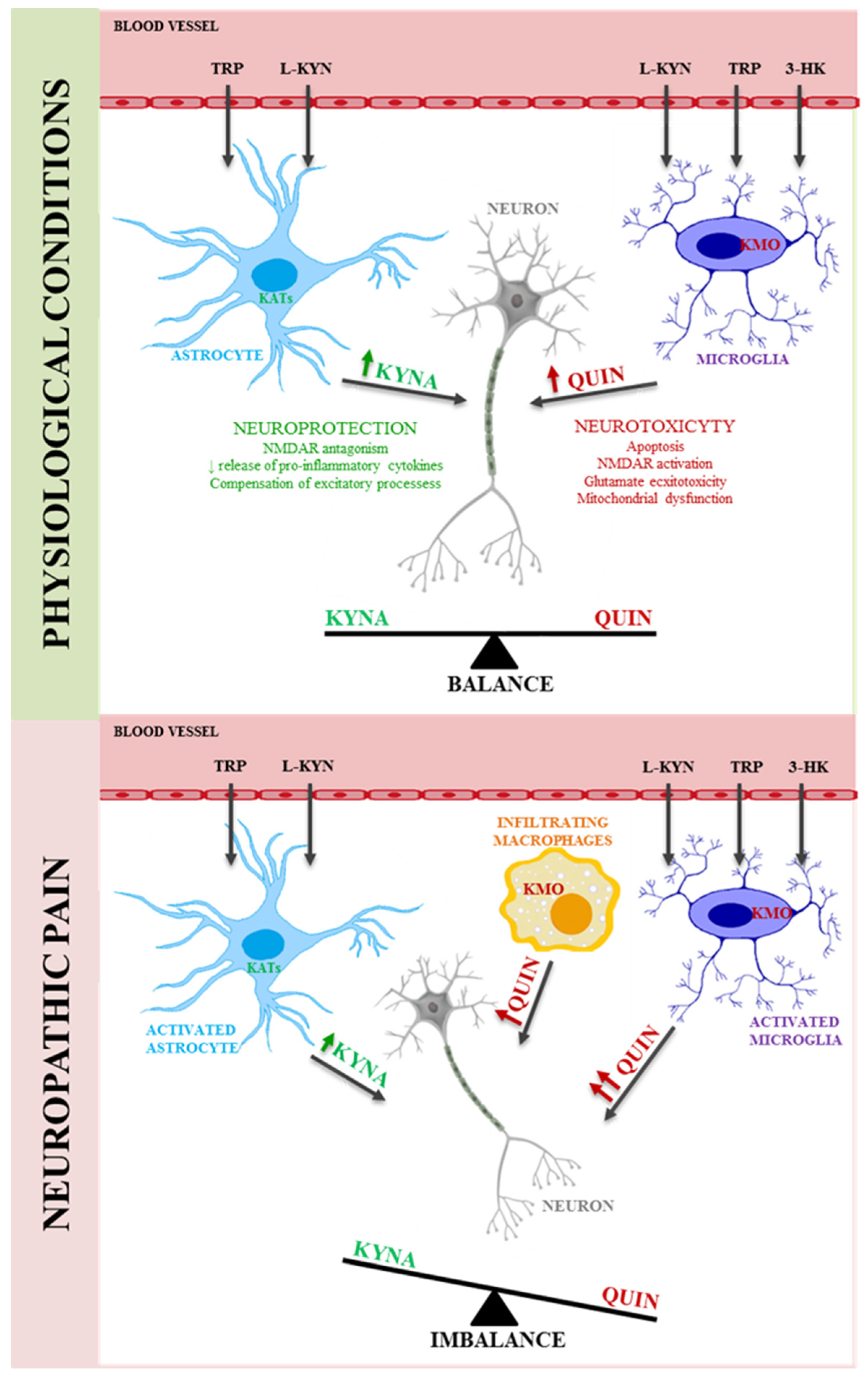
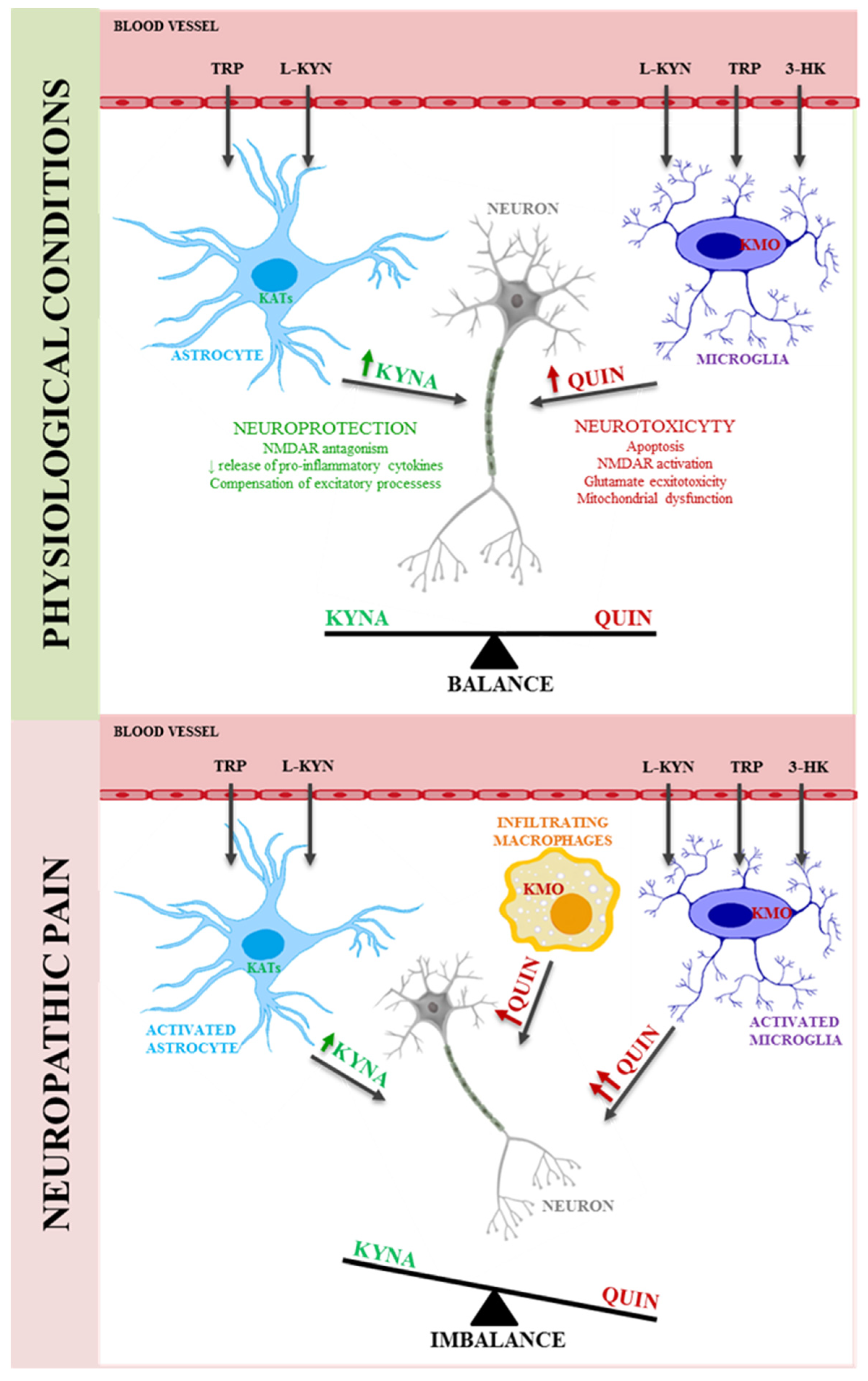
5. Kynurenine Pathway in Neuropathic Pain
6. Therapeutic Potential of the Kynurenine Pathway in the Treatment of Neuropathic Pain
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1-M-d-T | 1-Methyl-D-tryptophan |
| 3-HANA | 3-hydroxyanthranilic acid |
| 3-HAOO | 3-hydroxyanthranilic oxidase |
| 3-HK-3 | hydroxykinurenine |
| AA | anthranilic acid |
| AhR | aryl hydrocarbon receptor |
| AMPA | α-amino-2,3-dihydro-5-methyl-3-oxo-isoxazolpropionic acid |
| APCs | antigen-presenting cells |
| BBB | blood–brain barrier |
| BH4 | tetrahydrobiopterin |
| C1q | complement component 1q |
| CCI | chronic constriction injury |
| CD40 | cluster of differentiation 40 |
| CNS | central nervous system |
| DCs | dendritic cells |
| DRG | dorsal root ganglia |
| GFAP | glial fibrillary acidic protein |
| GPR35 | G-protein coupled receptor 35 |
| HCN channels | hyperpolarization-activated cyclic nucleotide–gated channels |
| HIV | human immunodeficiency virus |
| IASP | The International Association for the Study of Pain |
| IBA-1 | allograft inflammatory factor 1 |
| IDO | indoleamine 2,3-dioxygenase |
| IFN-1β | interferon beta -1 |
| IFN-6 | interferon-6 |
| IFN-gamma | interferon gamma |
| IL-1 | interleukin 1 |
| IL-10 | interleukin-10 |
| IL-1β | interleukin 1 beta |
| IL-4 | interleukin-4 |
| IL-6 | interleukin-6 |
| KAR | kainic acid receptor |
| KATs | kynurenine aminotransferases |
| KM | Michaelis constant |
| KMO | kynurenine-3-monooxygenase |
| KP | kynurenine pathway |
| KYNA | kynureinc acid |
| KYNU | kynureninase |
| L-KYN | L-kynurenine |
| L-KYN | L-kynurenine |
| MAO-B | B monoamine oxidase |
| NAD | nicotinamide adenine dinucleotide |
| NMDA | N-methyl-D-aspartate |
| NOS2 | Nitric oxide synthase 2 |
| QUIN | quinolinic acid |
| ROS | reactive oxygen species |
| SC | spinal cord |
| SNI | sciatic nerve injury |
| SNL | spinal nerve ligation |
| SNRIs | serotonin-noradrenaline reuptake inhibitors |
| SOD | superoxide dismutase |
| TCAs | tricyclic antidepressants |
| TDO | tryptophan 2,3-dioxygenase |
| TNF-α | tumor necrosis alpha |
| TRP | tryptophan |
| UPF648 | (1S,2S)-2-(3,4-Dichlorobenzoyl)cyclopropanecarboxylic acid |
| XA | xanturenic acid |
References
- Loeser, J.D.; Treede, R.D. The Kyoto protocol of IASP Basic Pain Terminology. Pain 2008, 137, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Aydede, M.; Shriver, A. Recently introduced definition of “nociplastic pain” by the International Association for the Study of Pain needs better formulation. Pain 2018, 159, 1176–1177. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Török, N.; Tóth, F.; Szabó, Á.; Vécsei, L. Co-players in chronic pain: Neuroinflammation and the tryptophan-kynurenine metabolic pathway. Biomedicines 2021, 9, 897. [Google Scholar] [CrossRef]
- Bouhassira, D. Neuropathic pain: Definition, assessment and epidemiology. Rev. Neurol. 2019, 175, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Prim. 2017, 3, 17002. [Google Scholar] [CrossRef] [Green Version]
- van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain 2014, 155, 654–662. [Google Scholar] [CrossRef]
- Cavalli, E.; Mammana, S.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The neuropathic pain: An overview of the current treatment and future therapeutic approaches. Int. J. Immunopathol. Pharmacol. 2019, 33, 2058738419838383. [Google Scholar] [CrossRef] [Green Version]
- Yawn, B.P.; Wollan, P.C.; Weingarten, T.N.; Watson, J.C.; Hooten, W.M.; Melton, L.J. The prevalence of neuropathic pain: Clinical evaluation compared with screening tools in a community population. Pain Med. 2009, 10, 586–593. [Google Scholar] [CrossRef] [Green Version]
- Butera, J.A. Current and emerging targets to treat neuropathic pain. J. Med. Chem. 2007, 50, 2543–2546. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Bates, D.; Carsten Schultheis, B.; Hanes, M.C.; Jolly, S.M.; Chakravarthy, K.V.; Deer, T.R.; Levy, R.M.; Hunter, C.W. A Comprehensive Algorithm for Management of Neuropathic Pain. Pain Med. 2019, 20, S2–S12. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, V.; Sharma, D.; Vaidya, S.; Shantanu, P.A.; Guan, Y.; Kalia, K.; Tiwari, V. Cellular and molecular mechanisms driving neuropathic pain: Recent advancements and challenges. Expert Opin. Ther. Targets 2018, 22, 131–142. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Kuner, R.; Jensen, T.S. Neuropathic pain: Frommechanisms to treatment. Physiol. Rev. 2021, 101, 259–301. [Google Scholar] [CrossRef] [PubMed]
- Meacham, K.; Shepherd, A.; Mohapatra, D.P.; Haroutounian, S. Neuropathic Pain: Central vs. Peripheral Mechanisms. Curr. Pain Headache Rep. 2017, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J. The kynurenine pathway: A finger in every pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A.B. Kynurenine pathway of tryptophan metabolism: Regulatory and functional aspects. Int. J. Tryptophan Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [Green Version]
- Höglund, E.; Øverli, Ø.; Winberg, S. Tryptophan metabolic pathways and brain serotonergic activity: A comparative review. Front. Endocrinol. 2019, 10, 158. [Google Scholar] [CrossRef]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef]
- Gulaj, E.; Pawlak, K.; Bien, B.; Pawlak, D. Kynurenine and its metabolites in Alzheimer’s disease patients. Adv. Med. Sci. 2010, 55, 204–211. [Google Scholar] [CrossRef]
- Tanaka, M.; Bohár, Z.; Vécsei, L. Are Kynurenines Accomplices or Principal Villains in Dementia? Maintenance of Kynurenine Metabolism. Molecules 2020, 25, 564. [Google Scholar] [CrossRef] [Green Version]
- Rojewska, E.; Piotrowska, A.; Makuch, W.; Przewlocka, B.; Mika, J. Pharmacological kynurenine 3-monooxygenase enzyme inhibition significantly reduces neuropathic pain in a rat model. Neuropharmacology 2016, 102, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Rojewska, E.; Ciapała, K.; Piotrowska, A.; Makuch, W.; Mika, J. Pharmacological Inhibition of Indoleamine 2,3-Dioxygenase-2 and Kynurenine 3-Monooxygenase, Enzymes of the Kynurenine Pathway, Significantly Diminishes Neuropathic Pain in a Rat Model. Front. Pharmacol. 2018, 9, 724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laumet, G.; Zhou, W.; Dantzer, R.; Edralin, J.D.; Huo, X.J.; Budac, D.P.; O’Connor, J.C.; Lee, A.W.; Heijnen, C.J.; Kavelaars, A. Upregulation of neuronal kynurenine 3-monooxygenase mediates depression-like behavior in a mouse model of neuropathic pain. Brain. Behav. Immun. 2017, 66, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, C.-M.; Han, R.; Wang, Z.-Z.; Gao, Y.-L.; Zhu, X.-Y.; Yu, X.; Du, G.-Y.; Wang, H.-B.; Tian, J.-W.; et al. PCC0208009, an indirect IDO1 inhibitor, alleviates neuropathic pain and co-morbidities by regulating synaptic plasticity of ACC and amygdala. Biochem. Pharmacol. 2020, 177, 113926. [Google Scholar] [CrossRef] [PubMed]
- Rojewska, E.; Ciapała, K.; Mika, J. Kynurenic acid and zaprinast diminished CXCL17-evoked pain-related behaviour and enhanced morphine analgesia in a mouse neuropathic pain model. Pharmacol. Rep. 2019, 71, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Pineda-Farias, J.B.; Pérez-Severiano, F.; González-Esquivel, D.F.; Barragán-Iglesias, P.; Bravo-Hernández, M.; Cervantes-Durán, C.; Aguilera, P.; Ríos, C.; Granados-Soto, V. The l -kynurenine-probenecid combination reduces neuropathic pain in rats. Eur. J. Pain 2013, 17, 1365–1373. [Google Scholar] [CrossRef]
- Reyes Ocampo, J.; Lugo Huitrón, R.; González-Esquivel, D.; Ugalde-Muñiz, P.; Jiménez-Anguiano, A.; Pineda, B.; Pedraza-Chaverri, J.; Ríos, C.; Pérez de la Cruz, V. Kynurenines with neuroactive and redox properties: Relevance to aging and brain diseases. Oxid. Med. Cell. Longev. 2014, 2014, 646909. [Google Scholar] [CrossRef]
- Schwarcz, R.; Stone, T.W. The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacology 2017, 112, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Saito, K. Species and cell types difference in tryptophan metabolism. Int. J. Tryptophan Res. 2013, 6, 47–54. [Google Scholar] [CrossRef]
- Heyes, M.P.; Saito, K.; Chen, C.Y.; Proescholdt, M.G.; Nowak, T.S.; Li, J.; Beagles, K.E.; Proescholdt, M.A.; Zito, M.A.; Kawai, K.; et al. Species heterogeneity between gerbils and rats: Quinolinate production by microgila and astrocytes and accumulations in response to ischemic brain injury and systemic immune activation. J. Neurochem. 1997, 69, 1519–1529. [Google Scholar] [CrossRef]
- Jovanovic, F.; Candido, K.D.; Knezevic, N.N. The role of the kynurenine signaling pathway in different chronic pain conditions and potential use of therapeutic agents. Int. J. Mol. Sci. 2020, 21, 6045. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, B.J. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv. Exp. Med. Biol. 2003, 527, 105–112. [Google Scholar]
- Fukui, S.; Schwarcz, R.; Rapoport, S.I.; Takada, Y.; Smith, Q.R. Blood-Brain Barrier Transport of Kynurenines: Implications for Brain Synthesis and Metabolism. J. Neurochem. 1991, 56, 2007–2017. [Google Scholar] [CrossRef]
- Kita, T.; Morrison, P.F.; Heyes, M.P.; Markey, S.P. Effects of systemic and central nervous system localized inflammation on the contributions of metabolic precursors to the l-kynurenine and quinolinic acid pools in brain. J. Neurochem. 2002, 82, 258–268. [Google Scholar] [CrossRef]
- Lapin, I.P. Depressor effect of kynurenine and its metabolites in rats. Life Sci. 1976, 19, 1479–1483. [Google Scholar] [CrossRef]
- Lapin, I.P. Stimulant and convulsive effects of kynurenines injected into brain ventricles in mice. J. Neural Transm. 1978, 42, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Pinelli, A.; Ossi, C.; Colombo, R.; Tofanetti, O.; Spazzi, L. Experimental convulsions in rats induced by intra ventricular administration of kynurenine and structurally related compounds. Neuropharmacology 1984, 23, 333–337. [Google Scholar] [CrossRef]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. 3-Hydroxykynurenine, an Endogenous Oxidative Stress Generator, Causes Neuronal Cell Death with Apoptotic Features and Region Selectivity. J. Neurochem. 2002, 70, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Peyrot, F.; Ducrocq, C. Potential role of tryptophan derivatives in stress responses characterized by the generation of reactive oxygen and nitrogen species. J. Pineal Res. 2008, 45, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Park, H.; Kim, Y.-S.; Kim, K.D.; Lee, H.-K.; Cho, D.-H.; Yang, J.-W.; Hur, D.Y. l-Kynurenine-induced apoptosis in human NK cells is mediated by reactive oxygen species. Int. Immunopharmacol. 2011, 11, 932–938. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Cuartero, M.I.; de la Parra, J.; García-Culebras, A.; Ballesteros, I.; Lizasoain, I.; Moro, M.Á. The Kynurenine Pathway in the Acute and Chronic Phases of Cerebral Ischemia. Curr. Pharm. Des. 2016, 22, 1060–1073. [Google Scholar] [CrossRef] [PubMed]
- Vamos, E.; Pardutz, A.; Klivenyi, P.; Toldi, J. The role of kynurenines in disorders of the central nervous system: Possibilities for neuroprotection. J. Neurol. Sci. 2009, 283, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Zádori, D.; Klivényi, P.; Vámos, E.; Fülöp, F.; Toldi, J.; Vécsei, L. Kynurenines in chronic neurodegenerative disorders: Future therapeutic strategies. J. Neural Transm. 2009, 116, 1403–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, M.N.; Stone, T.W. An iontophoretic investigation of the actions of convulsant kynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res. 1982, 247, 184–187. [Google Scholar] [CrossRef]
- Hilmas, C.; Pereira, E.F.R.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The Brain Metabolite Kynurenic Acid Inhibits α7 Nicotinic Receptor Activity and Increases Non-α7 Nicotinic Receptor Expression: Physiopathological Implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef]
- Stone, T.W. Does kynurenic acid act on nicotinic receptors? An assessment of the evidence. J. Neurochem. 2020, 152, 627–649. [Google Scholar] [CrossRef] [Green Version]
- Carpenedo, R.; Chiarugi, A.; Russi, P.; Lombardi, G.; Carlà, V.; Pellicciari, R.; Mattoli, L.; Moroni, F. Inhibitors of kynurenine hydroxylase and kynureninase increase cerebral formation of kynurenate and have sedative and anticonvulsant activities. Neuroscience 1994, 61, 237–244. [Google Scholar] [CrossRef]
- Cozzi, A.; Carpenedo, R.; Moroni, F. Kynurenine hydroxylase inhibitors reduce ischemic brain damage: Studies with (m-nitrobenzoyl)-alanine (mNBA) and 3,4-dimethoxy-[-N-4- (nitrophenyl)thiazol-2YL]-benzenesulfonamide (Ro 61-8048) in models of focal or global brain ischemia. J. Cereb. Blood Flow Metab. 1999, 19, 771–777. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic Acid as a Ligand for Orphan G Protein-coupled Receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef] [Green Version]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vecsei, L. Kynurenine metabolism in multiple sclerosis. Acta Neurol. Scand. 2005, 112, 93–96. [Google Scholar] [CrossRef]
- Forrest, C.M.; Gould, S.R.; Darlington, L.G.; Stone, T.W. Levels of purine, kynurenine and lipid peroxidation products in patients with inflammatory bowel disease. Adv. Exp. Med. Biol. 2003, 527, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Oxenkrug, G.F. Increased Plasma Levels of Xanthurenic and Kynurenic Acids in Type 2 Diabetes. Mol. Neurobiol. 2015, 52, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymona, K.; Zdzisińska, B.; Karakuła-Juchnowicz, H.; Kocki, T.; Kandefer-Szerszeń, M.; Flis, M.; Rosa, W.; Urbańska, E.M. Correlations of Kynurenic Acid, 3-Hydroxykynurenine, sIL-2R, IFN-α, and IL-4 with Clinical Symptoms During Acute Relapse of Schizophrenia. Neurotox. Res. 2017, 32, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Hartai, Z.; Juhász, A.; Rimanóczy, Á.; Janáky, T.; Donkó, T.; Dux, L.; Penke, B.; Tóth, G.K.; Janka, Z.; Kálmán, J. Decreased serum and red blood cell kynurenic acid levels in Alzheimer’s disease. Neurochem. Int. 2007, 50, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Curto, M.; Lionetto, L.; Negro, A.; Capi, M.; Perugino, F.; Fazio, F.; Giamberardino, M.A.; Simmaco, M.; Nicoletti, F.; Martelletti, P. Altered serum levels of kynurenine metabolites in patients affected by cluster headache. J. Headache Pain 2016, 17, 27. [Google Scholar] [CrossRef] [Green Version]
- Curto, M.; Lionetto, L.; Negro, A.; Capi, M.; Fazio, F.; Giamberardino, M.A.; Simmaco, M.; Nicoletti, F.; Martelletti, P. Altered kynurenine pathway metabolites in serum of chronic migraine patients. J. Headache Pain 2015, 17, 47. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F.; Matson, W.R.; Storey, E.; Milbury, P.; Ryan, E.A.; Ogawa, T.; Bird, E.D. Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J. Neurol. Sci. 1992, 108, 80–87. [Google Scholar] [CrossRef]
- Parada-Turska, J.; Zgrajka, W.; Majdan, M.; Lisse, J.; Westlund, K.N. Kynurenic acid in synovial fluid and serum of patients with rheumatoid arthritis, spondyloarthropathy, and osteoarthritis. J. Rheumatol. 2013, 40, 903–909. [Google Scholar] [CrossRef]
- Spekker, E.; Laborc, K.F.; Bohár, Z.; Nagy-Grócz, G.; Fejes-Szabó, A.; Szűcs, M.; Vécsei, L.; Párdutz, Á. Effect of dural inflammatory soup application on activation and sensitization markers in the caudal trigeminal nucleus of the rat and the modulatory effects of sumatriptan and kynurenic acid. J. Headache Pain 2021, 22, 17. [Google Scholar] [CrossRef]
- Eastman, C.L.; Guilarte, T.R. Cytotoxicity of 3-hydroxykynurenine in a neuronal hybrid cell line. Brain Res. 1989, 495, 225–231. [Google Scholar] [CrossRef]
- Chiarugi, A.; Meli, E.; Moroni, F. Similarities and differences in the neuronal death processes activated by 3OH-kynurenine and quinolinic acid. J. Neurochem. 2001, 77, 1310–1318. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.E.; Leopold, M.C.; Huang, X.; Atwood, C.S.; Saunders, A.J.; Hartshorn, M.; Lim, J.T.; Faget, K.Y.; Muffat, J.A.; Scarpa, R.C.; et al. 3-Hydroxykynurenine and 3-Hydroxyanthranilic Acid Generate Hydrogen Peroxide and Promote α-Crystallin Cross-Linking by Metal Ion Reduction. Biochemistry 2000, 39, 7266–7275. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Ocampo, J.; Ramírez-Ortega, D.; Vázquez Cervantes, G.I.; Pineda, B.; Montes de Oca Balderas, P.; González-Esquivel, D.; Sánchez-Chapul, L.; Lugo-Huitrón, R.; Silva-Adaya, D.; Ríos, C.; et al. Mitochondrial dysfunction related to cell damage induced by 3-hydroxykynurenine and 3-hydroxyanthranilic acid: Non-dependent-effect of early reactive oxygen species production. Neurotoxicology 2015, 50, 81–91. [Google Scholar] [CrossRef]
- Colín-González, A.L.; Maldonado, P.D.; Santamaría, A. 3-Hydroxykynurenine: An intriguing molecule exerting dual actions in the Central Nervous System. Neurotoxicology 2013, 34, 189–204. [Google Scholar] [CrossRef]
- Giles, G.I.; Collins, C.A.; Stone, T.W.; Jacob, C. Electrochemical and in vitro evaluation of the redox-properties of kynurenine species. Biochem. Biophys. Res. Commun. 2003, 300, 719–724. [Google Scholar] [CrossRef]
- Bonda, D.J.; Mailankot, M.; Stone, J.G.; Garrett, M.R.; Staniszewska, M.; Castellani, R.J.; Siedlak, S.L.; Zhu, X.; Lee, H.; Perry, G.; et al. Indoleamine 2,3-dioxygenase and 3-hydroxykynurenine modifications are found in the neuropathology of Alzheimer’s disease. Redox Rep. 2010, 15, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, S.J.; Reynolds, G.P. Increased brain concentrations of a neurotoxin, 3-hydroxykynurenine, in Huntington’s disease. Neurosci. Lett. 1992, 144, 199–201. [Google Scholar] [CrossRef]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef]
- Schwarcz, R.; Whetsell, W.O.; Mangano, R.M. Quinolinic acid: An endogenous metabolite that produces axon-sparing lesions in rat brain. Science 1983, 219, 316–318. [Google Scholar] [CrossRef]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-De La Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxid. Med. Cell. Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-De La Cruz, V.; Konigsberg, M.; Pedraza-Chaverri, J.; Herrera-Mundo, N.; Díaz-Muñoz, M.; Morán, J.; Fortoul-Van Der Goes, T.; Rondán-Zárate, A.; Maldonado, P.D.; Ali, S.F.; et al. Cytoplasmic calcium mediates oxidative damage in an excitotoxic/energetic deficit synergic model in rats. Eur. J. Neurosci. 2008, 27, 1075–1085. [Google Scholar] [CrossRef]
- Stipek, S.; Stastny, F.; Platenik, J.; Crkovska, J.; Zima, T. The effect of quinolinate on rat brain lipid peroxidation is dependent on iron. Neurochem. Int. 1997, 30, 233–237. [Google Scholar] [CrossRef]
- Naoi, M.; Ishiki, R.; Nomura, Y.; Hasegawa, S.; Nagatsu, T. Quinolinic acid: An endogenous inhibitor specific for type B monoamine oxidase in human brain synaptosomes. Neurosci. Lett. 1987, 74, 232–236. [Google Scholar] [CrossRef]
- Braidy, N.; Grant, R.; Brew, B.J.; Adams, S.; Jayasena, T.; Guillemin, G.J. Effects of kynurenine pathway metabolites on intracellular NAD+ synthesis and cell death in human primary astrocytes and neurons. Int. J. Tryptophan Res. 2009, 2, 61–69. [Google Scholar] [CrossRef]
- Guidetti, P.; Luthi-Carter, R.E.; Augood, S.J.; Schwarcz, R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease. Neurobiol. Dis. 2004, 17, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Sorgdrager, F.J.H.; Vermeiren, Y.; Faassen, M.; Ley, C.; Nollen, E.A.A.; Kema, I.P.; De Deyn, P.P. Age- and disease-specific changes of the kynurenine pathway in Parkinson’s and Alzheimer’s disease. J. Neurochem. 2019, 151, 656–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiner, J.; Walter, M.; Gos, T.; Guillemin, G.J.; Bernstein, H.G.; Sarnyai, Z.; Mawrin, C.; Brisch, R.; Bielau, H.; zu Schwabedissen, L.M.; et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: Evidence for an immune-modulated glutamatergic neurotransmission? J. Neuroinflammation 2011, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Stankovic, R.; Cullen, K.M.; Meininger, V.; Garner, B.; Coggan, S.; Grant, R.; Brew, B.J.; Guillemin, G.J. The kynurenine pathway and inflammation in amyotrophic lateral sclerosis. Neurotox. Res. 2010, 18, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Heyes, M.P.; Saito, K.; Lackner, A.; Wiley, C.A.; Achim, C.L.; Markey, S.P. Sources of the neurotoxin quinolinic acid in the brain of HIV-1-infected patients and retrovirus-infected macaques. FASEB J. 1998, 12, 881–896. [Google Scholar] [CrossRef]
- Stone, T.W. Endogenous neurotoxins from tryptophan. Toxicon 2001, 39, 61–73. [Google Scholar] [CrossRef]
- Gunn, J.; Hill, M.M.; Cotton, B.M.; Deer, T.R. An analysis of biomarkers in patients with chronic pain. Pain Physician 2020, 23, E41–E49. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.; Whiter, F.; Gallop, K.; Veronese, N.; Solmi, M.; Newton, P.; Stubbs, B. NMDA receptor antagonists and pain relief: A meta-analysis of experimental trials. Neurology 2019, 92, E1652–E1662. [Google Scholar] [CrossRef]
- Johnson, T.S.; Munn, D.H. Host indoleamine 2,3-dioxygenase: Contribution to systemic acquired tumor tolerance. Immunol. Investig. 2012, 41, 765–797. [Google Scholar] [CrossRef]
- Huang, Y.S.; Ogbechi, J.; Clanchy, F.I.; Williams, R.O.; Stone, T.W. IDO and Kynurenine Metabolites in Peripheral and CNS Disorders. Front. Immunol. 2020, 11, 388. [Google Scholar] [CrossRef] [Green Version]
- Kashi, A.A.; Davis, R.W.; Phair, R.D. The IDO Metabolic Trap Hypothesis for the Etiology of ME/CFS. Diagnostics 2019, 9, 82. [Google Scholar] [CrossRef] [Green Version]
- Merlo, L.M.F.; DuHadaway, J.B.; Montgomery, J.D.; Peng, W.-D.; Murray, P.J.; Prendergast, G.C.; Caton, A.J.; Muller, A.J.; Mandik-Nayak, L. Differential Roles of IDO1 and IDO2 in T and B Cell Inflammatory Immune Responses. Front. Immunol. 2020, 11, 1861. [Google Scholar] [CrossRef]
- Bilir, C.; Sarisozen, C. Indoleamine 2,3-dioxygenase (IDO): Only an enzyme or a checkpoint controller? J. Oncol. Sci. 2017, 3, 52–56. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widner, B.; Leblhuber, F.; Walli, J.; Tilz, G.P.; Demel, U.; Fuchs, D. Degradation of tryptophan in neurodegenerative disorders. Adv. Exp. Med. Biol. 1999, 467, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; DuHadaway, J.B.; Donover, P.S.; Sutanto-Ward, E.; Prendergast, G.C. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005, 11, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Darlington, L.G.; Mackay, G.M.; Forrest, C.M.; Stoy, N.; George, C.; Stone, T.W. Altered kynurenine metabolism correlates with infarct volume in stroke. Eur. J. Neurosci. 2007, 26, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- Opitz, C.A.; Wick, W.; Steinman, L.; Platten, M. Tryptophan degradation in autoimmune diseases. Cell. Mol. Life Sci. 2007, 64, 2542–2563. [Google Scholar] [CrossRef]
- Zelante, T.; Fallarino, F.; Bistoni, F.; Puccetti, P.; Romani, L. Indoleamine 2,3-dioxygenase in infection: The paradox of an evasive strategy that benefits the host. Microbes Infect. 2009, 11, 133–141. [Google Scholar] [CrossRef]
- Sage, L.K.; Fox, J.M.; Mellor, A.L.; Tompkins, S.M.; Tripp, R.A. Indoleamine 2,3-Dioxygenase (IDO) Activity During the Primary Immune Response to Influenza Infection Modifies the Memory T Cell Response to Influenza Challenge. Viral Immunol. 2014, 27, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Mazarei, G.; Leavitt, B.R. Indoleamine 2,3 Dioxygenase as a Potential Therapeutic Target in Huntington’s Disease. J. Huntingtons. Dis. 2015, 4, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laugeray, A.; Launay, J.M.; Callebert, J.; Surget, A.; Belzung, C.; Barone, P.R. Peripheral and cerebral metabolic abnormalities of the tryptophan-kynurenine pathway in a murine model of major depression. Behav. Brain Res. 2010, 210, 84–91. [Google Scholar] [CrossRef]
- Tang, K.; Wu, Y.H.; Song, Y.; Yu, B. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 68. [Google Scholar] [CrossRef]
- Lu, Y.; Shao, M.; Wu, T. Kynurenine-3-monooxygenase: A new direction for the treatment in different diseases. Food Sci. Nutr. 2020, 8, 711–719. [Google Scholar] [CrossRef]
- Courtney, S.; Scheel, A. Modulation of the Kynurenine Pathway for the Potential Treatment of Neurodegenerative Diseases; Springer: Berlin/Heidelberg, Germany, 2010; pp. 149–176. [Google Scholar]
- Tanaka, M.; Tóth, F.; Polyák, H.; Szabó, Á.; Mándi, Y.; Vécsei, L. Immune Influencers in Action: Metabolites and Enzymes of the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 734. [Google Scholar] [CrossRef]
- Wilson, K.; Auer, M.; Binnie, M.; Zheng, X.; Pham, N.T.; Iredale, J.P.; Webster, S.P.; Mole, D.J. Overexpression of human kynurenine-3-monooxygenase protects against 3-hydroxykynurenine-mediated apoptosis through bidirectional nonlinear feedback. Cell Death Dis. 2016, 7, e2197. [Google Scholar] [CrossRef] [Green Version]
- Mithaiwala, M.N.; Santana-Coelho, D.; Porter, G.A.; O’Connor, J.C. Neuroinflammation and the Kynurenine Pathway in CNS Disease: Molecular Mechanisms and Therapeutic Implications. Cells 2021, 10, 1548. [Google Scholar] [CrossRef]
- Forrest, C.M.; Kennedy, P.G.E.; Rodgers, J.; Dalton, R.N.; Turner, C.; Darlington, L.G.; Cobb, S.R.; Stone, T.W. Kynurenine pathway metabolism following prenatal KMO inhibition and in Mecp2+/− mice, using liquid chromatography-tandem mass spectrometry. Neurochem. Int. 2016, 100, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Collier, M.E.W.; Heyes, D.J.; Giorgini, F.; Scrutton, N.S. Advantages of brain penetrating inhibitors of kynurenine-3-monooxygenase for treatment of neurodegenerative diseases. Arch. Biochem. Biophys. 2021, 697, 108702. [Google Scholar] [CrossRef]
- Zhang, S.; Sakuma, M.; Deora, G.S.; Levy, C.W.; Klausing, A.; Breda, C.; Read, K.D.; Edlin, C.D.; Ross, B.P.; Wright Muelas, M.; et al. A brain-permeable inhibitor of the neurodegenerative disease target kynurenine 3-monooxygenase prevents accumulation of neurotoxic metabolites. Commun. Biol. 2019, 2, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, T.W.; Darlington, L.G. Endogenous kynurenines as targets for drug discovery and development. Nat. Rev. Drug Discov. 2002, 1, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Nematollahi, A.; Sun, G.; Jayawickrama, G.S.; Church, W.B. Kynurenine Aminotransferase Isozyme Inhibitors: A Review. Int. J. Mol. Sci. 2016, 17, 946. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Cai, T.; Tagle, D.A.; Li, J. Structure, expression, and function of kynurenine aminotransferases in human and rodent brains. Cell. Mol. Life Sci. 2010, 67, 353–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, Y.; Fujigaki, H.; Kato, K.; Yamazaki, K.; Fujigaki, S.; Kunisawa, K.; Yamamoto, Y.; Mouri, A.; Oda, A.; Nabeshima, T.; et al. Selective and competitive inhibition of kynurenine aminotransferase 2 by glycyrrhizic acid and its analogues. Sci. Rep. 2019, 9, 10243. [Google Scholar] [CrossRef] [Green Version]
- Alberati-Giani, D.; Ricciardi-Castagnoli, P.; Köhler, C.; Cesura, A.M. Regulation of the kynurenine metabolic pathway by interferon-gamma in murine cloned macrophages and microglial cells. J. Neurochem. 1996, 66, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Heyes, M.P.; Saito, K.; Major, E.O.; Milstien, S.; Markey, S.P.; Vickers, J.H. A mechanism of quinolinic acid formation by brain in inflammatory neurological disease: Attenuation of synthesis from L-tryptophan by 6-chlorotryptophan and 4-chloro-3-hydroxyanthranilate. Brain 1993, 116, 1425–1450. [Google Scholar] [CrossRef] [PubMed]
- Harden, J.L.; Lewis, S.M.; Lish, S.R.; Suárez-Fariñas, M.; Gareau, D.; Lentini, T.; Johnson-Huang, L.M.; Krueger, J.G.; Lowes, M.A. The tryptophan metabolism enzyme L-kynureninase is a novel inflammatory factor in psoriasis and other inflammatory diseases. J. Allergy Clin. Immunol. 2016, 137, 1830–1840. [Google Scholar] [CrossRef] [Green Version]
- Al-Mansoob, M.; Gupta, I.; Stefan Rusyniak, R.; Ouhtit, A. KYNU, a novel potential target that underpins CD44-promoted breast tumour cell invasion. J. Cell. Mol. Med. 2021, 25, 2309–2314. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Feng, X.; Lai, J.; Yi, W.; Yang, J.; Du, T.; Long, X.; Zhang, Y.; Xiao, Y. A novel role of kynureninase in the growth control of breast cancer cells and its relationships with breast cancer. J. Cell. Mol. Med. 2019, 23, 6700. [Google Scholar] [CrossRef] [Green Version]
- Rojewska, E.; Korostynski, M.; Przewlocki, R.; Przewlocka, B.; Mika, J. Expression profiling of genes modulated by minocycline in a rat model of neuropathic pain. Mol. Pain 2014, 10, 1744–8069. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Dantzer, R.; Budac, D.P.; Walker, A.K.; Mao-Ying, Q.L.; Lee, A.W.; Heijnen, C.J.; Kavelaars, A. Peripheral indoleamine 2,3-dioxygenase 1 is required for comorbid depression-like behavior but does not contribute to neuropathic pain in mice. Brain. Behav. Immun. 2015, 46, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Resta, F.; Masi, A.; Sili, M.; Laurino, A.; Moroni, F.; Mannaioni, G. Kynurenic acid and zaprinast induce analgesia by modulating HCN channels through GPR35 activation. Neuropharmacology 2016, 108, 136–143. [Google Scholar] [CrossRef]
- Costantino, G. Inhibitors of quinolinic acid synthesis: New weapons in the study of neuroinflammatory diseases. Future Med. Chem. 2014, 6, 841–843. [Google Scholar] [CrossRef]
- Varga, N.; Csapó, E.; Majláth, Z.; Ilisz, I.; Krizbai, I.A.; Wilhelm, I.; Knapp, L.; Toldi, J.; Vécsei, L.; Dékány, I. Targeting of the kynurenic acid across the blood–brain barrier by core-shell nanoparticles. Eur. J. Pharm. Sci. 2016, 86, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szűcs, E.; Stefanucci, A.; Dimmito, M.P.; Zádor, F.; Pieretti, S.; Zengin, G.; Vécsei, L.; Benyhe, S.; Nalli, M.; Mollica, A. Discovery of Kynurenines Containing Oligopeptides as Potent Opioid Receptor Agonists. Biomolecules 2020, 10, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Török, N.; Vécsei, L. Are 5-HT 1 receptor agonists effective anti-migraine drugs? Expert Opin. Pharmacother. 2021, 22, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Negro, A.; Martelletti, P. Novel synthetic treatment options for migraine. Expert Opin. Pharmacother. 2021, 22, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Staats Pires, A.; Tan, V.X.; Heng, B.; Guillemin, G.J.; Latini, A. Kynurenine and Tetrahydrobiopterin Pathways Crosstalk in Pain Hypersensitivity. Front. Neurosci. 2020, 14, 620. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciapała, K.; Mika, J.; Rojewska, E. The Kynurenine Pathway as a Potential Target for Neuropathic Pain Therapy Design: From Basic Research to Clinical Perspectives. Int. J. Mol. Sci. 2021, 22, 11055. https://doi.org/10.3390/ijms222011055
Ciapała K, Mika J, Rojewska E. The Kynurenine Pathway as a Potential Target for Neuropathic Pain Therapy Design: From Basic Research to Clinical Perspectives. International Journal of Molecular Sciences. 2021; 22(20):11055. https://doi.org/10.3390/ijms222011055
Chicago/Turabian StyleCiapała, Katarzyna, Joanna Mika, and Ewelina Rojewska. 2021. "The Kynurenine Pathway as a Potential Target for Neuropathic Pain Therapy Design: From Basic Research to Clinical Perspectives" International Journal of Molecular Sciences 22, no. 20: 11055. https://doi.org/10.3390/ijms222011055
APA StyleCiapała, K., Mika, J., & Rojewska, E. (2021). The Kynurenine Pathway as a Potential Target for Neuropathic Pain Therapy Design: From Basic Research to Clinical Perspectives. International Journal of Molecular Sciences, 22(20), 11055. https://doi.org/10.3390/ijms222011055