
Role of Mitochondria in Radiation Responses: Epigenetic, Metabolic, and Signaling Impacts

Abstract
1. Introduction
2. Mitochondria Structure and Function in Normal and Cancer Cells
2.1. Mitochondrial Metabolism and ROS Generation
2.2. Mitochondrial Dynamics
2.3. Mitochondrial Genome
Mitochondrial DNA Repair and Mutation
2.4. Role of Mitochondria in Epigenetic Processes
2.4.1. IR Interferes with Methylation of DNA
2.4.2. IR-Induced Epigenetic Changes Depend on Radiation Quality
2.4.3. IR-Induced Epigenetic Effects via microRNAs
2.4.4. IR-Induced Epigenetics and Genomic Instability
2.5. Mitochondria and the Specific Bioenergetics and Metabolism of Cancer Cells
2.5.1. Mitochondrial Hyperactivity of Cancer Cells Can Create DSBs in DNA
2.5.2. Epithelial-to-Mesenchymal Transition (EMT) and Metabolic Reprogramming of Cancer Cells
2.5.3. Adaptation to Hypoxia and the Role of HIF-1α in Cancer Cells
3. Ionizing Radiation (IR) Effects Involving Mitochondria
3.1. Induction of ROS by IR, Mitochondrial Dysfunction and Generation of Mitochondria-Mediated ROS
3.2. Low-Dose IR Responses and Signaling Involving Mitochondria
3.2.1. Hormesis
3.2.2. Radioadaptive Responses
3.2.3. Low-Dose Radiation Hypersensitivity (HRS)
- (1)
- (2)
- (3)
- (4)
- Mitochondrial metabolism involving ATP [259].
- (5)
- (6)
- Change in the balance between the induction of HRS and subsequently induced radioresistance (IRR) depending on cell type.
3.3. High-Dose IR Responses and Signaling Involving Mitochondria
3.3.1. IR-Induced DNA Damage and DNA Damage Response (DDR) Signaling
3.3.2. Involvement of IR-Induced ROS Production in G2/M Cell Cycle Arrests, MN Formation and Persistent Oxidative Stress
3.3.3. Mitochondria-Mediated Programmed Cell Death
Intrinsic Mitochondrial Apoptotic Pathway
Extrinsic Mitochondrial Apoptotic Pathway
The Ceramide-Dependent Pathway
3.3.4. Autophagy
3.3.5. Mitophagy
3.4. Impacts of Low- and High-LET IR (Heavy Ions and C-ions) on the Induction of Apoptosis, Autophagy/Mitophagy and Anastasis
- (1)
- IR-induced decrease in mitochondrial membrane potential (MMP)
- (2)
- IR-induced activation of Bax and Bak and the release of cytochrome c
- (3)
- IR-induced MOMPs.
- (4)
- IR-induced effects on ATP production
- (5)
- Mitochondrial fragmentation
- (6)
- IR induction of caspase activity and PARP cleavage
- (7)
- Involvement of SMAC in IR-induced apoptosis
- (8)
- IR-induced release of AIF, endoG and CAD
3.4.1. Partial Circumvention of IR-Induced Cell Death by Anastasis
3.4.2. Modulation of Cell Death through Autophagy/Mitophagy Induced by Low and High LET IR
3.4.3. Outcomes of High-Dose Low-LET and High-LET IR Effects on Cell Survival: Differences in Efficacy
Confirmation of the High Biological Effectiveness of High-LET IR
3.5. Involvement of Mitochondria in Low- and High-LET IR-Induced Bystander and Non-Targeted Effects
3.5.1. Involvement of IR-Induced Mitochondria-Driven Responses in Bystander and NTE Effects
3.5.2. Involvement of IR-Induced Mitochondria-Mediated Apoptosis in Bystander and Non-targeted Effects
4. Involvement of Mitochondria in Innate and Adaptive Immune Responses after IR
5. Discussion and Conclusions
5.1. Low-Dose Considerations
5.2. High-Dose Considerations
5.3. High-LET Considerations and Radiotherapeutic Outcomes
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karam, P.A. Inconstant sun: How solar evolution has affected cosmic and ultraviolet radiation exposure over the history of life on Earth. Health Phys. 2003, 84, 322–333. [Google Scholar] [CrossRef]
- Ferrari, F.; Szuszkiewicz, E. Cosmic rays: A review for astrobiologists. Astrobiology 2009, 9, 413–436. [Google Scholar] [CrossRef]
- Maalouf, M.; Durante, M.; Foray, N. Biological effects of space radiation on human cells: History, advances and outcomes. J. Radiat. Res. 2011, 52, 126–146. [Google Scholar] [CrossRef][Green Version]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis, 2nd ed.; ASM Press: Washington, DC, USA, 2006; pp. 1–1118. [Google Scholar]
- Harper, J.W.; Elledge, S.J. The DNA damage response: Ten years after. Mol. Cell. 2007, 28, 739–745. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell. 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Goodhead, D.T. Initial events in the cellular effects of ionizing radiations: Clustered damage in DNA. Int. J. Radiat. Biol. 1994, 65, 7–17. [Google Scholar]
- Azzam, E.I.; Jay-Guerin, J.-P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 32, 48–60. [Google Scholar] [CrossRef]
- Kawamura, K.; Qi, F.; Kobayashi, J. Potential relationship between the biological effects of low-dose irradiation and mitochondrial ROS production. J. Radiat. Res. 2018, 59 (Suppl. S2), ii91–ii97. [Google Scholar] [CrossRef]
- Hirano, S.I.; Ichikawa, Y.; Sato, B.; Yamamoto, H.; Takefuji, Y.; Satoh, F. Molecular Hydrogen as a Potential Clinically Applicable Radioprotective Agent. Int. J. Mol. Sci. 2021, 22, 4566. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.F. DNA damage produced by ionizing radiation in mammalian cells: Identities, mechanisms of formation, and reparability. Prog. Nucleic Acid Res. Mol. Biol. 1988, 35, 95–125. [Google Scholar] [CrossRef]
- Ward, J.F. Biochemistry of DNA lesions. Radiat. Res. Suppl. 1985, 8, S103–S111. [Google Scholar]
- Georgakilas, A.G. From chemistry of DNA damage to repair and biological significance. Comprehending the future. Mutat. Res. 2011, 711, 1–2. [Google Scholar] [CrossRef]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef]
- Szumiel, I. Ionizing radiation-induced oxidative stress, epigenetic changes and genomic instability: The pivotal role of mitochondria. Int. J. Radiat. Biol. 2015, 91, 1–12. [Google Scholar] [CrossRef]
- Stimpfel, M.; Jancar, N.; Virant-Klun, I. New challenge: Mitochondrial epigenetics? Stem Cell. Rev. Rep. 2018, 14, 13–26. [Google Scholar] [CrossRef]
- Sharma, N.; Sampath, H. Mitochondrial DNA integrity: Role in health and disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef]
- Dong, Z.; Tu, W.; He, M.; Fu, J.; Kobayashi, A.; Konishi, T.; Shao, C. Role of endoplasmic reticulum and mitochondrion in proton microbeam radiation-induced bystander effect. Radiat. Res. 2020, 193, 63–72. [Google Scholar] [CrossRef]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M., 3rd; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell. Biol. 2015, 25, 158–170. [Google Scholar] [CrossRef]
- Ippolito, L.; Giannoni, E.; Chiarugi, P.; Parri, M. Mitochondrial redox hubs as promising targets for anticancer therapy. Front. Oncol. 2020, 10, 256. [Google Scholar] [CrossRef]
- Nugent, S.M.; Mothersill, C.E.; Seymour, C.; McClean, B.; Lyng, F.M.; Murphy, J.E. Increased mitochondrial mass in cells with functionally compromised mitochondria after exposure to both direct gamma radiation and bystander factors. Radiat. Res. 2007, 168, 134–142. [Google Scholar] [CrossRef]
- Gray, M.W.; Burger, G.; Lang, B.F. The origin and early evolution of mitochondria. Genome Biol. 2001, 2, 1018. [Google Scholar] [CrossRef]
- Lang, B.F.; Gray, M.W.; Burger, G. Mitochondrial genome evolution and the origin of eukaryotes. Annu. Rev. Genet. 1999, 33, 351–397. [Google Scholar] [CrossRef]
- Fogg, V.C.; Lanning, N.J.; Mackeigan, J.P. Mitochondria in cancer: At the crossroads of life and death. Chin. J. Cancer 2011, 30, 526–539. [Google Scholar] [CrossRef]
- Garcia, I.; Aldaregia, J.; Marjanovic Vicentic, J.; Aldaz, P.; Moreno-Cugnon, L.; Torres-Bayona, S.; Carrasco-Garcia, E.; Garros-Regulez, L.; Egaña, L.; Rubio, A.; et al. Oncogenic activity of SOX1 in glioblastoma. Sci. Rep. 2017, 7, 46575. [Google Scholar] [CrossRef]
- Xie, L.L.; Shi, F.; Tan, Z.; Li, Y.; Bode, A.M.; Cao, Y. Mitochondrial network structure homeostasis and cell death. Cancer Sci. 2018, 109, 3686–3694. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Onishi, M.; Okamoto, K. Mitochondrial clearance: Mechanisms and roles in cellular fitness. FEBS Lett. 2021, 595, 1239–1263. [Google Scholar] [CrossRef]
- Wang, W.; Osenbroch, P.; Skinnes, R.; Esbensen, Y.; Bjørås, M.; Eide, L. Mitochondrial DNA integrity is essential for mitochondrial maturation during differentiation of neural stem cells. Stem Cells 2010, 28, 2195–2204. [Google Scholar] [CrossRef]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Khacho, M.; Slack, R.S. Mitochondrial dynamics in the regulation of neurogenesis: From development to the adult brain. Dev. Dyn. 2018, 247, 47–53. [Google Scholar] [CrossRef]
- Giachin, G.; Jessop, M.; Bouverot, R.; Acajjaoui, S.; Saïdi, M.; Chretien, A.; Bacia-Verloop, M.; Signor, L.; Mas, P.J.; Favier, A.; et al. Assembly of the Mitochondrial Complex I Assembly Complex Suggests a Regulatory Role for Deflavination. Angew. Chem. Int. Ed. Engl. 2021, 60, 4689–4697. [Google Scholar] [CrossRef]
- Ambrose, M.; Gatti, R.A. Pathogenesis of ataxia-telangiectasia: The next generation of ATM functions. Blood 2013, 121, 4036–4045. [Google Scholar] [CrossRef]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Banoth, B.; Cassel, S. Mitochondria in innate immune signaling. Transl. Res. 2018, 202, 52–68. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Bennett, B.; Zielonka, J. Teaching the basics of reactive oxygen species and their relevance to cancer biology: Mitochondrial reactive oxygen species detection, redox signaling, and targeted therapies. Redox Biol. 2018, 15, 347–362. [Google Scholar] [CrossRef]
- Zielonka, J.; Kalyanaraman, B. Small-molecule luminescent probes for the detection of cellular oxidizing and nitrating species. Free Radic. Biol. Med. 2018, 128, 3–22. [Google Scholar] [CrossRef]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef]
- Venditti, P.; Di Meo, S. The role of reactive oxygen species in the life cycle of the mitochondrion. Int. J. Mol. Sci. 2020, 21, 2173. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Kageyama, S.; Gudmundsson, S.R.; Sou, Y.S.; Ichimura, Y.; Tamura, N.; Kazuno, S.; Ueno, T.; Miura, Y.; Noshiro, D.; Abe, M.; et al. p62/SQSTM1-droplet serves as a platform for autophagosome formation and anti-oxidative stress response. Nat. Commun. 2021, 2, 16. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell. Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.A. Regulators of mitochondrial dynamics in cancer. Curr. Opin. Cell. Biol. 2016, 39, 43–52. [Google Scholar] [CrossRef]
- Yoo, S.M.; Jung, Y.K. A molecular approach to mitophagy and mitochondrial dynamics. Mol. Cells 2018, 41, 18–26. [Google Scholar] [CrossRef]
- Saki, M.; Prakash, A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radic. Biol. Med. 2017, 107, 216–227. [Google Scholar] [CrossRef]
- Schon, E.A.; Manfredi, G. Neuronal degeneration and mitochondrial dysfunction. J. Clin. Invest. 2003, 111, 303–312. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondrial disease. Lancet 2006, 368, 70–82. [Google Scholar] [CrossRef]
- Becker, T.; Böttinger, L.; Pfanner, N. Mitochondrial protein import: From transport pathways to an integrated network. Trends Biochem. Sci. 2012, 37, 85–91. [Google Scholar] [CrossRef]
- Fu, Y.; Tigano, M.; Sfeir, A. Safeguarding mitochondrial genomes in higher eukaryotes. Nat. Struct. Mol. Biol. 2020, 27, 687–695. [Google Scholar] [CrossRef]
- Tigano, M.; Vargas, D.C.; Tremblay-Belzile, S.; Fu, Y.; Sfeir, A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 2021, 591, 477–481. [Google Scholar] [CrossRef]
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and epigenetics—Crosstalk in homeostasis and stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef]
- Kam, W.W.; Banati, R.B. Effects of ionizing radiation on mitochondria. Free Radic. Biol. Med. 2013, 65, 607–619. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Castegna, A.; Infantino, V.; Andria, G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol. Genet. Metab. 2013, 110, 25–34. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef]
- Garrido, N.; Griparic, L.; Jokitalo, E.; Wartiovaara, J.; van der Bliek, A.M.; Spelbrink, J.N. Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell. 2003, 14, 1583–1596. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Liang, S.; Zhong, Z. Emerging role of mitochondrial DNA as a major driver of inflammation and disease progression. Trends Immunol. 2019, 40, 1120–1133. [Google Scholar] [CrossRef]
- Kang, D.; Kim, S.H.; Hamasaki, N. Mitochondrial transcription factor A (TFAM): Roles in maintenance of mtDNA and cellular functions. Mitochondrion 2007, 7, 39–44. [Google Scholar] [CrossRef]
- Muftuoglu, M.; Mori, M.P.; de Souza-Pinto, N.C. Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion 2014, 17, 164–181. [Google Scholar] [CrossRef]
- Stein, A.; Sia, E.A. Mitochondrial DNA repair and damage tolerance. Front. Biosci. 2017, 22, 920–943. [Google Scholar] [CrossRef]
- Zinovkina, L.A. Mechanisms of mitochondrial DNA repair in mammals. Biochemistry 2018, 83, 233–249. [Google Scholar]
- Tadi, S.K.; Sebastian, R.; Dahal, S.; Babu, R.K.; Choudhary, B.; Raghavan, S.C. Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions. Mol. Biol. Cell. 2016, 27, 223–235. [Google Scholar] [CrossRef]
- Boguszewska, K.; Szewczuk, M.; Kaźmierczak-Barańska, J.; Karwowski, B.T. The similarities between human mitochondria and bacteria in the context of structure, genome, and base excision repair system. Molecules. 2020, 25, 2857. [Google Scholar] [CrossRef]
- Phillips, A.F.; Millet, A.R.; Tigano, M.; Dubois, S.M.; Crimmins, H.; Babin, L.; Charpentier, M.; Piganeau, M.; Brunet, E.; Sfeir, A. Single-molecule analysis of mtDNA replication uncovers the basis of the common deletion. Mol. Cell. 2017, 65, 527–538.e6. [Google Scholar] [CrossRef]
- Nissanka, N.; Minczuk, M.; Moraes, C.T. Mechanisms of mitochondrial DNA deletion formation. Trends Genet. 2019, 35, 235–244. [Google Scholar] [CrossRef]
- Fontana, G.A.; Gahlon, H.L. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020, 48, 11244–11258. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, H.; Ye, W. Mitochondrial DNA mutations induced by carbon ions radiation: A preliminary study. Dose Response 2018, 16, 1–4. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, M.; Gao, Y.; Cao, G.; Yang, Y.; Lu, D.; Li, W. A genome-wide view of mutations in respiration-deficient mutants of Saccharomyces cerevisiae selected following carbon ion beam irradiation. Appl. Microbiol. Biotechnol. 2019, 103, 1851–1864. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef]
- Belli, M.; Indovina, L. The Response of Living Organisms to Low Radiation Environment and Its Implications in Radiation Protection. Front. Public Health 2020, 8, 601711. [Google Scholar] [CrossRef]
- Cavalcante, G.C.; Schaan, A.P.; Cabral, G.F.; Santana-da-Silva, M.N.; Pinto, P.; Vidal, A.F.; Ribeiro-Dos-Santos, Â. A cell’s fate: An overview of the molecular biology and genetics of apoptosis. Int. J. Mol. Sci. 2019, 20, 4133. [Google Scholar] [CrossRef]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef]
- Miousse, I.R.; Kutanzi, K.R.; Koturbash, I. Effects of ionizing radiation on DNA methylation: From experimental biology to clinical applications. Int. J. Radiat. Biol. 2017, 93, 457–469. [Google Scholar] [CrossRef]
- Madugundu, G.S.; Cadet, J.; Wagner, J.R. Hydroxyl-radical-induced oxidation of 5-methylcytosine in isolated and cellular DNA. Nucleic Acids Res. 2014, 42, 7450–7460. [Google Scholar] [CrossRef]
- Le, D.D.; Fujimori, D.G. Protein and nucleic acid methylating enzymes: Mechanisms and regulation. Curr. Opin. Chem. Biol. 2012, 16, 507–515. [Google Scholar] [CrossRef]
- Baulch, J.E. Radiation-induced genomic instability, epigenetic mechanisms and the mitochondria: A dysfunctional ménage à trois? Int. J. Radiat. Biol. 2019, 95, 516–525. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Christmann, M.; Kaina, B. Epigenetic regulation of DNA repair genes and implications for tumor therapy. Mutat. Res. 2019, 780, 15–28. [Google Scholar] [CrossRef]
- Kumar, A.; Rai, P.S.; Upadhya, R.; Vishwanatha, P.K.S.; Rao, B.S.; Satyamoorthy, K. γ-radiation induces cellular sensitivity and aberrant methylation in human tumor cell lines. Int. J. Radiat. Biol. 2011, 87, 1086–1096. [Google Scholar] [CrossRef]
- Averbeck, N.B.; Durante, M. Protein acetylation within the cellular response to radiation. J. Cell Physiol. 2011, 226, 962–967. [Google Scholar] [CrossRef]
- Sato, M.; Sato, K. Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochim. Biophys. Acta 2013, 1833, 1979–1984. [Google Scholar] [CrossRef]
- Kietzmann, T.; Petry, A.; Shvetsova, A.; Gerhold, J.M.; Görlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 2017, 174, 1533–1554. [Google Scholar] [CrossRef]
- Tamminga, J.; Kovalchuk, O. Role of DNA damage and epigenetic DNA methylation changes in radiation-induced genomic instability and bystander effects in germline in vivo. Curr. Mol. Pharmacol. 2011, 4, 115–125. [Google Scholar] [CrossRef]
- Goetz, W.; Morgan, M.N.; Baulch, J.E. The effect of radiation quality on genomic DNA methylation profiles in irradiated human cell lines. Radiat. Res. 2011, 175, 575–587. [Google Scholar] [CrossRef]
- Chen, X.; Liu, L.; Mims, J.; Punska, E.C.; Williams, K.E.; Zhao, W.; Arcaro, K.F.; Tsang, A.W.; Zhou, X.; Furdui, C.M. Analysis of DNA methylation and gene expression in radiation-resistant head and neck tumors. Epigenetics 2015, 10, 545–561. [Google Scholar] [CrossRef]
- Kennedy, E.M.; Powell, D.R.; Li, Z.; Bell, J.S.K.; Barwick, B.G.; Feng, H.; McCrary, M.R.; Dwivedi, B.; Kowalski, J.; Dynan, W.S.; et al. Galactic cosmic radiation induces persistent epigenome alterations relevant to human lung cancer. Sci. Rep. 2018, 8, 6709. [Google Scholar] [CrossRef]
- Cavalcante, G.C.; Magalhães, L.; Ribeiro-Dos-Santos, Â.; Vidal, A.F. Mitochondrial epigenetics: Non-coding RNAs as a novel layer of complexity. Int. J. Mol. Sci. 2020, 21, 1838. [Google Scholar] [CrossRef]
- Bandiera, S.; Rüberg, S.; Girard, M.; Cagnard, N.; Hanein, S.; Chrétien, D.; Munnich, A.; Lyonnet, S.; Henrion-Caude, A. Nuclear outsourcing of RNA interference components to human mitochondria. PLoS ONE 2011, 6, e20746. [Google Scholar] [CrossRef]
- Marta, G.N.; Garicochea, B.; Carvalho, A.L.; Real, J.M.; Kowalski, L.P. MicroRNAs, cancer and ionizing radiation: Where are we? Rev. Assoc. Med. Bras. 2015, 61, 275–281. [Google Scholar] [CrossRef]
- Rodrigues, S.C.; Cardoso, R.M.S.; Duarte, F.V. Mitochondrial microRNAs: A putative role in tissue regeneration. Biology 2020, 9, 486. [Google Scholar] [CrossRef]
- Jusic, A.; Devaux, Y. EU-CardioRNA COST Action (CA17129). Mitochondrial noncoding RNA-regulatory network in cardiovascular disease. Basic. Res. Cardiol. 2020, 115, 23. [Google Scholar] [CrossRef]
- D’Aquila, P.; Bellizzi, D.; Passarino, G. Mitochondria in health, aging and diseases: The epigenetic perspective. Biogerontology 2015, 16, 569–585. [Google Scholar] [CrossRef]
- Lambertini, L.; Byun, H.M. Mitochondrial epigenetics and environmental exposure. Curr. Environ. Health Rep. 2016, 3, 214–224. [Google Scholar] [CrossRef]
- Wallace, D.C.; Fan, W. Energetics, epigenetics, mitochondrial genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef]
- Wang, M.; Huang, T.; Luo, G.; Huang, C.; Xiao, X.Y.; Wang, L.; Jiang, G.S.; Zeng, F.Q. Long non-coding RNA MEG3 induces renal cell carcinoma cells apoptosis by activating the mitochondrial pathway. J. Huazhong Univ. Sci. Technolog. Med. Sci. 2015, 35, 541–545. [Google Scholar] [CrossRef]
- Borgna, V.; Villegas, J.; Burzio, V.A.; Belmar, S.; Araya, M.; Jeldes, E.; Lobos-González, L.; Silva, V.; Villota, C.; Oliveira-Cruz, L.; et al. Mitochondrial ASncmtRNA-1 and ASncmtRNA-2 as potent targets to inhibit tumor growth and metastasis in the RenCa murine renal adenocarcinoma model. Oncotarget 2017, 8, 43692–43708. [Google Scholar] [CrossRef]
- Chaudhry, M.A.; Sachdeva, H.; Omaruddin, R.A. Radiation-induced micro-RNA modulation in glioblastoma cells differing in DNA-repair pathways. DNA Cell Biol. 2010, 29, 553–561. [Google Scholar] [CrossRef]
- Chaudhry, M.A.; Omaruddin, R.A. Differential DNA methylation alterations in radiation-sensitive and -resistant cells. DNA Cell Biol. 2012, 31, 908–916. [Google Scholar] [CrossRef]
- Mao, A.; Liu, Y.; Zhang, H.; Di, C.; Sun, C. microRNA expression and biogenesis in cellular response to ionizing radiation. DNA Cell Biol. 2014, 33, 667–679. [Google Scholar] [CrossRef]
- Mao, A.; Tang, J.; Tang, D.; Wang, F.; Liao, S.; Yuan, H.; Tian, C.; Sun, C.; Si, J.; Zhang, H.; et al. MicroRNA-29b-3p enhances radiosensitivity through modulating WISP1-mediated mitochondrial apoptosis in prostate cancer cells. J. Cancer 2020, 11, 6356–6364. [Google Scholar] [CrossRef]
- Nguyen, L.; Schilling, D.; Dobiasch, S.; Raulefs, S.; Santiago Franco, M.; Buschmann, D.; Pfaffl, M.W.; Schmid, T.E.; Combs, S.E. The emerging role of miRNAs for the radiation treatment of pancreatic cancer. Cancers 2020, 12, 3703. [Google Scholar] [CrossRef]
- Wei, W.; He, J.; Wang, J.; Ding, N.; Wang, B.; Lin, S.; Zhang, X.; Hua, J.; Li, H.; Hu, B. Serum microRNAs as early indicators for estimation of exposure degree in response to ionizing irradiation. Radiat. Res. 2017, 188, 342–354. [Google Scholar] [CrossRef]
- He, Y.; Zhang, Y.; Li, H.; Zhang, H.; Li, Z.; Xiao, L.; Hu, J.; Ma, Y.; Zhang, Q.; Zhao, X. Comparative profiling of microRNAs reveals the underlying toxicological mechanism in mice testis following carbon ion radiation. Dose Response 2018, 16, 1559325818778633. [Google Scholar] [CrossRef]
- Jin, X.; Yuan, L.; Liu, B.; Kuang, Y.; Li, H.; Li, L.; Zhao, X.; Li, F.; Bing, Z.; Chen, W.; et al. Integrated analysis of circRNA-miRNA-mRNA network reveals potential prognostic biomarkers for radiotherapies with X-rays and carbon ions in non-small cell lung cancer. Ann. Transl. Med. 2020, 8, 1373. [Google Scholar] [CrossRef]
- Khan, M.T.; Yang, L.; More, E.; Irlam-Jones, J.J.; Valentine, H.R.; Hoskin, P.; Choudhury, A.; West, C.M.L. Developing tumor radiosensitivity signatures using lncRNAs. Radiat. Res. 2021, 195, 324–333. [Google Scholar] [CrossRef]
- Chen, Y.; Cui, J.; Gong, Y.; Wei, S.; Wei, Y.; Yi, L. MicroRNA: A novel implication for damage and protection against ionizing radiation. Environ. Sci. Pollut. Res. Int. 2021, 28, 15584–15596. [Google Scholar] [CrossRef]
- Nagasawa, H.; Little, J.B. Induction of sister chromatid exchanges by extremely low doses of alpha-particles. Cancer Res. 1992, 52, 6394–6396. [Google Scholar]
- Nagasawa, H.; Little, J.B. Unexpected sensitivity to the induction of mutations by very low doses of alpha-particle radiation: Evidence for a bystander effect. Radiat. Res. 1999, 152, 552–557. [Google Scholar]
- Kadhim, M.A.; Macdonald, D.A.; Goodhead, D.T.; Lorimore, S.A.; Marsden, S.J.; Wright, E.G. Transmission of chromosomal instability after plutonium alpha-particle irradiation. Nature 1992, 355, 738–740. [Google Scholar] [CrossRef]
- Sabatier, L.; Dutrillaux, B.; Martin, M.B. Chromosomal instability. Nature 1992, 357, 548. [Google Scholar] [CrossRef]
- Huang, L.; Snyder, A.R.; Morgan, W.F. Radiation-induced genomic instability and its implications for radiation carcinogenesis. Oncogene 2003, 22, 5848–5854. [Google Scholar] [CrossRef]
- Lorimore, S.A.; Kadhim, M.A.; Pocock, D.A.; Papworth, D.; Stevens, D.L.; Goodhead, D.T.; Wright, E.G. Chromosomal instability in the descendants of unirradiated surviving cells after alpha-particle irradiation. Proc. Natl. Acad. Sci. USA 1998, 95, 5730–5733. [Google Scholar] [CrossRef]
- Morgan, W.F. Non-targeted and delayed effects of exposure to ionizing radiation: I. Radiation-induced genomic instability and bystander effects in vitro. Radiat. Res. 2003, 159, 567–580. [Google Scholar] [CrossRef]
- Morgan, W.F. Non-targeted and delayed effects of exposure to ionizing radiation: II. Radiation-induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational effects. Radiat. Res. 2003, 159, 581–596. [Google Scholar] [CrossRef]
- Morgan, W.F. Radiation-induced genomic instability. Health Phys. 2011, 100, 280–281. [Google Scholar] [CrossRef]
- Spitz, D.R.; Azzam, E.I.; Li, J.J.; Gius, D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: A unifying concept in stress response biology. Cancer Metastasis Rev. 2004, 23, 311–322. [Google Scholar] [CrossRef]
- Spitz, D.R.; Hauer-Jensen, M. Ionizing radiation-induced responses: Where free radical chemistry meets redox biology and medicine. Antioxid. Redox Signal. 2014, 20, 1407–1409. [Google Scholar] [CrossRef]
- Miousse, I.R.; Tobacyk, J.; Melnyk, S.; James, S.J.; Cheema, A.K.; Boerma, M.; Hauer-Jensen, M.; Koturbash, I. One-carbon metabolism and ionizing radiation: A multifaceted interaction. Biomol. Concepts 2017, 8, 83–92. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kim, G.J.; Fiskum, G.M.; Morgan, W.F. A role for mitochondrial dysfunction in perpetuating radiation-induced genomic instability. Cancer Res. 2006, 66, 10377–10383. [Google Scholar] [CrossRef]
- Dayal, D.; Martin, S.M.; Owens, K.M.; Aykin-Burns, N.; Zhu, Y.; Boominathan, A.; Pain, D.; Limoli, C.L.; Goswami, P.C.; Domann, F.E.; et al. Mitochondrial complex II dysfunction can contribute significantly to genomic instability after exposure to ionizing radiation. Radiat. Res. 2009, 172, 737–745. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef]
- Shimura, T.; Sasatani, M.; Kawai, H.; Kamiya, K.; Kobayashi, J.; Komatsu, K.; Kunugita, N. A comparison of radiation-induced mitochondrial damage between neural progenitor stem cells and differentiated cells. Cell Cycle 2017, 16, 565–573. [Google Scholar] [CrossRef]
- Shimura, T.; Kunugita, N. Mitochondrial reactive oxygen species-mediated genomic instability in low-dose irradiated human cells through nuclear retention of cyclin D1. Cell Cycle 2016, 15, 1410–1414. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Lu, J.; Tan, M.; Cai, Q. The Warburg effect in tumor progression: Mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015, 356, 156–164. [Google Scholar] [CrossRef]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. The Warburg effect: Historical dogma versus current rationale. Adv. Exp. Med. Biol. 2021, 1269, 169–177. [Google Scholar] [CrossRef]
- Giampazolias, E.; Tait, S.W. Mitochondria and the hallmarks of cancer. FEBS J. 2016, 283, 803–814. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003, 348, 2656–2668. [Google Scholar] [CrossRef]
- Wilson, R.B.; Solass, W.; Archid, R.; Weinreich, F.J.; Königsrainer, A.; Reymond, M.A. Resistance to anoikis in transcoelomic shedding: The role of glycolytic enzymes. Pleura Peritoneum 2019, 4, 20190003. [Google Scholar] [CrossRef]
- Chen, Z.; Lu, W.; Garcia-Pieto, C.; Huang, P. The Warburg effect and its cancer therapeutic implication. J. Bioenerget. Biomembr. 2007, 39, 267–274. [Google Scholar]
- Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria in cancer cells: What is so special about them? Trends Cell. Biol. 2008, 18, 165–173. [Google Scholar] [CrossRef]
- Schlaff, C.D.; Krauze, A.; Belard, A.; O’Connell, J.J.; Camphausen, K.A. Bringing the heavy: Carbon ion therapy in the radiobiological and clinical context. Radiat. Oncol. 2014, 9, 88. [Google Scholar] [CrossRef]
- Masunaga, S.; Sakurai, Y.; Tanaka, H.; Hirayama, R.; Matsumoto, Y.; Uzawa, A.; Suzuki, M.; Kondo, N.; Narabayashi, M.; Maruhashi, A.; et al. Radiosensitivity of pimonidazole-unlabelled intratumour quiescent cell population to γ-rays, accelerated carbon ion beams and boron neutron capture reaction. Br. J. Radiol. 2013, 86, 20120302. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Lopez, M.; Joseph, J.; Zielonka, J.; Dwinell, M.B. A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: Therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox Biol. 2018, 14, 316–327, Erratum in (Redox Biol. 2018, 14, 316–327, doi:10.1016/j.redox.2018.03.001). [Google Scholar] [CrossRef]
- Kim, J.; Yu, L.; Chen, W.; Xu, Y.; Wu, M.; Todorova, D.; Tang, Q.; Feng, B.; Jiang, L.; He, J.; et al. Wild-Type p53 promotes cancer metabolic switch by inducing PUMA-dependent suppression of oxidative phosphorylation. Cancer Cell. 2019, 35, 191–203.e8. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Koukourakis, M.I.; Giatromanolaki, A. Warburg effect, lactate dehydrogenase, and radio/chemo-therapy efficacy. Int. J. Radiat. Biol. 2019, 95, 408–426. [Google Scholar] [CrossRef]
- Jin, L.; Chun, J.; Pan, C.; Alesi, G.N.; Li, D.; Magliocca, K.R.; Kang, Y.; Chen, Z.G.; Shin, D.M.; Khuri, F.R.; et al. Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis. Oncogene 2017, 36, 3797–3806. [Google Scholar] [CrossRef]
- Colen, C.B.; Seraji-Bozorgzad, N.; Marples, B.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic remodeling of malignant gliomas for enhanced sensitization during radiotherapy: An in vitro study. Neurosurgery 2006, 59, 1313–1324. [Google Scholar] [CrossRef]
- Feng, Y.; Xiong, Y.; Qiao, T.; Li, X.; Jia, L.; Han, Y. Lactate dehydrogenase A: A key player in carcinogenesis and potential target in cancer therapy. Cancer Med. 2018, 7, 6124–6136. [Google Scholar] [CrossRef]
- Conlon, G.A.; Murray, G.I. Recent advances in understanding the roles of matrix metalloproteinases in tumour invasion and metastasis. J. Pathol. 2019, 247, 629–640. [Google Scholar] [CrossRef]
- Bassiouni, W.; Ali, M.A.M.; Schulz, R. Multifunctional intracellular matrix metalloproteinases: Implications in disease. FEBS J. 2021. [Google Scholar] [CrossRef]
- Liu, X.; Li, F.; Huang, Q.; Zhang, Z.; Zhou, L.; Deng, Y.; Zhou, M.; Fleenor, D.E.; Wang, H.; Kastan, M.B.; et al. Self-inflicted DNA double-strand breaks sustain tumorigenicity and stemness of cancer cells. Cell Res. 2017, 27, 764–783. [Google Scholar] [CrossRef]
- O’Malley, J.; Kumar, R.; Inigo, J.; Yadava, N.; Chandra, D. Mitochondrial stress response and cancer. Trends Cancer 2020, 6, 688–701. [Google Scholar] [CrossRef]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef]
- Guerra, F.; Guaragnella, N.; Arbini, A.A.; Bucci, C.; Giannattasio, S.; Moro, L. Mitochondrial dysfunction: A novel potential driver of epithelial-to-mesenchymal transition in cancer. Front. Oncol. 2017, 7, 295. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Terentiev, A.A. Metabolic heterogeneity of cancer cells: An interplay between HIF-1, GLUTs, and AMPK. Cancers 2020, 12, 862. [Google Scholar] [CrossRef]
- Wu, H.; Ying, M.; Hu, X. Lactic acidosis switches cancer cells from aerobic glycolysis back to dominant oxidative phosphorylation. Oncotarget 2016, 7, 40621–40629. [Google Scholar] [CrossRef]
- Sun, C.; Liu, X.; Wang, B.; Wang, Z.; Liu, Y.; Di, C.; Si, J.; Li, H.; Wu, Q.; Xu, D.; et al. Endocytosis-mediated mitochondrial transplantation: Transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 2019, 9, 3595–3607. [Google Scholar] [CrossRef]
- Gu, Q.; Zhao, L.; Ma, Y.P.; Liu, J.D. Contribution of mitochondrial function to exercise-induced attenuation of renal dysfunction in spontaneously hypertensive rats. Mol. Cell Biochem. 2015, 406, 217–225. [Google Scholar] [CrossRef]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell. Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Lukyanova, L.D.; Kirova, Y.I. Mitochondria-controlled signaling mechanisms of brain protection in hypoxia. Front. Neurosci. 2015, 9, 320. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology 2009, 24, 97–106. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef]
- Meijer, T.W.; Kaanders, J.H.; Span, P.N.; Bussink, J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin. Cancer Res. 2012, 18, 5585–5594. [Google Scholar] [CrossRef]
- Leung, E.; Cairns, R.A.; Chaudary, N.; Vellanki, R.N.; Kalliomaki, T.; Moriyama, E.H.; Mujcic, H.; Wilson, B.C.; Wouters, B.G.; Hill, R.; et al. Metabolic targeting of HIF-dependent glycolysis reduces lactate, increases oxygen consumption and enhances response to high-dose single-fraction radiotherapy in hypoxic solid tumors. BMC Cancer 2017, 17, 418. [Google Scholar] [CrossRef]
- Grasso, D.; Medeiros, H.C.D.; Zampieri, L.X.; Bol, V.; Danhier, P.; van Gisbergen, M.W.; Bouzin, C.; Brusa, D.; Grégoire, V.; Smeets, H.; et al. Fitter mitochondria are associated with radioresistance in human head and heck SQD9 cancer cells. Front. Pharmacol. 2020, 11, 263. [Google Scholar] [CrossRef]
- Averbeck, D. Non-targeted effects as a paradigm breaking evidence. Mutat. Res. 2010, 687, 7–12. [Google Scholar] [CrossRef]
- Averbeck, D.; Salomaa, S.; Bouffler, S.; Ottolenghi, A.; Smyth, V.; Sabatier, L. Progress in low dose health risk research: Novel effects and new concepts in low dose radiobiology. Mutat. Res. 2018, 776, 46–69. [Google Scholar] [CrossRef]
- Riley, P.A. Free radicals in biology: Oxidative stress and the effects of ionizing radiation. Int. J. Radiat. Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef]
- Meesungnoen, J.; Jay-Gerin, J.P. High-LET ion radiolysis of water: Oxygen production in tracks. Radiat. Res. 2009, 171, 379–386. [Google Scholar] [CrossRef]
- Gebicki, J.M. Oxidative stress, free radicals and protein peroxides. Arch. Biochem. Biophys. 2016, 595, 33–39. [Google Scholar] [CrossRef]
- Goodhead, D.T. Trackstucture and the Quality Factor for Space Radiation Cancer Risk (REID). 2018. Available online: https://three.jsc.nasa.gov/articles/Track_QF_Goodhead.pdf (accessed on 11 March 2021).
- Wozny, A.S.; Vares, G.; Alphonse, G.; Lauret, A.; Monini, C.; Magné, N.; Cuerq, C.; Fujimori, A.; Monboisse, J.C.; Beuve, M.; et al. ROS production and distribution: A new paradigm to explain the differential effects of x-ray and carbon ion irradiation on cancer stem cell migration and invasion. Cancers 2019, 11, 468. [Google Scholar] [CrossRef]
- McFadden, C.H.; Rahmanian, S.; Flint, D.B.; Bright, S.J.; Yoon, D.S.; O’Brien, D.J.; Asaithamby, A.; Abdollahi, A.; Greilich, S.; Sawakuchi, G.O. Isolation of time-dependent DNA damage induced by energetic carbon ions and their fragments using fluorescent nuclear track detectors. Med. Phys. 2020, 47, 272–281. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef]
- Yahyapour, R.; Motevaseli, E.; Rezaeyan, A.; Abdollahi, H.; Farhood, B.; Cheki, M.; Rezapoor, S.; Shabeeb, D.; Musa, A.E.; Najafi, M.; et al. Reduction-oxidation (redox) system in radiation-induced normal tissue injury: Molecular mechanisms and implications in radiation therapeutics. Clin. Transl. Oncol. 2018, 20, 975–988. [Google Scholar] [CrossRef]
- Kim, E.M.; Yang, H.S.; Kang, S.W.; Ho, J.N.; Lee, S.B.; Um, H.D. Amplification of the gamma-irradiation-induced cell death pathway by reactive oxygen species in human U937 cells. Cell Signal. 2008, 20, 916–924. [Google Scholar] [CrossRef]
- Kobashigawa, S.; Suzuki, K.; Yamashita, S. Ionizing radiation accelerates Drp1-dependent mitochondrial fission, which involves delayed mitochondrial reactive oxygen species production in normal human fibroblast-like cells. Biochem. Biophys. Res. Commun. 2011, 414, 795–800. [Google Scholar] [CrossRef]
- Yamamori, T.; Yasui, H.; Yamazumi, M.; Wada, Y.; Nakamura, Y.; Nakamura, H.; Inanami, O. Ionizing radiation induces mitochondrial reactive oxygen species production accompanied by upregulation of mitochondrial electron transport chain function and mitochondrial content under control of the cell cycle checkpoint. Free Radic. Biol. Med. 2012, 53, 260–270. [Google Scholar] [CrossRef]
- Laurent, C.; Leduc, A.; Pottier, I.; Prévost, V.; Sichel, F.; Lefaix, J.L. Dramatic increase in oxidative stress in carbon-irradiated normal human skin fibroblasts. PLoS ONE 2013, 8, e85158. [Google Scholar] [CrossRef]
- Dettmering, T.; Zahnreich, S.; Colindres-Rojas, M.; Durante, M.; Taucher-Scholz, G.; Fournier, C. Increased effectiveness of carbon ions in the production of reactive oxygen species in normal human fibroblasts. J. Radiat. Res. 2015, 56, 67–76. [Google Scholar] [CrossRef]
- Jabbari, N.; Nawaz, M.; Rezaie, J. Ionizing radiation increases the activity of exosomal secretory pathway in MCF-7 human breast cancer cells: A possible way to communicate resistance against radiotherapy. Int. J. Mol. Sci. 2019, 20, E3649. [Google Scholar] [CrossRef]
- Sokolov, M.; Neumann, R. Global gene expression alterations as a crucial constituent of human cell response to low doses of ionizing radiation exposure. Int. J. Mol. Sci. 2015, 17, 55. [Google Scholar] [CrossRef]
- Franco, N.; Lamartine, J.; Frouin, V.; Le Minter, P.; Petat, C.; Leplat, J.J.; Libert, F.; Gidrol, X.; Martin, M.T. Low-dose exposure to gamma rays induces specific gene regulations in normal human keratinocytes. Radiat. Res. 2005, 163, 623–635. [Google Scholar] [CrossRef]
- Amundson, S.A. Gene expression studies for the development of particle therapy. Int. J. Part.Ther. 2018, 5, 49–59. [Google Scholar] [CrossRef]
- Schirrmacher, V. Less can be more: The hormesis theory of stress adaptation in the global biosphere and its implications. Biomedicines 2021, 9, 293. [Google Scholar] [CrossRef]
- Checa, J.; Aran, J.M. Reactive oxygen species: Drivers of physiological and pathological processes. J. Inflamm. Res. 2020, 13, 1057–1073. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Baldwin, L.A. The marginalization of hormesis. Hum. Exp. Toxicol. 2000, 19, 32–40. [Google Scholar] [CrossRef]
- Calabrese, E.J. Hormetic mechanisms. Crit. Rev. Toxicol. 2013, 43, 580–606. [Google Scholar] [CrossRef]
- Feinendegen, L.E. Evidence for beneficial low level radiation effects and radiation hormesis. Br. J. Radiol. 2005, 78, 3–7. [Google Scholar] [CrossRef]
- Szumiel, I. Radiation hormesis: Autophagy and other cellular mechanisms. Int. J. Radiat. Biol. 2012, 88, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.R.; Loke, W.K. Molecular mechanisms of low dose ionizing radiation-induced hormesis, adaptive responses, radioresistance, bystander effects, and genomic instability. Int. J. Radiat. Biol. 2015, 91, 13–27. [Google Scholar] [CrossRef]
- Hamada, R.; Kaminaga, K.; Suzuki, K.; Yokoya, A. Mitochondria membrane potential, morphology and ATP production in mammalian cells exposed to X-rays. Radiat. Prot. Dosim. 2019, 183, 98–101. [Google Scholar] [CrossRef]
- Calabrese, E.J. Flaws in the LNT single-hit model for cancer risk: An historical assessment. Environ. Res. 2017, 158, 773–788. [Google Scholar] [CrossRef]
- Sthijns, M.M.; Weseler, A.R.; Bast, A.; Haenen, G.R. Time in Redox Adaptation Processes: From Evolution to Hormesis. Int. J. Mol. Sci. 2016, 17, 1649. [Google Scholar] [CrossRef]
- Murray, D.; Mirzayans, R. Non linearities in the cellular response to ionizing radiation and the role of p53 therein. Int. J. Radiat. Biol. 2020, 97, 1088–1098. [Google Scholar] [CrossRef]
- Rossnerova, A.; Izzotti, A.; Pulliero, A.; Bast, A.; Rattan, S.I.S.; Rossner, P. The molecular mechanisms of adaptive response related to environmental stress. Int. J. Mol. Sci. 2020, 21, 7053. [Google Scholar] [CrossRef]
- Vaiserman, A.M. Radiation hormesis: Historical perspective and implications for low-dose cancer risk assessment. Dose Response 2010, 8, 172–191. [Google Scholar] [CrossRef]
- Feinendegen, L.E. Quantification of adaptive protection following low-dose irradiation. Health Phys. 2016, 110, 276–280. [Google Scholar] [CrossRef]
- Sies, H.; Feinendegen, L.E. Radiation hormesis: The link to nanomolar hydrogen peroxide. Antioxid. Redox Signal. 2017, 27, 596–598. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Frey, B.; Rückert, M.; Deloch, L.; Rühle, P.F.; Derer, A.; Fietkau, R.; Gaipl, U.S. Immunomodulation by ionizing radiation-impact for design of radio-immunotherapies and for treatment of inflammatory diseases. Immunol. Rev. 2017, 280, 231–248. [Google Scholar] [CrossRef]
- Bartolini, D.; Tew, K.D.; Marinelli, R.; Galli, F.; Wang, G.Y. Nrf2-modulation by seleno-hormetic agents and its potential for radiation protection. Biofactors 2020, 46, 239–245. [Google Scholar] [CrossRef]
- Yu, H.S.; Liu, Z.M.; Yu, X.Y.; Song, A.Q.; Liu, N.; Wang, H. Low-dose radiation induces antitumor effects and erythrocyte system hormesis. Asian Pac. J. Cancer Prev. 2013, 14, 4121–4126. [Google Scholar] [CrossRef]
- Bernal, A.J.; Dolinoy, D.C.; Huang, D.; Skaar, D.A.; Weinhouse, C.; Jirtle, R.L. Adaptive radiation-induced epigenetic alterations mitigated by antioxidants. FASEB J. 2013, 27, 665–671. [Google Scholar] [CrossRef]
- Qi, Z.; Guo, S.; Li, C.; Wang, Q.; Li, Y.; Wang, Z. Integrative analysis for the roles of lncRNAs in the immune responses of mouse PBMC exposed to low-dose ionizing radiation. Dose Response 2020, 18, 1559325820913800. [Google Scholar] [CrossRef]
- Shin, E.; Lee, S.; Kang, H.; Kim, J.; Kim, K.; Youn, H.; Jin, Y.W.; Seo, S.; Youn, B. Organ-specific effects of low dose radiation exposure: A comprehensive review. Front. Genet. 2020, 11, 566244. [Google Scholar] [CrossRef]
- Vaiserman, A.; Cuttler, J.M.; Socol, Y. Low-dose ionizing radiation as a hormetin: Experimental observations and therapeutic perspective for age-related disorders. Biogerontology 2021, 22, 145–164. [Google Scholar] [CrossRef]
- Tang, F.R.; Loganovsky, K. Low dose or low dose rate ionizing radiation-induced health effect in the human. J. Environ. Radioact. 2018, 192, 32–47. [Google Scholar] [CrossRef]
- Squillaro, T.; Galano, G.; De Rosa, R.; Peluso, G.; Galderisi, U. Concise review: The effect of low-dose ionizing radiation on stem cell biology: A contribution to radiation risk. Stem Cells 2018, 36, 1146–1153. [Google Scholar] [CrossRef]
- Scott, B.R.; Tharmalingam, S. The LNT model for cancer induction is not supported by radiobiological data. Chem. Biol. Interact. 2019, 301, 34–53. [Google Scholar] [CrossRef]
- Kataoka, T. Study of antioxidative effects and anti-inflammatory effects in mice due to low-dose X-irradiation or radon inhalation. J. Radiat. Res. 2013, 54, 587–596. [Google Scholar] [CrossRef]
- Kojima, S.; Matsuki, O.; Nomura, T.; Shimura, N.; Kubodera, A.; Yamaoka, K.; Tanooka, H.; Wakasugi, H.; Honda, Y.; Honda, S.; et al. Localization of glutathione and induction of glutathione synthesis-related proteins in mouse brain by low doses of gamma-rays. Brain Res. 1998, 808, 262–269. [Google Scholar] [CrossRef]
- Tharmalingam, S.; Sreetharan, S.; Kulesza, A.V.; Boreham, D.R.; Tai, T.C. Low-dose ionizing radiation exposure, oxidative stress and epigenetic programing of health and disease. Radiat. Res. 2017, 188, 525–538. [Google Scholar] [CrossRef]
- Vojta, A.; Zoldoš, V. Adaptation or malignant transformation: The two faces of epigenetically mediated response to stress. Biomed. Res. Int. 2013, 2013, 954060. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Dhawan, G.; Kapoor, R.; Iavicoli, I.; Calabrese, V. What is hormesis and its relevance to healthy aging and longevity? Biogerontology 2015, 16, 693–707. [Google Scholar] [CrossRef]
- Amundson, S.A.; Lee, R.A.; Koch-Paiz, C.A.; Bittner, M.L.; Meltzer, P.; Trent, J.M.; Fornace, A.J., Jr. Differential responses of stress genes to low dose-rate gamma irradiation. Mol. Cancer Res. 2003, 1, 445–452. [Google Scholar]
- Redpath, J.L.; Liang, D.; Taylor, T.H.; Christie, C.; Elmore, E. The shape of the dose-response curve for radiation-induced neoplastic transformation in vitro: Evidence for an adaptive response against neoplastic transformation at low doses of low-LET radiation. Radiat. Res. 2001, 156, 700–707. [Google Scholar] [CrossRef]
- Liu, G.; Gong, P.; Bernstein, L.R.; Bi, Y.; Gong, S.; Cai, L. Apoptotic cell death induced by low-dose radiation in male germ cells: Hormesis and adaptation. Crit. Rev. Toxicol. 2007, 37, 587–605. [Google Scholar] [CrossRef]
- Scott, B.R. Radiation-hormesis phenotypes, the related mechanisms and implications for disease prevention and therapy. J. Cell Commun. Signal. 2014, 8, 341–352. [Google Scholar] [CrossRef]
- Bauer, G. HOCl-dependent singlet oxygen and hydroxyl radical generation modulate and induce apoptosis of malignant cells. Anticancer Res. 2013, 33, 3589–3602. [Google Scholar]
- Temme, J.; Bauer, G. Low-dose gamma irradiation enhances superoxide anion production by nonirradiated cells through TGF-β1-dependent bystander signaling. Radiat. Res. 2013, 179, 422–432. [Google Scholar] [CrossRef]
- Frey, B.; Hehlgans, S.; Rödel, F.; Gaipl, U.S. Modulation of inflammation by low and high doses of ionizing radiation: Implications for benign and malign diseases. Cancer Lett. 2015, 368, 230–237. [Google Scholar] [CrossRef]
- Liu, S.Z. Nonlinear dose-response relationship in the immune system following exposure to ionizing radiation: Mechanisms and implications. Nonlinearity Biol. Toxicol. Med. 2003, 1, 71–92. [Google Scholar] [CrossRef]
- Cheda, A.; Wrembel-Wargocka, J.; Lisiak, E.; Nowosielska, E.M.; Marciniak, M.; Janiak, M.K. Single low doses of X rays inhibit the development of experimental tumor metastases and trigger the activities of NK cells in mice. Radiat. Res. 2004, 161, 335–340. [Google Scholar] [CrossRef]
- Hashimoto, S.; Shirato, H.; Hosokawa, M.; Nishioka, T.; Kuramitsu, Y.; Matushita, K.; Kobayashi, M.; Miyasaka, K. The suppression of metastases and the change in host immune response after low-dose total-body irradiation in tumor-bearing rats. Radiat. Res. 1999, 151, 717–724. [Google Scholar]
- Takahashi, M.; Kojima, S. Suppression of atopic dermatitis and tumor metastasis in mice by small amounts of radon. Radiat. Res. 2006, 165, 337–342. [Google Scholar] [CrossRef]
- Wozny, A.S.; Alphonse, G.; Battiston-Montagne, P.; Simonet, S.; Poncet, D.; Testa, E.; Guy, J.B.; Rancoule, C.; Magné, N.; Beuve, M.; et al. Influence of dose rate on the cellular response to low- and high-LET radiations. Front. Oncol. 2016, 6, 58. [Google Scholar] [CrossRef]
- Sykes, P.J. Until there is a resolution of the pro-LNT/anti-LNT debate, we should head toward a more sensible graded approach for protection from low-dose ionizing radiation. Dose Response 2020, 18, 1559325820921651. [Google Scholar] [CrossRef]
- Waltar, A.; Feinendegen, L. The double threshold: Consequences for identifying low-dose radiation effects. Dose Response 2020, 18, 1559325820949729. [Google Scholar] [CrossRef]
- Olivieri, G.; Bodycote, J.; Wolff, S. Adaptive response of human lymphocytes to low concentrations of radioactive thymidine. Science 1984, 223, 594–597. [Google Scholar] [CrossRef]
- Wolff, S. Aspects of the adaptive response to very low doses of radiation and other agents. Mutat. Res. 1996, 358, 135–142. [Google Scholar] [CrossRef]
- Paraswani, N.; Thoh, M.; Bhilwade, H.N.; Ghosh, A. Early antioxidant responses via the concerted activation of NF-κB and Nrf2 characterize the gamma-radiation-induced adaptive response in quiescent human peripheral blood mononuclear cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2018, 831, 50–61. [Google Scholar] [CrossRef]
- Bravard, A.; Luccioni, C.; Moustacchi, E.; Rigaud, O. Contribution of antioxidant enzymes to the adaptive response to ionizing radiation of human lymphoblasts. Int. J. Radiat. Biol. 1999, 75, 639–645. [Google Scholar] [CrossRef]
- Azzam, E.I.; Raaphorst, G.P.; Mitchel, R.E. Radiation-induced adaptive response for protection against micronucleus formation and neoplastic transformation in C3H 10T1/2 mouse embryo cells. Radiat. Res. 1994, 138, S28–S31. [Google Scholar]
- Rigaud, O.; Moustacchi, E. Radioadaptation for gene mutation and the possible molecular mechanisms of the adaptive response. Mutat. Res. 1996, 358, 127–134. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, Y.; Jeong, K.; Yoo, S.Y.; Cho, C.K.; Lee, Y.S. Different induction of adaptive response to ionizing radiation in normal and neoplastic cells. Cell. Biol. Toxicol. 1999, 15, 111–119. [Google Scholar] [CrossRef]
- Hauptmann, M.; Haghdoost, S.; Gomolka, M.; Sarioglu, H.; Ueffing, M.; Dietz, A.; Kulka, U.; Unger, K.; Babini, G.; Harms-Ringdahl, M.; et al. Differential response and priming dose effect on the proteome of human fibroblast and stem cells induced by exposure to low doses of ionizing radiation. Radiat. Res. 2016, 185, 299–312. [Google Scholar] [CrossRef]
- Wang, Z.; Lv, M.Y.; Huang, Y.X. Effects of low-dose x-ray on cell growth, membrane permeability, DNA damage and gene transfer efficiency. Dose Response 2020, 18, 1559325820962615. [Google Scholar] [CrossRef]
- Jiang, N.Y.; Huang, Z.; Zhao, Q.; Feng, W.; Belikova, N.A.; Kagan, V.E. Interplay between bax, reactive oxygen species production, and cardiolipin oxidation during apoptosis. Biochem. Biophys. Res. Commun. 2008, 368, 145–150. [Google Scholar] [CrossRef]
- Yu, H.; Liu, N.; Wang, H.; Shang, Q.; Jiang, P.; Zhang, Y. Different responses of tumor and normal cells to low-dose radiation. Contemp. Oncol. 2013, 17, 356–362. [Google Scholar] [CrossRef]
- Varès, G.; Wang, B.; Tanaka, K.; Kakimoto, A.; Eguchi-Kasai, K.; Nenoi, M. Mutagenic adaptive response to high-LET radiation in human lymphoblastoid cells exposed to X-rays. Mutat. Res. 2011, 706, 46–52. [Google Scholar] [CrossRef]
- Wang, B.; Ninomiya, Y.; Tanaka, K.; Maruyama, K.; Varès, G.; Eguchi-Kasai, K.; Nenoi, M. Adaptive response of low linear energy transfer X-rays for protection against high linear energy transfer accelerated heavy ion-induced teratogenesis. Birth Defects Res. B Dev. Reprod. Toxicol. 2012, 95, 379–385. [Google Scholar] [CrossRef]
- Katsube, T.; Wang, B.; Tanaka, K.; Ninomiya, Y.; Hirakawa, H.; Liu, C.; Maruyama, K.; Vares, G.; Liu, Q.; Kito, S.; et al. Synergistic Effects of Chronic Restraint-Induced Stress and Low-Dose 56Fe-particle Irradiation on Induction of Chromosomal Aberrations in Trp53-Heterozygous Mice. Radiat Res. 2021, 196, 100–112. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, X.; Li, H.; Niu, C.; Yu, D.; Yang, G.; Liang, X.; Wen, X.; Li, M.; Cui, J. Validating the pivotal role of the immune system in low-dose radiation-induced tumor inhibition in Lewis lung cancer-bearing mice. Cancer Med. 2018, 7, 1338–1348. [Google Scholar] [CrossRef]
- Guéguen, Y.; Bontemps, A.; Ebrahimian, T.G. Adaptive responses to low doses of radiation or chemicals: Their cellular and molecular mechanisms. Cell. Mol. Life Sci. 2019, 76, 1255–1273. [Google Scholar] [CrossRef]
- Kabilan, U.; Graber, T.E.; Alain, T.; Klokov, D. Ionizing Radiation and Translation Control: A Link to Radiation Hormesis? Int. J. Mol. Sci. 2020, 21, 6650. [Google Scholar] [CrossRef]
- Joiner, M.C.; Lambin, P.; Malaise, E.P.; Robson, T.; Arrand, J.E.; Skov, K.A.; Marples, B. Hypersensitivity to very-low single radiation doses: Its relationship to the adaptive response and induced radioresistance. Mutat. Res. 1996, 358, 171–183. [Google Scholar] [CrossRef]
- Joiner, M.C.; Lambin, P.; Marples, B. Adaptive response and induced resistance. C. R. Acad. Sci. III 1999, 322, 167–175. [Google Scholar] [CrossRef]
- Joiner, M.C.; Marples, B.; Lambin, P.; Short, S.C.; Turesson, I. Low-dose hypersensitivity: Current status and possible mechanisms. Int. J. Radiat. Oncol. Biol. Phys. 2001, 49, 379–389. [Google Scholar] [CrossRef]
- Marples, B.; Wouters, B.G.; Collis, S.J.; Chalmers, A.J.; Joiner, M.C. Low-dose hyper-radiosensitivity: A consequence of ineffective cell cycle arrest of radiation-damaged G2-phase cells. Radiat. Res. 2004, 161, 247–255. [Google Scholar] [CrossRef]
- Krueger, S.A.; Wilson, G.D.; Piasentin, E.; Joiner, M.C.; Marples, B. The effects of G2-phase enrichment and checkpoint abrogation on low-dose hyper-radiosensitivity. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 1509–1517. [Google Scholar] [CrossRef]
- Olobatuyi, O.; de Vries, G.; Hillen, T. Effects of G2-checkpoint dynamics on low-dose hyperradiosensitivity. J. Math. Biol. 2018, 77, 1969–1997. [Google Scholar] [CrossRef]
- Marples, B.; Collis, S.J. Low-dose hyper-radiosensitivity: Past, present, and future. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 1310–1318. [Google Scholar] [CrossRef]
- Fernet, M.; Mégnin-Chanet, F.; Hall, J.; Favaudon, V. Control of the G2/M checkpoints after exposure to low doses of ionising radiation: Implications for hyper-radiosensitivity. DNA Repair 2010, 9, 48–57. [Google Scholar] [CrossRef]
- Ye, F.; Ning, J.; Liu, X.; Jin, X.; Wang, T.; Li, Q. The influence of non-DNA-targeted effects on carbon ion-induced low-dose hyper-radiosensitivity in MRC-5 cells. J. Radiat. Res. 2016, 57, 103–109. [Google Scholar] [CrossRef]
- Cherubini, R.; De Nadal, V.; Gerardi, S. Hyper-radiosensitivity and induced radioresistance and bystander effects in rodent and human cells as a function of radiation quality. Radiat. Prot. Dosim. 2015, 166, 137–141. [Google Scholar] [CrossRef]
- Nuta, O.; Darroudi, F. The impact of the bystander effect on the low-dose hypersensitivity phenomenon. Radiat. Environ. Biophys. 2008, 47, 265–274. [Google Scholar] [CrossRef]
- Fernandez-Palomo, C.; Seymour, C.; Mothersill, C. Inter-relationship between low-dose hyper-radiosensitivity and radiation-induced bystander effects in the human T98G glioma and the epithelial HaCaT cell line. Radiat. Res. 2016, 185, 124–133. [Google Scholar] [CrossRef]
- Ryan, L.A.; Seymour, C.B.; Joiner, M.C.; Mothersill, C.E. Radiation-induced adaptive response is not seen in cell lines showing a bystander effect but is seen in lines showing HRS/IRR response. Int. J. Radiat. Biol. 2009, 85, 87–95. [Google Scholar] [CrossRef]
- Dionet, C.; Müller-Barthélémy, M.; Marceau, G.; Denis, J.M.; Averbeck, D.; Gueulette, J.; Sapin, V.; Pereira, B.; Tchirkov, A.; Chautard, E.; et al. Different dose rate-dependent responses of human melanoma cells and fibroblasts to low dose fast neutrons. Int. J. Radiat. Biol. 2016, 92, 527–535. [Google Scholar] [CrossRef][Green Version]
- Xue, L.; Yu, D.; Furusawa, Y.; Okayasu, R.; Tong, J.; Cao, J.; Fan, S. Regulation of ATM in DNA double strand break repair accounts for the radiosensitivity in human cells exposed to high linear energy transfer ionizing radiation. Mutat. Res. 2009, 670, 15–23. [Google Scholar] [CrossRef]
- Xue, L.; Furusawa, Y.; Yu, D. ATR signaling cooperates with ATM in the mechanism of low dose hypersensitivity induced by carbon ion beam. DNA Repair 2015, 34, 1–8. [Google Scholar] [CrossRef]
- Enns, L.; Rasouli-Nia, A.; Hendze, L.M.; Marples, B.; Weinfeld, M. Association of ATM activation and DNA repair with induced radioresistance after low-dose irradiation. Radiat. Prot. Dosimetry 2015, 166, 131–136. [Google Scholar] [CrossRef][Green Version]
- Burdak-Rothkamm, S.; Rothkamm, K.; Prise, K.M. ATM acts downstream of ATR in the DNA damage response signaling of bystander cells. Cancer Res. 2008, 68, 7059–7065. [Google Scholar] [CrossRef]
- Heuskin, A.C.; Michiels, C.; Lucas, S. Low dose hypersensitivity following in vitro cell irradiation with charged particles: Is the mechanism the same as with X-ray radiation? Int. J. Radiat. Biol. 2014, 90, 81–89. [Google Scholar] [CrossRef]
- Maeda, M.; Usami, N.; Kobayashi, K. Low-dose hypersensitivity in nucleus-irradiated V79 cells studied with synchrotron X-ray microbeam. J. Radiat. Res. 2008, 49, 171–180. [Google Scholar] [CrossRef][Green Version]
- Maeda, M.; Tomita, M.; Usami, N.; Kobayashi, K. Bystander cell death is modified by sites of energy deposition within cells irradiated with a synchrotron X-ray microbeam. Radiat. Res. 2010, 174, 37–45. [Google Scholar] [CrossRef]
- Jin, S.; Cordes, N. ATM controls DNA repair and mitochondria transfer between neighboring cells. Cell Commun. Signal. 2019, 17, 144. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Furusawa, Y.; Aoki-Nakano, M.; Matsufuji, N.; Hirayama, R.; Kanai, T.; Ando, K.; Sakurai, H. Estimation of RBE values for carbon-ion beams in the wide dose range using multicellular spheroids. Radiat. Prot. Dosim. 2019, 183, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Hartfiel, S.; Häfner, M.; Perez, R.L.; Rühle, A.; Trinh, T.; Debus, J.; Huber, P.E.; Nicolay, N.H. Differential response of esophageal cancer cells to particle irradiation. Radiat. Oncol. 2019, 14, 119. [Google Scholar] [CrossRef]
- Nakajima, N.I.; Brunton, H.; Watanabe, R.; Shrikhande, A.; Hirayama, R.; Matsufuji, N.; Fujimori, A.; Murakami, T.; Okayasu, R.; Jeggo, P.; et al. Visualisation of γH2AX foci caused by heavy ion particle traversal; distinction between core track versus non-track damage. PLoS ONE 2013, 8, e70107. [Google Scholar] [CrossRef]
- Averbeck, N.B.; Ringel, O.; Herrlitz, M. DNA end resection is needed for the repair of complex lesions in G1-phase human cells. Cell Cycle 2014, 13, 2509–2516. [Google Scholar]
- Yajima, H.; Xue, L. DNA repair processes and checkpoint pathways in human cells exposed to heavy ion beams. Int. J. Part. Ther. 2016, 2, 439–446. [Google Scholar] [CrossRef]
- Niimi, A.; Yamauchi, M.; Limsirichaikul, S.; Sekine, R.; Oike, T.; Sato, H.; Suzuki, K.; Held, K.D.; Nakano, T.; Shibata, A. Identification of DNA double strand breaks at chromosome boundaries along the track of particle irradiation. Genes Chromosomes Cancer 2016, 55, 650–660. [Google Scholar] [CrossRef]
- Hagiwara, Y.; Niimi, A.; Isono, M.; Yamauchi, M.; Yasuhara, T.; Limsirichaikul, S.; Oike, T.; Sato, H.; Held, K.D.; Nakano, T.; et al. 3D-structured illumination microscopy reveals clustered DNA double-strand break formation in widespread γH2AX foci after high LET heavy-ion particle radiation. Oncotarget 2017, 8, 109370–109381. [Google Scholar] [CrossRef]
- Hagiwara, Y.; Oike, T.; Niimi, A.; Yamauchi, M.; Sato, H.; Limsirichaikul, S.; Held, K.D.; Nakano, T.; Shibata, A. Clustered DNA double-strand break formation and the repair pathway following heavy-ion irradiation. J. Radiat. Res. 2019, 60, 69–79. [Google Scholar] [CrossRef]
- Carusillo, A.; Mussolino, C. DNA damage: From threat to reatment. Cells 2020, 9, 1665. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Sharma, N.; Taylor, L. Clustered DNA double-strand breaks: Biological effects and relevance to cancer radiotherapy. Genes 2020, 11, 99. [Google Scholar] [CrossRef]
- Okayasu, R. Repair of DNA damage induced by accelerated heavy ions--a mini review. Int. J. Cancer 2012, 130, 991–1000. [Google Scholar] [CrossRef]
- Sage, E.; Shikazono, N. Radiation-induced clustered DNA lesions: Repair and mutagenesis. Free Radic. Biol. Med. 2017, 107, 125–135. [Google Scholar] [CrossRef]
- Mavragani, I.V.; Nikitaki, Z.; Souli, M.P.; Aziz, A.; Nowsheen, S.; Aziz, K.; Rogakou, E.; Georgakilas, A.G. Complex DNA damage: A route to radiation-induced genomic instability and carcinogenesis. Cancers 2017, 9, 91. [Google Scholar] [CrossRef]
- Lopez Perez, R.; Nicolay, N.H.; Wolf, J.C.; Frister, M.; Schmezer, P.; Weber, K.-J.; Huber, P.E. DNA damage response of clinical carbon ion versus photon radiation in human glioblastoma cells. Radiother. Oncol. 2019, 133, 77–86. [Google Scholar] [CrossRef]
- Meyer, B.; Voss, K.O.; Tobias, F.; Jakob, B.; Durante, M.; Taucher-Scholz, G. Clustered DNA damage induces pan-nuclear H2AX phosphorylation mediated by ATM and DNA-PK. Nucleic Acids Res. 2013, 41, 6109–6118. [Google Scholar] [CrossRef]
- Timm, S.; Lorat, Y.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Clustered DNA damage concentrated in particle trajectories causes persistent large-scale rearrangements in chromatin architecture. Radiother. Oncol. 2018, 129, 600–610. [Google Scholar] [CrossRef]
- Liu, F.; Wang, Z.; Li, W.; Wei, Y. Transcriptional response of murine bone marrow cells to total-body carbon-ion irradiation. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2019, 839, 49–58. [Google Scholar] [CrossRef]
- Ghosh, S.; Narang, H.; Sarma, A.; Krishna, M. DNA damage response signaling in lung adenocarcinoma A549 cells following gamma and carbon beam irradiation. Mutat. Res. 2011, 716, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Dokic, I.; Mairani, A.; Brons, S.; Schoell, B.; Jauch, A.; Krunic, D.; Debus, J.; Régnier-Vigouroux, A.; Weber, K.-J. High resistance to X-rays and therapeutic carbon ions in glioblastoma cells bearing dysfunctional ATM associates with intrinsic chromosomal instability. Int. J. Radiat. Biol. 2015, 91, 157–165. [Google Scholar] [CrossRef]
- Maalouf, M.; Granzotto, A.; Devic, C.; Bodgi, L.; Ferlazzo, M.; Peaucelle, C.; Bajard, M.; Giraud, J.Y.; Balosso, J.; Hérault, J.; et al. Influence of linear energy transfer on the nucleo-shuttling of the ATM protein: A novel biological interpretation relevant for particles and radiation. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 709–718. [Google Scholar] [CrossRef]
- Wozny, A.S.; Gauthier, A.; Alphonse, G.; Malésys, C.; Varoclier, V.; Beuve, M.; Brichart-Vernos, D.; Magné, N.; Vial, N.; Ardail, D.; et al. Involvement of HIF-1α in the Detection, Signaling, and Repair of DNA Double-Strand Breaks after Photon and Carbon-Ion Irradiation. Cancers 2021, 13, 3833. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.S.; Steinauer, K.K.; Hornung, B.; Irish, J.M.; Lecane, P.; Birrel, G.W.; Peehl, D.M.; Knox, S.J. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 2002, 9, 252–263. [Google Scholar] [CrossRef]
- Hanot, M.; Boivin, A.; Malésys, C. Glutathione depletion and carbon ion radiation potentiate clustered DNA lesions, cell death and prevent chromosomal changes in cancer cells progeny. PLoS ONE 2012, 7, e44367. [Google Scholar] [CrossRef]
- Suetens, A.; Konings, K.; Moreels, M.; Quintens, R.; Versiegers, M.; Soors, E.; Tabury, K.; Grégoire, V.; Baatout, S. Higher initial DNA damage and persistent cell cycle arrest after carbon ion irradiation compared to x-irradiation in prostate and colon cancer cells. Front. Oncol. 2016, 6, 87. [Google Scholar] [CrossRef]
- Zhang, Q.; Kong, Y.; Yang, Z.; Liu, Y.; Liu, R.; Geng, Y.; Luo, H.; Zhang, H.; Li, H.; Feng, S.; et al. Preliminary study on radiosensitivity to carbon ions in human breast cancer. J. Radiat. Res. 2020, 61, 399–409. [Google Scholar]
- Habermehl, D.; Ilicic, K.; Dehne, S.; Rieken, S.; Orschiedt, L.; Brons, S.; Haberer, T.; Weber, K.J.; Debus, J.; Combs, S.E. The relative biological effectiveness for carbon and oxygen ion beams using the raster-scanning technique in hepatocellular carcinoma cell lines. PLoS ONE 2014, 9, e113591. [Google Scholar] [CrossRef]
- Matsui, A.; Kobayashi, J.; Kanno, S.I.; Hashiguchi, K.; Miyaji, M.; Yoshikawa, Y.; Yasui, A.; Zhang-Akiyama, Q.M. Oxidation resistance 1 prevents genome instability through maintenance of G2/M arrest in gamma-ray-irradiated cells. J. Radiat. Res. 2020, 61, 1–13. [Google Scholar] [CrossRef]
- Lee, J.H.; Paull, T.T. Mitochondria at the crossroads of ATM-mediated stress signaling and regulation of reactive oxygen species. Redox Biol. 2020, 32, 101511. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell. Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H.E. Molecular mechanisms of radiation-induced cancer cell death: A primer. Front. Cell. Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef]
- Livingston, K.; Schlaak, R.A.; Puckett, L.L.; Bergom, C. The role of mitochondrial dysfunction in radiation-induced heart disease: From bench to bedside. Front. Cardiovasc. Med. 2020, 7, 20. [Google Scholar] [CrossRef]
- Rongvaux, A. Innate immunity and tolerance toward mitochondria. Mitochondrion 2018, 41, 14–20. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Mohammed, J.N.; Gelles, J.D.; Chen, Y. Mechanistic connections between mitochondrial biology and regulated cell death. Dev. Cell. 2021, 56, 1221–1233. [Google Scholar] [CrossRef]
- Lei, G.; Zhang, Y.; Hong, T.; Zhang, X.; Liu, X.; Mao, C.; Yan, Y.; Koppula, P.; Cheng, W.; Sood, A.K.; et al. Ferroptosis as a mechanism to mediate p53 function in tumor radiosensitivity. Oncogene 2021, 40, 3533–3547. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2020, 66, 89–100. [Google Scholar] [CrossRef]
- Toyokuni, S.; Yanatori, I.; Kong, Y.; Zheng, H.; Motooka, Y.; Jiang, L. Ferroptosis at the crossroads of infection, aging and cancer. Cancer Sci. 2020, 111, 2665–2671. [Google Scholar] [CrossRef]
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.M.; Monian, P.; Thompson, C.B.; Jiang, X. Role of mitochondria in ferroptosis. Mol. Cell. 2019, 73, 354–363.e3. [Google Scholar] [CrossRef]
- Ashrafizadeh, M.; Mohammadinejad, R.; Tavakol, S.; Ahmadi, Z.; Roomiani, S.; Katebi, M. Autophagy, anoikis, ferroptosis, necroptosis, and endoplasmic reticulum stress: Potential applications in melanoma therapy. J. Cell. Physiol. 2019, 234, 19471–19479. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target Ther. 2021, 6, 128. [Google Scholar] [CrossRef]
- Xiao, J.; Wang, C.; Yao, J.C.; Alippe, Y.; Yang, T.; Kress, D.; Sun, K.; Kostecki, K.L.; Monahan, J.B.; Veis, D.J.; et al. Radiation causes tissue damage by dysregulating inflammasome-gasdermin D signaling in both host and transplanted cells. PLoS Biol. 2020, 18, e3000807. [Google Scholar] [CrossRef]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef]
- Golden, E.B.; Pellicciotta, I.; Demaria, S.; Barcellos-Hoff, M.H.; Formenti, S.C. The convergence of radiation and immunogenic cell death signaling pathways. Front. Oncol. 2012, 2, 88. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondrial regulation of cell death. Cold Spring Harb. Perspect. Biol. 2013, 5, a008706. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef]
- Reap, E.A.; Roof, K.; Maynor, K.; Borrero, M.; Booker, J.; Cohen, P.L. Radiation and stress-induced apoptosis: A role for Fas/Fas ligand interactions. Proc. Natl. Acad. Sci. USA 1997, 94, 5750–5755. [Google Scholar] [CrossRef]
- Das, A.; McDonald, D.G.; Dixon-Mah, Y.N.; Jacqmin, D.J.; Samant, V.N.; Vandergrift, W.A., 3rd; Lindhorst, S.M.; Cachia, D.; Varma, A.K.; Vanek, K.N.; et al. RIP1 and RIP3 complex regulates radiation-induced programmed necrosis in glioblastoma. Tumour Biol. 2016, 37, 7525–7534. [Google Scholar] [CrossRef]
- Sheard, M.A.; Vojtesek, B.; Janakova, L.; Kovarik, J.; Zaloudik, J. Up-regulation of Fas (CD95) in human p53wild-type cancer cells treated with ionizing radiation. Int. J. Cancer 1997, 73, 757–762. [Google Scholar] [CrossRef]
- Abdulkarim, B.; Sabri, S.; Deutsch, E.; Vaganay, S.; Marangoni, E.; Vainchenker, W.; Bongrand, P.; Busson, P.; Bourhis, J. Radiation-induced expression of functional Fas ligand in EBV-positive human nasopharyngeal carcinoma cells. Int. J. Cancer 2000, 86, 229–237. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, K.H.; Lee, S.J. Ionizing radiation utilizes c-Jun N-terminal kinase for amplification of mitochondrial apoptotic cell death in human cervical cancer cells. FEBS J. 2008, 275, 2096–2108. [Google Scholar] [CrossRef]
- Tan, J.X.; Finkel, T. Mitochondria as intracellular signaling platforms in health and disease. J. Cell. Biol. 2020, 219, e202002179. [Google Scholar] [CrossRef]
- Ardail, D.; Maalouf, M.; Boivin, A.; Chapet, O.; Bodennec, J.; Rousson, R.; Rodriguez-Lafrasse, C. Diversity and complexity of ceramide generation after exposure of jurkat leukemia cells to irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef]
- Alphonse, G.; Aloy, M.T.; Broquet, P.; Gerard, J.P.; Louisot, P.; Rousson, R.; Rodriguez-Lafrasse, C. Ceramide induces activation of the mitochondrial/caspases pathway in Jurkat and SCC61 cells sensitive to gamma-radiation but activation of this sequence is defective in radioresistant SQ20B cells. Int. J. Radiat. Biol. 2002, 78, 821–835. [Google Scholar] [CrossRef]
- Alphonse, G.; Maalouf, M.; Battisa-Montagne, P.; Ardail, D.; Beuve, M.; Rousson, R.; Taucher-Scholz, G.; Fournier, C.; Rodriguez-Lafrasse, C. p53-independent early and late apoptosis is mediated by ceramide after exposure of tumor cells to photon or carbon ion irradiation. BMC Cancer 2013, 13, 151. [Google Scholar] [CrossRef]
- Ferranti, C.S.; Cheng, J.; Thompson, C.; Zhang, J.; Rotolo, J.A.; Buddaseth, S.; Fuks, Z.; Kolesnick, R.N. Fusion of lysosomes to plasma membrane initiates radiation-induced apoptosis. J. Cell. Biol. 2020, 219, e201903176. [Google Scholar] [CrossRef]
- Hu, L.; Wang, H.; Huang, L.; Zhao, Y.; Wang, J. Crosstalk between autophagy and intracellular radiation response (Review). Int. J. Oncol. 2016, 49, 2217–2226. [Google Scholar] [CrossRef]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Jin, M.; Liu, X.; Klionsky, D.J. SnapShot: Selective autophagy. Cell 2013, 152, 368–368.e2. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyuri, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef]
- Anding, A.L.; Baehrecke, E.H. Cleaning house: Selective autophagy of organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Marinković, M.; Šprung, M.; Novak, I. Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 2021, 17, 1232–1243. [Google Scholar] [CrossRef]
- Poole, L.P.; Macleod, K.F. Mitophagy in tumorigenesis and metastasis. Cell. Mol. Life. Sci. 2021, 78, 3817–3851. [Google Scholar] [CrossRef]
- Doblado, L.; Lueck, C.; Rey, C.; Samhan-Arias, A.K.; Prieto, I.; Stacchiotti, A.; Monsalve, M. Mitophagy in human diseases. Int. J. Mol. Sci. 2021, 22, 3903. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell. Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Kao, S.Y. DNA damage induces nuclear translocation of parkin. J. Biomed. Sci. 2009, 16, 67. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Ichim, G.; Tait, S.W.; Passos, J.F. Depletion of mitochondria in mammalian cells through enforced mitophagy. Nat. Protoc. 2017, 12, 183–194. [Google Scholar] [CrossRef]
- Stagni, V.; Cirotti, C.; Barilà, D. Ataxia-telangiectasia mutated kinase in the control of oxidative stress, mitochondria, and autophagy in cancer: A maestro with a large orchestra. Front. Oncol. 2018, 8, 73. [Google Scholar] [CrossRef]
- Sarkar, A.; Gandhi, V. Activation of ATM kinase by ROS generated during ionophore-induced mitophagy in human T and B cell malignancies. Mol. Cell. Biochem. 2021, 476, 417–423. [Google Scholar] [CrossRef]
- Mariño, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell. Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef]
- Melser, S.; Chatelain, E.H.; Lavie, J.; Mahfouf, W.; Jose, C.; Obre, E.; Goorden, S.; Priault, M.; Elgersma, Y.; Rezvani, H.R.; et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab. 2013, 17, 719–730. [Google Scholar] [CrossRef]
- Dan, X.; Babbar, M.; Moore, A.; Wechter, N.; Tian, J.; Mohanty, J.G.; Croteau, D.L.; Bohr, V.A. DNA damage invokes mitophagy through a pathway involving Spata18. Nucleic Acids Res. 2020, 48, 6611–6623. [Google Scholar] [CrossRef]
- Maguire, J.J.; Tyurina, Y.Y.; Mohammadyani, D.; Kapralov, A.; Anthonymuthu, T.S.; Qu, F.; Amoscato, A.A.; Sparvero, L.J.; Tyurin, V.A.; Planas-Iglesias, J.; et al. Known unknowns of cardiolipin signaling: The best is yet to come. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids. 2017, 1862, 8–24. [Google Scholar] [CrossRef]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell. Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef]
- Shimura, T.; Kobayashi, J.; Komatsu, K.; Kunugita, N. Severe mitochondrial damage associated with low-dose radiation sensitivity in ATM- and NBS1-deficient cells. Cell Cycle 2016, 15, 1099–1107. [Google Scholar] [CrossRef]
- Jin, X.; Zheng, X.; Li, F.; Liu, B.; Li, H.; Hirayama, R.; Li, P.; Liu, X.; Shen, G.; Li, Q. Fragmentation level determines mitochondrial damage response and subsequently the fate of cancer cells exposed to carbon ions. Radiother. Oncol. 2018, 129, 75–83. [Google Scholar] [CrossRef]
- Song, J.; Herrmann, J.M.; Becker, T. Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell. Biol. 2021, 22, 54–70. [Google Scholar] [CrossRef]
- Wu, H.; Chen, Q. Hypoxia activation of mitophagy and its role in disease pathogenesis. Antioxid. Redox Signal 2015, 22, 1032–1046. [Google Scholar] [CrossRef]
- Rios, C.I.; Cassatt, D.R.; Hollingsworth, B.A.; Satyamitra, M.M.; Tadesse, Y.S.; Taliaferro, L.P.; Winters, T.A.; DiCarlo, A.L. Commonalities between COVID-19 and radiation injury. Radiat. Res. 2021, 195, 1–24. [Google Scholar] [CrossRef]
- Prasanna, P.G.; Woloschak, G.E.; DiCarlo, A.L.; Buchsbaum, J.C.; Schaue, D.; Chakravarti, A.; Cucinotta, F.A.; Formenti, S.C.; Guha, C.; Hu, D.J.; et al. Low-dose radiation therapy (LDRT) for COVID-19: Benefits or risks? Radiat. Res. 2020, 194, 452–464. [Google Scholar] [CrossRef]
- Tapio, S.; Little, M.P.; Kaiser, J.C.; Impens, N.; Hamada, N.; Georgakilas, A.G.; Simar, D.; Salomaa, S. Ionizing radiation-induced circulatory and metabolic diseases. Environ. Int. 2021, 146, 106235. [Google Scholar] [CrossRef]
- Lee, J.M.; Bernstein, A. p53 mutations increase resistance to ionizing radiation. Proc. Natl. Acad. Sci. USA 1993, 90, 5742–5746. [Google Scholar] [CrossRef]
- Tsuboi, K.; Tsuchida, Y.; Nose, T.; Ando, K. Cytotoxic effect of accelerated carbon beams on glioblastoma cell lines with p53 mutation: Clonogenic survival and cell-cycle analysis. Int. J. Radiat. Biol. 1998, 74, 71–79. [Google Scholar] [CrossRef]
- McIlwrath, A.J.; Vasey, P.A.; Ross, G.M.; Brown, R. Cell cycle arrests and radiosensitivity of human tumor cell lines: Dependence on wild-type p53 for radiosensitivity. Cancer Res. 1994, 54, 3718–3722. [Google Scholar]
- Mori, E.; Takahashi, A.; Yamakawa, N.; Kirita, T.; Ohnishi, T. High LET Heavy Ion radiation induces p53-independent apoptosis. J. Radiat. Res. 2009, 50, 37–42. [Google Scholar] [CrossRef]
- Ferrandon, S.; Magné, N.; Battiston-Montagne, P.; Hau-Desbat, N.H.; Diaz, O.; Beuve, M.; Constanzo, J.; Chargari, C.; Poncet, D.; Chautard, E.; et al. Cellular and molecular portrait of eleven human glioblastoma cell lines under photon and carbon ion irradiation. Cancer Lett. 2015, 360, 10–16. [Google Scholar] [CrossRef]
- Takahashi, T.; Mitsuhashi, N.; Furuta, M.; Hasegawa, M.; Ohno, T.; Saito, Y.; Sakurai, H.; Nakano, T.; Niibe, H. Apoptosis induced by heavy ion (carbon) irradiation of two human tumours with different radiosensitivities in vivo: Relative biological effectiveness (RBE) of carbon beam. Anticancer Res. 1998, 18, 253–256. [Google Scholar]
- Takahashi, A.; Matsumoto, H.; Yuki, K.; Yasumoto, J.; Kajiwara, A.; Aoki, M.; Furusawa, Y.; Ohnishi, K.; Ohnishi, T. High-LET radiation enhanced apoptosis but not necrosis regardless of p53 status. Int. J. Radiat. Oncol. Biol. Phys. 2004, 60, 591–597. [Google Scholar] [CrossRef]
- Takahashi, A.; Matsumoto, H.; Furusawa, Y.; Ohnishi, K.; Ishioka, N.; Ohnishi, T. Apoptosis induced by high-LET radiations is not affected by cellular p53 gene status. Int. J. Radiat. Biol. 2005, 81, 581–589. [Google Scholar] [CrossRef]
- Yamakawa, N.; Takahashi, A.; Mori, E.; Imai, Y.; Furusawa, Y.; Ohnishi, K.; Kirita, T.; Ohnishi, T. High LET radiation enhances apoptosis in mutated p53 cancer cells through Caspase-9 activation. Cancer Sci. 2008, 99, 1455–1460. [Google Scholar] [CrossRef]
- Tsuboi, K.; Moritake, T.; Tsuchida, Y.; Tokuuye, K.; Matsumura, A.; Ando, K. Cell cycle checkpoint and apoptosis induction in glioblastoma cells and fibroblasts irradiated with carbon beam. J. Radiat. Res. 2007, 48, 317–325. [Google Scholar] [CrossRef]
- Aninditha, K.P.; Weber, K.J.; Brons, S.; Debus, J.; Hauswald, H. In vitro sensitivity of malignant melanoma cells lines to photon and heavy ion radiation. Clin. Transl. Radiat. Oncol. 2019, 17, 51–56. [Google Scholar] [CrossRef]
- Maalouf, M.; Alphonse, G.; Colliaux, A.; Beuve, M.; Trajkovic-Bodennec, S.; Battiston-Montagne, P.; Testard, I.; Chapet, O.; Bajard, M.; Taucher-Scholz, G.; et al. Different mechanisms of cell death in radiosensitive and radioresistant p53 mutated head and neck squamous cell carcinoma cell lines exposed to carbon ions and x-rays. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 200–209. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Takahashi, A.; Kajihara, A.; Yamakawa, N.; Imai, Y.; Ota, I.; Okamoto, N.; Mori, E.; Noda, T.; Furusawa, Y.; et al. Depression of p53-independent Akt survival signals in human oral cancer cells bearing mutated p53 gene after exposure to high-LET radiation. Biochem. Biophys. Res. Commun. 2012, 423, 654–660. [Google Scholar] [CrossRef]
- Combs, S.E.; Zipp, L.; Rieken, S.; Habermehl, D.; Brons, S.; Winter, M.; Haberer, T.; Debus, J.; Weber, K.J. In vitro evaluation of photon and carbon ion radiotherapy in combination with chemotherapy in glioblastoma cells. Radiat. Oncol. 2012, 7, 9. [Google Scholar] [CrossRef]
- Schlaich, F.; Brons, S.; Haberer, T.; Debus, J.; Combs, S.E.; Weber, K.J. Comparison of the effects of photon versus carbon ion irradiation when combined with chemotherapy in vitro. Radiat. Oncol. 2013, 8, 260. [Google Scholar] [CrossRef]
- Qin, J.; Li, S.; Zhang, C.; Gao, D.W.; Li, Q.; Zhang, H.; Jin, X.D.; Liu, Y. Apoptosis and injuries of heavy ion beam and x-ray radiation on malignant melanoma cell. Exp. Biol. Med. 2017, 242, 953–960. [Google Scholar] [CrossRef]
- Zheng, X.; Jin, X.; Li, F.; Liu, X.; Liu, Y.; Ye, F.; Li, P.; Zhao, T.; Li, Q. Inhibiting autophagy with chloroquine enhances the anti-tumor effect of high-LET carbon ions via ER stress-related apoptosis. Med. Oncol. 2017, 34, 25. [Google Scholar] [CrossRef]
- Jin, X.; Li, F.; Liu, B.; Zheng, X.; Li, H.; Ye, F.; Chen, W.; Li, Q. Different mitochondrial fragmentation after irradiation with X-rays and carbon ions in HeLa cells and its influence on cellular apoptosis. Biochem. Biophys. Res. Commun. 2018, 500, 958–965. [Google Scholar] [CrossRef]
- Jelena, Ž.; Lela, K.; Otilija, K.; Danijela, T.; Cirrone Giuseppe, A.P.; Francesco, R.; Giacomo, C.; Ivan, P.; Aleksandra, R.F. Carbon ions of different linear energy transfer (LET) values induce apoptosis & G2 cell cycle arrest in radio-resistant melanoma cells. Indian J. Med. Res. 2016, 143, S120–S128. [Google Scholar] [CrossRef]
- Held, K.D.; Kawamura, H.; Kaminuma, T.; Paz, A.E.; Yoshida, Y.; Liu, Q.; Willers, H.; Takahashi, A. Effects of charged particles on human tumor cells. Front. Oncol. 2016, 6, 23. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Herr, I.; Debatin, K.M. Cellular stress response and apoptosis in cancer therapy. Blood 2001, 98, 2603–2614. [Google Scholar] [CrossRef]
- Saenko, Y.; Cieslar-Pobuda, A.; Skonieczna, M.; Rzeszowska-Wolny, J. Changes of reactive oxygen and nitrogen species and mitochondrial functioning in human K562 and HL60 cells exposed to ionizing radiation. Radiat. Res. 2013, 180, 360–366. [Google Scholar] [CrossRef]
- Sun, C.; Wang, Z.; Liu, Y.; Liu, Y.; Li, H.; Di, C.; Wu, Z.; Gan, L.; Zhang, H. Carbon ion beams induce hepatoma cell death by NADPH oxidase-mediated mitochondrial damage. J. Cell. Physiol. 2014, 229, 100–107. [Google Scholar] [CrossRef]
- Walsh, D.W.M.; Siebenwirth, C.; Greubel, C.; Ilicic, K.; Reindl, J.; Girst, S.; Muggiolu, G.; Simon, M.; Barberet, P.; Seznec, H.; et al. Live cell imaging of mitochondria following targeted irradiation in situ reveals rapid and highly localized loss of membrane potential. Sci. Rep. 2017, 7, 46684. [Google Scholar] [CrossRef]
- Yan, J.; Xie, Y.; Wang, F.; Chen, Y.; Zhang, J.; Dou, Z.; Gan, L.; Li, H.; Si, J.; Sun, C.; et al. Carbon ion combined with tigecycline inhibits lung cancer cell proliferation by inducing mitochondrial dysfunction. Life Sci. 2020, 263, 118586. [Google Scholar] [CrossRef]
- Zhang, B.; Li, L.; Li, Z.; Liu, Y.; Zhang, H.; Wang, J. Carbon ion-irradiated hepatoma cells exhibit coupling interplay between apoptotic signaling and morphological and mechanical remodeling. Sci. Rep. 2016, 6, 35131. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, J.; Sun, C.; Li, G.; Li, S.; Zhang, L.; Di, C.; Gan, L.; Wang, Y.; Zhou, R.; et al. Ameliorating mitochondrial dysfunction restores carbon ion-induced cognitive deficits via co-activation of NRF2 and PINK1 signaling pathway. Redox Biol. 2018, 17, 143–157. [Google Scholar] [CrossRef]
- Wang, J.; Konishi, T. Nuclear factor (erythroid-derived 2)-like 2 antioxidative response mitigates cytoplasmic radiation-induced DNA double-strand breaks. Cancer Sci. 2019, 110, 686–696. [Google Scholar] [CrossRef]
- Anuranjani, B.M. Concerted action of Nrf2-ARE pathway, MRN complex, HMGB1 and inflammatory cytokines—Implication in modification of radiation damage. Redox Biol. 2014, 2, 832–846. [Google Scholar] [CrossRef]
- Zhang, L.; Yan, J.; Liu, Y.; Zhao, Q.; Di, C.; Chao, S.; Jie, L.; Liu, Y.; Zhang, H. Contribution of caspase-independent pathway to apoptosis in malignant glioma induced by carbon ion beams. Oncol. Rep. 2017, 37, 2994–3000. [Google Scholar] [CrossRef][Green Version]
- Wang, M.; Saha, J.; Hada, M.; Anderson, J.A.; Pluth, J.M.; O’Neill, P.; Cucinotta, F.A. Novel Smad proteins localize to IR-induced double-strand breaks: Interplay between TGFβ and ATM pathways. Nucleic Acids Res. 2013, 41, 933–942. [Google Scholar] [CrossRef]
- Kim, C.S.; Kim, J.M.; Nam, S.Y.; Yang, K.H.; Jeong, M.; Kim, H.S.; Lim, Y.K.; Kim, C.S.; Jin, Y.W.; Kim, J. Low-dose of ionizing radiation enhances cell proliferation via transient ERK1/2 and p38 activation in normal human lung fibroblasts. J. Radiat. Res. 2007, 48, 407–415. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Wang, X.Y.; Wei, Q.Y.; Xu, Y.M.; Lau, A.T.Y. Potency and selectivity of SMAC/DIABLO mimetics in solid tumor therapy. Cells 2020, 9, 1012. [Google Scholar] [CrossRef]
- Fandy, T.E.; Shankar, S.; Srivastava, R.K. Smac/DIABLO enhances the therapeutic potential of chemotherapeutic drugs and irradiation, and sensitizes TRAIL-resistant breast cancer cells. Mol. Cancer 2008, 7, 60. [Google Scholar] [CrossRef]
- Zhang, R.; Dang, X.; Zhang, Z.; Yuan, Y.; Ren, Y.; Duan, Z.; Zuo, Y. Comparison of transcriptional profiles in human lymphocyte cells irradiated with (12)C ion beams at 0-2.0 Gy. Cancer Manag. Res. 2019, 11, 2363–2369. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lu, H.; Jiang, Z.; Pastuszyn, A.; Hu, C.A. Apolipoprotein l6, a novel proapoptotic Bcl-2 homology 3-only protein, induces mitochondria-mediated apoptosis in cancer cells. Mol. Cancer Res. 2005, 3, 21–31. [Google Scholar]
- Tait, S.W.; Ichim, G.; Green, D.R. Die another way--non-apoptotic mechanisms of cell death. J. Cell. Sci. 2014, 127, 2135–2144. [Google Scholar] [CrossRef]
- Lartigue, L.; Kushnareva, Y.; Seong, Y.; Lin, H.; Faustin, B.; Newmeyer, D.D. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol. Biol. Cell. 2009, 20, 4871–4884. [Google Scholar] [CrossRef]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001, 412, 95–99. [Google Scholar] [CrossRef]
- Er, E.; Oliver, L.; Cartron, P.F.; Juin, P.; Manon, S.; Vallette, F.M. Mitochondria as the target of the pro-apoptotic protein Bax. Biochim. Biophys. Acta. 2006, 1757, 1301–1311. [Google Scholar] [CrossRef]
- Saelens, X.; Festjens, N.; Vande Walle, L.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar] [CrossRef]
- Liu, X.; He, Y.; Li, F.; Huang, Q.; Kato, T.A.; Hall, R.P.; Li, C.Y. Caspase-3 promotes genetic instability and carcinogenesis. Mol. Cell. 2015, 58, 284–296. [Google Scholar] [CrossRef]
- Bao, X.; Liu, X.; Li, F.; Li, C.Y. Limited MOMP, ATM, and their roles in carcinogenesis and cancer treatment. Cell Biosci. 2020, 10, 81. [Google Scholar] [CrossRef]
- Pandya, V.; Githaka, J.M.; Patel, N.; Veldhoen, R.; Hugh, J.; Damaraju, S.; McMullen, T.; Mackey, J.; Goping, I.S. BIK drives an aggressive breast cancer phenotype through sublethal apoptosis and predicts poor prognosis of ER-positive breast cancer. Cell. Death Dis. 2020, 11, 448. [Google Scholar] [CrossRef]
- Sun, G.; Montell, D.J. Cellular neardeath experiences-what is anastasis? BMC Biol. 2017, 15, 92. [Google Scholar] [CrossRef]
- Tang, H.M.; Fung, M.C.; Tang, H.L. Detecting anastasis in vivo by caspasetracker biosensor. J. Vis. Exp. 2018, 132, 54107. [Google Scholar] [CrossRef]
- Tait, S.W.; Parsons, M.J.; Llambi, F.; Bouchier-Hayes, L.; Connell, S.; Muñoz-Pinedo, C.; Green, D.R. Resistance to caspase-independent cell death requires persistence of intact mitochondria. Dev. Cell. 2010, 18, 802–813. [Google Scholar] [CrossRef]
- Tang, H.M.; Tang, H.L. Anastasis: Recovery from the brink of cell death. R. Soc. Open Sci. 2018, 5, 180442. [Google Scholar] [CrossRef]
- Gong, Y.N.; Crawford, J.C.; Heckmann, B.L.; Green, D.R. To the edge of cell death and back. FEBS J. 2019, 286, 430–440. [Google Scholar] [CrossRef]
- Tang, H.M.; Talbot, C.C., Jr.; Fung, M.C.; Tang, H.L. Molecular signature of anastasis for reversal of apoptosis. F1000Research 2017, 6, 43. [Google Scholar] [CrossRef]
- Sun, G.; Guzman, E.; Balasanyan, V.; Conner, C.M.; Wong, K.; Zhou, H.R.; Kosik, K.S.; Montell, D.J. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J. Cell. Biol. 2017, 216, 3355–3368. [Google Scholar] [CrossRef]
- Kaur, E.; Rajendra, J.; Jadhav, S.; Shridhar, E.; Goda, J.S.; Moiyadi, A.; Dutt, S. Radiation-induced homotypic cell fusions of innately resistant glioblastoma cells mediate their sustained survival and recurrence. Carcinogenesis 2015, 36, 685–695. [Google Scholar] [CrossRef]
- Ichim, G.; Lopez, J.; Ahmed, S.U.; Muthalagu, N.; Giampazolias, E.; Delgado, M.E.; Haller, M.; Riley, J.S.; Mason, S.M.; Athineos, D.; et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell. 2015, 57, 860–872. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Kumar, P.; Murray, D. Multinucleated giant cancer cells produced in response to ionizing radiation retain viability and replicate their genome. Int. J. Mol. Sci. 2017, 18, 360. [Google Scholar] [CrossRef]
- Mirzayans, R.; Murray, D. Intratumor heterogeneity and therapy resistance: Contributions of dormancy, apoptosis reversal (Anastasis) and cell fusion to disease recurrence. Int. J. Mol. Sci. 2020, 21, 1308. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Kaakati, R.; Lee, A.K.; Liu, X.; Li, F.; Li, C.Y. Novel roles of apoptotic caspases in tumor repopulation, epigenetic reprogramming, carcinogenesis, and beyond. Cancer Metastasis Rev. 2018, 37, 227–236. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, Y.; Zhao, Y.; He, S.; Zhao, R.; Song, Y.; Cheng, J.; Gong, Y.; Xie, J.; Wang, Y.; et al. Caspase-3 knockout attenuates radiation-induced tumor repopulation via impairing the ATM/p53/Cox-2/PGE2 pathway in non-small cell lung cancer. Aging 2020, 12, 21758–21776. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, Y.; Han, W.; Chiu, S.K.; Zhu, L.; Wu, L.; Yu, K.N. Rescue effects in radiobiology: Unirradiated bystander cells assist irradiated cells through intercellular signal feedback. Mutat. Res. 2011, 706, 59–64. [Google Scholar] [CrossRef]
- Yu, K.N. Radiation-induced rescue effect. J. Radiat. Res. 2019, 60, 163–170. [Google Scholar] [CrossRef]
- Kong, E.Y.; Cheng, S.H.; Yu, K.N. Induction of autophagy and interleukin 6 secretion in bystander cells: Metabolic cooperation for radiation-induced rescue effect? J. Radiat. Res. 2018, 59, 129–140. [Google Scholar] [CrossRef]
- Pathikonda, S.; Cheng, S.H.; Yu, K.N. Role of PARP1 regulation in radiation-induced rescue effect. J. Radiat. Res. 2020, 61, 352–367. [Google Scholar] [CrossRef]
- Durante, M. New challenges in high-energy particle radiobiology. Br. J. Radiol. 2014, 87, 20130626. [Google Scholar] [CrossRef]
- Yokota, Y.; Wada, Y.; Funayama, T. Distinct modes of death in human neural stem and glioblastoma cells irradiated with carbon-ion radiation and gamma-rays. Int. J. Radiat. Biol. 2020, 96, 172–178. [Google Scholar] [CrossRef]
- Durante, M.; Orecchia, R.; Loeffler, J.S. Charged-particle therapy in cancer: Clinical uses and future perspectives. Nat. Rev. Clin. Oncol. 2017, 14, 483–495. [Google Scholar] [CrossRef]
- Tinganelli, W.; Durante, M. Carbon ion radiobiology. Cancers 2020, 12, 3022. [Google Scholar] [CrossRef]
- Belyakov, O.V.; Mitchell, S.A.; Parikh, D.; Randers-Pehrson, G.; Marino, S.A.; Amundson, S.A.; Geard, C.R.; Brenner, D.J. Biological effects in unirradiated human tissue induced by radiation damage up to 1 mm away. Proc. Natl. Acad. Sci. USA 2005, 102, 14203–14208. [Google Scholar] [CrossRef]
- Hu, B.; Wu, L.; Han, W.; Zhang, L.; Chen, S.; Xu, A.; Hei, T.K.; Yu, Z. The time and spatial effects of bystander response in mammalian cells induced by low dose radiation. Carcinogenesis 2006, 27, 245–251. [Google Scholar] [CrossRef]
- Tartier, L.; Gilchrist, S.; Burdak-Rothkamm, S.; Folkard, M.; Prise, K.M. Cytoplasmic irradiation induces mitochondrial-dependent 53BP1 protein relocalization in irradiated and bystander cells. Cancer Res. 2007, 67, 5872–5879. [Google Scholar] [CrossRef]
- Choi, K.M.; Kang, C.M.; Cho, E.S.; Kang, S.M.; Lee, S.B.; Um, H.D. Ionizing radiation-induced micronucleus formation is mediated by reactive oxygen species that are produced in a manner dependent on mitochondria, Nox1, and JNK. Oncol. Rep. 2007, 17, 1183–1188. [Google Scholar] [PubMed]
- Murphy, J.E.; Nugent, S.; Seymour, C.; Mothersill, C. Mitochondrial DNA point mutations and a novel deletion induced by direct low-LET radiation and by medium from irradiated cells. Mutat. Res. 2005, 585, 127–136. [Google Scholar] [CrossRef]
- Zhou, H.; Ivanov, V.N.; Lien, Y.C.; Davidson, M.; Hei, T.K. Mitochondrial function and nuclear factor-kappaB-mediated signaling in radiation-induced bystander effects. Cancer Res. 2008, 68, 2233–2240. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, Y.; Han, W.; Zhao, G.; Zhu, L.; Wang, J.; Bao, L.; Jiang, E.; Xu, A.; Hei, T.K.; et al. Mitochondria-dependent signalling pathway are involved in the early process of radiation-induced bystander effects. Br. J. Cancer 2008, 98, 1839–1844. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Chen, S.; Zhu, L.; Huang, P.; Tong, L.; Zhao, Y.; Zhao, G.; Wang, J.; Mei, T.; et al. Mitochondrial dysfunction resulting from loss of cytochrome c impairs radiation-induced bystander effect. Br. J. Cancer 2009, 100, 1912–1916. [Google Scholar] [CrossRef][Green Version]
- Burdak-Rothkamm, S.; Rothkamm, K. Radiation-induced bystander and systemic effects serve as a unifying model system for genotoxic stress responses. Mutat. Res. 2018, 778, 13–22. [Google Scholar] [CrossRef]
- Huang, L.; Kim, P.M.; Nickoloff, J.A.; Morgan, W.F. Targeted and nontargeted effects of low-dose ionizing radiation on delayed genomic instability in human cells. Cancer Res. 2007, 67, 1099–1104. [Google Scholar] [CrossRef]
- Wang, H.; Yu, K.N.; Hou, J.; Liu, Q.; Han, W. Radiation-induced bystander effect: Early process and rapid assessment. Cancer Lett. 2015, 356, 137–144. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, Y.; Zhao, G.; Han, W.; Bao, L.; Yu, K.N.; Wu, L. Up-regulation of ROS by mitochondria-dependent bystander signaling contributes to genotoxicity of bystander effects. Mutat. Res. 2009, 666, 68–73. [Google Scholar] [CrossRef]
- Furlong, H.; Mothersill, C.; Lyng, F.M.; Howe, O. Apoptosis is signalled early by low doses of ionising radiation in a radiation-induced bystander effect. Mutat. Res. 2013, 741–742, 35–43. [Google Scholar] [CrossRef]
- Portess, D.I.; Bauer, G.; Hill, M.A.; O’Neill, P. Low-dose irradiation of nontransformed cells stimulates the selective removal of precancerous cells via intercellular induction of apoptosis. Cancer Res. 2007, 67, 1246–1253. [Google Scholar] [CrossRef]
- Prise, K.M.; O’Sullivan, J.M. Radiation-induced bystander signalling in cancer therapy. Nat. Rev. Cancer 2009, 9, 351–360. [Google Scholar] [CrossRef]
- Heeran, A.B.; Berrigan, H.P.; O’Sullivan, J. The radiation-induced bystander effect (RIBE) and its connections with the hallmarks of cancer. Radiat. Res. 2019, 192, 668–679. [Google Scholar] [CrossRef]
- Autsavapromporn, N.; Suzuki, M.; Funayama, T.; Usami, N.; Plante, I.; Yokota, Y.; Mutou, Y.; Ikeda, H.; Kobayashi, K.; Kobayashi, Y.; et al. Gap junction communication and the propagation of bystander effects induced by microbeam irradiation in human fibroblast cultures: The impact of radiation quality. Radiat. Res. 2013, 180, 367–375. [Google Scholar] [CrossRef]
- Autsavapromporn, N.; Plante, I.; Liu, C.; Konishi, T.; Usami, N.; Funayama, T.; Azzam, E.I.; Murakami, T.; Suzuki, M. Genetic changes in progeny of bystander human fibroblasts after microbeam irradiation with X-rays, protons or carbon ions: The relevance to cancer risk. Int. J. Radiat. Biol. 2015, 91, 62–70. [Google Scholar] [CrossRef]
- Dong, C.; He, M.; Ren, R.; Xie, Y.; Yuan, D.; Dang, B.; Li, W.; Shao, C. Role of the MAPK pathway in the observed bystander effect in lymphocytes co-cultured with macrophages irradiated with γ-rays or carbon ions. Life Sci. 2015, 127, 19–25. [Google Scholar] [CrossRef]
- Dong, C.; He, M.; Tu, W.; Konishi, T.; Liu, W.; Xie, Y.; Dang, B.; Li, W.; Uchihori, Y.; Hei, T.K.; et al. The differential role of human macrophage in triggering secondary bystander effects after either gamma-ray or carbon beam irradiation. Cancer Lett. 2015, 363, 92–100. [Google Scholar] [CrossRef]
- Harada, K.; Nonaka, T.; Hamada, N.; Sakurai, H.; Hasegawa, M.; Funayama, T.; Kakizaki, T.; Kobayashi, Y.; Nakano, T. Heavy-ion-induced bystander killing of human lung cancer cells: Role of gap junctional intercellular communication. Cancer Sci. 2009, 100, 684–688. [Google Scholar] [CrossRef]
- Tu, W.; Dong, C.; Fu, J.; Pan, Y.; Kobayashi, A.; Furusawa, Y.; Konishi, T.; Shao, C. Both irradiated and bystander effects link with DNA repair capacity and the linear energy transfer. Life Sci. 2019, 222, 228–234. [Google Scholar] [CrossRef]
- Buonanno, M.; de Toledo, S.M.; Pain, D.; Azzam, E.I. Long-term consequences of radiation-induced bystander effects depend on radiation quality and dose and correlate with oxidative stress. Radiat. Res. 2011, 175, 405–415. [Google Scholar] [CrossRef]
- Jelonek, K.; Wojakowska, A.; Marczak, L.; Muer, A.; Tinhofer-Keilholz, I.; Lysek-Gladysinska, M.; Widlak, P.; Pietrowska, M. Ionizing radiation affects protein composition of exosomes secreted in vitro from head and neck squamous cell carcinoma. Acta Biochim. Pol. 2015, 62, 265–272. [Google Scholar] [CrossRef]
- Ariyoshi, K.; Miura, T.; Kasai, K.; Fujishima, Y.; Nakata, A.; Yoshida, M. Radiation-induced bystander effect is mediated by mitochondrial DNA in exosome-like vesicles. Sci. Rep. 2019, 9, 9103. [Google Scholar] [CrossRef]
- Shan, Z.; Wang, H.; Zhang, Y.; Min, W. The role of tumor-derived exosomes in the abscopal effect and immunotherapy. Life 2021, 11, 381. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, J.; Zhou, L.; Hao, S.; Ding, Z.; Xiao, L.; Zhou, M. Exosomal small RNA sequencing Uncovers Dose-Specific MiRNA Markers for Ionizing Radiation Exposure. Dose Response 2020, 18, 1559325820926735. [Google Scholar] [CrossRef]
- Xu, S.; Wang, J.; Ding, N.; Hu, W.; Zhang, X.; Wang, B.; Hua, J.; Wei, W.; Zhu, Q. Exosome-mediated microRNA transfer plays a role in radiation-induced bystander effect. RNA Biol. 2015, 12, 1355–1363. [Google Scholar] [CrossRef]
- Yin, X.; Tian, W.; Wang, L.; Wang, J.; Zhang, S.; Cao, J.; Yang, H. Radiation quality-dependence of bystander effect in unirradiated fibroblasts is associated with TGF-β1-Smad2 pathway and miR-21 in irradiated keratinocytes. Sci. Rep. 2015, 5, 11373. [Google Scholar] [CrossRef]
- Elbakrawy, E.M.; Mayah, A.; Hill, M.A.; Kadhim, M. Induction of genomic instability in a primary human fibroblast cell line following low-dose alpha-particle exposure and the potential role of exosomes. Biology 2020, 10, 11. [Google Scholar] [CrossRef]
- Tan, W.; Zhang, Y.; Li, M.; Zhu, X.; Yang, X.; Wang, J.; Zhang, S.; Zhu, W.; Cao, J.; Yang, H.; et al. MiR-27a-containing exosomes secreted by irradiated skin keratinocytes delayed the migration of unirradiated skin fibroblasts. Int. J. Biol. Sci. 2019, 15, 2240–2255. [Google Scholar] [CrossRef]
- Yu, Q.; Li, P.; Weng, M.; Wu, S.; Zhang, Y.; Chen, X.; Zhang, Q.; Shen, G.; Ding, X.; Fu, S. Nano-vesicles are a potential tool to monitor therapeutic efficacy of carbon ion radiotherapy in prostate cancer. J. Biomed. Nanotechnol. 2018, 14, 168–178. [Google Scholar] [CrossRef]
- He, C.; Li, L.; Wang, L.; Meng, W.; Hao, Y.; Zhu, G. Exosome-mediated cellular crosstalk within the tumor microenvironment upon irradiation. Cancer Biol. Med. 2021, 18, 21–33. [Google Scholar] [CrossRef]
- Diegeler, S.; Hellweg, C.E. Intercellular Communication of Tumor Cells and Immune Cells after Exposure to Different Ionizing Radiation Qualities. Front. Immunol. 2017, 8, 664–678. [Google Scholar] [CrossRef]
- Daguenet, E.; Louati, S.; Wozny, A.S.; Vial, N.; Gras, M.; Guy, J.B.; Vallard, A.; Rodriguez-Lafrasse, C.; Magné, N. Radiation-induced bystander and abscopal effects: Important lessons from preclinical models. Br. J. Cancer 2020, 123, 339–348. [Google Scholar] [CrossRef]
- Demaria, S.; Formenti, S.C. The abscopal effect 67 years later: From a side story to center stage. Br. J. Radiol. 2020, 93, 1–8. [Google Scholar] [CrossRef]
- Demaria, S.; Ng, B.; Devitt, M.L.; Babb, J.S.; Kawashima, N.; Liebes, L.; Formenti, S.C. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 862–870. [Google Scholar] [CrossRef]
- Ogata, T.; Teshima, T.; Kagawa, K.; Hishikawa, Y.; Takahashi, Y.; Kawaguchi, A.; Suzumoto, Y.; Nojima, K.; Furusawa, Y.; Matsuura, N. Particle irradiation suppresses metastatic potential of cancer cells. Cancer Res. 2005, 65, 113–120. [Google Scholar]
- Kong, L.; Gao, J.; Hu, J.; Lu, R.; Yang, J.; Qiu, X.; Hu, W.; Lu, J.J. Carbon ion radiotherapy boost in the treatment of glioblastoma: A randomized phase I/III clinical trial. Cancer Commun. 2019, 39, 5. [Google Scholar] [CrossRef]
- Ando, K.; Fujita, H.; Hosoi, A.; Ma, L.; Wakatsuki, M.; Seino, K.I. Intravenous dendritic cell administration enhances suppression of lung metastasis induced by carbon-ion irradiation. J. Radiat. Res. 2017, 58, 446–455. [Google Scholar]
- Matsunaga, A.; Ueda, Y.; Yamada, S.; Harada, Y.; Shimada, H.; Hasegawa, M. Carbon-ion beam treatment induces systemic antitumor immunity against murine squamous cell carcinoma. Cancer 2010, 116, 3740–3748. [Google Scholar] [CrossRef]
- Yoshimoto, Y.; Kono, K.; Suzuki, Y. Anti-tumor immune responses induced by radiotherapy: A review. Fukushima J. Med. Sci. 2015, 61, 13–22. [Google Scholar] [CrossRef]
- Fernandez-Gonzalo, R.; Baatout, S.; Moreels, M. Impact of particle irradiation on the immune system: From the clinic to Mars. Front. Immunol. 2017, 8, 177. [Google Scholar] [CrossRef]
- Carvalho, H.A.; Villar, R.C. Radiotherapy and immune response: The systemic effects of a local treatment. Clinics 2018, 73, e557s. [Google Scholar] [CrossRef]
- Weinberg, S.E.; Sena, L.A.; Chandel, N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity 2015, 42, 406–417. [Google Scholar] [CrossRef]
- Lartigue, L.; Faustin, B. Mitochondria: Metabolic regulators of innate immune responses to pathogens and cell stress. Int. J. Biochem. Cell. Biol. 2013, 45, 2052–2056. [Google Scholar] [CrossRef]
- Breda, C.N.S.; Davanzo, G.G.; Basso, P.J.; Saraiva Câmara, N.O.; Moraes-Vieira, P.M.M. Mitochondria as central hub of the immune system. Redox Biol. 2019, 26, 101255. [Google Scholar] [CrossRef]
- Storozynsky, Q.; Hitt, M.M. The impact of radiation-induced DNA damage on cGAS-STING-mediated immune responses to cancer. Int. J. Mol. Sci. 2020, 21, 8877. [Google Scholar] [CrossRef]
- Hu, B.; Jin, C.; Li, H.B.; Tong, J.; Ouyang, X.; Cetinbas, N.M.; Zhu, S.; Strowig, T.; Lam, F.C.; Zhao, C.; et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 2016, 354, 765–768. [Google Scholar] [CrossRef]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; de Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.A.; Kuan, C.Y.; et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.C.; Herold, M.J.; van Delft, M.F.; Bedoui, S.; Lessene, G.; Ritchie, M.E.; et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef]
- Santa, P.; Garreau, A.; Serpas, L.; Ferriere, A.; Blanco, P.; Soni, C.; Sisirak, V. The role of nucleases and nucleic acid editing enzymes in the regulation of self-nucleic acid sensing. Front. Immunol. 2021, 12, 629922. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Cai, X.; Chiu, Y.H.; Chen, Z.J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell. 2014, 54, 289–296. [Google Scholar] [CrossRef]
- Zierhut, C.; Yamaguchi, N.; Paredes, M.; Luo, J.D.; Carroll, T.; Funabiki, H. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell 2019, 178, 302–315.e23. [Google Scholar] [CrossRef]
- Fang, C.; Mo, F.; Liu, L.; Du, J.; Luo, M.; Men, K.; Na, F.; Wang, W.; Yang, H.; Wei, X. Oxidized mitochondrial DNA sensing by STING signaling promotes the antitumor effect of an irradiated immunogenic cancer cell vaccine. Cell. Mol. Immunol. 2020, 12, 1–13. [Google Scholar] [CrossRef]
- Yamazaki, T.; Galluzzi, L. Mitochondrial control of innate immune signaling by irradiated cancer cells. Oncoimmunology 2020, 9, 1797292. [Google Scholar] [CrossRef]
- Rodriguez-Ruiz, M.E.; Vitale, I.; Harrington, K.J.; Melero, I.; Galluzzi, L. Immunological impact of cell death signaling driven by radiation on the tumor microenvironment. Nat. Immunol. 2020, 21, 120–134. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Lindqvist, L.M.; Frank, D.; McArthur, K.; Dite, T.A.; Lazarou, M.; Oakhill, J.S.; Kile, B.T.; Vaux, D.L. Autophagy induced during apoptosis degrades mitochondria and inhibits type I interferon secretion. Cell Death Differ. 2018, 25, 784–796. [Google Scholar] [CrossRef]
- Murray, D.; Mirzayans, R.; McBride, W.H. Defenses against pro-oxidant forces—Maintenance of cellular and genomic integrity and longevity. Radiat. Res. 2018, 190, 331–349. [Google Scholar] [CrossRef]
- Suzuki, M.; Yasuda, N.; Kitamura, H. Lethal and mutagenic bystander effects in human fibroblast cell cultures subjected to low-energy-carbon ions. Int. J. Radiat. Biol. 2020, 96, 179–186. [Google Scholar] [CrossRef]
- Rombouts, C.; Aerts, A.; Quintens, R.; Baselet, B.; El-Saghire, H.; Harms-Ringdahl, M.; Haghdoost, S.; Janssen, A.; Michaux, A.; Yentrapalli, R.; et al. Transcriptomic profiling suggests a role for IGFBP5 in premature senescence of endothelial cells after chronic low dose rate irradiation. Int. J. Radiat. Biol. 2014, 90, 560–574. [Google Scholar] [CrossRef]
- Redpath, J.L.; Elmore, E. Radiation-induced neoplastic transformation in vitro, hormesis and risk assessment. Dose Response 2006, 5, 123–130. [Google Scholar] [CrossRef]
- Song, K.H.; Oh, S.J.; Kim, S.; Cho, H.; Lee, H.J.; Song, J.S.; Chung, J.Y.; Cho, E.; Lee, J.; Jeon, S.; et al. HSP90A inhibition promotes anti-tumor immunity by reversing multi-modal resistance and stem-like property of immune-refractory tumors. Nat. Commun. 2020, 11, 562. [Google Scholar] [CrossRef]
- Tazat, K.; Reshetnyak, O.; Shtraus, N.; Sayag, I.; Mabjeesh, N.J.; Amir, S. Delivery of radiation at the lowest dose rate by a modern linear accelerator is most effective in inhibiting prostate cancer growth. Technol. Cancer Res. Treat. 2020, 19, 1533033820935525. [Google Scholar] [CrossRef]
- Yang, G.; Yu, D.; Li, W.; Zhao, Y.; Wen, X.; Liang, X.; Zhang, X.; Zhou, L.; Hu, J.; Niu, C.; et al. Distinct biological effects of low-dose radiation on normal and cancerous human lung cells are mediated by ATM signaling. Oncotarget 2016, 7, 71856–71872. [Google Scholar] [CrossRef]
- Kabakov, A.; Yakimova, A.; Matchuk, O. Molecular Chaperones in Cancer Stem Cells: Determinants of Stemness and Potential Targets for Antitumor Therapy. Cells 2020, 9, 892. [Google Scholar] [CrossRef]
- Konkova, M.; Abramova, M.; Kalianov, A.; Ershova, E.; Dolgikh, O.; Umriukhin, P.; Izhevskaya, V.; Kutsev, S.; Veiko, N.; Kostyuk, S. Mesenchymal stem cells early response to low-dose ionizing radiation. Front. Cell. Dev. Biol. 2020, 8, 584497. [Google Scholar] [CrossRef]
- Asuthkar, S.; Velpula, K.K.; Chetty, C.; Gorantla, B.; Rao, J.S. Epigenetic regulation of miRNA-211 by MMP-9 governs glioma cell apoptosis, chemosensitivity and radiosensitivity. Oncotarget 2012, 3, 1439–1454. [Google Scholar] [CrossRef]
- Tubiana, M.; Aurengo, A.; Averbeck, D.; Masse, R. The debate on the use of linear no threshold for assessing the effects of low doses. J. Radiol. Prot. 2006, 26, 317–324. [Google Scholar] [CrossRef]
- Feinendegen, L.E.; Cuttler, J.M. Biological effects from low doses and dose rates of ionizing radiation: Science in the service of protecting humans, a synopsis. Health Phys. 2018, 114, 623–626. [Google Scholar] [CrossRef]
- Reda, M.; Bagley, A.F.; Zaidan, H.Y.; Yantasee, W. Augmenting the therapeutic window of radiotherapy: A perspective on molecularly targeted therapies and nanomaterials. Radiother. Oncol. 2020, 150, 225–235. [Google Scholar] [CrossRef]
- Tharmalingam, S.; Sreetharan, S.; Brooks, A.L.; Boreham, D.R. Re-evaluation of the linear no-threshold (LNT) model using new paradigms and modern molecular studies. Chem. Biol. Interact. 2019, 301, 54–67. [Google Scholar] [CrossRef]
- Ebner, D.K.; Kamada, T. The emerging role of carbon-ion radiotherapy. Front. Oncol. 2016, 6, 140. [Google Scholar] [CrossRef]
- Iuchi, T.; Dai, M.; Sanada, H.; Okuwa, M.; Nakatani, T.; Sugama, J. Associations between the treatments and outcomes of patients with upper and lower lymphoedema in Japan: A cross-sectional observational study. Int. J. Nurs. Stud. 2015, 52, 913–919. [Google Scholar] [CrossRef]
- Schulz-Ertner, D.; Tsujii, H. Particle radiation therapy using proton and heavier ion beams. J. Clin. Oncol. 2007, 25, 953–964. [Google Scholar] [CrossRef]
- Lu, V.M.; Welby, J.P.; Laack, N.N.; Mahajan, A.; Daniels, D.J. Pseudoprogression after radiation therapies for low grade glioma in children and adults: A systematic review and meta-analysis. Radiother. Oncol. 2020, 142, 36–42. [Google Scholar] [CrossRef]
- Hayashi, K.; Yamamoto, N.; Nakajima, M.; Nomoto, A.; Tsuji, H.; Ogawa, K.; Kamada, T. Clinical outcomes of carbon-ion radiotherapy for locally advanced non-small-cell lung cancer. Cancer Sci. 2019, 110, 734–741. [Google Scholar] [CrossRef]
- Anzai, M.; Yamamoto, N.; Hayashi, K.; Nakajima, M.; Nomoto, A.; Ogawa, K.; Tsuji, H. Safety and efficacy of carbon-ion radiotherapy alone for stage III non-small cell lung cancer. Anticancer Res. 2020, 40, 379–386. [Google Scholar] [CrossRef]
- Spychalski, P.; Kobiela, J.; Antoszewska, M.; Błażyńska-Spychalska, A.; Jereczek-Fossa, B.A.; Høyer, M. Patient specific outcomes of charged particle therapy for hepatocellular carcinoma—A systematic review and quantitative analysis. Radiother. Oncol. 2019, 132, 127–134. [Google Scholar] [CrossRef]
- Yamada, S.; Takiyama, H.; Isozaki, Y.; Shinoto, M.; Makishima, H.; Yamamoto, N.; Tsuji, H. Carbon-ion radiotherapy for colorectal cancer. J. Anus Rectum Colon. 2021, 5, 113–120. [Google Scholar] [CrossRef]
- Kawamura, H.; Kubo, N.; Sato, H.; Mizukami, T.; Katoh, H.; Ishikawa, H.; Ohno, T.; Matsui, H.; Ito, K.; Suzuki, K.; et al. Moderately hypofractionated carbon ion radiotherapy for prostate cancer; a prospective observational study “GUNMA0702”. BMC Cancer 2020, 20, 75. [Google Scholar] [CrossRef]
- Marcus, D.; Lieverse, R.I.Y.; Klein, C.; Abdollahi, A.; Lambin, P.; Dubois, L.J.; Yaromina, A. Charged particle and conventional radiotherapy: Current implications as partner for immunotherapy. Cancers 2021, 13, 1468. [Google Scholar] [CrossRef]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Cleary, J.M.; Aguirre, A.J.; Shapiro, G.I.; D’Andrea, A.D. Biomarker-guided development of DNA tepair inhibitors. Mol. Cell. 2020, 78, 1070–1085. [Google Scholar] [CrossRef]
- Keisari, Y.; Kelson, I. The potentiation of anti-tumor immunity by tumor abolition with alpha particles, protons, or carbon ion radiation and its enforcement by combination with immunoadjuvants or inhibitors of immune suppressor cells and checkpoint molecules. Cells 2021, 10, 228. [Google Scholar] [CrossRef]
- Procureur, A.; Simonaggio, A.; Bibault, J.E.; Oudard, S.; Vano, Y.A. Enhance the immune checkpoint inhibitors efficacy with radiotherapy induced immunogenic cell death: A comprehensive review and latest developments. Cancers 2021, 13, 678. [Google Scholar] [CrossRef]
- Sato, H.; Demaria, S.; Ohno, T. The role of radiotherapy in the age of immunotherapy. Jpn. J. Clin. Oncol. 2021, 51, 513–522. [Google Scholar] [CrossRef]
- Favaudon, V.; Fouillade, C.; Vozenin, M.C. Radiothérapie « flash » à très haut débit de dose: Un moyen d’augmenter l’indice thérapeutique par minimisation des dommages aux tissus sains? [Ultrahigh dose-rate, “flash” irradiation minimizes the side-effects of radiotherapy]. Cancer Radiother. 2015, 19, 526–531. [Google Scholar] [CrossRef]
- Bourhis, J.; Montay-Gruel, P.; Gonçalves Jorge, P.; Bailat, C.; Petit, B.; Ollivier, J.; Jeanneret-Sozzi, W.; Ozsahin, M.; Bochud, F.; Moeckli, R.; et al. Clinical translation of FLASH radiotherapy: Why and how? Radiother. Oncol. 2019, 139, 11–17. [Google Scholar] [CrossRef]
- De Kruijff, R.M. FLASH radiotherapy: Ultra-high dose rates to spare healthy tissue. Int. J. Radiat. Biol. 2020, 96, 419–423. [Google Scholar] [CrossRef]
- Zakaria, A.M.; Colangel, N.W.; Meesungnoen, J.; Azzam, E.I.; Plourde, M.É.; Jay-Gerin, J.P. Ultra-high dose-rate, pulsed (FLASH) radiotherapy with carbon Ions: Generation of early, transient, highly oxygenated conditions in the tumor environment. Radiat. Res. 2020, 194, 587–593. [Google Scholar] [CrossRef]
- Colangelo, N.W.; Azzam, E.I. The importance and clinical implications of FLASH ultra-high dose-rate studies for proton and heavy ion radiotherapy. Radiat. Res. 2020, 193, 1–4. [Google Scholar] [CrossRef]
- Mazal, A.; Prezado, Y.; Ares, C.; de Marzi, L.; Patriarca, A.; Miralbell, R.; Favaudon, V. FLASH and minibeams in radiation therapy: The effect of microstructures on time and space and their potential application to protontherapy. Br. J. Radiol. 2020, 93, 20190807. [Google Scholar] [CrossRef]





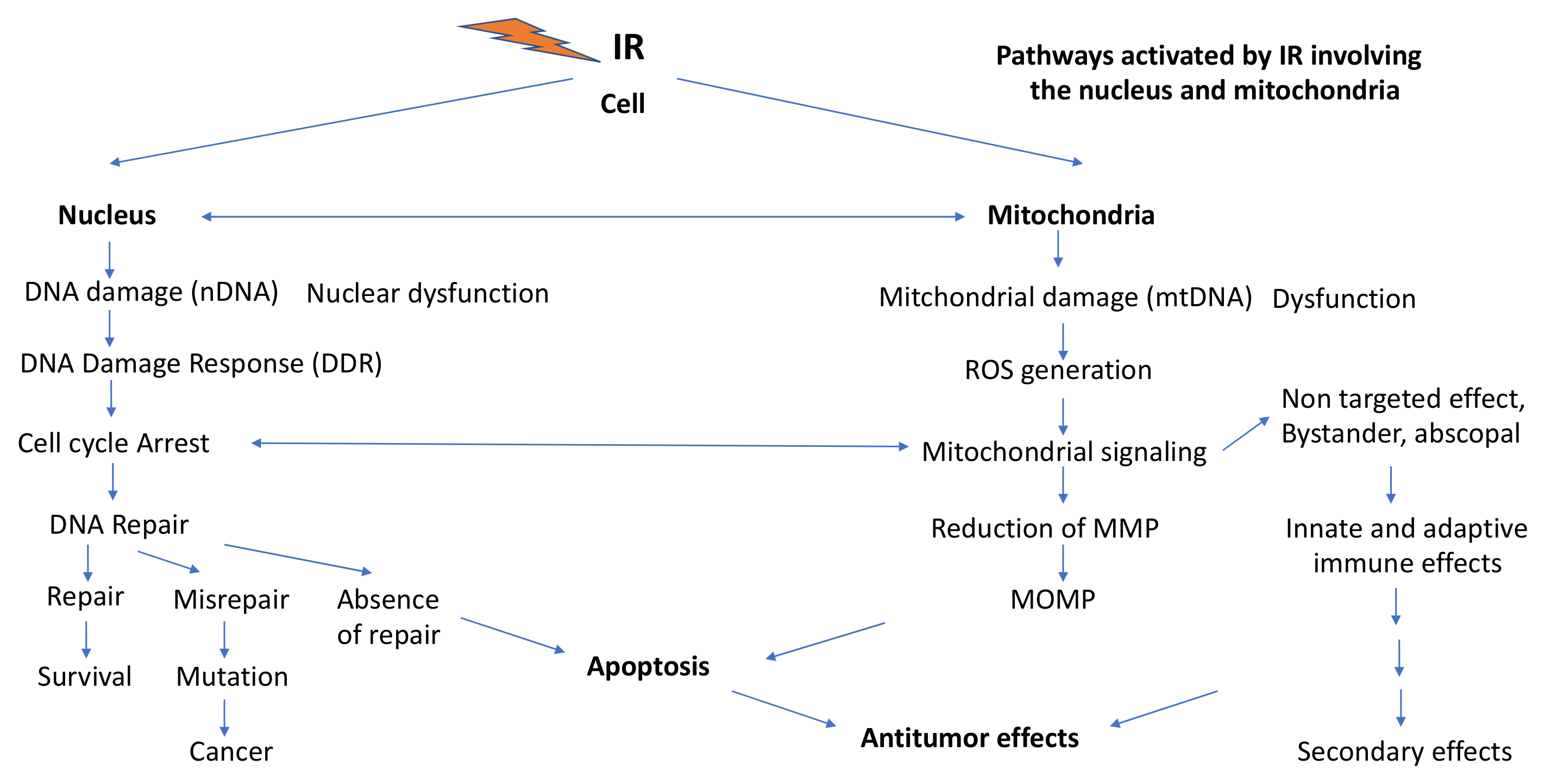{kind=link}
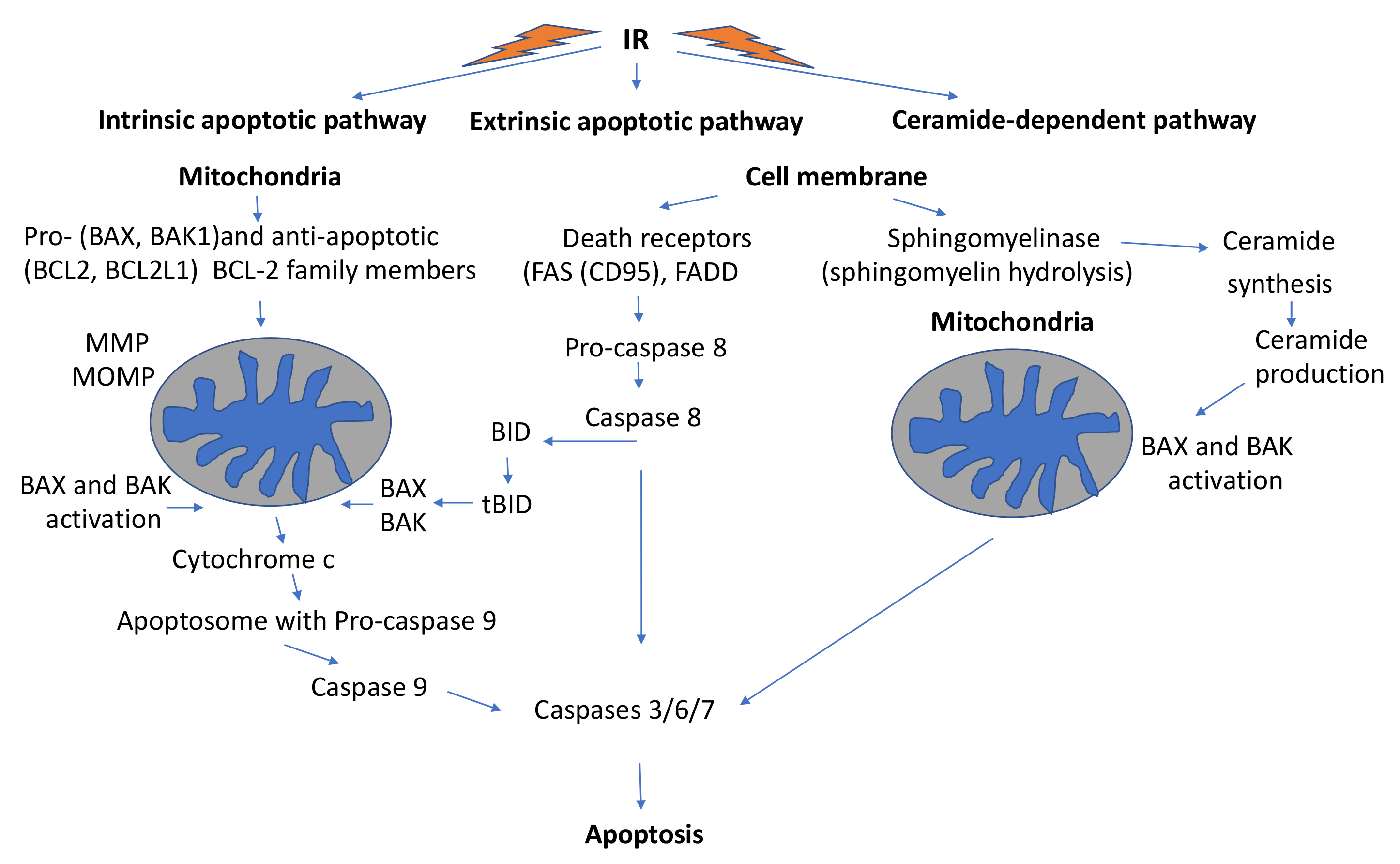{kind=link}
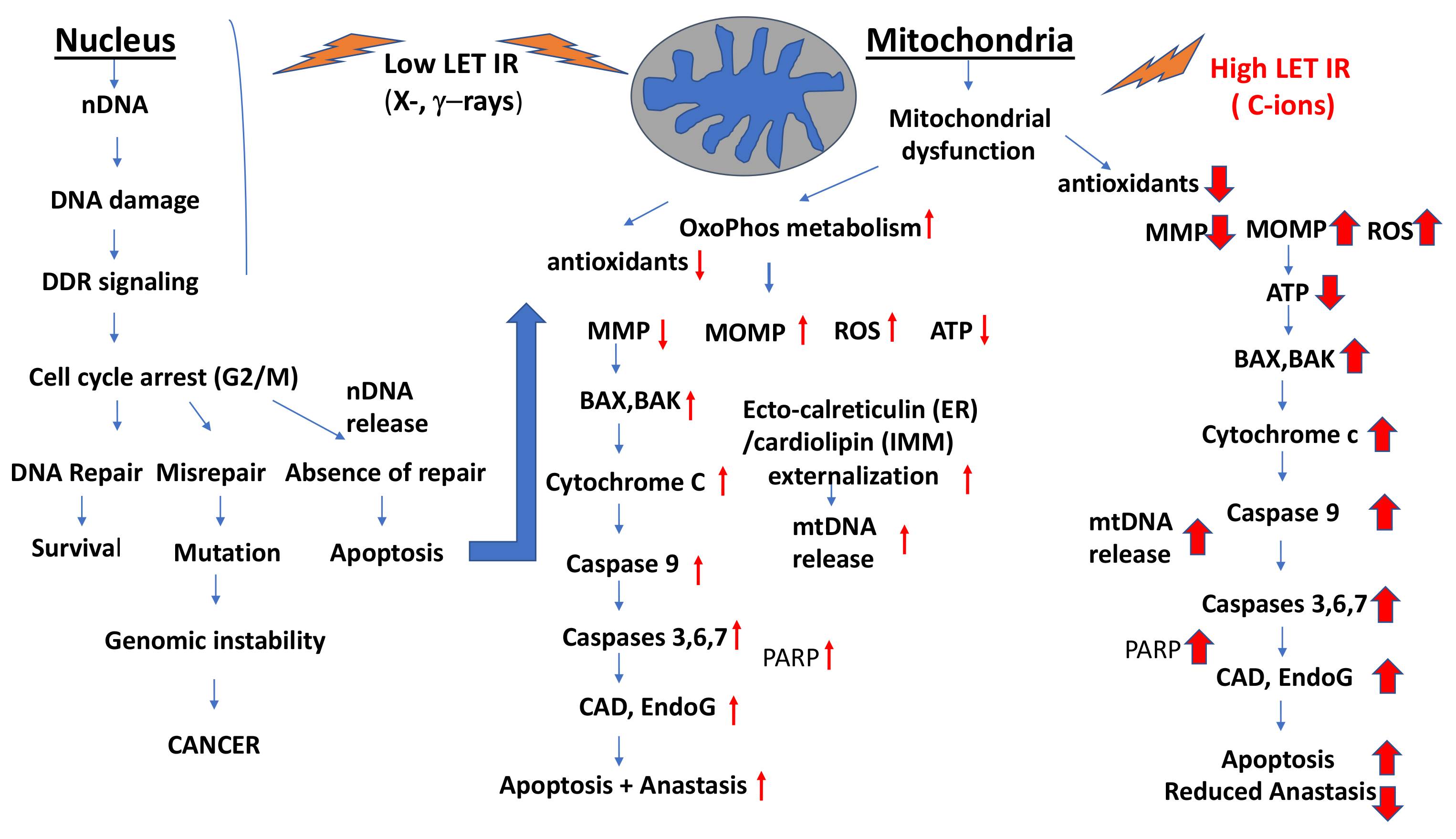{kind=link}
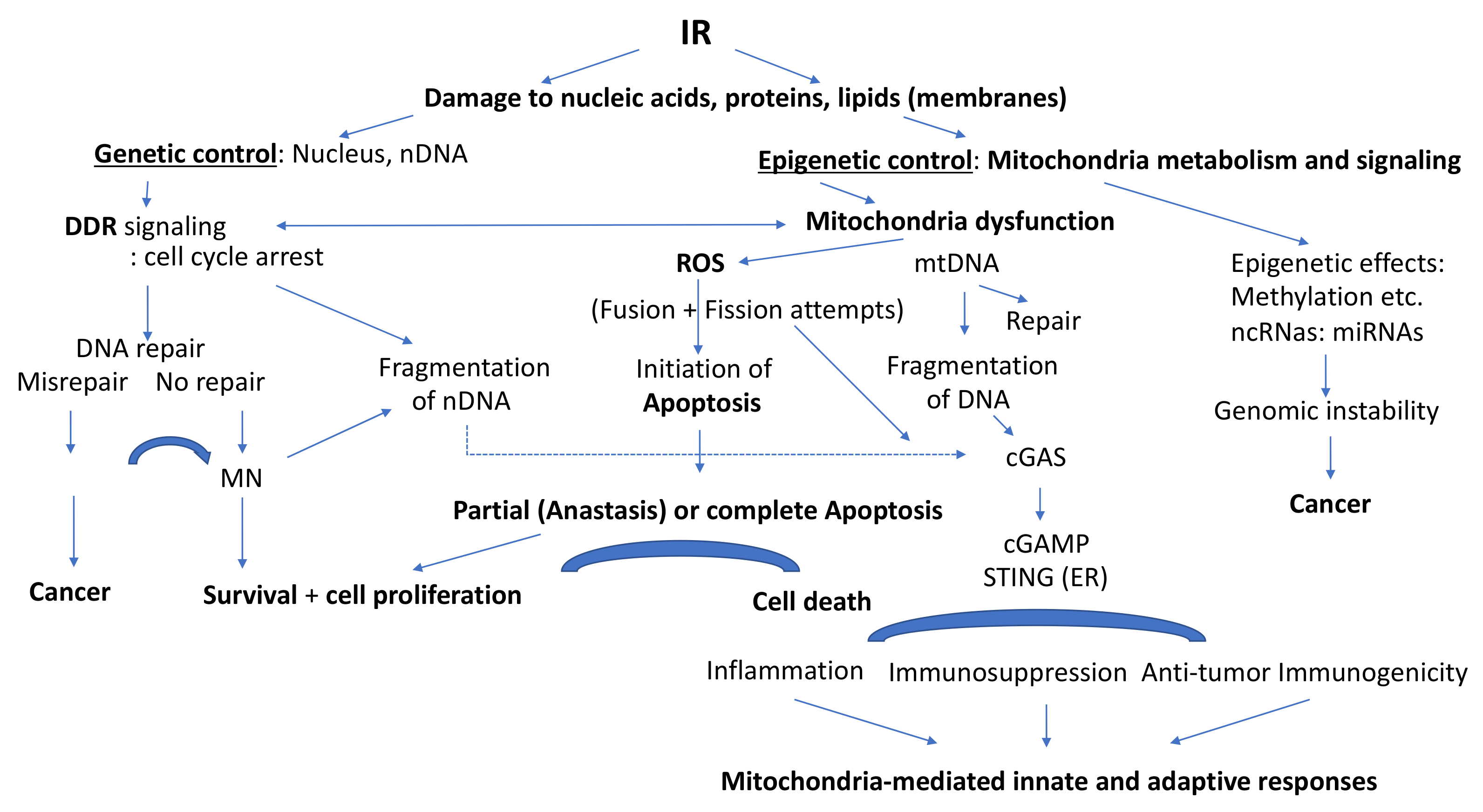{kind=link}
| Radiation | Cell System | Apoptosis | References |
|---|---|---|---|
| γ-rays/C-ions (105 keV/μm) | Glioblastoma cells (p53 wt/p53mutant) | C-ions are more effective than γ-rays. p53 mutants are more resistant than p53wt, higher G2/M block in p53mutants than in p53wt. | [353] |
| γ-rays/C-ions (20–80 keV/μm) Dose: 10 Gy | Glioblastoma cells (U87MG, p53 wt) and normal human fibroblasts (FB) (NB1RGB) | High induction of apoptosis by C-ions in normal and especially in glioblastoma cells. Fibroblasts show less apoptosis than glioblastoma cells. | [361] |
| X-rays/C-ions (13–100 keV/μm)/56Fe ions (200 keV/μm) | Human gingival cells (Ca9-99) | C-ions and 56Fe ions were more effective than X-rays in inducing apoptosis. | [360] |
| X-rays/C-ions (33.4 and 184 keV/μm) | Head and neck squamous cell carcinoma cells (HNSCC): SCC61 (radiosensitive) and SQ20B(radioresistant) | Induction of early (p53-independent) apoptosis: higher effectiveness of C-ions than X-rays. | [363] |
| γ-rays/C-ions (290 keV/μm) | Lung adenocarcinoma A549 cells | Early and more effective apoptosis induction by C-ions compared to γ-rays. C-ions induced predominantly pro-apoptotic signals: JNK transiently, but not ERK1, although ERK1 was activated by γ-rays (1 Gy). | [285] |
| X-rays/56Fe ions (200 keV/μm) | Human gingival cancer cells (Ca9-22) | 56Fe ions were about 8 times more effective than X-rays in apoptosis induction | [364] |
| Photons/C-ions (103 keV/μm) | Human glioblastoma (U87) | Higher efficacy of C-ions than photons (RBE: 3.3–3.9) | [365] |
| X-rays/C-ions (33.6 and 184 keV/μm) Dose: 10Gy | Head and neck squamous cell carcinoma cells (HNSCC): SCC61 (radiosensitive) and SQ20B (radioresistant) | Late apoptosis > 120 h post-irradiation was independent of p53 status, higher after C-ions than after X-rays, involving ceramide synthesis | [322] |
| X-rays/C-ions (keV103/μm) | Human cancer cells (A549, LN-229, PANC-1) | C-ions much more effective than photons for induction of apoptosis | [366] |
| X-rays/C-ions (33.6 keV/μm) | Human glioblastoma cell lines | Ceramide-dependent late apoptosis, higher after C-ions than after X-rays | [356] |
| X-rays/C-ions | Prostate cancer cells (PC3) | Induction of a persistent G2/M cell cycle arrest with C-ions | [291] |
| Low-LET photons/12C-ions/16O-ions | Hepatocarcinoma cell lines | Increased growth arrest (RBE: 1.9–3.3) | [293] |
| Low-LET photons/C-ions | Mice: malignant melanoma cells BF16F10 and tumors | C-ions very effectively increased apoptosis and inhibited cell proliferation. In vivo, tumor growth was inhibited by C-ions to 95% and by X-rays to 37.5%. | [367] |
| 50 kVp X-rays/C-ions (75 keV/μm) Dose: 2 Gy | Mouse sarcoma 180 cells | C-ions were significantly more effective than X-rays in inducing intrinsic apoptosis in vitro and in vivo. | [368] |
| C-ions (75keV/μm) Doses: 0.5 and 3 Gy | Human breast cancer cells (MDA-MB-231) | Considerable increase in LC3 expression (autophagy), cleaved caspase-3 expression and strong apoptosis (strong cell killing) | [346] |
| X-rays/C-ions (70 keV/μm) | Human cervical cancer cells (HeLa) | Significantly increased cytochrome c release and apoptosis at 3 Gy; both could be inhibited by an inhibitor of cytochrome c release (Mivi-1) | [369] |
| X-rays/12C-ions (101 keV/μm), 16O -ions (141 keV/μm) | Human malignant melanoma cell lines WM115, WM266-4 (metastatic) | Photon RT (2–6 Gy) resulted in G2/M arrests comparable to 0.5–2 Gy with 12C-and 16O-ions. Heavy ions were up to 4 times more effective than RT photons. | [362] |
| Radiation | Cell System | Autophagy/Mitophagy | Reference |
|---|---|---|---|
| IR | Mammalian cells | Induction of auto- phagy | [9] |
| Low-LET IR | Mammalian cells | Regulation of autophagy by mTOR/complex Akt/PI3K can be blocked by IR | [296] |
| Low-LET IR | Mammalian cells | The DDR proteins p53, ATM, PARP1, FOXO3a, mTOR and SIRT1 are involved in the regulation of autophagy. | [324] |
| Low-LET IR | Mammalian cells | mTORC1 activity is induced by low levels of ROS, inhibited by higher ROS levels and mitochondria mass reduced via mitophagy | [41] |
| C-ions (75 keV/μm) Dose: 2 Gy | Sarcoma cell line S180 | High ER stress with subsequent IRE1 signaling, autophagy (via IRE1/INK/p-Bcl-2/Beclin-1 signaling axis) and apoptosis. Apoptosis could be further increased with chloroquine 10 μg 4 h treatment | [368] |
| X-rays/C-ions (75 keV/μm) | Human cervical cancer cells (HeLa) | Mitochondrial fusion occurred at low dose X-rays (0.5 Gy) and ERK1/2 -mediated phosphorylation of DRP1 at higher doses (> 1 Gy Gy). C-ions induce effective mitochondria fragmentation, fusion (increased DRP and MFN expression) and apoptosis | [369] |
| C-ions (75 keV/μm) | Human breast cancer cells (MCF-7, MDA-MB-231) | At low dose (0.5 Gy) of C-ions damaged mitochondria were eliminated by mitophagy. High-dose 3 Gy exposure resulted in high BAX expression, cytochrome release and apoptosis. Autophagy inhibition led to mitophagy and increased survival. | [346] |
| Protons (microbeam) | Normal human lung fibroblast WI-38 cells (Cytoplasmic irradiation) | Mitochondrial fragmentation, induction of ROS and DSBs, localization of NRF to the nucleus, down-regulation of p53. | [380] |
| C-ions 300 MeV/u Dose: 4 Gy | Mouse hippocampus | Changes in LC3II/LC3I ratio, co-localization of LC3 with COX IV (indicative for the presence of mitophagosomes), PINK1 and Parkin levels were markedly reduced;overexpression of NRF2 and PINK1 in mouse hippocampus HT122 cells and apoptosis attenuated. | [379] |
| Low-LET IR (γ-rays) | Human normal fetal lung fibroblasts | Mitophagy involves the Spata 18 pathway and accumulation of lysosome-like organelles in mitochondria | [342] |
| Proton microbeam | Human lung fibroblasts (WI-38) | Increased ROS, DSB induction, Nrf2 activation, mitochondria fragmentation and down-regulation of p53 | [380] |
| X-rays (10 mGy, 50 mGy fractions, total doses 460 mGy, 2.3 Gy): low dose long-term exposures (31 days) | Human fibroblasts, ATM (+/+), ATM-defective (AT5BIVA), NBS1+/+, NBS -defective (GM7166) | Low-dose long-term X-irradiation increased mitochondrial mass and ROS in ATM (+/+) and NBS(+/+) cells leading to mitophagy involving Parkin1. In ATM and NBS-defective cells defective mitophagy was induced leading to mitochondrial fragmentation, decreased mitochondrial membrane potential and apoptosis. | [345] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Averbeck, D.; Rodriguez-Lafrasse, C. Role of Mitochondria in Radiation Responses: Epigenetic, Metabolic, and Signaling Impacts. Int. J. Mol. Sci. 2021, 22, 11047. https://doi.org/10.3390/ijms222011047
Averbeck D, Rodriguez-Lafrasse C. Role of Mitochondria in Radiation Responses: Epigenetic, Metabolic, and Signaling Impacts. International Journal of Molecular Sciences. 2021; 22(20):11047. https://doi.org/10.3390/ijms222011047
Chicago/Turabian StyleAverbeck, Dietrich, and Claire Rodriguez-Lafrasse. 2021. "Role of Mitochondria in Radiation Responses: Epigenetic, Metabolic, and Signaling Impacts" International Journal of Molecular Sciences 22, no. 20: 11047. https://doi.org/10.3390/ijms222011047
APA StyleAverbeck, D., & Rodriguez-Lafrasse, C. (2021). Role of Mitochondria in Radiation Responses: Epigenetic, Metabolic, and Signaling Impacts. International Journal of Molecular Sciences, 22(20), 11047. https://doi.org/10.3390/ijms222011047