
SH3-Binding Glutamic Acid Rich-Deficiency Augments Apoptosis in Neonatal Rat Cardiomyocytes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. SH3BGR Is Confined to Striated Muscle and Upregulated in Cardiac Hypertrophy
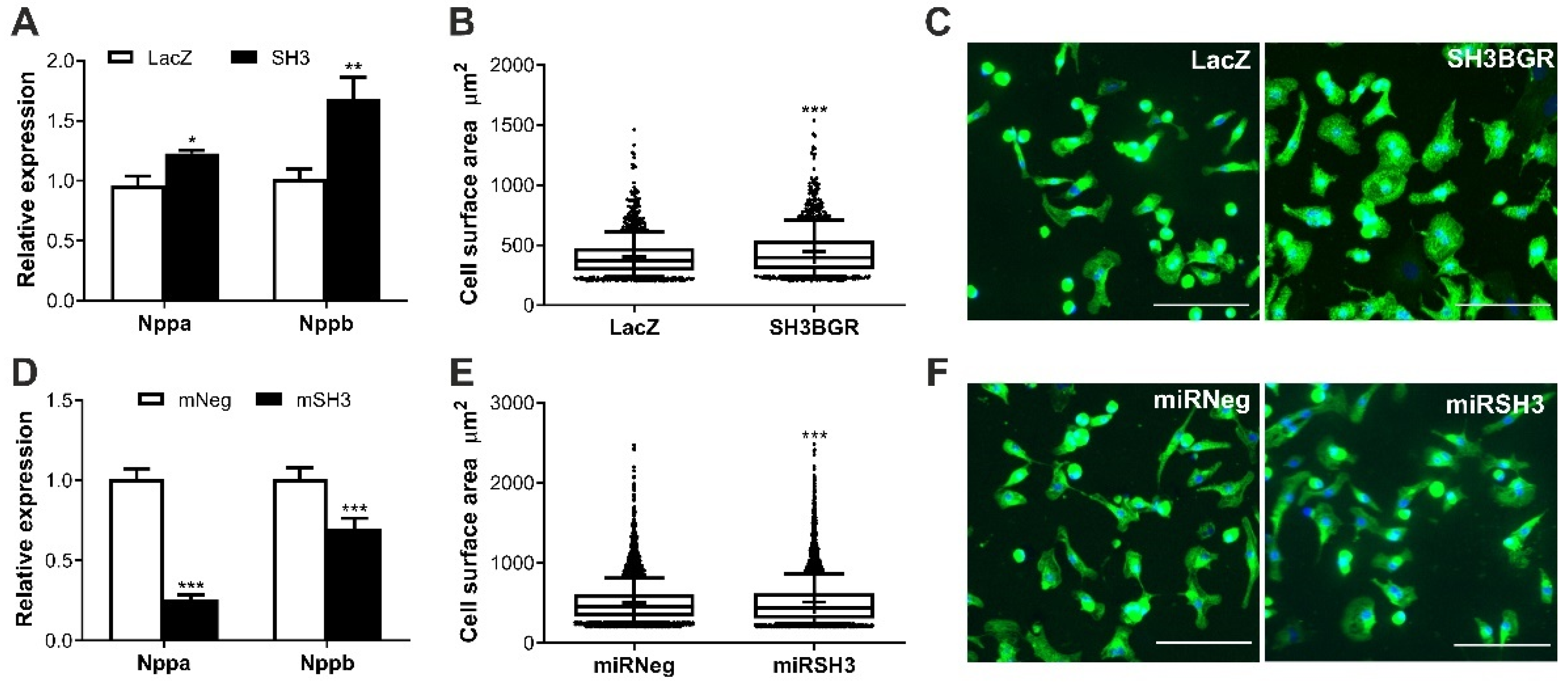
2.2. SH3BGR Induces Cellular Hypertrophy in NRVCMs
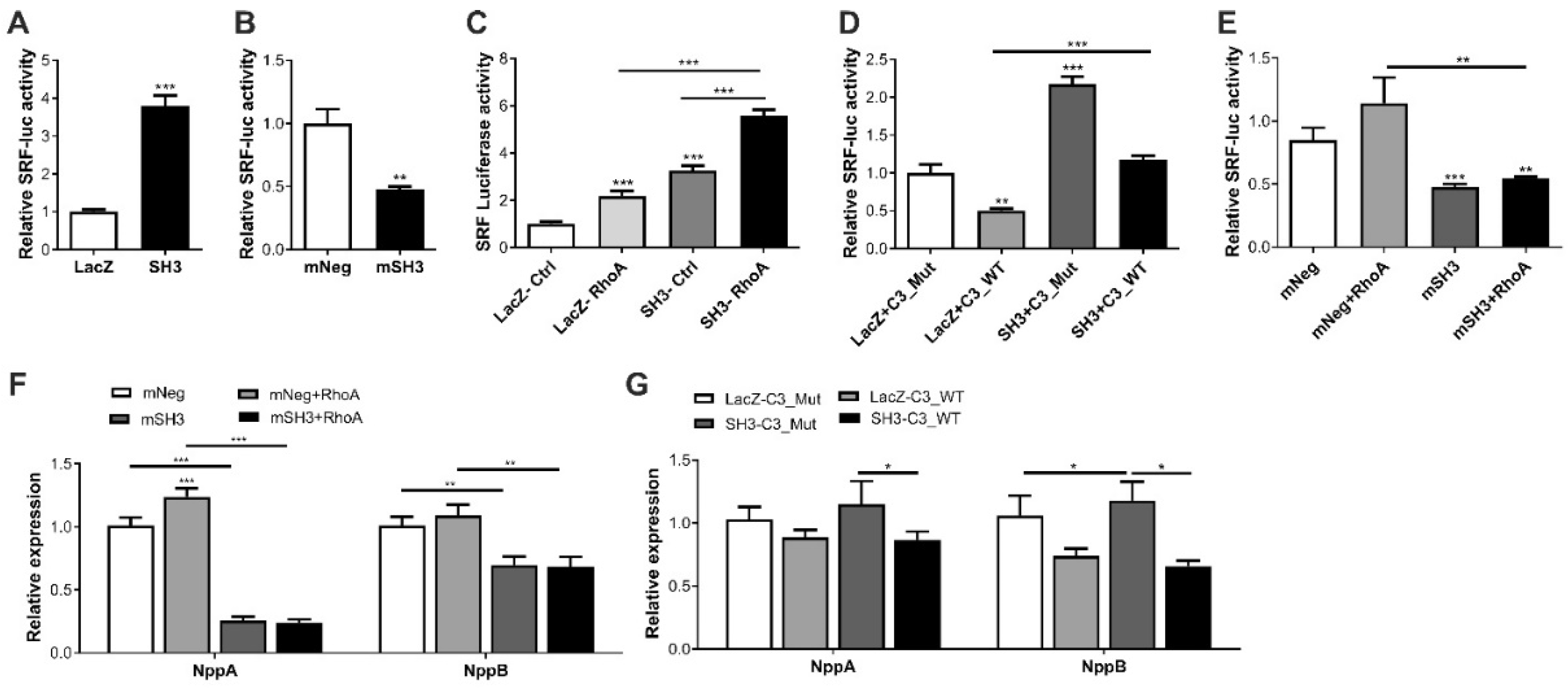
2.3. SH3BGR Regulates RhoA–SRF Signaling in NRVCMs
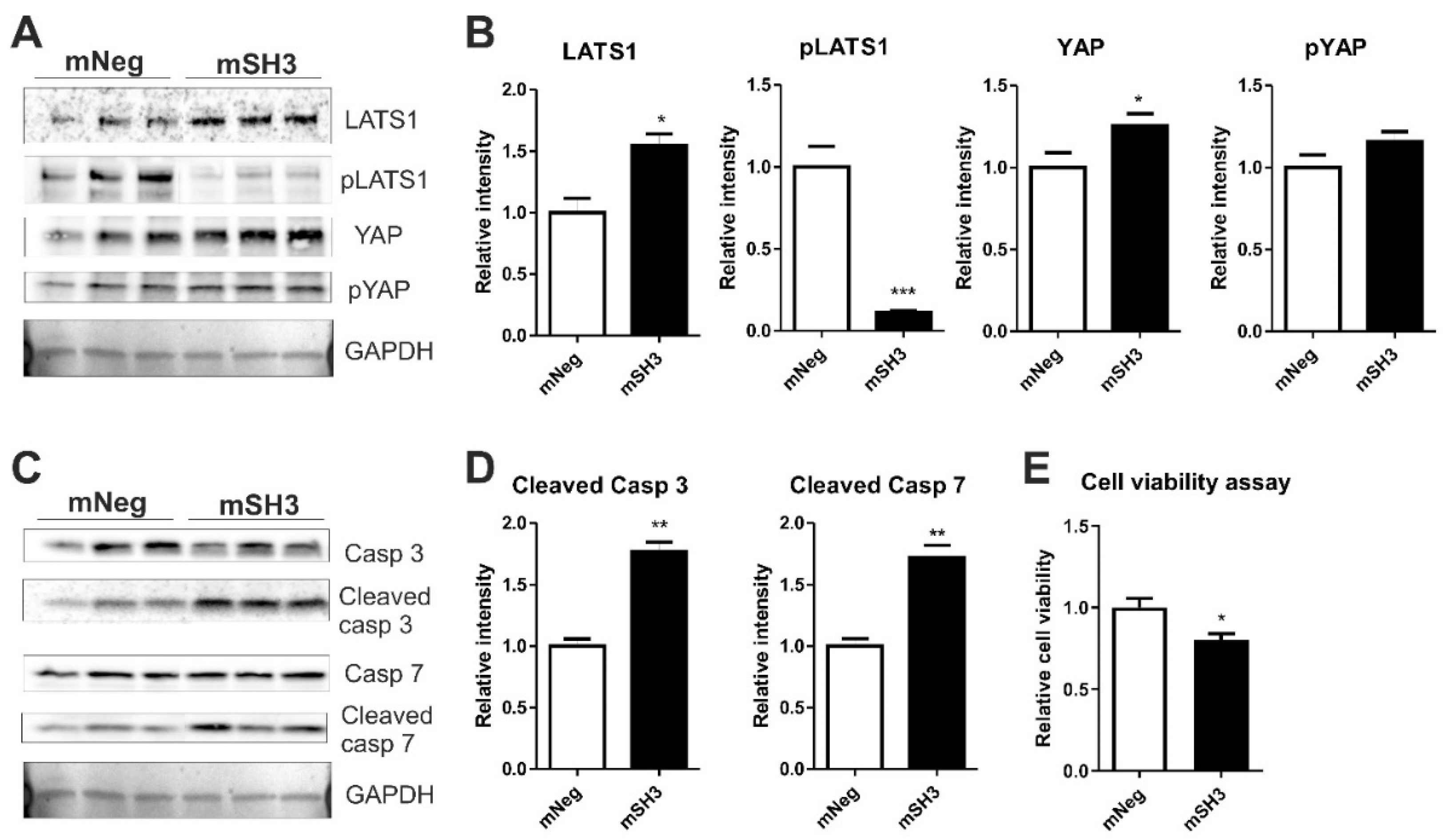
2.4. SH3BGR Knockdown Affects NRVCM-Viability and Induces Apoptosis via HIPPO Signaling
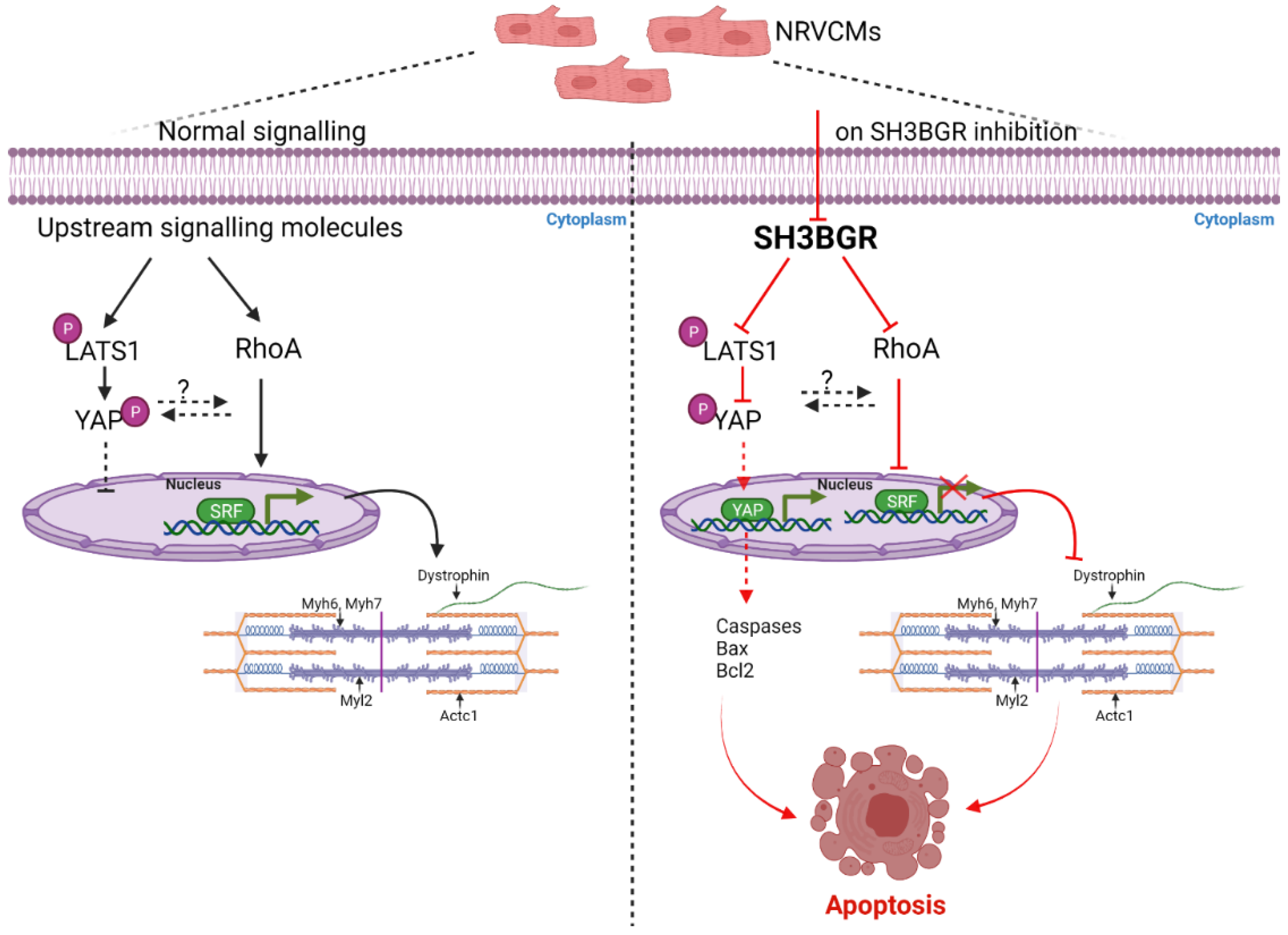
3. Discussion
4. Materials and Methods
4.1. Cloning of SH3BGR Vectors
4.2. Antibodies
4.3. Isolation of NRVCMs
4.4. Co-Localization Analysis of SH3BGR with α-Actinin
4.5. Immunofluorescence Microscopy for Cell Size Measurement
4.6. MTT Assay for Cell Viability
4.7. RNA Isolation and qRT-PCR
4.8. Protein Preparation and Immunoblotting
4.9. Human Heart Samples
4.10. SRF Luciferase Assay
4.11. RhoA Inhibitor Usage
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CHD | Congenital heart disease |
| DS | Down’s syndrome |
| SH3BGR | SH3-binding glutamic acid rich |
| Nppa | Natriuretic peptide A |
| Nppb | Natriuretic peptide B |
| NRVCM | Neonatal rat ventricular cardiomyocytes |
| RhoA | Ras Homolog family member A |
| SRF | Serum Response Factor |
| YAP | Yes associated protein |
References
- Crawford, D.; Dearmun, A. Down’s syndrome. Nurs. Child. Young People 2016, 28, 17. [Google Scholar] [CrossRef]
- Antonarakis, S.E.; Skotko, B.G.; Rafii, M.S.; Strydom, A.; Pape, S.E.; Bianchi, D.W.; Sherman, S.L.; Reeves, R.H. Down syndrome. Nat. Rev. Dis. Primers 2020, 6, 9. [Google Scholar] [CrossRef] [PubMed]
- Benhaourech, S.; Drighil, A.; Hammiri, A.E. Congenital heart disease and Down syndrome: Various aspects of a confirmed association. Cardiovasc. J. Afr. 2016, 27, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Asim, A.; Kumar, A.; Muthuswamy, S.; Jain, S.; Agarwal, S. Down syndrome: An insight of the disease. J. Biomed. Sci. 2015, 22, 41. [Google Scholar] [CrossRef] [PubMed]
- Shore, P.; Sharrocks, A.D. The MADS-box family of transcription factors. Eur. J. Biochem. 1995, 229, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Miano, J.M. Role of serum response factor in the pathogenesis of disease. Lab. Investig. A J. Tech. Methods Pathol. 2010, 90, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Nishikimi, T.; Kuwahara, K.; Nakao, K. Current biochemistry, molecular biology, and clinical relevance of natriuretic peptides. J. Cardiol. 2011, 57, 131–140. [Google Scholar] [CrossRef]
- Rangrez, A.Y.; Bernt, A.; Poyanmehr, R.; Harazin, V.; Boomgaarden, I.; Kuhn, C.; Rohrbeck, A.; Frank, D.; Frey, N. Dysbindin is a potent inducer of RhoA-SRF-mediated cardiomyocyte hypertrophy. J. Cell Biol. 2013, 203, 643–656. [Google Scholar] [CrossRef]
- Foster, C.T.; Gualdrini, F.; Treisman, R. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. 2017, 31, 2361–2375. [Google Scholar] [CrossRef]
- Xiao, Y.; Hill, M.C.; Zhang, M.; Martin, T.J.; Morikawa, Y.; Wang, S.; Moise, A.R.; Wythe, J.D.; Martin, J.F. Hippo Signaling Plays an Essential Role in Cell State Transitions during Cardiac Fibroblast Development. Dev. Cell 2018, 45, 153–169.e6. [Google Scholar] [CrossRef]
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 672–684. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, L.; Zhao, B.; Guan, K.L. The hippo pathway in heart development, regeneration, and diseases. Circ. Res. 2015, 116, 1431–1447. [Google Scholar] [CrossRef]
- Del Re, D.P.; Matsuda, T.; Zhai, P.; Maejima, Y.; Jain, M.R.; Liu, T.; Li, H.; Hsu, C.P.; Sadoshima, J. Mst1 promotes cardiac myocyte apoptosis through phosphorylation and inhibition of Bcl-xL. Mol. Cell 2014, 54, 639–650. [Google Scholar] [CrossRef]
- Matsuda, T.; Zhai, P.; Sciarretta, S.; Zhang, Y.; Jeong, J.I.; Ikeda, S.; Park, J.; Hsu, C.P.; Tian, B.; Pan, D.; et al. NF2 Activates Hippo Signaling and Promotes Ischemia/Reperfusion Injury in the Heart. Circ. Res. 2016, 119, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Sadoshima, J. Regulation of Myocardial Cell Growth and Death by the Hippo Pathway. Circ. J. Off. J. Jpn. Circ. Soc. 2016, 80, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Del Re, D.P.; Yang, Y.; Nakano, N.; Cho, J.; Zhai, P.; Yamamoto, T.; Zhang, N.; Yabuta, N.; Nojima, H.; Pan, D.; et al. Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte survival and growth to protect against myocardial ischemic injury. J. Biol. Chem. 2013, 288, 3977–3988. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Zhai, P.; Del Re, D.P.; Sciarretta, S.; Yabuta, N.; Nojima, H.; Lim, D.S.; Pan, D.; Sadoshima, J. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat. Commun. 2014, 5, 3315. [Google Scholar] [CrossRef]
- Eden, M.; Meder, B.; Volkers, M.; Poomvanicha, M.; Domes, K.; Branchereau, M.; Marck, P.; Will, R.; Bernt, A.; Rangrez, A.; et al. Myoscape controls cardiac calcium cycling and contractility via regulation of L-type calcium channel surface expression. Nat. Commun. 2016, 7, 11317. [Google Scholar] [CrossRef]
- Rangrez, A.Y.; Eden, M.; Poyanmehr, R.; Kuhn, C.; Stiebeling, K.; Dierck, F.; Bernt, A.; Lullmann-Rauch, R.; Weiler, H.; Kirchof, P.; et al. Myozap Deficiency Promotes Adverse Cardiac Remodeling via Differential Regulation of Mitogen-activated Protein Kinase/Serum-response Factor and beta-Catenin/GSK-3beta Protein Signaling. J. Biol. Chem. 2016, 291, 4128–4143. [Google Scholar] [CrossRef]
- Frank, D.; Rangrez, A.Y.; Poyanmehr, R.; Seeger, T.S.; Kuhn, C.; Eden, M.; Stiebeling, K.; Bernt, A.; Grund, C.; Franke, W.W.; et al. Mice with cardiac-restricted overexpression of Myozap are sensitized to biomechanical stress and develop a protein-aggregate-associated cardiomyopathy. J. Mol. Cell. Cardiol. 2014, 72, 196–207. [Google Scholar] [CrossRef]
- Seeger, T.S.; Frank, D.; Rohr, C.; Will, R.; Just, S.; Grund, C.; Lyon, R.; Luedde, M.; Koegl, M.; Sheikh, F.; et al. Myozap, a novel intercalated disc protein, activates serum response factor-dependent signaling and is required to maintain cardiac function in vivo. Circ. Res. 2010, 106, 880–890. [Google Scholar] [CrossRef]
- Dierck, F.; Kuhn, C.; Rohr, C.; Hille, S.; Braune, J.; Sossalla, S.; Molt, S.; van der Ven, P.F.M.; Furst, D.O.; Frey, N. The novel cardiac z-disc protein CEFIP regulates cardiomyocyte hypertrophy by modulating calcineurin signaling. J. Biol. Chem. 2017, 292, 15180–15191. [Google Scholar] [CrossRef]
- Scartezzini, P.; Egeo, A.; Colella, S.; Fumagalli, P.; Arrigo, P.; Nizetic, D.; Taramelli, R.; Rasore-Quartino, A. Cloning a new human gene from chromosome 21q22.3 encoding a glutamic acid-rich protein expressed in heart and skeletal muscle. Hum. Genet. 1997, 99, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Egeo, A.; Di Lisi, R.; Sandri, C.; Mazzocco, M.; Lapide, M.; Schiaffino, S.; Scartezzini, P. Developmental expression of the SH3BGR gene, mapping to the Down syndrome heart critical region. Mech. Dev. 2000, 90, 313–316. [Google Scholar] [CrossRef]
- Sandri, C.; Di Lisi, R.; Picard, A.; Argentini, C.; Calabria, E.; Myklak, K.; Scartezzini, P.; Schiaffino, S. Heart morphogenesis is not affected by overexpression of the Sh3bgr gene mapping to the Down syndrome heart critical region. Hum. Genet. 2004, 114, 517–519. [Google Scholar] [CrossRef]
- Mazzocco, M.; Maffei, M.; Egeo, A.; Vergano, A.; Arrigo, P.; Di Lisi, R.; Ghiotto, F.; Scartezzini, P. The identification of a novel human homologue of the SH3 binding glutamic acid-rich (SH3BGR) gene establishes a new family of highly conserved small proteins related to Thioredoxin Superfamily. Gene 2002, 291, 233–239. [Google Scholar] [CrossRef]
- Guo, Y.; Cao, Y.; Jardin, B.D.; Sethi, I.; Ma, Q.; Moghadaszadeh, B.; Troiano, E.C.; Mazumdar, N.; Trembley, M.A.; Small, E.M.; et al. Sarcomeres regulate murine cardiomyocyte maturation through MRTF-SRF signaling. Proc. Natl. Acad. Sci. USA 2021, 118, e2008861118. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016, 2016, baw100. [Google Scholar] [CrossRef] [PubMed]
- Kilian, L.S.; Voran, J.; Frank, D.; Rangrez, A.Y. RhoA: A dubious molecule in cardiac pathophysiology. J. Biomed. Sci. 2021, 28, 33. [Google Scholar] [CrossRef]
- De Kreuk, B.J.; Hordijk, P.L. Control of Rho GTPase function by BAR-domains. Small GTPases 2012, 3, 45–52. [Google Scholar] [CrossRef]
- Yin, L.; Li, W.; Xu, A.; Shi, H.; Wang, K.; Yang, H.; Wang, R.; Peng, B. SH3BGRL2 inhibits growth and metastasis in clear cell renal cell carcinoma via activating hippo/TEAD1-Twist1 pathway. EBioMedicine 2020, 51, 102596. [Google Scholar] [CrossRef]
- Vidal-Taboada, J.M.; Bergonon, S.; Scartezzini, P.; Egeo, A.; Nizetic, D.; Oliva, R. High-resolution physical map and identification of potentially regulatory sequences of the human SH3BGR located in the Down syndrome chromosomal region. Biochem. Biophys. Res. Commun. 1997, 241, 321–326. [Google Scholar] [CrossRef]
- Mazzocco, M.; Arrigo, P.; Egeo, A.; Maffei, M.; Vergano, A.; Di Lisi, R.; Ghiotto, F.; Ciccone, E.; Cinti, R.; Ravazzolo, R.; et al. A novel human homologue of the SH3BGR gene encodes a small protein similar to Glutaredoxin 1 of Escherichia coli. Biochem. Biophys. Res. Commun. 2001, 285, 540–545. [Google Scholar] [CrossRef]
- Egeo, A.; Mazzocco, M.; Arrigo, P.; Vidal-Taboada, J.M.; Oliva, R.; Pirola, B.; Giglio, S.; Rasore-Quartino, A.; Scartezzini, P. Identification and characterization of a new human gene encoding a small protein with high homology to the proline-rich region of the SH3BGR gene. Biochem. Biophys. Res. Commun. 1998, 247, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Huang, J.; Jiang, M.; Lin, H. Signal transducer and activator of transcription 2 (STAT2) metabolism coupling postmitotic outgrowth to visual and sound perception network in human left cerebrum by biocomputation. J. Mol. Neurosci. 2012, 47, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Majid, S.M.; Liss, A.S.; You, M.; Bose, H.R. The suppression of SH3BGRL is important for v-Rel-mediated transformation. Oncogene 2006, 25, 756–768. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Borlepawar, A.; Rangrez, A.Y.; Bernt, A.; Christen, L.; Sossalla, S.; Frank, D.; Frey, N. TRIM24 protein promotes and TRIM32 protein inhibits cardiomyocyte hypertrophy via regulation of dysbindin protein levels. J. Biol. Chem. 2017, 292, 10180–10196. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deshpande, A.; Borlepawar, A.; Rosskopf, A.; Frank, D.; Frey, N.; Rangrez, A.Y. SH3-Binding Glutamic Acid Rich-Deficiency Augments Apoptosis in Neonatal Rat Cardiomyocytes. Int. J. Mol. Sci. 2021, 22, 11042. https://doi.org/10.3390/ijms222011042
Deshpande A, Borlepawar A, Rosskopf A, Frank D, Frey N, Rangrez AY. SH3-Binding Glutamic Acid Rich-Deficiency Augments Apoptosis in Neonatal Rat Cardiomyocytes. International Journal of Molecular Sciences. 2021; 22(20):11042. https://doi.org/10.3390/ijms222011042
Chicago/Turabian StyleDeshpande, Anushka, Ankush Borlepawar, Alexandra Rosskopf, Derk Frank, Norbert Frey, and Ashraf Yusuf Rangrez. 2021. "SH3-Binding Glutamic Acid Rich-Deficiency Augments Apoptosis in Neonatal Rat Cardiomyocytes" International Journal of Molecular Sciences 22, no. 20: 11042. https://doi.org/10.3390/ijms222011042
APA StyleDeshpande, A., Borlepawar, A., Rosskopf, A., Frank, D., Frey, N., & Rangrez, A. Y. (2021). SH3-Binding Glutamic Acid Rich-Deficiency Augments Apoptosis in Neonatal Rat Cardiomyocytes. International Journal of Molecular Sciences, 22(20), 11042. https://doi.org/10.3390/ijms222011042