
Disrupted Calcium Homeostasis in Duchenne Muscular Dystrophy: A Common Mechanism behind Diverse Consequences




{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Genetics of Duchenne Muscular Dystrophy
1.2. Dystrophin Structure and Presumed Functions
2. Objectives of This Review
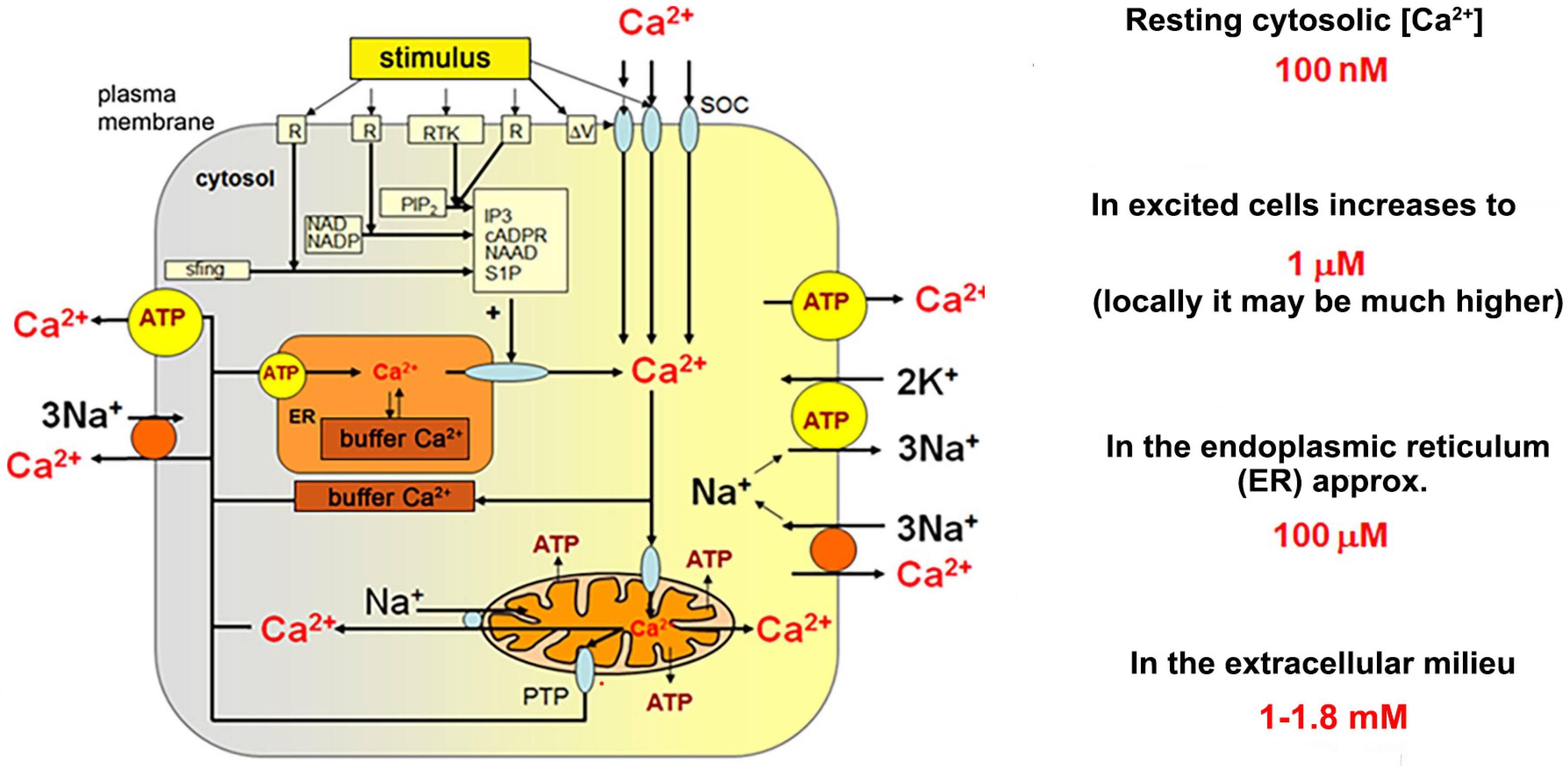
3. Mechanisms of Calcium Homeostasis
Calcium Signalling Toolkit
4. Abnormal Calcium Homeostasis in DMD
5. Calcium Dysregulation in DMD Skeletal Muscle
Dystrophin, the Scaffold
6. Calcium Abnormalities in DMD Myogenic and Non-Muscle Cells
The Loss of the Absent
7. Calcium Abnormalities in DMD Cardiac Muscle
Muscles That Get Away
8. Calcium Abnormalities in the Dystrophic CNS
9. Dystrophic Mitochondria and Altered Calcium Homeostasis
9.1. Cardiac Mitochondria
9.2. Skeletal Muscle Mitochondria
9.3. Structural Alterations
10. Impact of Therapies on Calcium Handling
11. Are There Calcium Abnormalities in BMD?
12. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AQP4 | Aquaporin isoform 4 |
| BMD | Becker muscular dystrophy |
| DMD | Duchenne muscular dystrophy |
| DAP | dystrophin-associated proteins complex |
| ER/SR | endoplasmic reticulum/sarcoplasmic reticulum |
| SOCE | store-operated calcium entry |
| NCX | Na+/Ca2+ exchanger |
| LTCC/DHPR | L-type calcium channel/dihydropiridine receptor |
| CICR | Calcium-induced calcium release |
| SERCA | sarco/endoplasmic calcium ATPase |
| PMCA | plasma membrane calcium ATPase |
| MCU | mitochondrial calcium uniporter |
| RyR | ryanodine receptor |
| MPTP | mitochondrial permeability transition pore |
References
- Toop, J.; Emery, A.E. Muscle histology in fetuses at risk for Duchenne muscular dystrophy. Clin. Genet. 1974, 5, 230–233. [Google Scholar] [CrossRef]
- Emery, A.E. Muscle histology and creatine kinase levels in the foetus in Duchenne muscular dystrophy. Nature 1977, 266, 472–473. [Google Scholar] [CrossRef]
- Vassilopoulos, D.; Emery, A.E. Muscle nuclear changes in fetuses at risk for Duchenne muscular dystrophy. J. Med. Genet. 1977, 14, 13–15. [Google Scholar] [CrossRef]
- Nguyen, F.; Cherel, Y.; Guigand, L.; Goubault-Leroux, I.; Wyers, M. Muscle lesions associated with dystrophin deficiency in neonatal golden retriever puppies. J. Comp. Pathol. 2002, 126, 100–108. [Google Scholar] [CrossRef]
- Bassett, D.I.; Bryson-Richardson, R.J.; Daggett, D.F.; Gautier, P.; Keenan, D.G.; Currie, P.D. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development 2003, 130, 5851–5860. [Google Scholar] [CrossRef] [PubMed]
- Merrick, D.; Stadler, L.K.; Larner, D.; Smith, J. Muscular dystrophy begins early in embryonic development deriving from stem cell loss and disrupted skeletal muscle formation. Dis. Models Mech. 2009, 2, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Mournetas, V.; Massouridès, E.; Dupont, J.B.; Kornobis, E.; Polvèche, H.; Jarrige, M.; Dorval, A.R.L.; Gosselin, M.R.F.; Manousopoulou, A.; Garbis, S.D.; et al. Myogenesis modelled by human pluripotent stem cells: A multi-omic study of Duchenne myopathy early onset. J. Cachexia Sarcopenia Muscle 2021, 12, 209–232. [Google Scholar] [CrossRef]
- Pescatori, M.; Broccolini, A.; Minetti, C.; Bertini, E.; Bruno, C.; D’Amico, A.; Bernardini, C.; Mirabella, M.; Silvestri, G.; Giglio, V.; et al. Gene expression profiling in the early phases of DMD: A constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007, 21, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- Van Dommelen, P.; van Dijk, O.; de Wilde, J.A.; Verkerk, P.H. Early developmental milestones in Duchenne muscular dystrophy. Dev. Med. Child Neurol. 2020, 62, 1198–1204. [Google Scholar] [CrossRef]
- Koeks, Z.; Bladen, C.L.; Salgado, D.; van Zwet, E.; Pogoryelova, O.; McMacken, G.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; et al. Clinical Outcomes in Duchenne Muscular Dystrophy: A Study of 5345 Patients from the TREAT-NMD DMD Global Database. J. Neuromuscul. Dis. 2017, 4, 293–306. [Google Scholar] [CrossRef]
- Crisafulli, S.; Sultana, J.; Fontana, A.; Salvo, F.; Messina, S.; Trifiro, G. Global epidemiology of Duchenne muscular dystrophy: An updated systematic review and meta-analysis. Orphanet. J. Rare Dis. 2020, 15, 141. [Google Scholar] [CrossRef] [PubMed]
- Juan-Mateu, J.; Gonzalez-Quereda, L.; Rodriguez, M.J.; Baena, M.; Verdura, E.; Nascimento, A.; Ortez, C.; Baiget, M.; Gallano, P. DMD Mutations in 576 Dystrophinopathy Families: A Step Forward in Genotype-Phenotype Correlations. PLoS ONE 2015, 10, e0135189. [Google Scholar] [CrossRef]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. 2003, 2, 731–740. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Desguerre, I.; Christov, C.; Mayer, M.; Zeller, R.; Becane, H.M.; Bastuji-Garin, S.; Leturcq, F.; Chiron, C.; Chelly, J.; Gherardi, R.K. Clinical heterogeneity of duchenne muscular dystrophy (DMD): Definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS ONE 2009, 4, e4347. [Google Scholar] [CrossRef]
- Masubuchi, N.; Shidoh, Y.; Kondo, S.; Takatoh, J.; Hanaoka, K. Subcellular localization of dystrophin isoforms in cardiomyocytes and phenotypic analysis of dystrophin-deficient mice reveal cardiac myopathy is predominantly caused by a deficiency in full-length dystrophin. Exp. Anim. 2013, 62, 211–217. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef]
- Taylor, P.J.; Betts, G.A.; Maroulis, S.; Gilissen, C.; Pedersen, R.L.; Mowat, D.R.; Johnston, H.M.; Buckley, M.F. Dystrophin gene mutation location and the risk of cognitive impairment in Duchenne muscular dystrophy. PLoS ONE 2010, 5, e8803. [Google Scholar] [CrossRef]
- Bello, L.; Pegoraro, E. The “Usual Suspects”: Genes for Inflammation, Fibrosis, Regeneration, and Muscle Strength Modify Duchenne Muscular Dystrophy. J. Clin. Med. 2019, 8, 649. [Google Scholar] [CrossRef]
- Zhao, J.; Kodippili, K.; Yue, Y.; Hakim, C.H.; Wasala, L.; Pan, X.; Zhang, K.; Yang, N.N.; Duan, D.; Lai, Y. Dystrophin contains multiple independent membrane-binding domains. Hum. Mol. Genet. 2016, 25, 3647–3653. [Google Scholar] [CrossRef] [PubMed]
- Dalkilic, I.; Kunkel, L.M. Muscular dystrophies: Genes to pathogenesis. Curr. Opin. Genet. Dev. 2003, 13, 231–238. [Google Scholar] [CrossRef]
- Gao, Q.Q.; McNally, E.M. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- Ghahramani Seno, M.M.; Graham, I.R.; Athanasopoulos, T.; Trollet, C.; Pohlschmidt, M.; Crompton, M.R.; Dickson, G. RNAi-mediated knockdown of dystrophin expression in adult mice does not lead to overt muscular dystrophy pathology. Hum. Mol. Genet. 2008, 17, 2622–2632. [Google Scholar] [CrossRef]
- Rader, E.P.; Turk, R.; Willer, T.; Beltrán, D.; Inamori, K.; Peterson, T.A.; Engle, J.; Prouty, S.; Matsumura, K.; Saito, F.; et al. Role of dystroglycan in limiting contraction-induced injury to the sarcomeric cytoskeleton of mature skeletal muscle. Proc. Natl. Acad. Sci. USA 2016, 113, 10992–10997. [Google Scholar] [CrossRef] [PubMed]
- Vieira, N.M.; Elvers, I.; Alexander, M.S.; Moreira, Y.B.; Eran, A.; Gomes, J.P.; Marshall, J.L.; Karlsson, E.K.; Verjovski-Almeida, S.; Lindblad-Toh, K.; et al. Jagged 1 Rescues the Duchenne Muscular Dystrophy Phenotype. Cell 2015, 163, 1204–1213. [Google Scholar] [CrossRef]
- Ferrari, D.; Munerati, M.; Melchiorri, L.; Hanau, S.; di Virgilio, F.; Baricordi, O.R. Responses to extracellular ATP of lymphoblastoid cell lines from Duchenne muscular dystrophy patients. Am. J. Physiol. 1994, 267, C886–C892. [Google Scholar] [CrossRef] [PubMed]
- Rog, J.; Oksiejuk, A.; Gosselin, M.R.F.; Brutkowski, W.; Dymkowska, D.; Nowak, N.; Robson, S.; Gorecki, D.C.; Zablocki, K. Dystrophic mdx mouse myoblasts exhibit elevated ATP/UTP-evoked metabotropic purinergic responses and alterations in calcium signalling. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1138–1151. [Google Scholar] [CrossRef]
- Yeung, D.; Zablocki, K.; Lien, C.F.; Jiang, T.; Arkle, S.; Brutkowski, W.; Brown, J.; Lochmuller, H.; Simon, J.; Barnard, E.A.; et al. Increased susceptibility to ATP via alteration of P2X receptor function in dystrophic mdx mouse muscle cells. FASEB J. 2006, 20, 610–620. [Google Scholar] [CrossRef]
- Onopiuk, M.; Brutkowski, W.; Young, C.; Krasowska, E.; Rog, J.; Ritso, M.; Wojciechowska, S.; Arkle, S.; Zablocki, K.; Gorecki, D.C. Store-operated calcium entry contributes to abnormal Ca2+ signalling in dystrophic mdx mouse myoblasts. Arch. Biochem. Biophys. 2015, 569, 1–9. [Google Scholar] [CrossRef]
- Blake, D.J.; Tinsley, J.M.; Davies, K.E. Utrophin: A structural and functional comparison to dystrophin. Brain Pathol. 1996, 6, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Huard, J.; Tremblay, J.P. Localization of dystrophin in the Purkinje cells of normal mice. Neurosci. Lett. 1992, 137, 105–108. [Google Scholar] [CrossRef]
- Jancsik, V.; Hajos, F. Differential distribution of dystrophin in postsynaptic densities of spine synapses. Neuroreport 1998, 9, 2249–2251. [Google Scholar] [CrossRef] [PubMed]
- Lidov, H.G.; Byers, T.J.; Watkins, S.C.; Kunkel, L.M. Localization of dystrophin to postsynaptic regions of central nervous system cortical neurons. Nature 1990, 348, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.; Seifi, M.; Górecki, D.C.; Swinny, J.D. Specific Dystrophins Selectively Associate with Inhibitory and Excitatory Synapses of the Mouse Cerebellum and their Loss Alters Expression of P2X7 Purinoceptors and Pro-Inflammatory Mediators. Cell. Mol. Neurobiol. 2021. online ahead of print. [Google Scholar] [CrossRef]
- Turner, P.R.; Fong, P.Y.; Denetclaw, W.F.; Steinhardt, R.A. Increased calcium influx in dystrophic muscle. J. Cell Biol. 1991, 115, 1701–1712. [Google Scholar] [CrossRef]
- Turner, P.R.; Westwood, T.; Regen, C.M.; Steinhardt, R.A. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature 1988, 335, 735–738. [Google Scholar] [CrossRef]
- Hopf, F.W.; Turner, P.R.; Denetclaw, W.F., Jr.; Reddy, P.; Steinhardt, R.A. A critical evaluation of resting intracellular free calcium regulation in dystrophic mdx muscle. Am. J. Physiol. 1996, 271, C1325–C1339. [Google Scholar] [CrossRef] [PubMed]
- Waugh, T.A.; Horstick, E.; Hur, J.; Jackson, S.W.; Davidson, A.E.; Li, X.; Dowling, J.J. Fluoxetine prevents dystrophic changes in a zebrafish model of Duchenne muscular dystrophy. Hum. Mol. Genet. 2014, 23, 4651–4662. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Lopez, J.R.; Alamo, L.; Caputo, C.; DiPolo, R.; Vergara, S. Determination of ionic calcium in frog skeletal muscle fibers. Biophys. J. 1983, 43, 1–4. [Google Scholar] [CrossRef]
- Lopez, J.R.; Kolster, J.; Uryash, A.; Estève, E.; Altamirano, F.; Adams, J.A. Dysregulation of Intracellular Ca2+ in Dystrophic Cortical and Hippocampal Neurons. Mol. Neurobiol. 2018, 55, 603–618. [Google Scholar] [CrossRef]
- Mijares, A.; Altamirano, F.; Kolster, J.; Adams, J.A.; Lopez, J.R. Age-dependent changes in diastolic Ca2+ and Na+ concentrations in dystrophic cardiomyopathy: Role of Ca2+ entry and IP3. Biochem. Biophys. Res. Commun. 2014, 452, 1054–1059. [Google Scholar] [CrossRef]
- Patel, S.; Docampo, R. Acidic calcium stores open for business: Expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010, 20, 277–286. [Google Scholar] [CrossRef]
- Murphy, S.; Ohlendieck, K. Mass spectrometric identification of dystrophin, the protein product of the Duchenne muscular dystrophy gene, in distinct muscle surface membranes. Int. J. Mol. Med. 2017, 40, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Gissel, H. The role of Ca2+ in muscle cell damage. Ann. N. Y. Acad. Sci. 2005, 1066, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Schnellmann, R.G. Calpains, mitochondria, and apoptosis. Cardiovasc. Res. 2012, 96, 32–37. [Google Scholar] [CrossRef]
- Vanlangenakker, N.; Vanden Berghe, T.; Krysko, D.V.; Festjens, N.; Vandenabeele, P. Molecular mechanisms and pathophysiology of necrotic cell death. Curr. Mol. Med. 2008, 8, 207–220. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Ong, W.Y.; Horrocks, L.A. Biochemical aspects of neurodegeneration in human brain: Involvement of neural membrane phospholipids and phospholipases A2. Neurochem. Res. 2004, 29, 1961–1977. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Randazzo, D.; Del Re, V.; Sorrentino, V.; Rossi, D. Organization of junctional sarcoplasmic reticulum proteins in skeletal muscle fibers. J. Muscle Res. Cell Motil. 2015, 36, 501–515. [Google Scholar] [CrossRef]
- Friedrich, O.; Both, M.; Gillis, J.M.; Chamberlain, J.S.; Fink, R.H. Mini-dystrophin restores L-type calcium currents in skeletal muscle of transgenic mdx mice. J. Physiol. 2004, 555, 251–265. [Google Scholar] [CrossRef]
- Balghi, H.; Sebille, S.; Mondin, L.; Cantereau, A.; Constantin, B.; Raymond, G.; Cognard, C. Mini-dystrophin expression down-regulates IP3-mediated calcium release events in resting dystrophin-deficient muscle cells. J. Gen. Physiol. 2006, 128, 219–230. [Google Scholar] [CrossRef]
- Constantin, B.; Sebille, S.; Cognard, C. New insights in the regulation of calcium transfers by muscle dystrophin-based cytoskeleton: Implications in DMD. J. Muscle Res. Cell Motil. 2006, 27, 375–386. [Google Scholar] [CrossRef]
- Allen, D.G.; Gervasio, O.L.; Yeung, E.W.; Whitehead, N.P. Calcium and the damage pathways in muscular dystrophy. Can. J. Physiol. Pharmacol. 2010, 88, 83–91. [Google Scholar] [CrossRef]
- Ascah, A.; Khairallah, M.; Daussin, F.; Bourcier-Lucas, C.; Godin, R.; Allen, B.G.; Petrof, B.J.; Des Rosiers, C.; Burelle, Y. Stress-induced opening of the permeability transition pore in the dystrophin-deficient heart is attenuated by acute treatment with sildenafil. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H144–H153. [Google Scholar] [CrossRef] [PubMed]
- Fanchaouy, M.; Polakova, E.; Jung, C.; Ogrodnik, J.; Shirokova, N.; Niggli, E. Pathways of abnormal stress-induced Ca2+ influx into dystrophic mdx cardiomyocytes. Cell Calcium 2009, 46, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Mehler, M.F. Brain dystrophin, neurogenetics and mental retardation. Brain Res. Brain Res. Rev. 2000, 32, 277–307. [Google Scholar] [CrossRef]
- Mokri, B.; Engel, A.G. Duchenne dystrophy: Electron microscopic findings pointing to a basic or early abnormality in the plasma membrane of the muscle fiber. Neurology 1975, 25, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Whitehead, N.P. Duchenne muscular dystrophy--what causes the increased membrane permeability in skeletal muscle? Int. J. Biochem. Cell Biol. 2011, 43, 290–294. [Google Scholar] [CrossRef]
- Hutter, O.F. The membrane hypothesis of Duchenne muscular dystrophy: Quest for functional evidence. J. Inherit. Metab. Dis. 1992, 15, 565–577. [Google Scholar] [CrossRef]
- Anderson, J.T.; Rogers, R.P.; Jarrett, H.W. Ca2+-calmodulin binds to the carboxyl-terminal domain of dystrophin. J. Biol. Chem. 1996, 271, 6605–6610. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Proteomic analysis of the sarcolemma-enriched fraction from dystrophic mdx-4cv skeletal muscle. J. Proteom. 2019, 191, 212–227. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.F.; Quinlivan, R. Calcium antagonists for Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2008, Cd004571. [Google Scholar] [CrossRef]
- Friedrich, O.; von Wegner, F.; Chamberlain, J.S.; Fink, R.H.; Rohrbach, P. L-type Ca2+ channel function is linked to dystrophin expression in mammalian muscle. PLoS ONE 2008, 3, e1762. [Google Scholar] [CrossRef]
- Hara, H.; Nolan, P.M.; Scott, M.O.; Bucan, M.; Wakayama, Y.; Fischbeck, K.H. Running endurance abnormality in mdx mice. Muscle Nerve 2002, 25, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Shoji, E.; Sakurai, H.; Nishino, T.; Nakahata, T.; Heike, T.; Awaya, T.; Fujii, N.; Manabe, Y.; Matsuo, M.; Sehara-Fujisawa, A. Early pathogenesis of Duchenne muscular dystrophy modelled in patient-derived human induced pluripotent stem cells. Sci. Rep. 2015, 5, 12831. [Google Scholar] [CrossRef]
- Ullrich, N.D.; Fanchaouy, M.; Gusev, K.; Shirokova, N.; Niggli, E. Hypersensitivity of excitation-contraction coupling in dystrophic cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1992–H2003. [Google Scholar] [CrossRef]
- Tanihata, J.; Nagata, T.; Ito, N.; Saito, T.; Nakamura, A.; Minamisawa, S.; Aoki, Y.; Ruegg, U.T.; Takeda, S. Truncated dystrophin ameliorates the dystrophic phenotype of mdx mice by reducing sarcolipin-mediated SERCA inhibition. Biochem. Biophys. Res. Commun. 2018, 505, 51–59. [Google Scholar] [CrossRef]
- Landi, N.; Nassi, P.; Liguri, G.; Bobbi, S.; Sbrilli, C.; Marconi, G. Sarcoplasmic reticulum Ca2+-ATPase and acylphosphatase activities in muscle biopsies from patients with Duchenne muscular dystrophy. Clin. Chim. Acta Int. J. Clin. Chem. 1986, 158, 245–251. [Google Scholar] [CrossRef]
- Gómez, J.; Neco, P.; DiFranco, M.; Vergara, J.L. Calcium release domains in mammalian skeletal muscle studied with two-photon imaging and spot detection techniques. J. Gen. Physiol. 2006, 127, 623–637. [Google Scholar] [CrossRef]
- Badalamente, M.A.; Stracher, A. Delay of muscle degeneration and necrosis in mdx mice by calpain inhibition. Muscle Nerve 2000, 23, 106–111. [Google Scholar] [CrossRef]
- Burr, A.R.; Molkentin, J.D. Genetic evidence in the mouse solidifies the calcium hypothesis of myofiber death in muscular dystrophy. Cell Death Differ. 2015, 22, 1402–1412. [Google Scholar] [CrossRef]
- Onopiuk, M.; Brutkowski, W.; Wierzbicka, K.; Wojciechowska, S.; Szczepanowska, J.; Fronk, J.; Lochmuller, H.; Gorecki, D.C.; Zablocki, K. Mutation in dystrophin-encoding gene affects energy metabolism in mouse myoblasts. Biochem. Biophys. Res. Commun. 2009, 386, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, M.R.F.; Mournetas, V.; Borczyk, M.; Bozycki, L.; Korostynski, M.; Robson, S.; Pinset, C.; Górecki, D.C. Loss of full-length dystrophin expression results in major cell-autonomous abnormalities in proliferating myoblasts. bioRxiv 2021. [Google Scholar] [CrossRef]
- Gonzalez, J.P.; Kyrychenko, S.; Kyrychenko, V.; Schneider, J.S.; Granier, C.J.; Himelman, E.; Lahey, K.C.; Zhao, Q.; Yehia, G.; Tao, Y.X.; et al. Small Fractions of Muscular Dystrophy Embryonic Stem Cells Yield Severe Cardiac and Skeletal Muscle Defects in Adult Mouse Chimeras. Stem Cells 2017, 35, 597–610. [Google Scholar] [CrossRef]
- Palladino, M.; Gatto, I.; Neri, V.; Straino, S.; Smith, R.C.; Silver, M.; Gaetani, E.; Marcantoni, M.; Giarretta, I.; Stigliano, E.; et al. Angiogenic impairment of the vascular endothelium: A novel mechanism and potential therapeutic target in muscular dystrophy. Arter. Thromb. Vasc. Biol. 2013, 33, 2867–2876. [Google Scholar] [CrossRef]
- Dabiré, H.; Barthélémy, I.; Blanchard-Gutton, N.; Sambin, L.; Sampedrano, C.C.; Gouni, V.; Unterfinger, Y.; Aguilar, P.; Thibaud, J.L.; Ghaleh, B.; et al. Vascular endothelial dysfunction in Duchenne muscular dystrophy is restored by bradykinin through upregulation of eNOS and nNOS. Basic Res. Cardiol. 2012, 107, 240. [Google Scholar] [CrossRef][Green Version]
- Kodippili, K.; Thorne, P.K.; Laughlin, M.H.; Duan, D. Dystrophin deficiency impairs vascular structure and function in the canine model of Duchenne muscular dystrophy. J. Pathol. 2021, 254, 589–605. [Google Scholar] [CrossRef]
- Tennyson, C.N.; Klamut, H.J.; Worton, R.G. The human dystrophin gene requires 16 hours to be transcribed and is cotranscriptionally spliced. Nat. Genet. 1995, 9, 184–190. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Neueder, A. RNA-Mediated Disease Mechanisms in Neurodegenerative Disorders. J. Mol. Biol. 2019, 431, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Kamieniarz-Gdula, K.; Proudfoot, N.J. Transcriptional Control by Premature Termination: A Forgotten Mechanism. Trends Genet. 2019, 35, 553–564. [Google Scholar] [CrossRef]
- Williams, I.A.; Allen, D.G. Intracellular calcium handling in ventricular myocytes from mdx mice. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H846–H855. [Google Scholar] [CrossRef] [PubMed]
- Kyrychenko, S.; Poláková, E.; Kang, C.; Pocsai, K.; Ullrich, N.D.; Niggli, E.; Shirokova, N. Hierarchical accumulation of RyR post-translational modifications drives disease progression in dystrophic cardiomyopathy. Cardiovasc. Res. 2013, 97, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Viola, H.M.; Adams, A.M.; Davies, S.M.K.; Fletcher, S.; Filipovska, A.; Hool, L.C. Impaired functional communication between the L-type calcium channel and mitochondria contributes to metabolic inhibition in the mdx heart. Proc. Natl. Acad. Sci. USA 2014, 111, E2905–E2914. [Google Scholar] [CrossRef] [PubMed]
- Podkalicka, P.; Mucha, O.; Dulak, J.; Loboda, A. Targeting angiogenesis in Duchenne muscular dystrophy. Cell. Mol. Life Sci. CMLS 2019, 76, 1507–1528. [Google Scholar] [CrossRef]
- Khurana, T.S.; Prendergast, R.A.; Alameddine, H.S.; Tomé, F.M.; Fardeau, M.; Arahata, K.; Sugita, H.; Kunkel, L.M. Absence of extraocular muscle pathology in Duchenne’s muscular dystrophy: Role for calcium homeostasis in extraocular muscle sparing. J. Exp. Med. 1995, 182, 467–475. [Google Scholar] [CrossRef]
- Murphy, S.; Zweyer, M.; Henry, M.; Meleady, P.; Mundegar, R.R.; Swandulla, D.; Ohlendieck, K. Label-free mass spectrometric analysis reveals complex changes in the brain proteome from the mdx-4cv mouse model of Duchenne muscular dystrophy. Clin. Proteom. 2015, 12, 27. [Google Scholar] [CrossRef]
- Aleman, V.; Osorio, B.; Chavez, O.; Rendon, A.; Mornet, D.; Martinez, D. Subcellular localization of Dp71 dystrophin isoforms in cultured hippocampal neurons and forebrain astrocytes. Histochem. Cell Biol. 2001, 115, 243–254. [Google Scholar] [CrossRef]
- Blake, D.J.; Hawkes, R.; Benson, M.A.; Beesley, P.W. Different dystrophin-like complexes are expressed in neurons and glia. J. Cell Biol. 1999, 147, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Lidov, H.G.; Byers, T.J.; Kunkel, L.M. The distribution of dystrophin in the murine central nervous system: An immunocytochemical study. Neuroscience 1993, 54, 167–187. [Google Scholar] [CrossRef]
- Sekiguchi, M. The role of dystrophin in the central nervous system: A mini review. Acta Myol. 2005, 24, 93–97. [Google Scholar] [PubMed]
- Souttou, S.; Benabdesselam, R.; Siqueiros-Marquez, L.; Sifi, M.; Deliba, M.; Vacca, O.; Charles-Messance, H.; Vaillend, C.; Rendon, A.; Guillonneau, X.; et al. Expression and localization of dystrophins and β-dystroglycan in the hypothalamic supraoptic nuclei of rat from birth to adulthood. Acta Histochem. 2019, 121, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Hopf, F.W.; Steinhardt, R.A. Regulation of intracellular free calcium in normal and dystrophic mouse cerebellar neurons. Brain Res. 1992, 578, 49–54. [Google Scholar] [CrossRef]
- Vaillend, C.; Billard, J.M. Facilitated CA1 hippocampal synaptic plasticity in dystrophin-deficient mice: Role for GABAA receptors? Hippocampus 2002, 12, 713–717. [Google Scholar] [CrossRef] [PubMed]
- Vaillend, C.; Billard, J.M.; Claudepierre, T.; Rendon, A.; Dutar, P.; Ungerer, A. Spatial discrimination learning and CA1 hippocampal synaptic plasticity in mdx and mdx3cv mice lacking dystrophin gene products. Neuroscience 1998, 86, 53–66. [Google Scholar] [CrossRef]
- Fritschy, J.M.; Schweizer, C.; Brunig, I.; Luscher, B. Pre- and post-synaptic mechanisms regulating the clustering of type A gamma-aminobutyric acid receptors (GABAA receptors). Biochem. Soc. Trans. 2003, 31, 889–892. [Google Scholar] [CrossRef]
- Knuesel, I.; Zuellig, R.A.; Schaub, M.C.; Fritschy, J.M. Alterations in dystrophin and utrophin expression parallel the reorganization of GABAergic synapses in a mouse model of temporal lobe epilepsy. Eur. J. Neurosci. 2001, 13, 1113–1124. [Google Scholar] [CrossRef]
- Sekiguchi, M.; Zushida, K.; Yoshida, M.; Maekawa, M.; Kamichi, S.; Yoshida, M.; Sahara, Y.; Yuasa, S.; Takeda, S.; Wada, K. A deficit of brain dystrophin impairs specific amygdala GABAergic transmission and enhances defensive behaviour in mice. Brain 2008, 132, 124–135. [Google Scholar] [CrossRef]
- Sumita, K.; Sato, Y.; Iida, J.; Kawata, A.; Hamano, M.; Hirabayashi, S.; Ohno, K.; Peles, E.; Hata, Y. Synaptic scaffolding molecule (S-SCAM) membrane-associated guanylate kinase with inverted organization (MAGI)-2 is associated with cell adhesion molecules at inhibitory synapses in rat hippocampal neurons. J. Neurochem. 2007, 100, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Tuckett, E.; Gosetti, T.; Hayes, A.; Rybalka, E.; Verghese, E. Increased calcium in neurons in the cerebral cortex and cerebellum is not associated with cell loss in the mdx mouse model of Duchenne muscular dystrophy. Neuroreport 2015, 26, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, S.; Viggiano, L.; Annese, T.; Rubolino, C.; Gerbino, A.; De Zio, R.; Corsi, P.; Tamma, R.; Ribatti, D.; Errede, M.; et al. DP71 and SERCA2 alteration in human neurons of a Duchenne muscular dystrophy patient. Stem Cell Res. Ther. 2019, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Tsurumi, F.; Baba, S.; Yoshinaga, D.; Umeda, K.; Hirata, T.; Takita, J.; Heike, T. The intracellular Ca2+ concentration is elevated in cardiomyocytes differentiated from hiPSCs derived from a Duchenne muscular dystrophy patient. PLoS ONE 2019, 14, e0213768. [Google Scholar] [CrossRef] [PubMed]
- Robin, G.; Berthier, C.; Allard, B. Sarcoplasmic reticulum Ca2+ permeation explored from the lumen side in mdx muscle fibers under voltage control. J. Gen. Physiol. 2012, 139, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.M.; Wierda, K.; Thorrez, L.; van Putten, M.; De Smedt, J.; Ribeiro, L.; Tricot, T.; Gajjar, M.; Duelen, R.; Van Damme, P.; et al. Dystrophin deficiency leads to dysfunctional glutamate clearance in iPSC derived astrocytes. Transl. Psychiatry 2019, 9, 200. [Google Scholar] [CrossRef]
- Del Tongo, C.; Carretta, D.; Fulgenzi, G.; Catini, C.; Minciacchi, D. Parvalbumin-positive GABAergic interneurons are increased in the dorsal hippocampus of the dystrophic mdx mouse. Acta Neuropathol. 2009, 118, 803–812. [Google Scholar] [CrossRef]
- Krasowska, E.; Zabłocki, K.; Górecki, D.C.; Swinny, J.D. Aberrant location of inhibitory synaptic marker proteins in the hippocampus of dystrophin-deficient mice: Implications for cognitive impairment in duchenne muscular dystrophy. PLoS ONE 2014, 9, e108364. [Google Scholar] [CrossRef]
- Chavas, J.; Forero, M.E.; Collin, T.; Llano, I.; Marty, A. Osmotic tension as a possible link between GABA(A) receptor activation and intracellular calcium elevation. Neuron 2004, 44, 701–713. [Google Scholar] [CrossRef]
- Nico, B.; Frigeri, A.; Nicchia, G.P.; Corsi, P.; Ribatti, D.; Quondamatteo, F.; Herken, R.; Girolamo, F.; Marzullo, A.; Svelto, M.; et al. Severe alterations of endothelial and glial cells in the blood-brain barrier of dystrophic mdx mice. Glia 2003, 42, 235–251. [Google Scholar] [CrossRef]
- Lien, C.F.; Mohanta, S.K.; Frontczak-Baniewicz, M.; Swinny, J.D.; Zablocka, B.; Gorecki, D.C. Absence of glial alpha-dystrobrevin causes abnormalities of the blood-brain barrier and progressive brain edema. J. Biol. Chem. 2012, 287, 41374–41385. [Google Scholar] [CrossRef]
- Nico, B.; Tamma, R.; Annese, T.; Mangieri, D.; De Luca, A.; Corsi, P.; Benagiano, V.; Longo, V.; Crivellato, E.; Salmaggi, A.; et al. Glial dystrophin-associated proteins, laminin and agrin, are downregulated in the brain of mdx mouse. Lab. Investig. J. Tech. Methods Pathol. 2010, 90, 1645–1660. [Google Scholar] [CrossRef][Green Version]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef]
- Balaban, R.S. The role of Ca2+ signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim. Biophys. Acta 2009, 1787, 1334–1341. [Google Scholar] [CrossRef]
- Berchtold, M.W.; Brinkmeier, H.; Müntener, M. Calcium ion in skeletal muscle: Its crucial role for muscle function, plasticity, and disease. Physiol. Rev. 2000, 80, 1215–1265. [Google Scholar] [CrossRef]
- Halestrap, A.P. Calcium, mitochondria and reperfusion injury: A pore way to die. Biochem. Soc. Trans. 2006, 34, 232–237. [Google Scholar] [CrossRef]
- Zhang, S.S.; Zhou, S.; Crowley-McHattan, Z.J.; Wang, R.Y.; Li, J.P. A Review of the Role of Endo/Sarcoplasmic Reticulum-Mitochondria Ca2+ Transport in Diseases and Skeletal Muscle Function. Int. J. Environ. Res. Public Health 2021, 18, 3874. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Mikheeva, I.B.; Belosludtsev, K.N. Transport of Ca2+ and Ca2+-dependent permeability transition in heart mitochondria in the early stages of Duchenne muscular dystrophy. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148250. [Google Scholar] [CrossRef]
- Kyrychenko, V.; Poláková, E.; Janíček, R.; Shirokova, N. Mitochondrial dysfunctions during progression of dystrophic cardiomyopathy. Cell Calcium 2015, 58, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Charge, S.B.; Rudnicki, M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.R.; Abu-Dief, E.E.; Kamel, N.F.; Mostafa, M.G. Steroid therapy is associated with decreased numbers of dendritic cells and fibroblasts, and increased numbers of satellite cells, in the dystrophic skeletal muscle. J. Clin. Pathol. 2010, 63, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Zulian, A.; Gualandi, F.; Manzati, E.; Merlini, L.; Michelini, M.E.; Benassi, L.; Marmiroli, S.; Ferlini, A.; Sabatelli, P.; et al. Melanocytes—A novel tool to study mitochondrial dysfunction in Duchenne muscular dystrophy. J. Cell. Physiol. 2013, 228, 1323–1331. [Google Scholar] [CrossRef] [PubMed]
- Law, M.L.; Cohen, H.; Martin, A.A.; Angulski, A.B.B.; Metzger, J.M. Dysregulation of Calcium Handling in Duchenne Muscular Dystrophy-Associated Dilated Cardiomyopathy: Mechanisms and Experimental Therapeutic Strategies. J. Clin. Med. 2020, 9, 520. [Google Scholar] [CrossRef]
- Meyers, T.A.; Townsend, D. Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2019, 20, 4098. [Google Scholar] [CrossRef] [PubMed]
- Mareedu, S.; Million, E.D.; Duan, D.; Babu, G.J. Abnormal Calcium Handling in Duchenne Muscular Dystrophy: Mechanisms and Potential Therapies. Front. Physiol. 2021, 12, 647010. [Google Scholar] [CrossRef]
- Angebault, C.; Panel, M.; Lacôte, M.; Rieusset, J.; Lacampagne, A.; Fauconnier, J. Metformin Reverses the Enhanced Myocardial SR/ER-Mitochondria Interaction and Impaired Complex I-Driven Respiration in Dystrophin-Deficient Mice. Front. Cell Dev. Biol. 2020, 8, 609493. [Google Scholar] [CrossRef]
- Dong, X.; Hui, T.; Chen, J.; Yu, Z.; Ren, D.; Zou, S.; Wang, S.; Fei, E.; Jiao, H.; Lai, X. Metformin Increases Sarcolemma Integrity and Ameliorates Neuromuscular Deficits in a Murine Model of Duchenne Muscular Dystrophy. Front. Physiol. 2021, 12, 642908. [Google Scholar] [CrossRef]
- Ljubicic, V.; Jasmin, B.J. Metformin increases peroxisome proliferator-activated receptor γ Co-activator-1α and utrophin a expression in dystrophic skeletal muscle. Muscle Nerve 2015, 52, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Dubinin, M.V.; Talanov, E.Y.; Tenkov, K.S.; Starinets, V.S.; Belosludtseva, N.V.; Belosludtsev, K.N. The Effect of Deflazacort Treatment on the Functioning of Skeletal Muscle Mitochondria in Duchenne Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 8763. [Google Scholar] [CrossRef]
- Yasuda, S.; Townsend, D.; Michele, D.E.; Favre, E.G.; Day, S.M.; Metzger, J.M. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 2005, 436, 1025–1029. [Google Scholar] [CrossRef]
- Terry, R.L.; Kaneb, H.M.; Wells, D.J. Poloxamer 188 has a deleterious effect on dystrophic skeletal muscle function. PLoS ONE 2014, 9, e91221, Erratum in 2015, 10, e0119252. [Google Scholar] [CrossRef]
- Quinlan, J.G.; Wong, B.L.; Niemeier, R.T.; McCullough, A.S.; Levin, L.; Emanuele, M. Poloxamer 188 failed to prevent exercise-induced membrane breakdown in mdx skeletal muscle fibers. Neuromuscul. Disord. 2006, 16, 855–864. [Google Scholar] [CrossRef]
- Vaillend, C.; Perronnet, C.; Ros, C.; Gruszczynski, C.; Goyenvalle, A.; Laroche, S.; Danos, O.; Garcia, L.; Peltekian, E. Rescue of a dystrophin-like protein by exon skipping in vivo restores GABAA-receptor clustering in the hippocampus of the mdx mouse. Mol. Ther. J. Am. Soc. Gene Ther. 2010, 18, 1683–1688. [Google Scholar] [CrossRef]
- Young, C.N.; Sinadinos, A.; Lefebvre, A.; Chan, P.; Arkle, S.; Vaudry, D.; Gorecki, D.C. A novel mechanism of autophagic cell death in dystrophic muscle regulated by P2RX7 receptor large-pore formation and HSP90. Autophagy 2015, 11, 113–130. [Google Scholar] [CrossRef]
- Browne, S.E. When too much ATP is a bad thing: A pivotal role for P2X(7) receptors in motor neuron degeneration. J. Neurochem. 2013, 126, 301–304. [Google Scholar] [CrossRef]
- Gazzerro, E.; Baldassari, S.; Assereto, S.; Fruscione, F.; Pistorio, A.; Panicucci, C.; Volpi, S.; Perruzza, L.; Fiorillo, C.; Minetti, C.; et al. Enhancement of Muscle T Regulatory Cells and Improvement of Muscular Dystrophic Process in mdx Mice by Blockade of Extracellular ATP/P2X Axis. Am. J. Pathol. 2015, 185, 3349–3360. [Google Scholar] [CrossRef]
- Al-Khalidi, R.; Panicucci, C.; Cox, P.; Chira, N.; Róg, J.; Young, C.N.J.; McGeehan, R.E.; Ambati, K.; Ambati, J.; Zabłocki, K.; et al. Zidovudine ameliorates pathology in the mouse model of Duchenne muscular dystrophy via P2RX7 purinoceptor antagonism. Acta Neuropathol. Commun. 2018, 6, 27. [Google Scholar] [CrossRef]
- Sinadinos, A.; Young, C.N.; Al-Khalidi, R.; Teti, A.; Kalinski, P.; Mohamad, S.; Floriot, L.; Henry, T.; Tozzi, G.; Jiang, T.; et al. P2RX7 purinoceptor: A therapeutic target for ameliorating the symptoms of duchenne muscular dystrophy. PLoS Med. 2015, 12, e1001888. [Google Scholar] [CrossRef] [PubMed]
- Young, C.N.; Brutkowski, W.; Lien, C.F.; Arkle, S.; Lochmuller, H.; Zablocki, K.; Gorecki, D.C. P2X7 purinoceptor alterations in dystrophic mdx mouse muscles: Relationship to pathology and potential target for treatment. J. Cell. Mol. Med. 2012, 16, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Arbeloa, J.; Perez-Samartin, A.; Gottlieb, M.; Matute, C. P2X7 receptor blockade prevents ATP excitotoxicity in neurons and reduces brain damage after ischemia. Neurobiol. Dis. 2012, 45, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Górecki, D.C. P2X7 purinoceptor as a therapeutic target in muscular dystrophies. Curr. Opin. Pharmacol. 2019, 47, 40–45. [Google Scholar] [CrossRef]
- Dabertrand, F.; Mironneau, J.; Henaff, M.; Macrez, N.; Morel, J.L. Comparison between gentamycin and exon skipping treatments to restore ryanodine receptor subtype 2 functions in mdx mouse duodenum myocytes. Eur. J. Pharm. 2010, 628, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Clemens, P.R.; Hoffman, E.P. Exon-Skipping in Duchenne Muscular Dystrophy. J. Neuromuscul. Dis. 2021, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Capitanio, D.; Moriggi, M.; Torretta, E.; Barbacini, P.; De Palma, S.; Viganò, A.; Lochmüller, H.; Muntoni, F.; Ferlini, A.; Mora, M.; et al. Comparative proteomic analyses of Duchenne muscular dystrophy and Becker muscular dystrophy muscles: Changes contributing to preserve muscle function in Becker muscular dystrophy patients. J. Cachexia Sarcopenia Muscle 2020, 11, 547–563. [Google Scholar] [CrossRef]
- Sato, M.; Shiba, N.; Miyazaki, D.; Shiba, Y.; Echigoya, Y.; Yokota, T.; Takizawa, H.; Aoki, Y.; Takeda, S.; Nakamura, A. Amelioration of intracellular Ca2+ regulation by exon-45 skipping in Duchenne muscular dystrophy-induced pluripotent stem cell-derived cardiomyocytes. Biochem. Biophys. Res. Commun. 2019, 520, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Vulin, A.; Barthélémy, I.; Goyenvalle, A.; Thibaud, J.L.; Beley, C.; Griffith, G.; Benchaouir, R.; le Hir, M.; Unterfinger, Y.; Lorain, S.; et al. Muscle function recovery in golden retriever muscular dystrophy after AAV1-U7 exon skipping. Mol. Ther. J. Am. Soc. Gene Ther. 2012, 20, 2120–2133. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zabłocka, B.; Górecki, D.C.; Zabłocki, K. Disrupted Calcium Homeostasis in Duchenne Muscular Dystrophy: A Common Mechanism behind Diverse Consequences. Int. J. Mol. Sci. 2021, 22, 11040. https://doi.org/10.3390/ijms222011040
Zabłocka B, Górecki DC, Zabłocki K. Disrupted Calcium Homeostasis in Duchenne Muscular Dystrophy: A Common Mechanism behind Diverse Consequences. International Journal of Molecular Sciences. 2021; 22(20):11040. https://doi.org/10.3390/ijms222011040
Chicago/Turabian StyleZabłocka, Barbara, Dariusz C. Górecki, and Krzysztof Zabłocki. 2021. "Disrupted Calcium Homeostasis in Duchenne Muscular Dystrophy: A Common Mechanism behind Diverse Consequences" International Journal of Molecular Sciences 22, no. 20: 11040. https://doi.org/10.3390/ijms222011040
APA StyleZabłocka, B., Górecki, D. C., & Zabłocki, K. (2021). Disrupted Calcium Homeostasis in Duchenne Muscular Dystrophy: A Common Mechanism behind Diverse Consequences. International Journal of Molecular Sciences, 22(20), 11040. https://doi.org/10.3390/ijms222011040