
Cyclophilin D Regulates the Nuclear Translocation of AIF, Cardiac Endothelial Cell Necroptosis and Murine Cardiac Transplant Injury


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
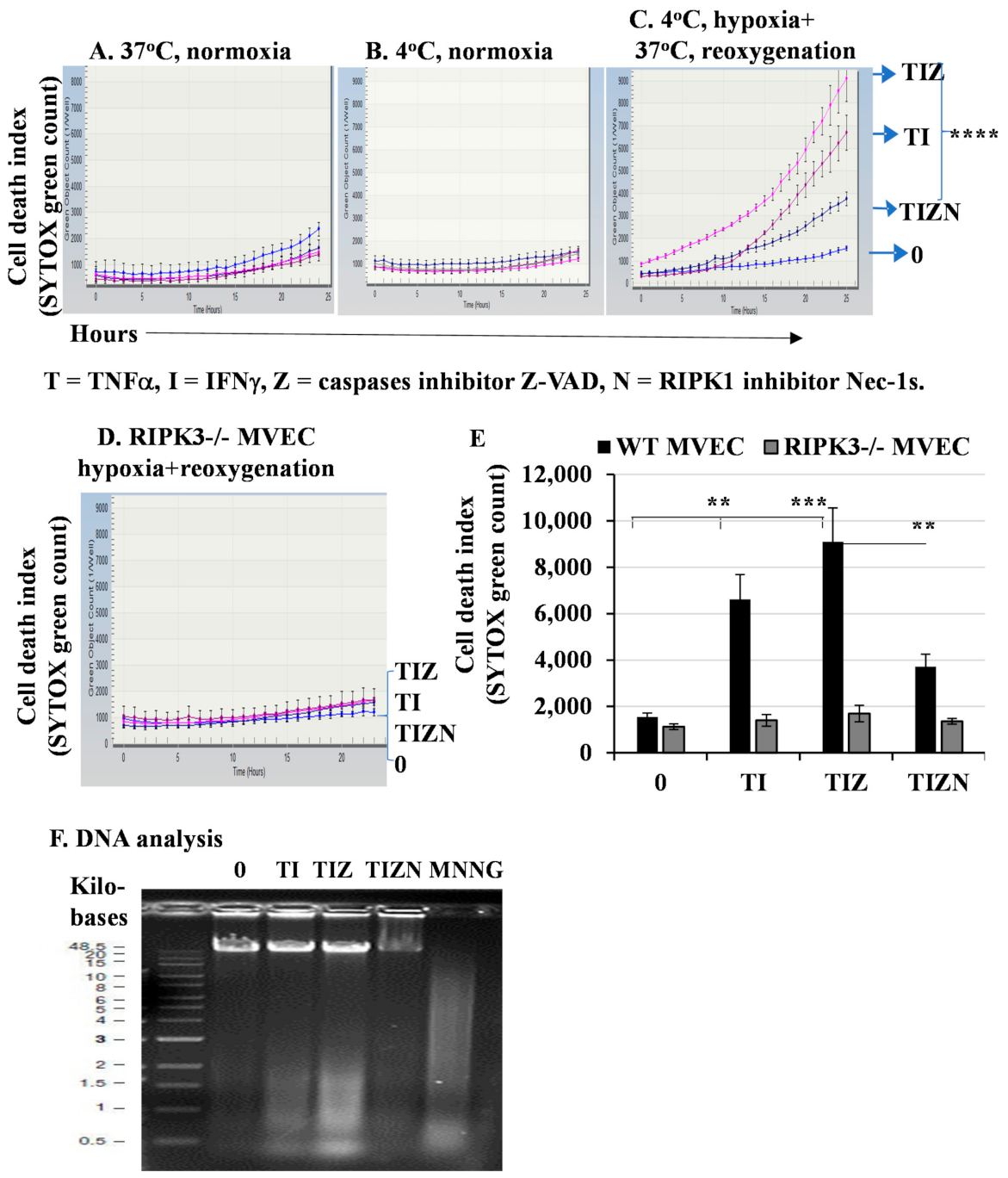
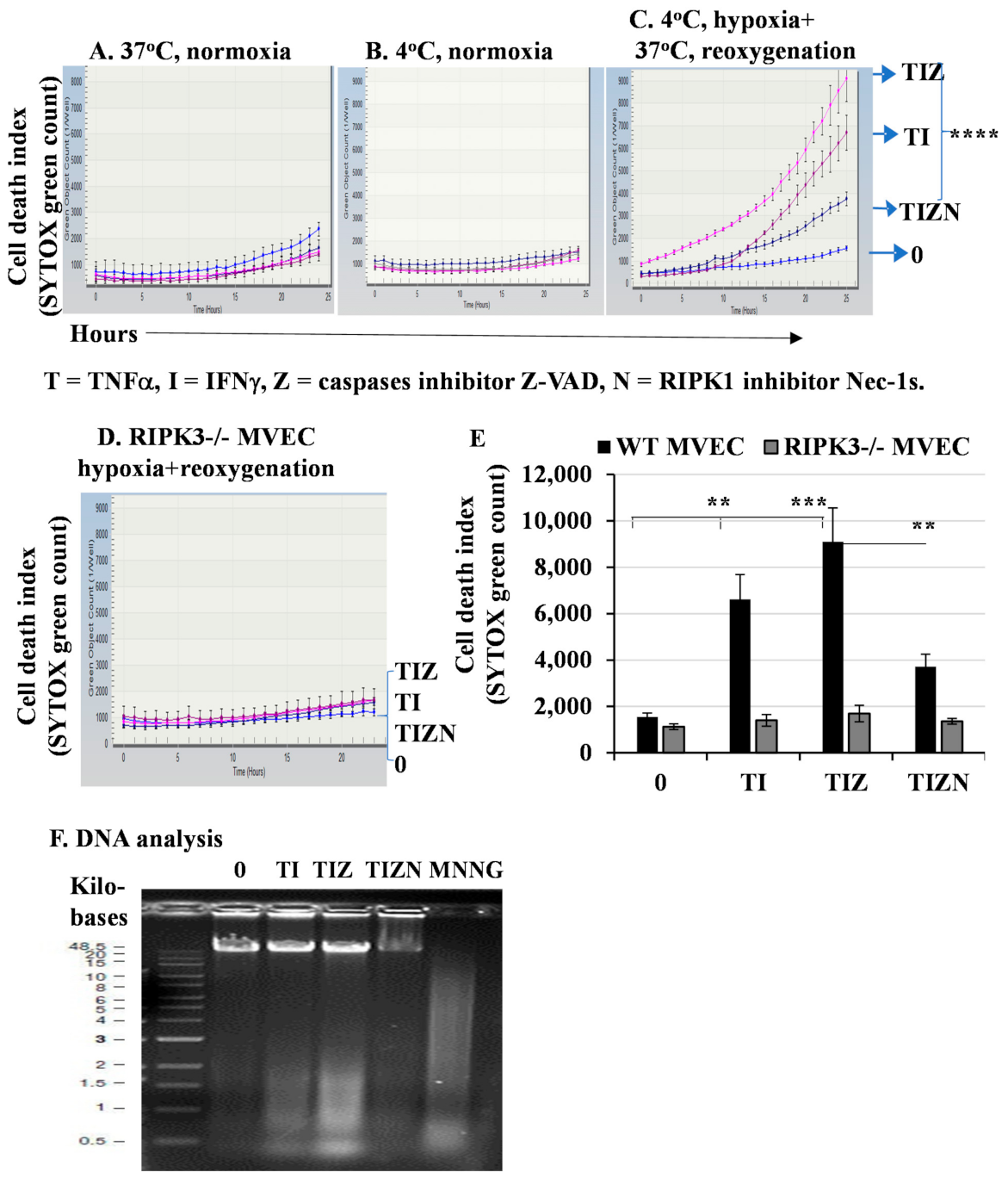
2.1. In Vitro Simulation of Ischemia and Reperfusion Promotes Necroptosis in MVECs
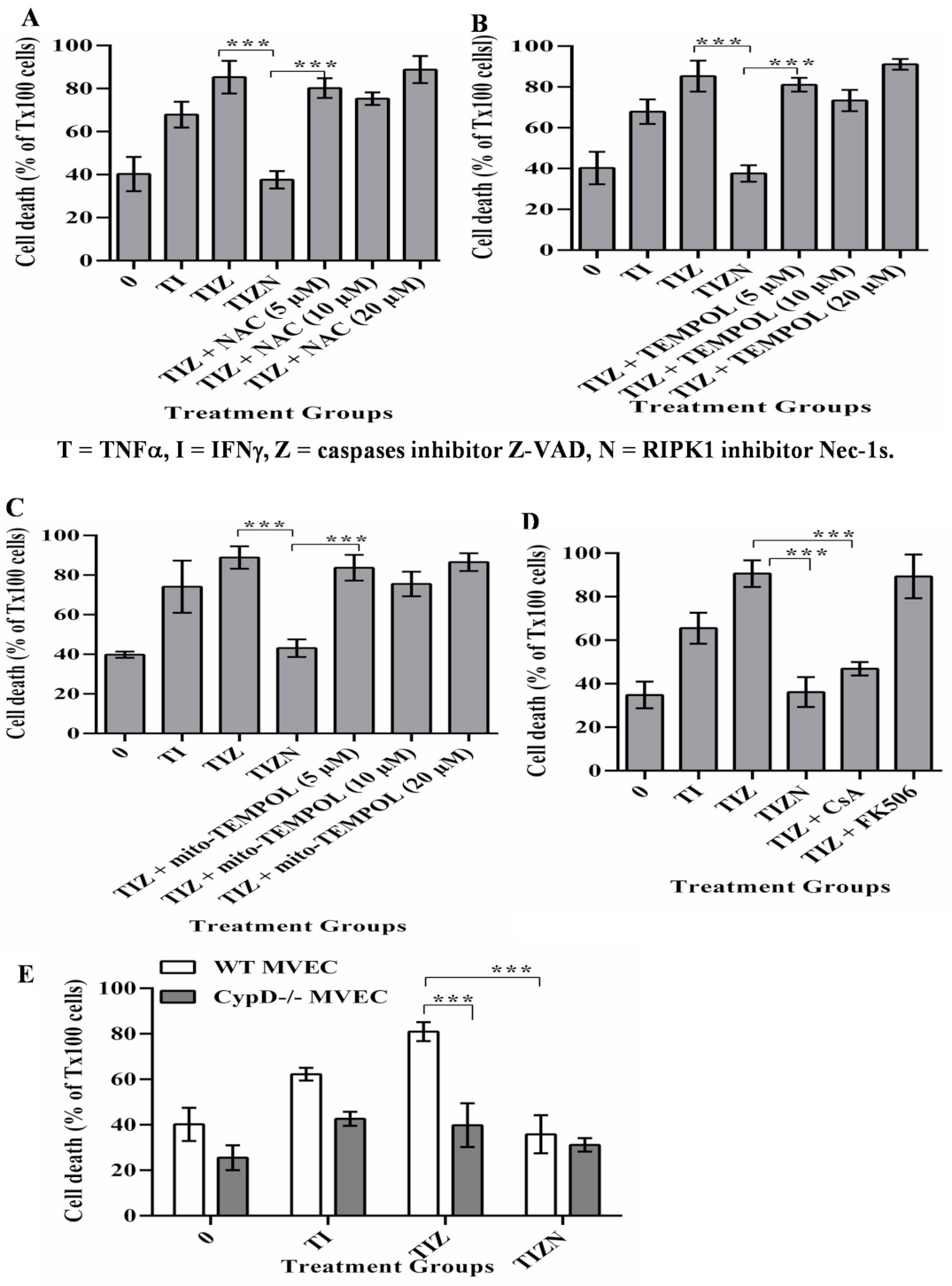
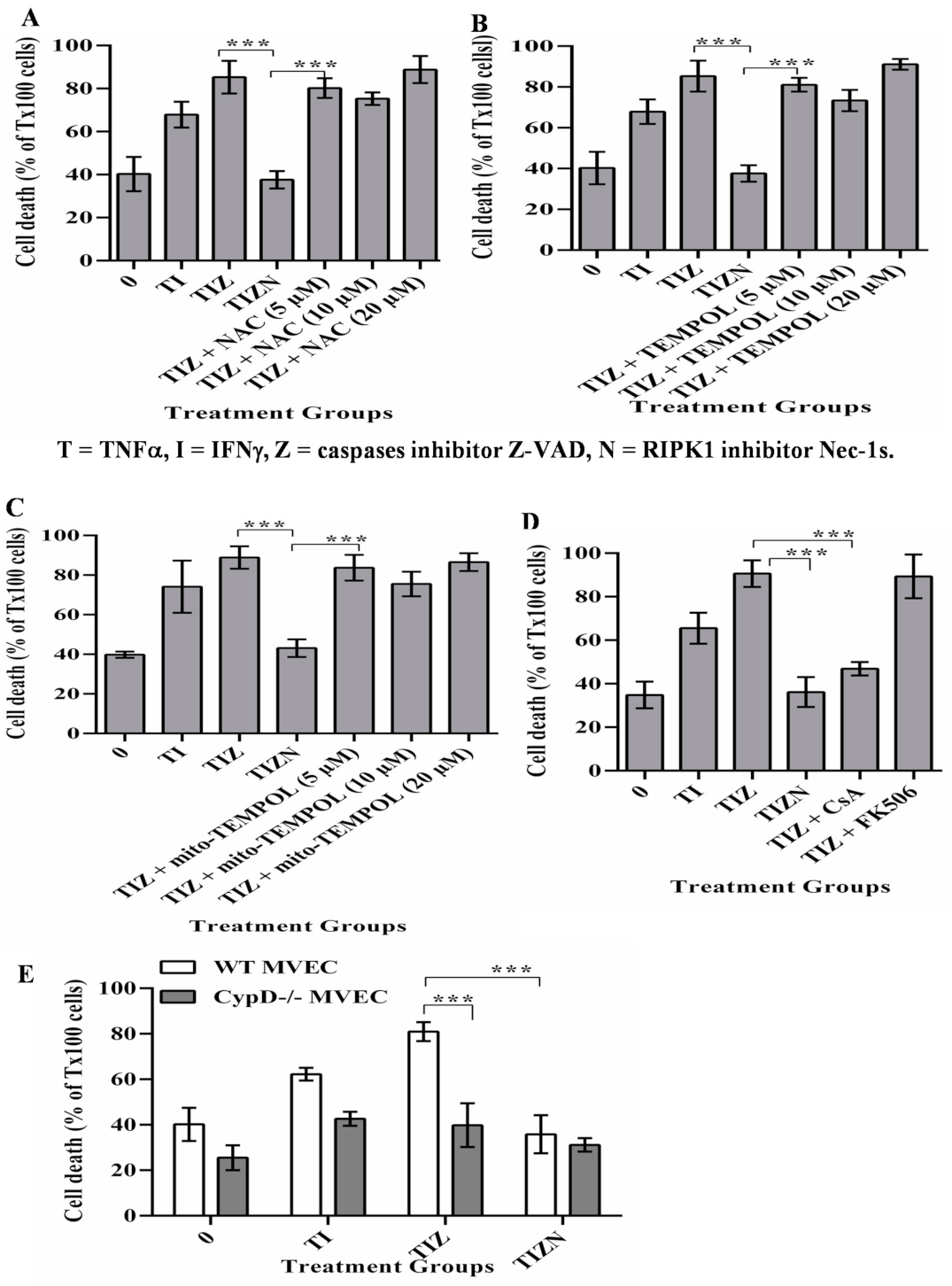
2.2. CypD, Not ROS, Contributes to MVECs Necroptosis under In Vitro Simulating IRI Condition
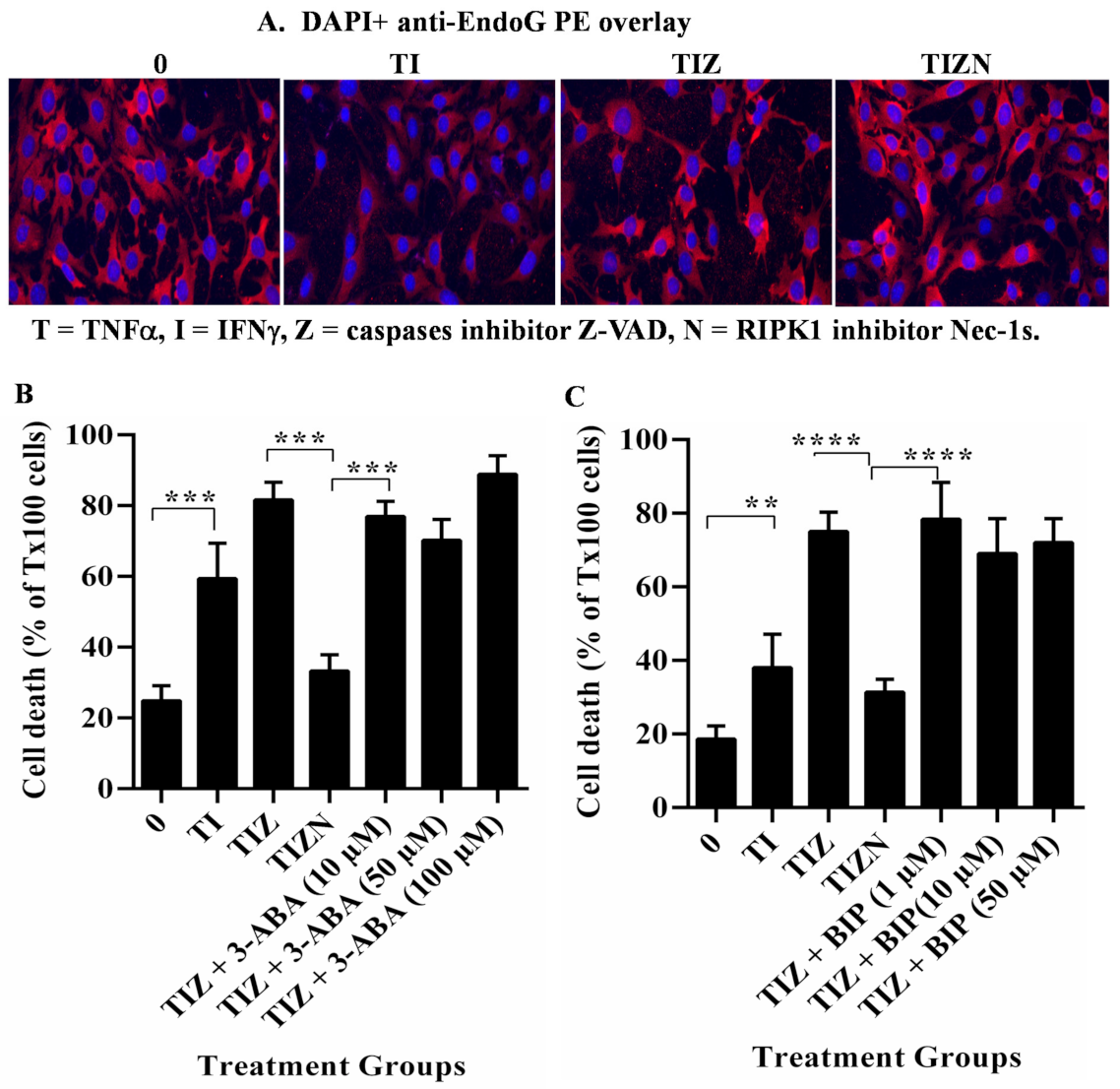
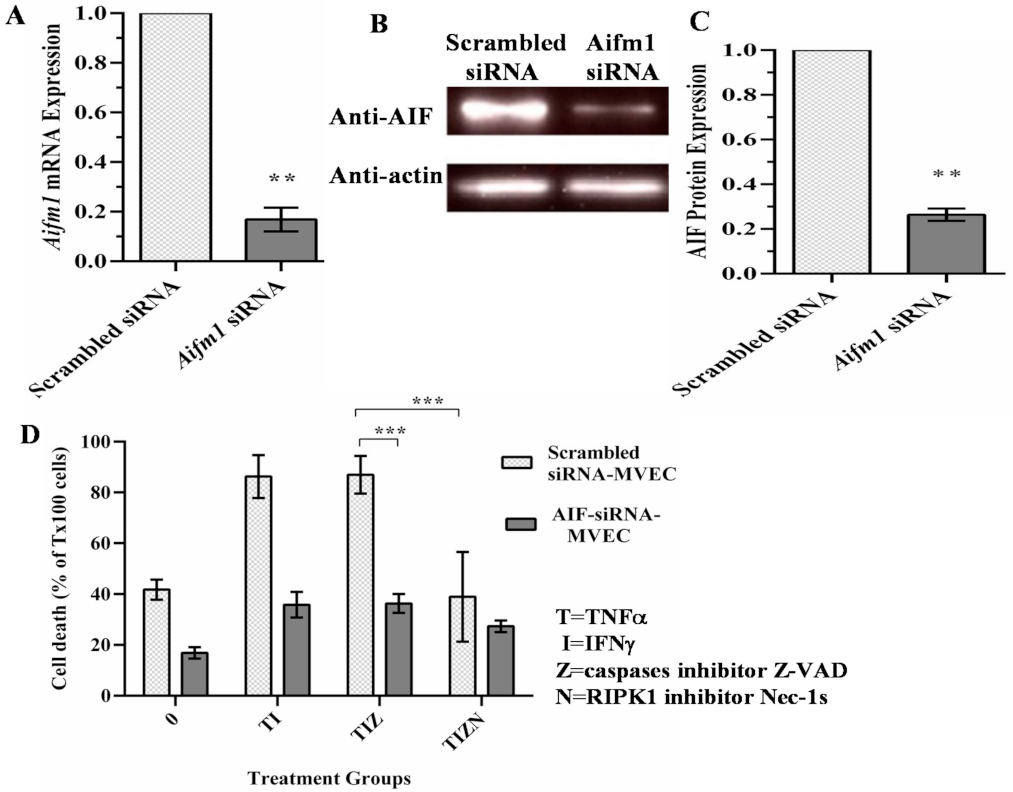
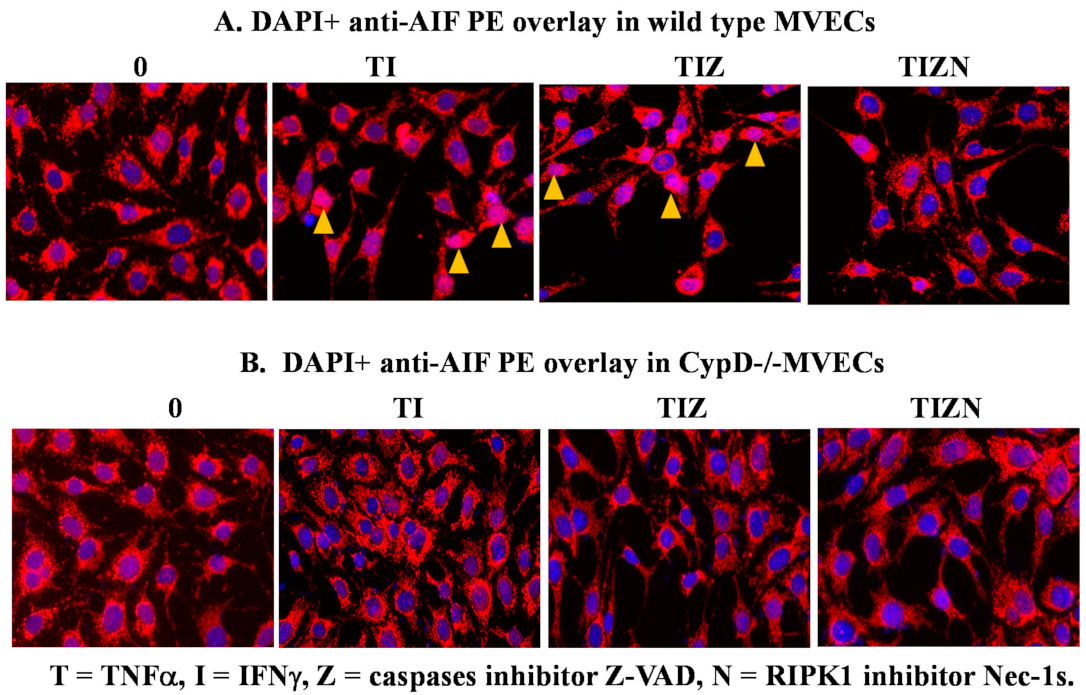
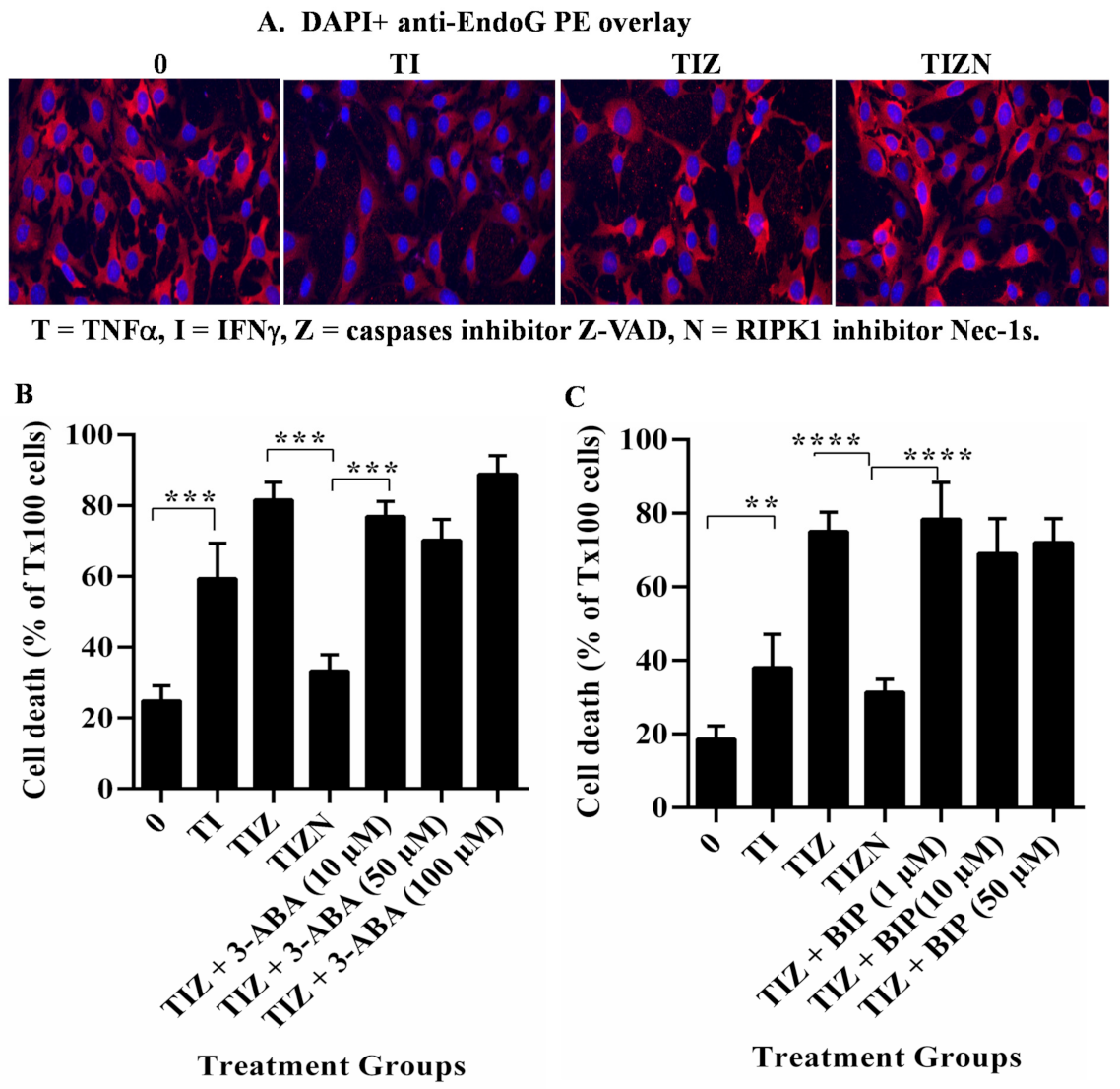
2.3. AIF Translocates to the Nucleus during Necroptosis under In Vitro Simulation of IRI Conditions
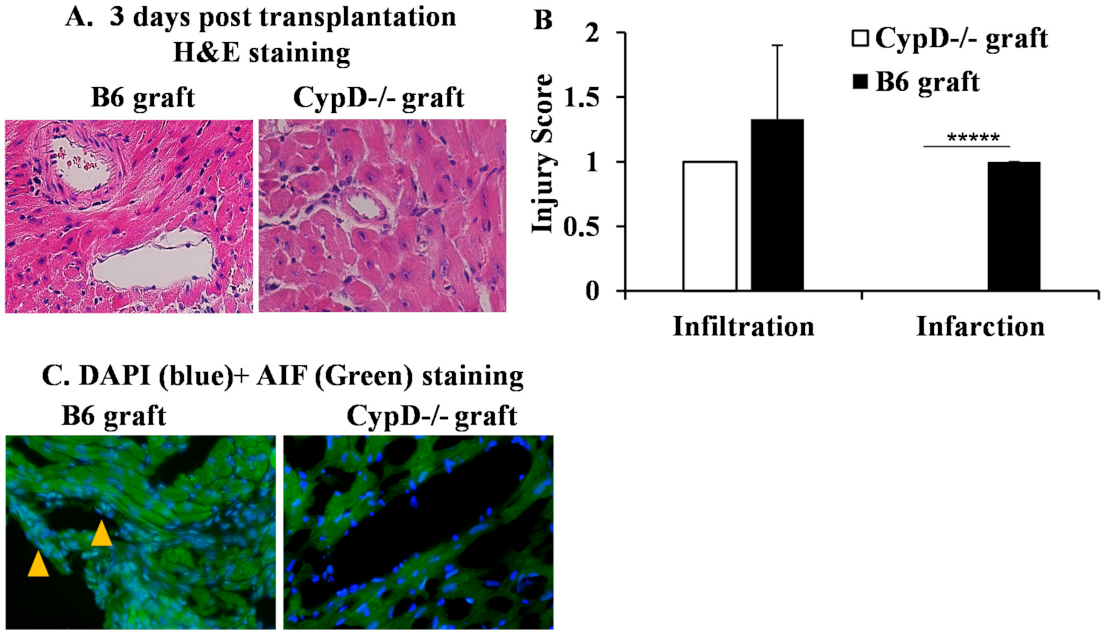
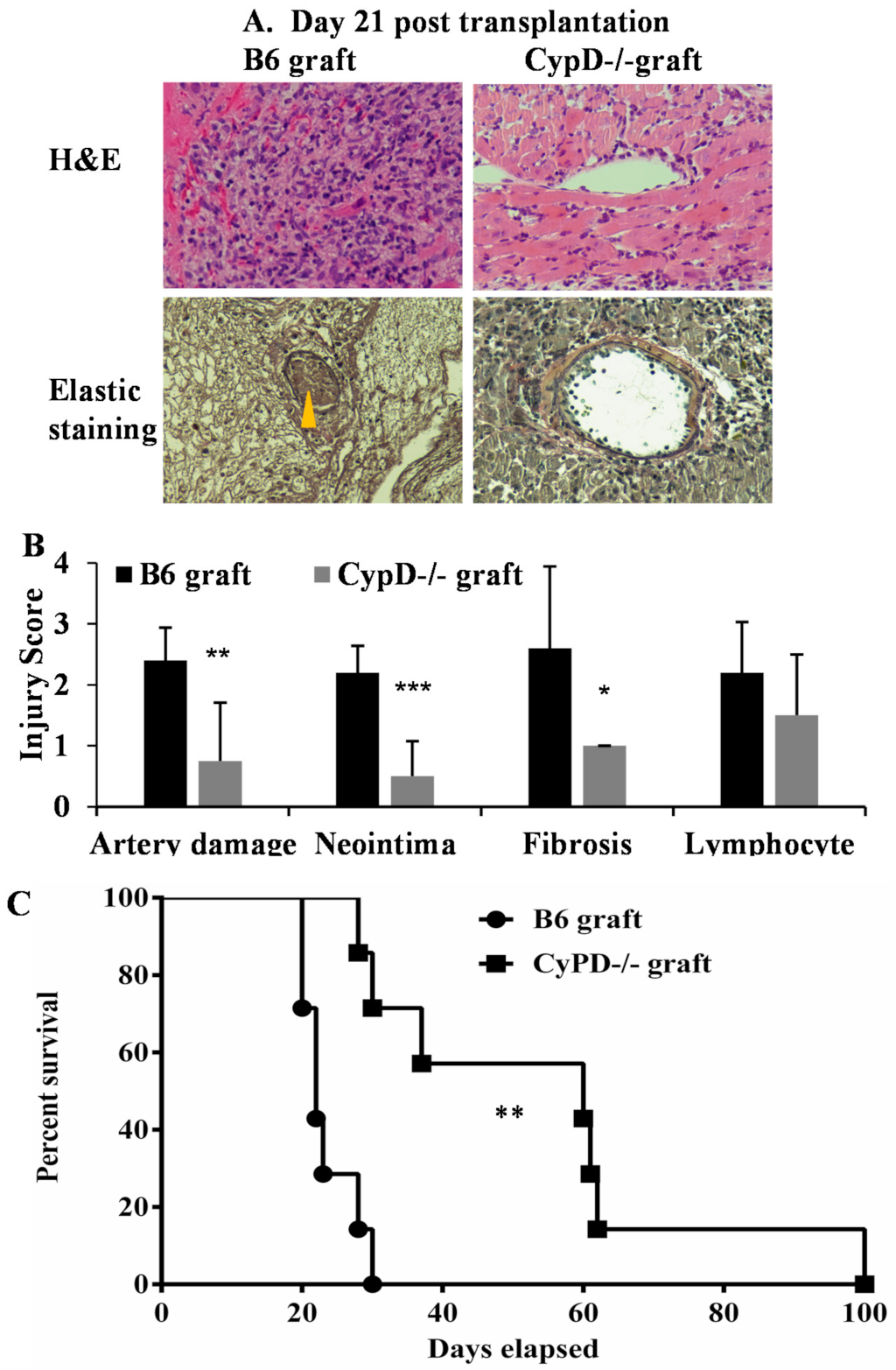
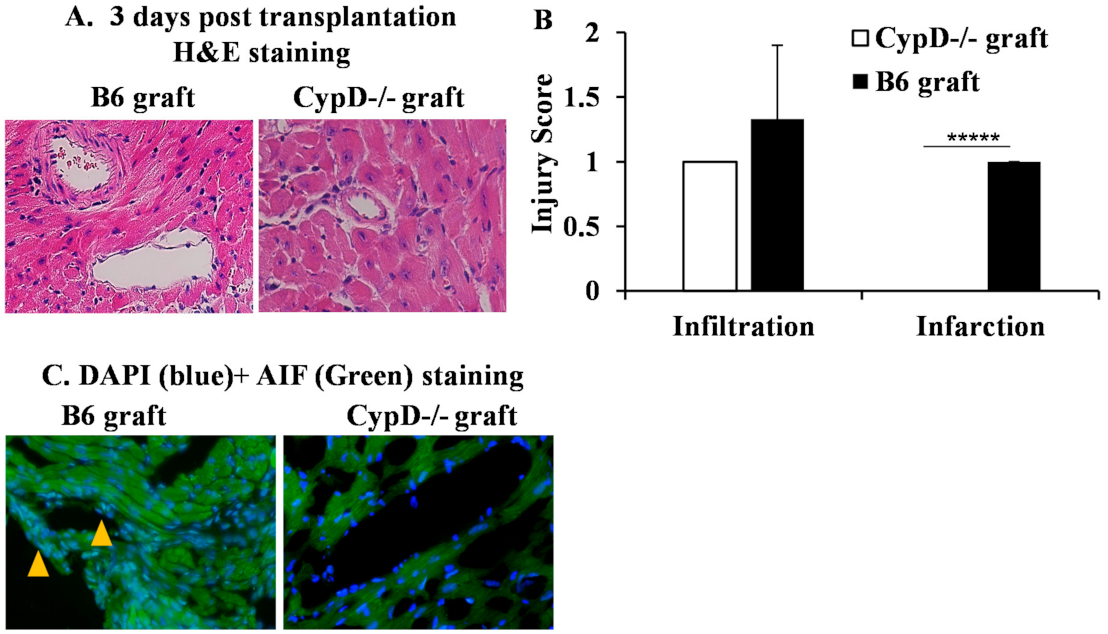
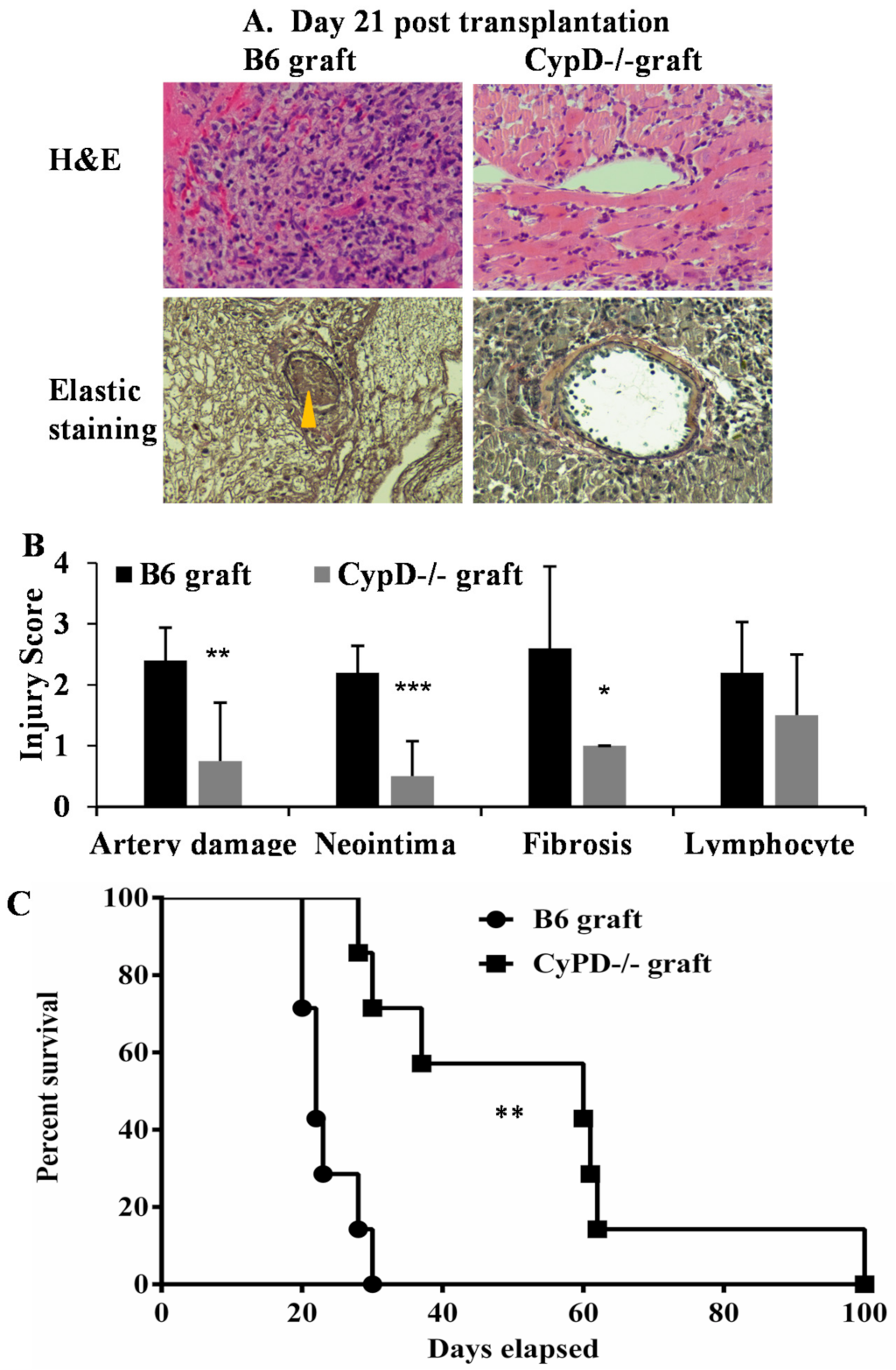
2.4. CypD Deficiency in Donor Heart Graft Attenuates Graft IRI, AIF Translocation and Rejection
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Ethic Statement
4.3. In Vitro Cold Hypoxia and Warm Reoxygenation
4.4. RNA Interference
4.5. PCR
4.6. Western Blots
4.7. Immunocytochemistry
4.8. DNA Analysis
4.9. Cardiac Transplantation
4.10. Histology and Immunohistochemistry
4.11. Inclusion and Exclusion Criteria
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-ABA | 3-aminobanzamide |
| BAX | Bcl-2-associated X. |
| BID | Bcl-2 Homology 3-interacting domain death agonist. |
| CsA | Cyclosporin A. |
| CypD | Cyclophilin D. |
| DAPI | Diamidino-2-phenylindole dihydrochloride |
| MVEC | mouse microvascular endothelial cells. |
| NAC | Nacetyl-L-cysteine. |
| Nec-1s | Necrostatin-1s. |
| PARP-1 | poly(ADP-ribose) polymerase 1 |
| PCR | polymerase chain reaction. |
| RIPK | receptor interacting protein kinase. |
| ROS | reactive oxygen species. |
| TEMPOL | tetramethylpiperidine-oxyl. |
| Z-VAD-fmk | Z-Val-Ala-Asp-fluoromethylketoe. |
References
- Khan, M.A.; Hashim, M.J.; Mustafa, H.; Baniyas, M.Y.; Al Suwaidi, S.; AlKatheeri, R.; Alblooshi, F.M.K.; Almatrooshi, M.; Alzaabi, M.E.H.; Al Darmaki, R.S.; et al. Global Epidemiology of Ischemic Heart Disease: Results from the Global Burden of Disease Study. Cureus 2020, 12, e9349. [Google Scholar] [CrossRef]
- Nowbar, A.N.; Gitto, M.; Howard, J.P.; Francis, D.P.; Al-Lamee, R. Mortality From Ischemic Heart Disease. Circ. Cardiovasc. Qual. Outcomes 2019, 12, e005375. [Google Scholar] [CrossRef]
- Jahania, M.S.; Sanchez, J.A.; Narayan, P.; Lasley, R.D.; Mentzer, R.M., Jr. Heart preservation for transplantation: Principles and strategies. Ann. Thorac. Surg. 1999, 68, 1983–1987. [Google Scholar] [CrossRef]
- Rosenbaum, D.H.; Peltz, M.; DiMaio, J.M.; Meyer, D.M.; Wait, M.A.; Merritt, M.E.; Ring, W.S.; Jessen, M.E. Perfusion preservation versus static preservation for cardiac transplantation: Effects on myocardial function and metabolism. J. Heart Lung Transplant. 2008, 27, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.K.; Kittleson, M.; Kobashigawa, J.A. Cardiac allograft rejection. Surgeon 2011, 9, 160–167. [Google Scholar] [CrossRef]
- Tonsho, M.; Michel, S.; Ahmed, Z.; Alessandrini, A.; Madsen, J.C. Heart transplantation: Challenges facing the field. Cold Spring Harb. Perspect. Med. 2014, 4, a015636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobashigawa, J.; Zuckermann, A.; Macdonald, P.; Leprince, P.; Esmailian, F.; Luu, M.; Mancini, D.; Patel, J.; Razi, R.; Reichenspurner, H.; et al. Report from a consensus conference on primary graft dysfunction after cardiac transplantation. J. Heart Lung Transplant. 2014, 33, 327–340. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, A.; Petrov, L.; Georgieva, A.; Kessiova, M.; Tzvetanova, E.; Kirkova, M.; Kukan, M. Effect of MG132 on proteasome activity and prooxidant/antioxidant status of rat liver subjected to ischemia/reperfusion injury. Hepatol Res. 2008, 38, 393–401. [Google Scholar] [CrossRef]
- Tsivilika, M.; Doumaki, E.; Stavrou, G.; Sioga, A.; Grosomanidis, V.; Meditskou, S.; Maranginos, A.; Tsivilika, D.; Stafylarakis, D.; Kotzampassi, K.; et al. The adaptive immune response in cardiac arrest resuscitation induced ischemia reperfusion renal injury. J. Biol. Res. 2020, 27, 15. [Google Scholar] [CrossRef]
- Bobinac, M.; Celic, T.; Vukelic, I.; Spanjol, J.; Rubinic, N.; Bobinac, D. Nuclear factor erythroid 2-related factor 2 and choline acetyltransferase co-expression in rat spinal cord neurons after ischemia-reperfusion injury. J. Biol. Regul. Homeost. Agents 2018, 32, 803–813. [Google Scholar]
- Rout, A.; Tantry, U.S.; Novakovic, M.; Sukhi, A.; Gurbel, P.A. Targeted pharmacotherapy for ischemia reperfusion injury in acute myocardial infarction. Expert Opin. Pharmacother. 2020, 21, 1851–1865. [Google Scholar] [CrossRef] [PubMed]
- Khush, K.K.; Menza, R.; Nguyen, J.; Zaroff, J.G.; Goldstein, B.A. Donor predictors of allograft use and recipient outcomes after heart transplantation. Circ. Heart Fail. 2013, 6, 300–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef]
- Chen, D.; Ding, Y.; Schroppel, B.; Zhang, N.; Fu, S.; Chen, D.; Zhang, H.; Bromberg, J.S. Differential chemokine and chemokine receptor gene induction by ischemia, alloantigen, and gene transfer in cardiac grafts. Am. J. Transplant. 2003, 3, 1216–1229. [Google Scholar] [CrossRef]
- Toledo-Pereyra, L.H.; Lopez-Neblina, F.; Toledo, A.H. Reactive oxygen species and molecular biology of ischemia/reperfusion. Ann. Transplant. 2004, 9, 81–83. [Google Scholar]
- Millar, T.M.; Phan, V.; Tibbles, L.A. ROS generation in endothelial hypoxia and reoxygenation stimulates MAP kinase signaling and kinase-dependent neutrophil recruitment. Free Radic. Biol. Med. 2007, 42, 1165–1177. [Google Scholar] [CrossRef]
- Yang, Q.; He, G.W.; Underwood, M.J.; Yu, C.M. Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: Perspectives and implications for postischemic myocardial protection. Am. J. Transl. Res. 2016, 8, 765–777. [Google Scholar] [PubMed]
- Rifle, G.; Mousson, C.; Herve, P. Endothelial cells in organ transplantation: Friends or foes? Transplantation 2006, 82, S4–S5. [Google Scholar] [CrossRef] [PubMed]
- Singhal, A.K.; Symons, J.D.; Boudina, S.; Jaishy, B.; Shiu, Y.T. Role of Endothelial Cells in Myocardial Ischemia-Reperfusion Injury. Vasc. Dis. Prev. 2010, 7, 1–14. [Google Scholar] [CrossRef]
- Duni, A.; Liakopoulos, V.; Koutlas, V.; Pappas, C.; Mitsis, M.; Dounousi, E. The Endothelial Glycocalyx as a Target of Ischemia and Reperfusion Injury in Kidney Transplantation-Where Have We Gone So Far? Int. J. Mol. Sci. 2021, 22, 2157. [Google Scholar] [CrossRef]
- Huck, V.; Niemeyer, A.; Goerge, T.; Schnaeker, E.M.; Ossig, R.; Rogge, P.; Schneider, M.F.; Oberleithner, H.; Schneider, S.W. Delay of acute intracellular pH recovery after acidosis decreases endothelial cell activation. J. Cell Physiol. 2007, 211, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, G.; Flather, M.D.; Bohm, M.; Cohen-Solal, A.; Murrone, A.; Mascagni, F.; Spinucci, G.; Conti, M.G.; van Veldhuisen, D.J.; Tavazzi, L.; et al. beta-blockade with nebivolol for prevention of acute ischaemic events in elderly patients with heart failure. Heart 2011, 97, 209–214. [Google Scholar] [CrossRef]
- Ross, R. The pathogenesis of atherosclerosis: A perspective for the 1990s. Nature 1993, 362, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.N. Graft vascular disease: Immune response meets the vessel wall. Annu. Rev. Pathol. 2009, 4, 19–47. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Hackl, M.J.; Kunzendorf, U.; Walczak, H.; Krautwald, S.; Jevnikar, A.M. Necroptosis in immunity and ischemia-reperfusion injury. Am. J. Transplant. 2013, 13, 2797–2804. [Google Scholar] [CrossRef]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef]
- Kwok, C.; Pavlosky, A.; Lian, D.; Jiang, J.; Huang, X.; Yin, Z.; Liu, W.; Haig, A.; Jevnikar, A.; Zhang, Z.X. Necroptosis is Involved in CD4+ T-cell Mediated Microvascular Endothelial Cell Death and Chronic Cardiac Allograft Rejection. Transplantation 2017, 101, 2026–2037. [Google Scholar] [CrossRef]
- Pavlosky, A.; Lau, A.; Su, Y.; Lian, D.; Huang, X.; Yin, Z.; Haig, A.; Jevnikar, A.M.; Zhang, Z.X. RIPK3-Mediated Necroptosis Regulates Cardiac Allograft Rejection. Am. J. Transplant. 2014, 14, 1778–1790. [Google Scholar] [CrossRef] [Green Version]
- Gan, I.; Jiang, J.; Lian, D.; Huang, X.; Fuhrmann, B.; Liu, W.; Haig, A.; Jevnikar, A.M.; Zhang, Z.X. Mitochondrial permeability regulates cardiac endothelial cell necroptosis and cardiac allograft rejection. Am. J. Transplant. 2019, 19, 686–698. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Gan, I.; Pavlosky, A.; Huang, X.; Fuhrmann, B.; Jevnikar, A.M. Intracellular pH Regulates TRAIL-Induced Apoptosis and Necroptosis in Endothelial Cells. J. Immunol. Res. 2017, 2017, 1503960. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.; Wang, S.; Jiang, J.; Haig, A.; Pavlosky, A.; Linkermann, A.; Zhang, Z.X.; Jevnikar, A.M. RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am. J. Transplant. 2013, 13, 2805–2818. [Google Scholar] [CrossRef]
- Marshall, K.D.; Baines, C.P. Necroptosis: Is there a role for mitochondria? Front. Physiol. 2014, 5, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Jaffer, T.; Eguchi, S.; Wang, Z.; Linkermann, A.; Ma, D. Role of necroptosis in the pathogenesis of solid organ injury. Cell Death Dis. 2015, 6, e1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Manning, G. Necroptosis and Inflammation. Annu. Rev. Biochem. 2016, 85, 743–763. [Google Scholar] [CrossRef]
- Kinnally, K.W.; Peixoto, P.M.; Ryu, S.Y.; Dejean, L.M. Is mPTP the gatekeeper for necrosis, apoptosis, or both? Biochim. Biophys. Acta 2011, 1813, 616–622. [Google Scholar] [CrossRef] [Green Version]
- Tait, S.W.; Oberst, A.; Quarato, G.; Milasta, S.; Haller, M.; Wang, R.; Karvela, M.; Ichim, G.; Yatim, N.; Albert, M.L.; et al. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 2013, 5, 878–885. [Google Scholar] [CrossRef]
- Ying, Y.; Padanilam, B.J. Regulation of necrotic cell death: p53, PARP1 and cyclophilin D-overlapping pathways of regulated necrosis? Cell Mol. Life Sci. 2016, 73, 2309–2324. [Google Scholar] [CrossRef]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Brasen, J.H.; Darding, M.; Jin, M.K.; Sanz, A.B.; Heller, J.O.; De, Z.F.; Weinlich, R.; Ortiz, A.; Walczak, H.; et al. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2013, 110, 12024–12029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Joza, N.; Susin, S.A.; Daugas, E.; Stanford, W.L.; Cho, S.K.; Li, C.Y.; Sasaki, T.; Elia, A.J.; Cheng, H.Y.; Ravagnan, L.; et al. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 2001, 410, 549–554. [Google Scholar] [CrossRef]
- Bano, D.; Prehn, J.H.M. Apoptosis-Inducing Factor (AIF) in Physiology and Disease: The Tale of a Repented Natural Born Killer. EBioMedicine 2018, 30, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001, 412, 95–99. [Google Scholar] [CrossRef]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef]
- Moubarak, R.S.; Yuste, V.J.; Artus, C.; Bouharrour, A.; Greer, P.A.; Menissier-de Murcia, J.; Susin, S.A. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol. Cell Biol. 2007, 27, 4844–4862. [Google Scholar] [CrossRef] [Green Version]
- Landshamer, S.; Hoehn, M.; Barth, N.; Duvezin-Caubet, S.; Schwake, G.; Tobaben, S.; Kazhdan, I.; Becattini, B.; Zahler, S.; Vollmar, A.; et al. Bid-induced release of AIF from mitochondria causes immediate neuronal cell death. Cell Death Differ. 2008, 15, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Culmsee, C.; Zhu, C.; Landshamer, S.; Becattini, B.; Wagner, E.; Pellecchia, M.; Blomgren, K.; Plesnila, N. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J. NeuroSci. 2005, 25, 10262–10272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vahsen, N.; Cande, C.; Briere, J.J.; Benit, P.; Joza, N.; Larochette, N.; Mastroberardino, P.G.; Pequignot, M.O.; Casares, N.; Lazar, V.; et al. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004, 23, 4679–4689. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.; Yu, B.D.; Joza, N.; Benit, P.; Meneses, J.; Firpo, M.; Rustin, P.; Penninger, J.M.; Martin, G.R. Loss of Aif function causes cell death in the mouse embryo, but the temporal progression of patterning is normal. Proc. Natl. Acad. Sci. USA 2006, 103, 9918–9923. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Lesnefsky, E.J. Heart mitochondria and calpain 1: Location, function, and targets. Biochim. Biophys. Acta 2015, 1852, 2372–2378. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.; Hu, Y.; Lesnefsky, E.J.; Chen, Q. Activation of mitochondrial calpain and increased cardiac injury: Beyond AIF release. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H376–H384. [Google Scholar] [CrossRef] [Green Version]
- Polster, B.M.; Basanez, G.; Etxebarria, A.; Hardwick, J.M.; Nicholls, D.G. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J. Biol. Chem. 2005, 280, 6447–6454. [Google Scholar] [CrossRef] [Green Version]
- Ozaki, T.; Tomita, H.; Tamai, M.; Ishiguro, S. Characteristics of mitochondrial calpains. J. Biochem. 2007, 142, 365–376. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, N.S.; Li, X.; Greer, P.A.; Koehler, R.C.; Dawson, V.L.; Dawson, T.M. Calpain activation is not required for AIF translocation in PARP-1-dependent cell death (parthanatos). J. Neurochem. 2009, 110, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, J.; Song, X.; Qu, L.; Wei, R.; He, F.; Wang, K.; Luo, B. RIP3 induces ischemic neuronal DNA degradation and programmed necrosis in rat via AIF. Sci. Rep. 2016, 6, 29362. [Google Scholar] [CrossRef]
- Delavallee, L.; Cabon, L.; Galan-Malo, P.; Lorenzo, H.K.; Susin, S.A. AIF-mediated caspase-independent necroptosis: A new chance for targeted therapeutics. IUBMB Life 2011, 63, 221–232. [Google Scholar] [CrossRef]
- Cabon, L.; Galan-Malo, P.; Bouharrour, A.; Delavallee, L.; Brunelle-Navas, M.N.; Lorenzo, H.K.; Gross, A.; Susin, S.A. BID regulates AIF-mediated caspase-independent necroptosis by promoting BAX activation. Cell Death Differ. 2012, 19, 245–256. [Google Scholar] [CrossRef]
- Fukui, M.; Choi, H.J.; Zhu, B.T. Rapid generation of mitochondrial superoxide induces mitochondrion-dependent but caspase-independent cell death in hippocampal neuronal cells that morphologically resembles necroptosis. Toxicol. Appl. Pharmacol. 2012, 262, 156–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; An, R.; Umanah, G.K.; Park, H.; Nambiar, K.; Eacker, S.M.; Kim, B.; Bao, L.; Harraz, M.M.; Chang, C.; et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science 2016, 354, aad6872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasola, A.; Sciacovelli, M.; Pantic, B.; Bernardi, P. Signal transduction to the permeability transition pore. FEBS Lett. 2010, 584, 1989–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basso, E.; Fante, L.; Fowlkes, J.; Petronilli, V.; Forte, M.A.; Bernardi, P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem. 2005, 280, 18558–18561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, L.; Raisky, O.; Chalabreysse, L.; Verschelde, C.; Bonnefoy-Berard, N.; Ovize, M. Link between immune cell infiltration and mitochondria-induced cardiomyocyte death during acute cardiac graft rejection. Am. J. Transplant. 2006, 6, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Raisky, O.; Gomez, L.; Chalabreysse, L.; Gateau-Roesch, O.; Loufouat, J.; Thivolet-Bejui, F.; Ninet, J.; Ovize, M. Mitochondrial permeability transition in cardiomyocyte apoptosis during acute graft rejection. Am. J. Transplant. 2004, 4, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qamar, A.; Zhao, J.; Xu, L.; McLeod, P.; Huang, X.; Jiang, J.; Liu, W.; Haig, A.; Zhang, Z.-X. Cyclophilin D Regulates the Nuclear Translocation of AIF, Cardiac Endothelial Cell Necroptosis and Murine Cardiac Transplant Injury. Int. J. Mol. Sci. 2021, 22, 11038. https://doi.org/10.3390/ijms222011038
Qamar A, Zhao J, Xu L, McLeod P, Huang X, Jiang J, Liu W, Haig A, Zhang Z-X. Cyclophilin D Regulates the Nuclear Translocation of AIF, Cardiac Endothelial Cell Necroptosis and Murine Cardiac Transplant Injury. International Journal of Molecular Sciences. 2021; 22(20):11038. https://doi.org/10.3390/ijms222011038
Chicago/Turabian StyleQamar, Adnan, Jianqi Zhao, Laura Xu, Patrick McLeod, Xuyan Huang, Jifu Jiang, Weihua Liu, Aaron Haig, and Zhu-Xu Zhang. 2021. "Cyclophilin D Regulates the Nuclear Translocation of AIF, Cardiac Endothelial Cell Necroptosis and Murine Cardiac Transplant Injury" International Journal of Molecular Sciences 22, no. 20: 11038. https://doi.org/10.3390/ijms222011038
APA StyleQamar, A., Zhao, J., Xu, L., McLeod, P., Huang, X., Jiang, J., Liu, W., Haig, A., & Zhang, Z.-X. (2021). Cyclophilin D Regulates the Nuclear Translocation of AIF, Cardiac Endothelial Cell Necroptosis and Murine Cardiac Transplant Injury. International Journal of Molecular Sciences, 22(20), 11038. https://doi.org/10.3390/ijms222011038