
PACAP and VIP Modulate LPS-Induced Microglial Activation and Trigger Distinct Phenotypic Changes in Murine BV2 Microglial Cells

 ,
,  , , ,
, , , 
Abstract
1. Introduction
2. Results
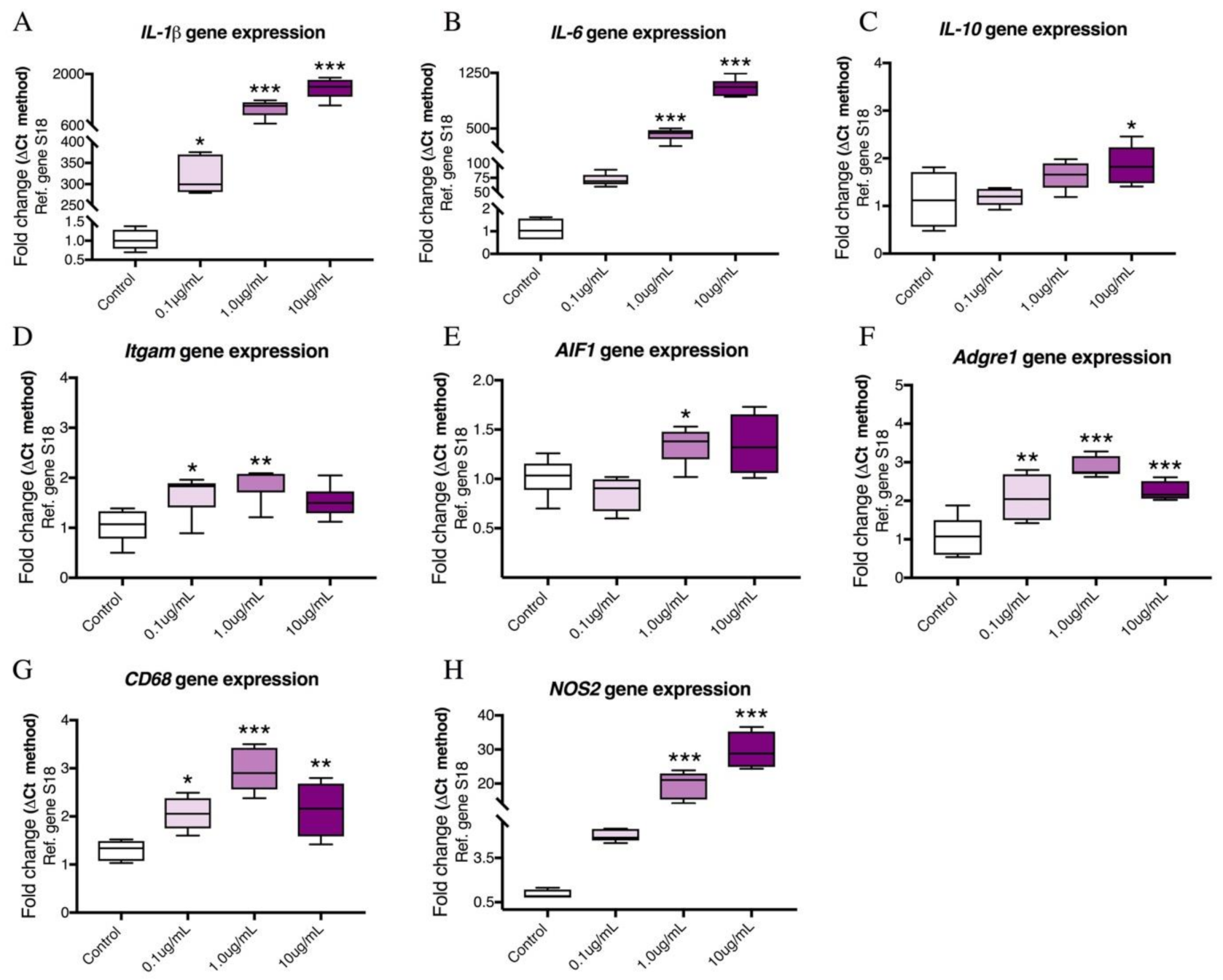
2.1. Dose–Response Effects of LPS on the mRNA Expression of Pro-Inflammatory Cytokines and Microglia Activation Markers in BV2 Cells
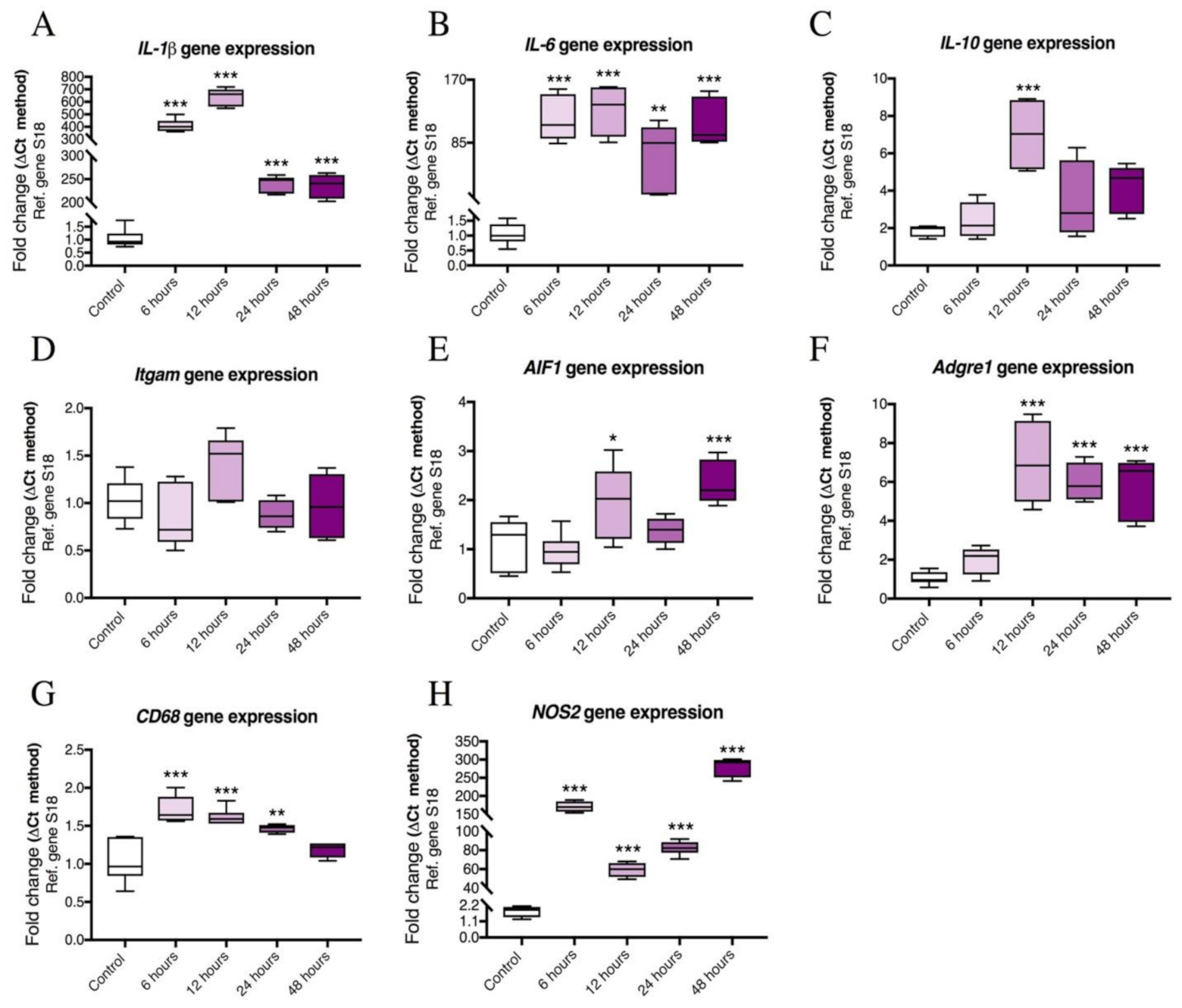
2.2. Time-Course Analyses Showing Temporal Gene Expression Changes of Pro-Inflammatory Cytokines and Microglial Activation Markers in LPS-treated BV2 Cells
2.3. Expression of PACAP, VIP and Receptor Transcripts in BV2 Cells Exposed to LPS
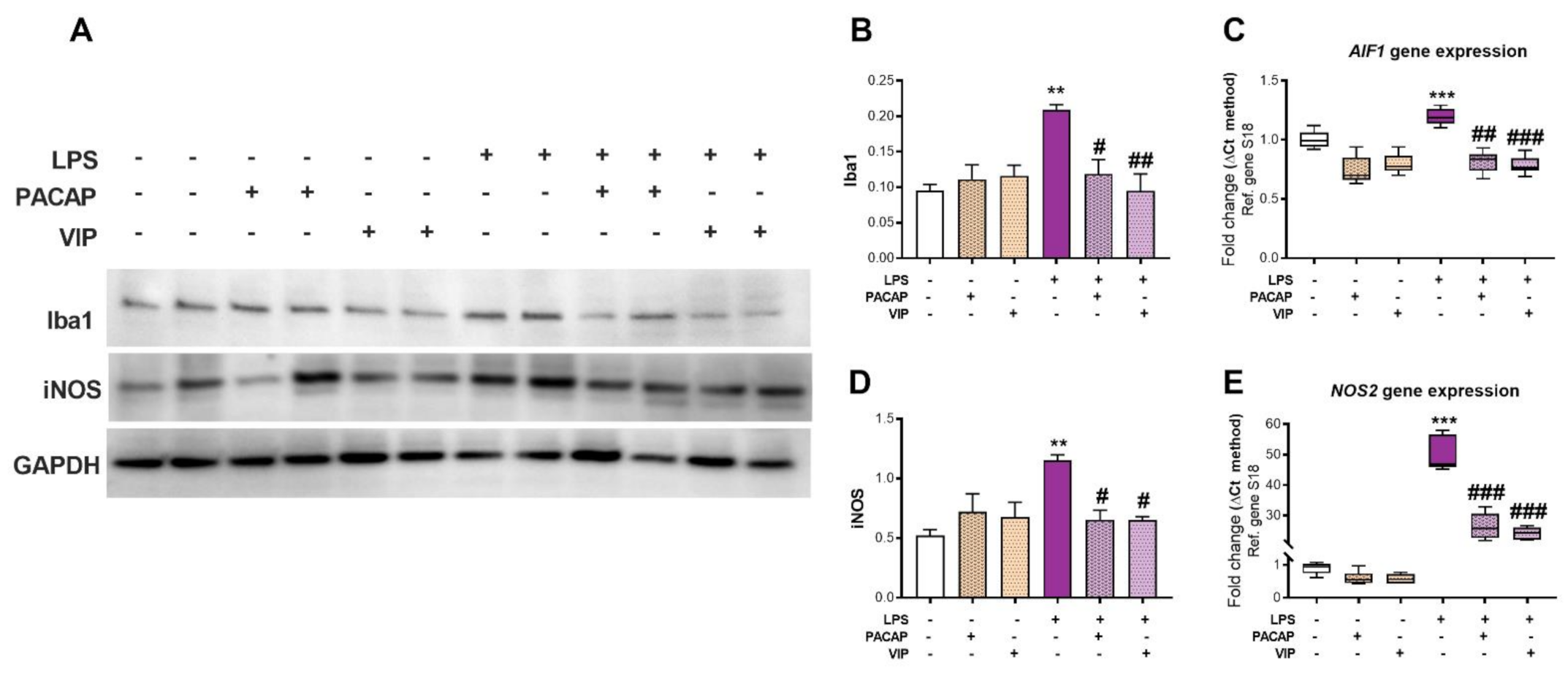
2.4. PACAP or VIP Treatment Rescue LPS-Induced AIF1/Iba1 and NOS2/iNOS Levels in BV2 Microglial Cells
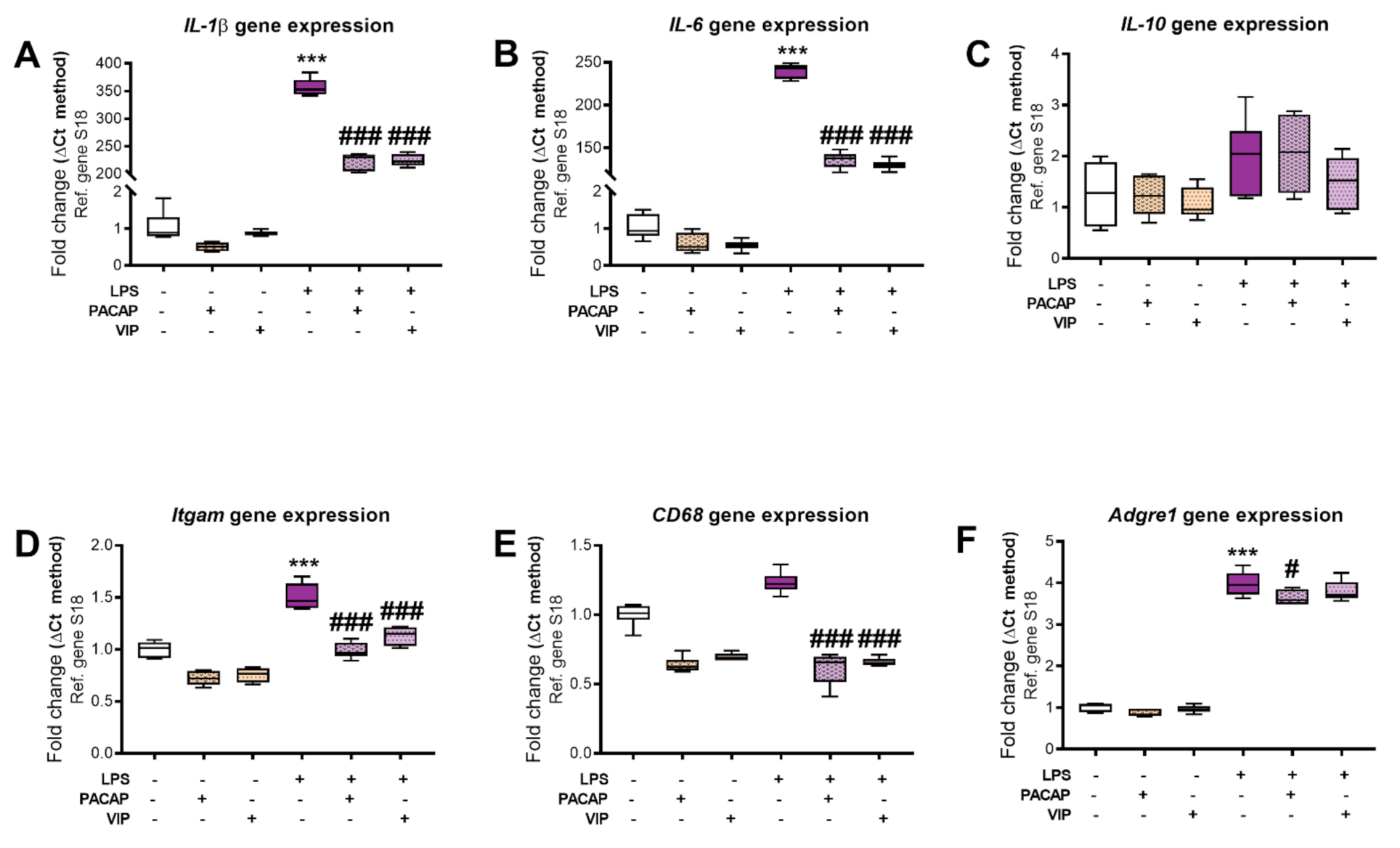
2.5. PACAP or VIP Treatment Reduce the Expression of pro-Inflammatory Cytokines in BV2 Cells Exposed to LPS
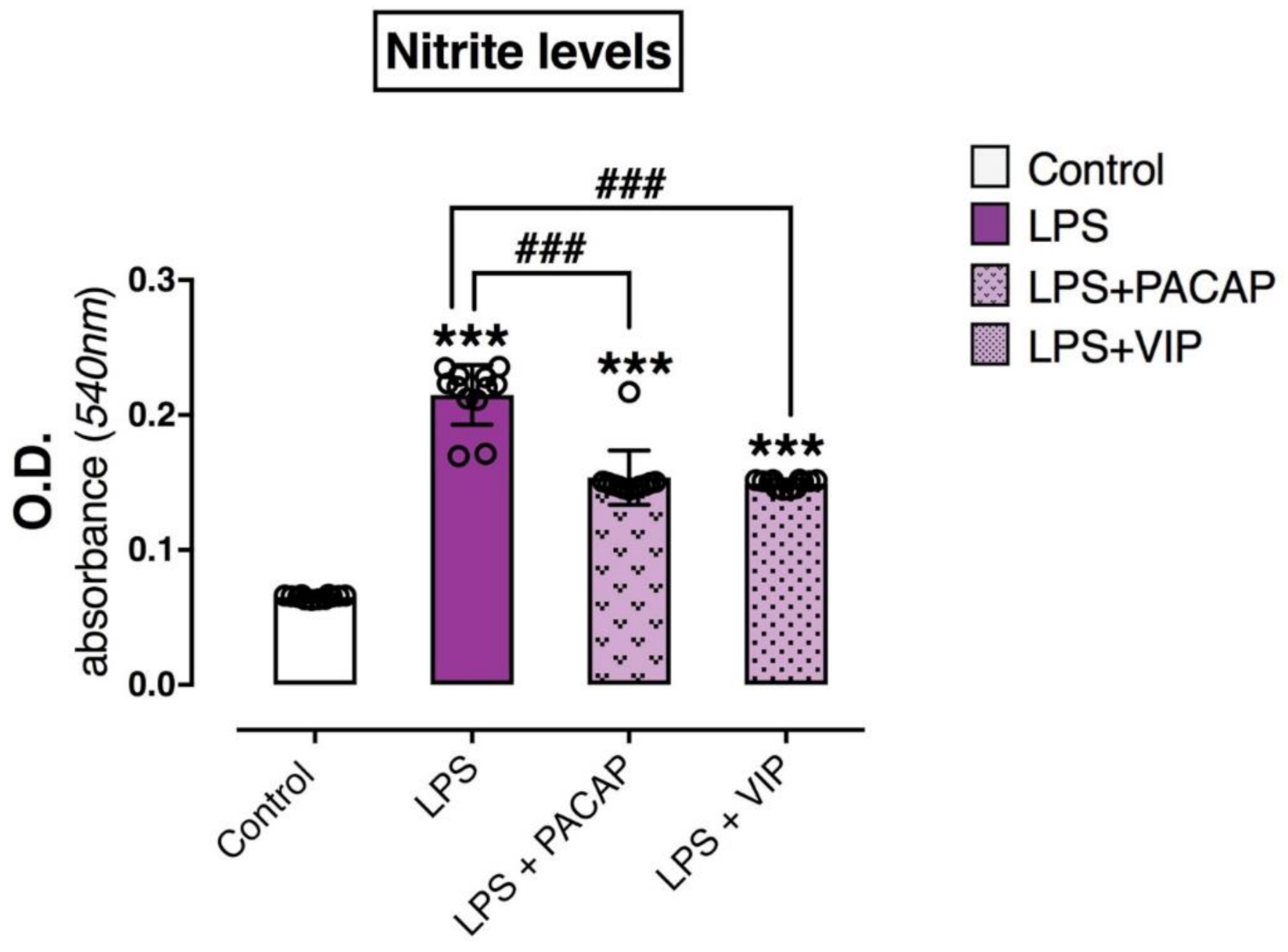
2.6. PACAP or VIP Treatment Reduce the Release of Nitrites in the Culture Media
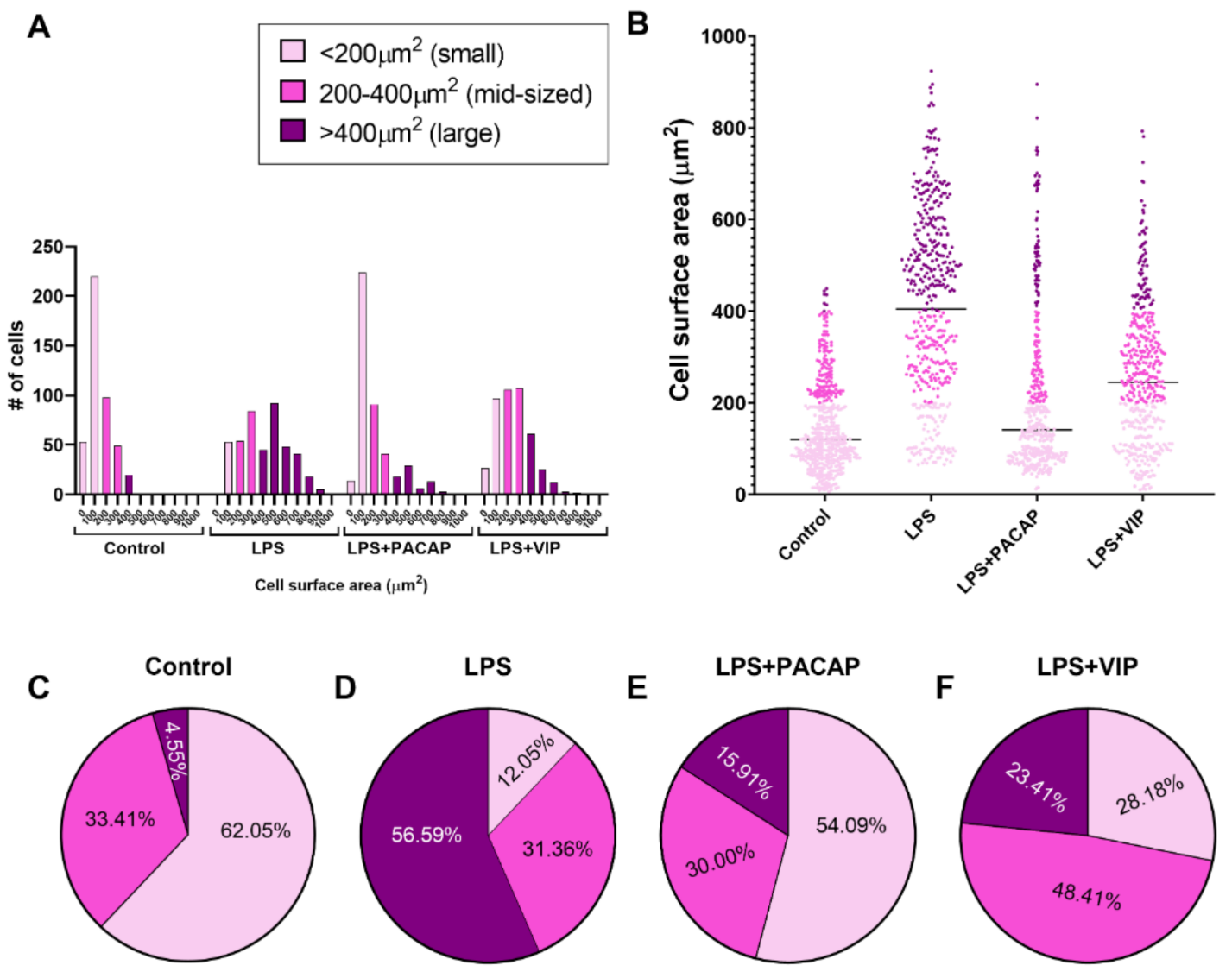
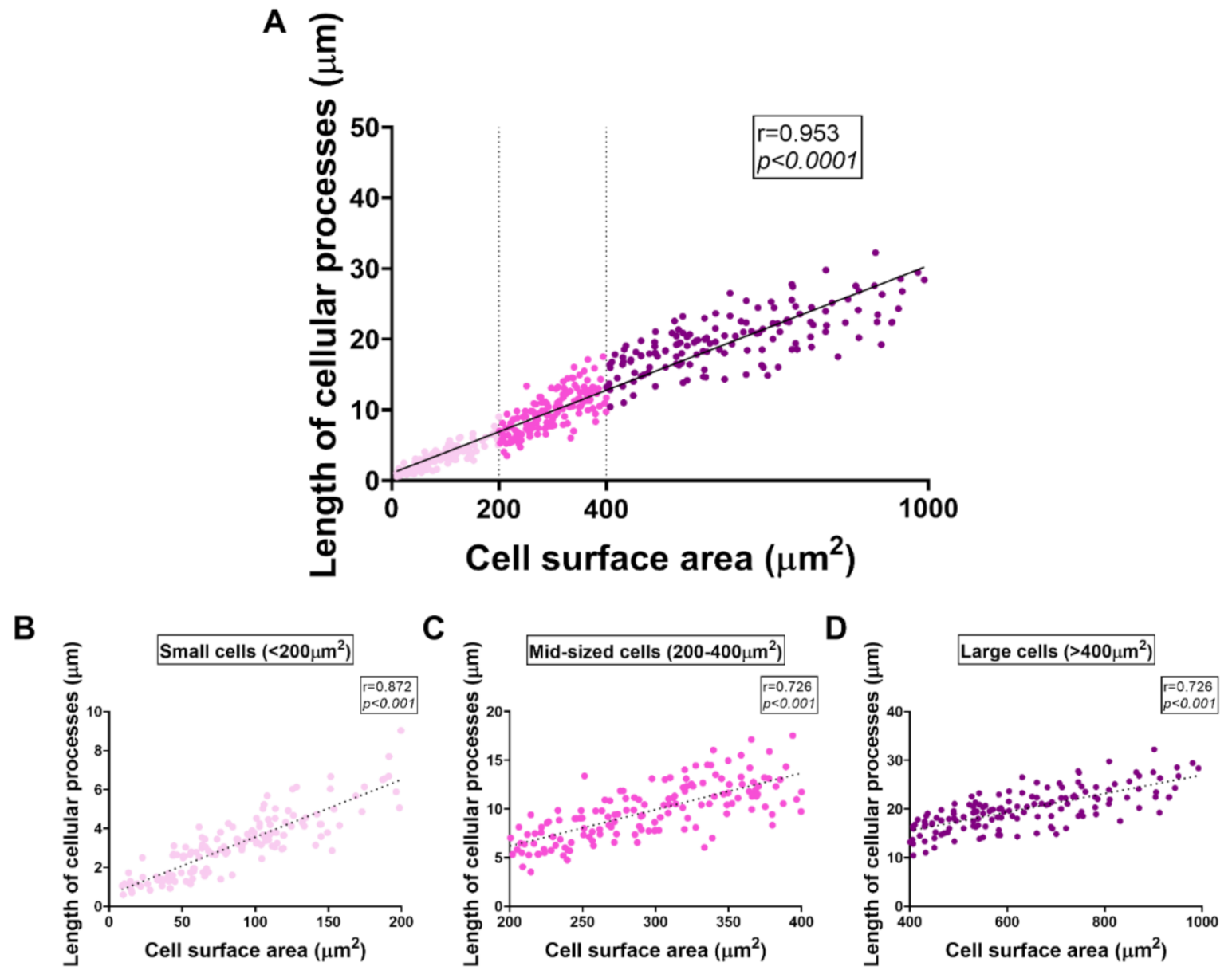
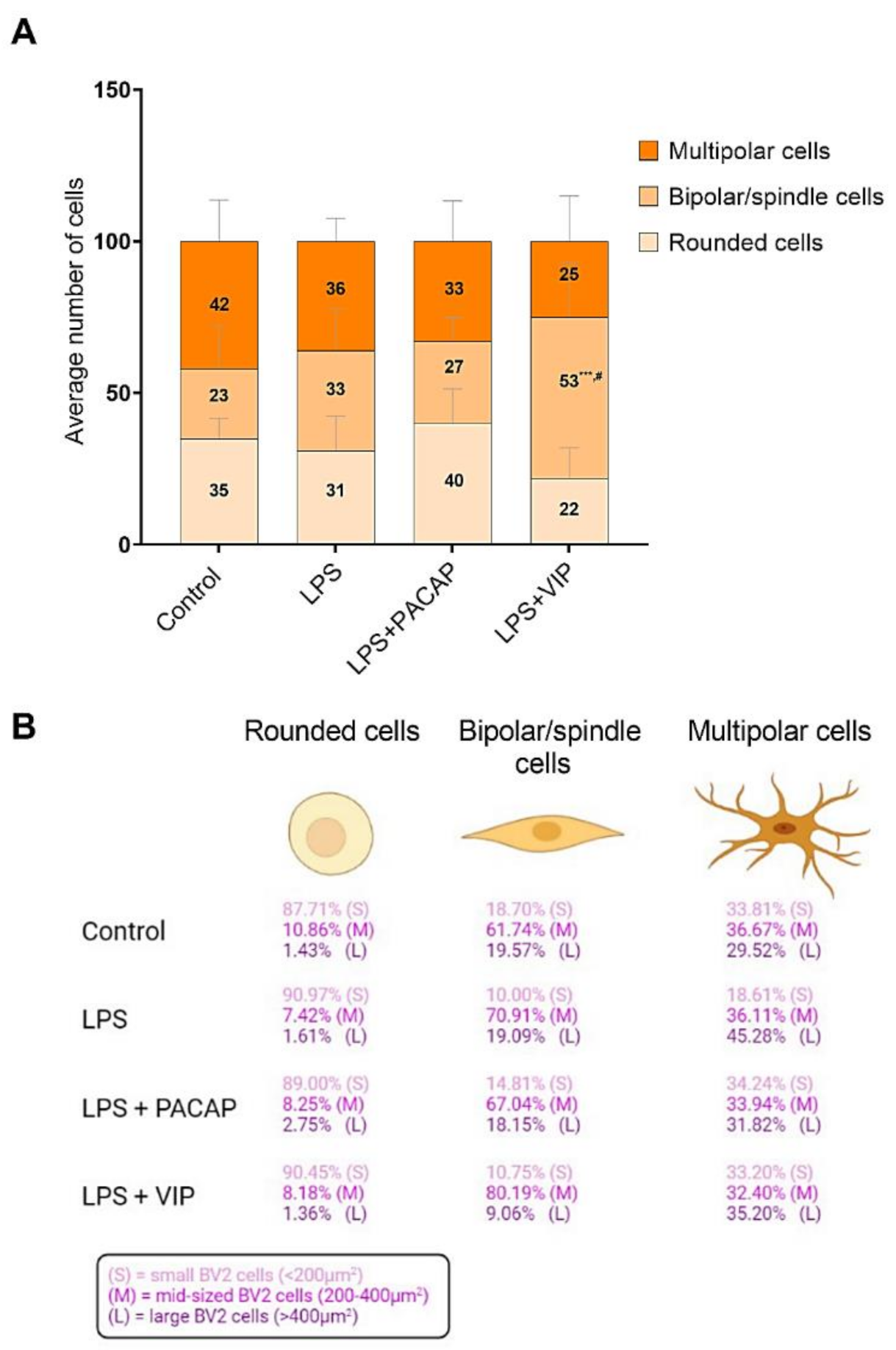
2.7. Distinct Effects of PACAP or VIP Treatment on LPS-Stimulated BV2 Cell Morphology
2.8. VIP but Not PACAP Treatment Causes the Phenotypic Shift of BV2 Cells towards Mid-Sized Spindle/Bipolar-Shaped Cells after LPS Challenge
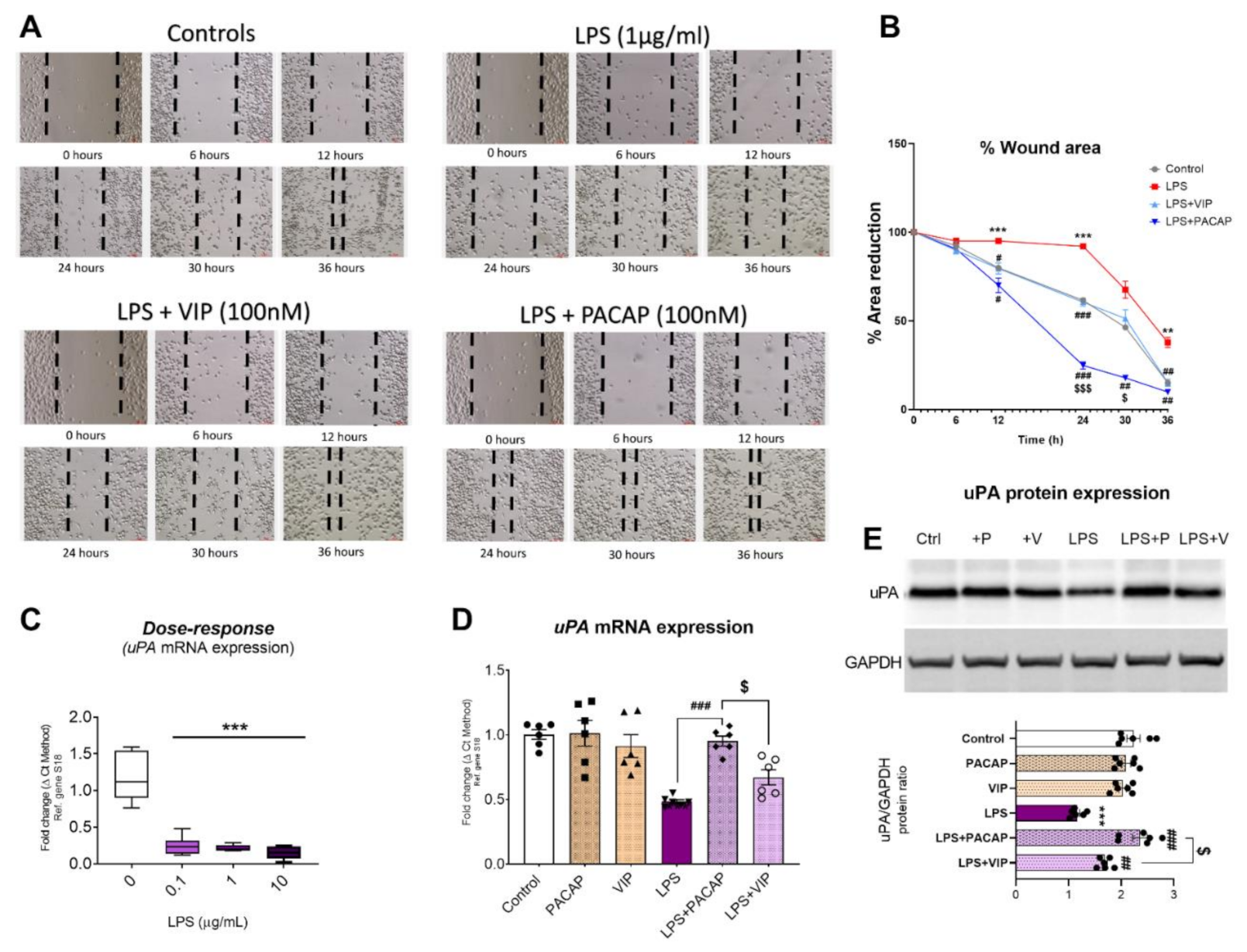
2.9. PACAP and VIP Differentially affect BV2 Cell Motility and Urokinase Plasminogen Activator (uPA) Expression after LPS Challenge
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Quantitative Real Time Polymerase Chain Reaction
4.3. Western Blot Analysis
4.4. Immunocytochemistry
4.5. Nitrite Assay
4.6. Morphological Analyses
4.7. Wound Healing Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nakagawa, Y.; Chiba, K. Role of microglial m1/m2 polarization in relapse and remission of psychiatric disorders and diseases. Pharmaceuticals 2014, 7, 1028–1048. [Google Scholar] [CrossRef] [PubMed]
- Thameem Dheen, S.; Kaur, C.; Ling, E.-A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Blander, J.M.; Medzhitov, R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature 2006, 440, 808. [Google Scholar] [CrossRef]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple tlrs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef] [PubMed]
- Caito, S.W.; Milatovic, D.; Hill, K.E.; Aschner, M.; Burk, R.F.; Valentine, W.M. Progression of neurodegeneration and morphologic changes in the brains of juvenile mice with selenoprotein p deleted. Brain Res. 2011, 1398, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gorman, A.M. Neuronal cell death in neurodegenerative diseases: Recurring themes around protein handling. J. Cell. Mol. Med. 2008, 12, 2263–2280. [Google Scholar] [CrossRef] [PubMed]
- Procaccini, C.; Santopaolo, M.; Faicchia, D.; Colamatteo, A.; Formisano, L.; De Candia, P.; Galgani, M.; De Rosa, V.; Matarese, G. Role of metabolism in neurodegenerative disorders. Metabolism 2016, 65, 1376–1390. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory response in the cns: Friend or foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef]
- Block, M.L.; Hong, J.-S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98. [Google Scholar] [CrossRef]
- Gao, H.-M.; Hong, J.-S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef]
- Carniglia, L.; Ramirez, D.; Durand, D.; Saba, J.; Turati, J.; Caruso, C.; Scimonelli, T.N.; Lasaga, M. Neuropeptides and microglial activation in inflammation, pain, and neurodegenerative diseases. Mediat. Inflamm. 2017, 2017, 23. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A.; Mott, R.T.; Sharpe, H.; Xu, Q.; Van Nostrand, W.E.; Vitek, M.P. Expression profiles for macrophage alternative activation genes in ad and in mouse models of ad. J. Neuroinflammation 2006, 3, 27. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hong, H.; Kim, B.S.; Im, H.-I. Pathophysiological role of neuroinflammation in neurodegenerative diseases and psychiatric disorders. Int. Neurourol. J. 2016, 20, S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Muccigrosso, M.M.; Godbout, J.P. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic cns injury, and neurodegenerative disease. Neuropharmacology 2015, 96, 29–41. [Google Scholar] [CrossRef]
- Ponomarev, E.D.; Maresz, K.; Tan, Y.; Dittel, B.N. Cns-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J. Neurosci. 2007, 27, 10714–10721. [Google Scholar] [CrossRef]
- Pott Godoy, M.C.; Tarelli, R.; Ferrari, C.C.; Sarchi, M.I.; Pitossi, F.J. Central and systemic il-1 exacerbates neurodegeneration and motor symptoms in a model of parkinson’s disease. Brain A J. Neurol. 2008, 131, 1880–1894. [Google Scholar] [CrossRef]
- Rojo, A.I.; Innamorato, N.G.; Martín-Moreno, A.M.; De Ceballos, M.L.; Yamamoto, M.; Cuadrado, A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental parkinson’s disease. Glia 2010, 58, 588–598. [Google Scholar] [CrossRef]
- Smith, P.F. Inflammation in parkinson’s disease: An update. Curr. Opin. Investig. Drugs 2008, 9, 478–484. [Google Scholar]
- Bernier, L.P.; Bohlen, C.J.; York, E.M.; Choi, H.B.; Kamyabi, A.; Dissing-Olesen, L.; Hefendehl, J.K.; Collins, H.Y.; Stevens, B.; Barres, B.A.; et al. Nanoscale surveillance of the brain by microglia via camp-regulated filopodia. Cell Rep. 2019, 27, 2895–2908. [Google Scholar] [CrossRef]
- Town, T.; Nikolic, V.; Tan, J. The microglial “activation” continuum: From innate to adaptive responses. J. Neuroinflamm. 2005, 2, 24. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential roles of m1 and m2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Stertz, L.; Magalhães, P.V.S.; Kapczinski, F. Is bipolar disorder an inflammatory condition? The relevance of microglial activation. Curr. Opin. Psychiatry 2013, 26, 19–26. [Google Scholar] [CrossRef]
- Colton, C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef]
- Laskin, D.L. Macrophages and inflammatory mediators in chemical toxicity: A battle of forces. Chem. Res. Toxicol. 2009, 22, 1376–1385. [Google Scholar] [CrossRef]
- Hernangómez, M.; Carrillo-Salinas, F.J.; Mecha, M.; Correa, F.; Mestre, L.; Loría, F.; Feliú, A.; Docagne, F.; Guaza, C. Brain innate immunity in the regulation of neuroinflammation: Therapeutic strategies by modulating cd200-cd200r interaction involve the cannabinoid system. Curr. Pharm. Des. 2014, 20, 4707–4722. [Google Scholar] [CrossRef]
- Bogie, J.F.; Stinissen, P.; Hellings, N.; Hendriks, J.J. Myelin-phagocytosing macrophages modulate autoreactive t cell proliferation. J. Neuroinflamm. 2011, 8, 85. [Google Scholar] [CrossRef]
- Cagnin, A.; Kassiou, M.; Meikle, S.; Banati, R. In vivo evidence for microglial activation in neurodegenerative dementia. Acta Neurol. Scand. 2006, 114, 107–114. [Google Scholar] [CrossRef]
- Trapp, B.D.; Peterson, J.; Ransohoff, R.M.; Rudick, R.; Mörk, S.; Bö, L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998, 338, 278–285. [Google Scholar] [CrossRef]
- Bai, L.; Zhang, X.; Li, X.; Liu, N.; Lou, F.; Ma, H.; Luo, X.; Ren, Y. Somatostatin prevents lipopolysaccharide-induced neurodegeneration in the rat substantia nigra by inhibiting the activation of microglia. Mol. Med. Rep. 2015, 12, 1002–1008. [Google Scholar] [CrossRef]
- Castorina, A.; Scuderi, S.; D’Amico, A.G.; Drago, F.; D’Agata, V. Pacap and vip increase the expression of myelin-related proteins in rat schwannoma cells: Involvement of pac1/vpac2 receptor-mediated activation of pi3k/akt signaling pathways. Exp. Cell Res. 2014, 322, 108–121. [Google Scholar] [CrossRef]
- Castorina, A.; Tiralongo, A.; Giunta, S.; Carnazza, M.L.; Rasi, G.; D’Agata, V. Pacap and vip prevent apoptosis in schwannoma cells. Brain Res. 2008, 1241, 29–35. [Google Scholar] [CrossRef]
- Dejda, A.; Sokolowska, P.; Nowak, J.Z. Neuroprotective potential of three neuropeptides pacap, vip and phi. Pharmacol. Rep. Pr. 2005, 57, 307–320. [Google Scholar]
- Delgado, M.; Jonakait, G.M.; Ganea, D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit chemokine production in activated microglia. Glia 2002, 39, 148–161. [Google Scholar] [CrossRef]
- Gozes, I.; Brenneman, D.E. Vip: Molecular biology and neurobiological function. Mol. Neurobiol. 1989, 3, 201–236. [Google Scholar] [CrossRef]
- Ringer, C.; Büning, L.-S.; Schäfer, M.K.; Eiden, L.E.; Weihe, E.; Schütz, B. Pacap signaling exerts opposing effects on neuroprotection and neuroinflammation during disease progression in the sod1 (g93a) mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2013, 54, 32–42. [Google Scholar] [CrossRef]
- Waschek, J. Vip and pacap: Neuropeptide modulators of cns inflammation, injury, and repair. Br. J. Pharmacol. 2013, 169, 512–523. [Google Scholar] [CrossRef]
- Nowak, J.Z.; Zawilska, J.B. Pacap in avians: Origin, occurrence, and receptors-pharmacological and functional considerations. Curr. Pharm. Des. 2003, 9, 467–481. [Google Scholar] [CrossRef]
- Sherwood, N.M.; Krueckl, S.L.; McRory, J.E. The origin and function of the pituitary adenylate cyclase-activating polypeptide (pacap)/glucagon superfamily. Endocr. Rev. 2000, 21, 619–670. [Google Scholar]
- Delgado, M.; Leceta, J.; Ganea, D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit the production of inflammatory mediators by activated microglia. J. Leukoc. Biol. 2003, 73, 155–164. [Google Scholar] [CrossRef]
- Grimaldi, M.; Cavallaro, S. Functional and molecular diversity of pacap/vip receptors in cortical neurons and type i astrocytes. Eur. J. Neurosci. 1999, 11, 2767–2772. [Google Scholar] [CrossRef]
- Lee, M.; Lelièvre, V.; Zhao, P.; Torres, M.; Rodriguez, W.; Byun, J.-Y.; Doshi, S.; Ioffe, Y.; Gupta, G.; De Los Monteros, A.E. Pituitary adenylyl cyclase-activating polypeptide stimulates DNA synthesis but delays maturation of oligodendrocyte progenitors. J. Neurosci. 2001, 21, 3849–3859. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Shi, Q.-D.; Song, T.-B.; Feng, G.-F.; Zang, W.-J.; Zong, C.-H.; Chang, L. Vasoactive intestinal peptide increases vegf expression to promote proliferation of brain vascular endothelial cells via the camp/pka pathway after ischemic insult in vitro. Peptides 2013, 42, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, D.L.; Lorton, D.; Brouxhon, S.; Felten, S.; Felten, D.L. The significance of vasoactive intestinal polypeptide (vip) in immunomodulation. Adv. Neuroimmunol. 1996, 6, 5–27. [Google Scholar] [CrossRef]
- De la Fuente, M.; Delgado, M.; Gomariz, R.P. Vip modulation of immune cell functions. Adv. Neuroimmunol. 1996, 6, 75–91. [Google Scholar] [CrossRef]
- Ganea, D. Regulatory effects of vasoactive intestinal peptide on cytokine production in central and peripheral lymphoid organs. Adv. Neuroimmunol. 1996, 6, 61–74. [Google Scholar] [CrossRef]
- Martínez, C.; Delgado, M.; Pozo, D.; Leceta, J.; Calvo, J.R.; Ganea, D.; Gomariz, R.P. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide modulate endotoxin-induced il-6 production by murine peritoneal macrophages. J. Leukoc. Biol. 1998, 63, 591–601. [Google Scholar] [CrossRef]
- Delgado, M.; Ganea, D. Neuroprotective effect of vasoactive intestinal peptide (vip) in a mouse model of parkinson’s disease by blocking microglial activation. FASEB J. 2003, 17, 944–946. [Google Scholar] [CrossRef]
- Delgado, M.; Ganea, D. Vasoactive intestinal peptide prevents activated microglia-induced neurodegeneration under inflammatory conditions: Potential therapeutic role in brain trauma. FASEB J. 2003, 17, 1922–1924. [Google Scholar] [CrossRef]
- Sokołowska, P.; Dejda, A.; Nowak, J. Neuroprotective role of pacap, vip, and phi in the central nervous system. Postepy Hig. I Med. Dosw. 2004, 58, 416–427. [Google Scholar]
- Henn, A.; Lund, S.; Hedtjärn, M.; Schrattenholz, A.; Pörzgen, P.; Leist, M. The suitability of bv2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. Altex Altern. Anim. Exp. 2009, 26, 83–94. [Google Scholar] [CrossRef]
- Chiocchio, I.; Prata, C.; Mandrone, M.; Ricciardiello, F.; Marrazzo, P.; Tomasi, P.; Angeloni, C.; Fiorentini, D.; Malaguti, M.; Poli, F.; et al. Leaves and spiny burs of castanea sativa from an experimental chestnut grove: Metabolomic analysis and anti-neuroinflammatory activity. Metabolites 2020, 10, 408. [Google Scholar] [CrossRef]
- An, J.; Chen, B.; Kang, X.; Zhang, R.; Guo, Y.; Zhao, J.; Yang, H. Neuroprotective effects of natural compounds on lps-induced inflammatory responses in microglia. Am. J. Transl. Res. 2020, 12, 2353–2378. [Google Scholar]
- Cheng, H.-H.; Ye, H.; Peng, R.-P.; Deng, J.; Ding, Y. Inhibition of retinal ganglion cell apoptosis: Regulation of mitochondrial function by pacap. Neural Regen. Res. 2018, 13, 923. [Google Scholar]
- Juhász, T.; Matta, C.; Katona, É.; Somogyi, C.; Takács, R.; Gergely, P.; Csernoch, L.; Panyi, G.; Tóth, G.; Reglődi, D. Pituitary adenylate cyclase activating polypeptide (pacap) signalling exerts chondrogenesis promoting and protecting effects: Implication of calcineurin as a downstream target. PLoS ONE 2014, 9, e91541. [Google Scholar] [CrossRef]
- Scuderi, S.; D’Amico, A.G.; Castorina, A.; Imbesi, R.; Carnazza, M.L.; D’Agata, V. Ameliorative effect of pacap and vip against increased permeability in a model of outer blood retinal barrier dysfunction. Peptides 2013, 39, 119–124. [Google Scholar] [CrossRef]
- Bolaños, J.P.; Almeida, A.; Stewart, V.; Peuchen, S.; Land, J.M.; Clark, J.B.; Heales, S.J. Nitric oxide-mediated mitochondrial damage in the brain: Mechanisms and implications for neurodegenerative diseases. J. Neurochem. 1997, 68, 2227–2240. [Google Scholar] [CrossRef]
- Brown, G.C.; Bal-Price, A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol. Neurobiol. 2003, 27, 325–355. [Google Scholar] [CrossRef]
- Hill, K.E.; Zollinger, L.V.; Watt, H.E.; Carlson, N.G.; Rose, J.W. Inducible nitric oxide synthase in chronic active multiple sclerosis plaques: Distribution, cellular expression and association with myelin damage. J. Neuroimmunol. 2004, 151, 171–179. [Google Scholar] [CrossRef]
- Tatsch, E.; Bochi, G.V.; da Silva Pereira, R.; Kober, H.; Agertt, V.A.; de Campos, M.M.A.; Gomes, P.; Duarte, M.M.M.F.; Moresco, R.N. A simple and inexpensive automated technique for measurement of serum nitrite/nitrate. Clin. Biochem. 2011, 44, 348–350. [Google Scholar] [CrossRef]
- Miller, E.B.; Zhang, P.; Ching, K.; Pugh, E.N., Jr.; Burns, M.E. In vivo imaging reveals transient microglia recruitment and functional recovery of photoreceptor signaling after injury. Proc. Natl. Acad. Sci. USA 2019, 116, 16603–16612. [Google Scholar] [CrossRef]
- Crippa, M.P. Urokinase-type plasminogen activator. Int. J. Biochem. Cell Biol. 2007, 39, 690–694. [Google Scholar] [CrossRef]
- Reglodi, D.; Tamas, A.; Somogyvari-Vigh, A.; Szanto, Z.; Kertes, E.; Lenard, L.; Arimura, A.; Lengvari, I. Effects of pretreatment with pacap on the infarct size and functional outcome in rat permanent focal cerebral ischemia. Peptides 2002, 23, 2227–2234. [Google Scholar] [CrossRef]
- Somogyvari-Vigh, A.; Reglodi, D. Pituitary adenylate cyclase activating polypeptide: A potential neuroprotective peptide. Curr. Pharm. Des. 2004, 10, 2861–2889. [Google Scholar] [CrossRef]
- Tanaka, K.; Hashimoto, H.; Shintani, N.; Yamamoto, A.; Baba, A. Reduced hypothermic and hypnotic responses to ethanol in pacap-deficient mice. Regul. Pept. 2004, 123, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Falktoft, B.; Georg, B.; Fahrenkrug, J. Signaling pathways in pacap regulation of vip gene expression in human neuroblastoma cells. Neuropeptides 2009, 43, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, M.; Miyakawa, H.; Wada, K.; Furuta, A. Activation of the vip/vpac2 system induces reactive astrocytosis associated with increased expression of glutamate transporters. Brain Res. 2011, 1383, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Girard, B.M.; May, V.; Bora, S.H.; Fina, F.; Braas, K.M. Regulation of neurotrophic peptide expression in sympathetic neurons: Quantitative analysis using radioimmunoassay and real-time quantitative polymerase chain reaction. Regul. Pept. 2002, 109, 89–101. [Google Scholar] [CrossRef]
- Martinez, C.; Abad, C.; Delgado, M.; Arranz, A.; Juarranz, M.G.; Rodriguez-Henche, N.; Brabet, P.; Leceta, J.; Gomariz, R.P. Anti-inflammatory role in septic shock of pituitary adenylate cyclase-activating polypeptide receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 1053–1058. [Google Scholar] [CrossRef]
- Zeng, N.; Kang, T.; LYU, R.M.; Wong, H.; Wen, Y.; Walsh, J.H.; Sachs, G.; Pisegna, J.R. The pituitary adenylate cyclase activating polypeptide type 1 receptor (pac1-r) is expressed on gastric ecl cells: Evidence by immunocytochemistry and rt-pcr. Ann. N. Y. Acad. Sci. 1998, 865, 147–156. [Google Scholar] [CrossRef]
- Kojima, M.; Ito, T.; Oono, T.; Hisano, T.; Igarashi, H.; Arita, Y.; Kawabe, K.; Coy, D.H.; Jensen, R.T.; Nawata, H. Vip attenuation of the severity of experimental pancreatitis is due to vpac1 receptor-mediated inhibition of cytokine production. Pancreas 2005, 30, 62–70. [Google Scholar]
- Tan, Y.-V.; Abad, C.; Wang, Y.; Lopez, R.; Waschek, J. Vpac2 (vasoactive intestinal peptide receptor type 2) receptor deficient mice develop exacerbated experimental autoimmune encephalomyelitis with increased th1/th17 and reduced th2/treg responses. Brain Behav. Immun. 2015, 44, 167–175. [Google Scholar] [CrossRef]
- Delgado, M.; Abad, C.; Martinez, C.; Juarranz, M.; Arranz, A.; Gomariz, R.; Leceta, J. Vasoactive intestinal peptide in the immune system: Potential therapeutic role in inflammatory and autoimmune diseases. J. Mol. Med. 2002, 80, 16–24. [Google Scholar] [CrossRef]
- Lowenstein, C.J.; Padalko, E. Inos (nos2) at a glance. J. Cell Sci. 2004, 117, 2865–2867. [Google Scholar] [CrossRef]
- Blasi, E.; Barluzzi, R.; Bocchini, V.; Mazzolla, R.; Bistoni, F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 1990, 27, 229–237. [Google Scholar] [CrossRef]
- Blasi, E.; Mathieson, B.J.; Varesio, L.; Cleveland, J.L.; Borchert, P.A.; Rapp, U.R. Selective immortalization of murine macrophages from fresh bone marrow by a raf/myc recombinant murine retrovirus. Nature 1985, 318, 667. [Google Scholar] [CrossRef]
- Kingham, P.; Cuzner, M.; Pocock, J. Apoptotic pathways mobilized in microglia and neurones as a consequence of chromogranin a-induced microglial activation. J. Neurochem. 1999, 73, 538–547. [Google Scholar] [CrossRef]
- Loihl, A.K.; Murphy, S. Expression of Nitric Oxide Synthase-2 in Glia Associated with CNS Pathology. Prog. Brain Res. 1998, 118, 253–267. [Google Scholar]
- Dehmer, T.; Lindenau, J.; Haid, S.; Dichgans, J.; Schulz, J.B. Deficiency of inducible nitric oxide synthase protects against mptp toxicity in vivo. J. Neurochem. 2000, 74, 2213–2216. [Google Scholar] [CrossRef]
- Parmentier, S.; Böhme, G.A.; Lerouet, D.; Damour, D.; Stutzmann, J.M.; Margaill, I.; Plotkine, M. Selective inhibition of inducible nitric oxide synthase prevents ischaemic brain injury. Br. J. Pharmacol. 1999, 127, 546–552. [Google Scholar] [CrossRef]
- Lowenstein, C.J.; Alley, E.W.; Raval, P.; Snowman, A.M.; Snyder, S.H.; Russell, S.W.; Murphy, W.J. Macrophage nitric oxide synthase gene: Two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc. Natl. Acad. Sci. USA 1993, 90, 9730–9734. [Google Scholar] [CrossRef]
- Xie, Q.; Whisnant, R.; Nathan, C. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J. Exp. Med. 1993, 177, 1779–1784. [Google Scholar] [CrossRef]
- Horvath, R.J.; Nutile-McMenemy, N.; Alkaitis, M.S.; DeLeo, J.A. Differential migration, lps-induced cytokine, chemokine, and no expression in immortalized bv-2 and hapi cell lines and primary microglial cultures. J. Neurochem. 2008, 107, 557–569. [Google Scholar] [CrossRef]
- Leceta, J.; Gomariz, R.P.; Martinez, C.; Abad, C.; Ganea, D.; Delgado, M. Receptors and transcriptional factors involved in the anti-inflammatory activity of vip and pacap. Ann. N. Y. Acad. Sci. 2000, 921, 92–102. [Google Scholar] [CrossRef]
- Delgado, M.; Munoz-Elias, E.; Gomariz, R.; Ganea, D. Vip and pacap prevent inducible nitric oxide synthase transcription in macrophages by inhibiting nf-κb and interferon regulatory factor 1 activation. J. Immunol. 1999, 162, 4685. [Google Scholar]
- Ito, D.; Imai, Y.; Ohsawa, K.; Nakajima, K.; Fukuuchi, Y.; Kohsaka, S. Microglia-specific localisation of a novel calcium binding protein, iba1. Mol. Brain Res. 1998, 57, 1–9. [Google Scholar] [CrossRef]
- Tao, X.; Li, N.; Liu, F.; Hu, Y.; Liu, J.; Zhang, Y.-M. In vitro examination of microglia-neuron crosstalk with bv2 cells, and primary cultures of glia and hypothalamic neurons. Heliyon 2018, 4, e00730. [Google Scholar] [CrossRef]
- Qu, Z.; Chen, Y.; Luo, Z.H.; Shen, X.L.; Hu, Y.J. 7-methoxyflavanone alleviates neuroinflammation in lipopolysaccharide-stimulated microglial cells by inhibiting tlr4/myd88/mapk signalling and activating the nrf2/nqo-1 pathway. J. Pharm. Pharmacol. 2020, 72, 385–395. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the cns. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Cunningham, C.L.; Martínez-Cerdeño, V.; Noctor, S.C. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J. Neurosci. 2013, 33, 4216–4233. [Google Scholar] [CrossRef]
- Cizkova, D.; Devaux, S.; Le Marrec-Croq, F.; Franck, J.; Slovinska, L.; Blasko, J.; Rosocha, J.; Spakova, T.; Lefebvre, C.; Fournier, I. Modulation properties of factors released by bone marrow stromal cells on activated microglia: An in vitro study. Sci. Rep. 2014, 4, 7514. [Google Scholar] [CrossRef]
- Pocock, J.M.; Kettenmann, H. Neurotransmitter receptors on microglia. Trends Neurosci. 2007, 30, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Aloisi, F. Immune function of microglia. Glia 2001, 36, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, A.; Patnala, R.; Jadhav, S.P.; Eng-Ang, L.; Dheen, S.T. Micrornas: Key players in microglia and astrocyte mediated inflammation in cns pathologies. Curr. Med. Chem. 2016, 23, 3528–3546. [Google Scholar] [CrossRef] [PubMed]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and microglia: In sickness and in health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef]
- Cunningham, O.; Campion, S.; Perry, V.H.; Murray, C.; Sidenius, N.; Docagne, F.; Cunningham, C. Microglia and the urokinase plasminogen activator receptor/upa system in innate brain inflammation. Glia 2009, 57, 1802–1814. [Google Scholar] [CrossRef]
- Castorina, A.; Waschek, J.A.; Marzagalli, R.; Cardile, V.; Drago, F. Pacap interacts with pac1 receptors to induce tissue plasminogen activator (tpa) expression and activity in schwann cell-like cultures. PLoS ONE 2015, 10, e0117799. [Google Scholar] [CrossRef]
- Castorina, A.; D’Amico, A.; Scuderi, S.; Leggio, G.; Drago, F.; D’Agata, V. Dopamine d3 receptor deletion increases tissue plasminogen activator (tpa) activity in prefrontal cortex and hippocampus. Neuroscience 2013, 250, 546–556. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Description | Process Diameter (μm) | Process Length (μm) ± SD | Surface Area (µm2) | Visual Example |
|---|---|---|---|---|---|
| Small | Mostly round or oval shaped cells (87–91%), with small soma and thin, short/absent processes | ˂2 or none | (min = 0, max = 9.3) mean 3.17 ± 1.56 | <200 |  |
| Mid-sized | Predominantly spindle-shaped cells (70–80%) with flat appearance, variable length and mainly thick processes | 2–4 | (min = 3.54, max = 17.54) mean 9.79 ± 2.88 | 200–400 |  |
| Large | Mixed multipolar (30-45%) or spindle-shape appearance (9–19%), flattened cells with hypertrophied soma, with thick, elongated processes | 2–4 | (min = 10.45, max = 32.24) mean 19.90 ± 4.09 | >400 |  |
| Gene | Forward | Reverse | Bp Length |
|---|---|---|---|
| Adcyap1 Acc# NM_009625.2 | CTGCGTGACGCTTACGCCCT | CCTAGGTTCTCCCCCGCGCC | 152 |
| Vip Acc# NM_011702.2 | TGGCAAACGAATCAGCAGCAGCA | AGCCATTTGCTTTCTGAGGCGGG | 106 |
| Adcyap1r1 Acc# NM_007407.3 | CAGTCCCCAGACATGGGAGGCA | AGCGGGCCAGCCGTAGAGTA | 139 |
| Vipr1 Acc# NM_011703.4 | TCAATGGCGAGGTGCAGGCAG | TGTGTGCTGCACGAGACGCC | 127 |
| Vipr2 Acc# NM_009511.2 | GCGTCGGTGGTGCTGACCTG | ACACCGCTGCAGGCTCTCTGAT | 155 |
| IL-1β Acc# NM_008361.4 | GCTACCTGTGTCTTTCCCGT | CATCTCGGAGCCTGTAGTGC | 164 |
| IL-6 Acc# NM_031168.2 | CCCCAATTTCCAATGCTCTCC | CGCACTAGGTTTGCCGAGTA | 141 |
| IL-10 Acc# NM_010548.2 | GCATGGCCCAGAAATCAAGG | GAGAAATCGATGACAGCGCC | 91 |
| Itgam Acc# NM_001082960.1 | GAGCAGGGGTCATTCGCTAC | GCTGGCTTAGATGCGATGGT | 94 |
| Adgre1 Acc# NM_001355722.1 | GCTTATGCCACCTGCACTGA | GGTGAGTCACTTTGAAGACATTCG | 143 |
| AIF1 Acc# NM_001361501.1 | GCTTTTGGACTGCTGAAGGC | GCTTCAAGTTTGGACGGCAG | 114 |
| CD68 Acc# NM_001291058.1 | CTCCCACCACAAATGGCACT | CTTGGACCTTGGACTAGGCG | 95 |
| NOS2 Acc# NM_010927.4 | AATCTTGGAGCGAGTTGTGG | CAGGAAGTAGGTGAGGGCTTG | 139 |
| uPA Acc# NM_008873.3 | CATCCATCCAGTCCTTGCGT | TTTCATGGTAGTGCCGCTGG | 87 |
| S18 Acc# NM_011296.2 | CCCTGAGAAGTTCCAGCACA | GGTGAGGTCGATGTCTGCTT | 145 |
| Antibody | Source | Predicted Band Size | Dilution |
|---|---|---|---|
| PLAU | LS-C193095 | 55 kDa | 1:1000 |
| Iba1 | GTX100042, GeneTex | 17 kDa | 1:300 |
| iNOS | GTX60599, GeneTex | 32 kDa | 1:1000 |
| GAPDH | VPA00187, Bio-Rad | 37 kDa | 1:1500 |
| Goat anti Rabbit IgG HRP (Secondary) | STAR208P, Bio-Rad | 1:20,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karunia, J.; Niaz, A.; Mandwie, M.; Thomas Broome, S.; Keay, K.A.; Waschek, J.A.; Al-Badri, G.; Castorina, A. PACAP and VIP Modulate LPS-Induced Microglial Activation and Trigger Distinct Phenotypic Changes in Murine BV2 Microglial Cells. Int. J. Mol. Sci. 2021, 22, 10947. https://doi.org/10.3390/ijms222010947
Karunia J, Niaz A, Mandwie M, Thomas Broome S, Keay KA, Waschek JA, Al-Badri G, Castorina A. PACAP and VIP Modulate LPS-Induced Microglial Activation and Trigger Distinct Phenotypic Changes in Murine BV2 Microglial Cells. International Journal of Molecular Sciences. 2021; 22(20):10947. https://doi.org/10.3390/ijms222010947
Chicago/Turabian StyleKarunia, Jocelyn, Aram Niaz, Mawj Mandwie, Sarah Thomas Broome, Kevin A. Keay, James A. Waschek, Ghaith Al-Badri, and Alessandro Castorina. 2021. "PACAP and VIP Modulate LPS-Induced Microglial Activation and Trigger Distinct Phenotypic Changes in Murine BV2 Microglial Cells" International Journal of Molecular Sciences 22, no. 20: 10947. https://doi.org/10.3390/ijms222010947
APA StyleKarunia, J., Niaz, A., Mandwie, M., Thomas Broome, S., Keay, K. A., Waschek, J. A., Al-Badri, G., & Castorina, A. (2021). PACAP and VIP Modulate LPS-Induced Microglial Activation and Trigger Distinct Phenotypic Changes in Murine BV2 Microglial Cells. International Journal of Molecular Sciences, 22(20), 10947. https://doi.org/10.3390/ijms222010947