TNAP as a New Player in Chronic Inflammatory Conditions and Metabolism


{kind=link}
{kind=link}
Abstract
1. Structure, Function, and Substrates of TNAP
2. Heritable TNAP Dysfunction: The Rare Disease Hypophosphatasia
2.1. General Information on HPP
2.2. Clinical Subtypes
2.3. Diagnosis
2.4. Treatment
3. TNAP as an Ectophosphatase—The Molecular Role of TNAP in Mineralization Processes
4. The Molecular Role of TNAP in Neuronal Biology and Neurotransmitter Metabolism
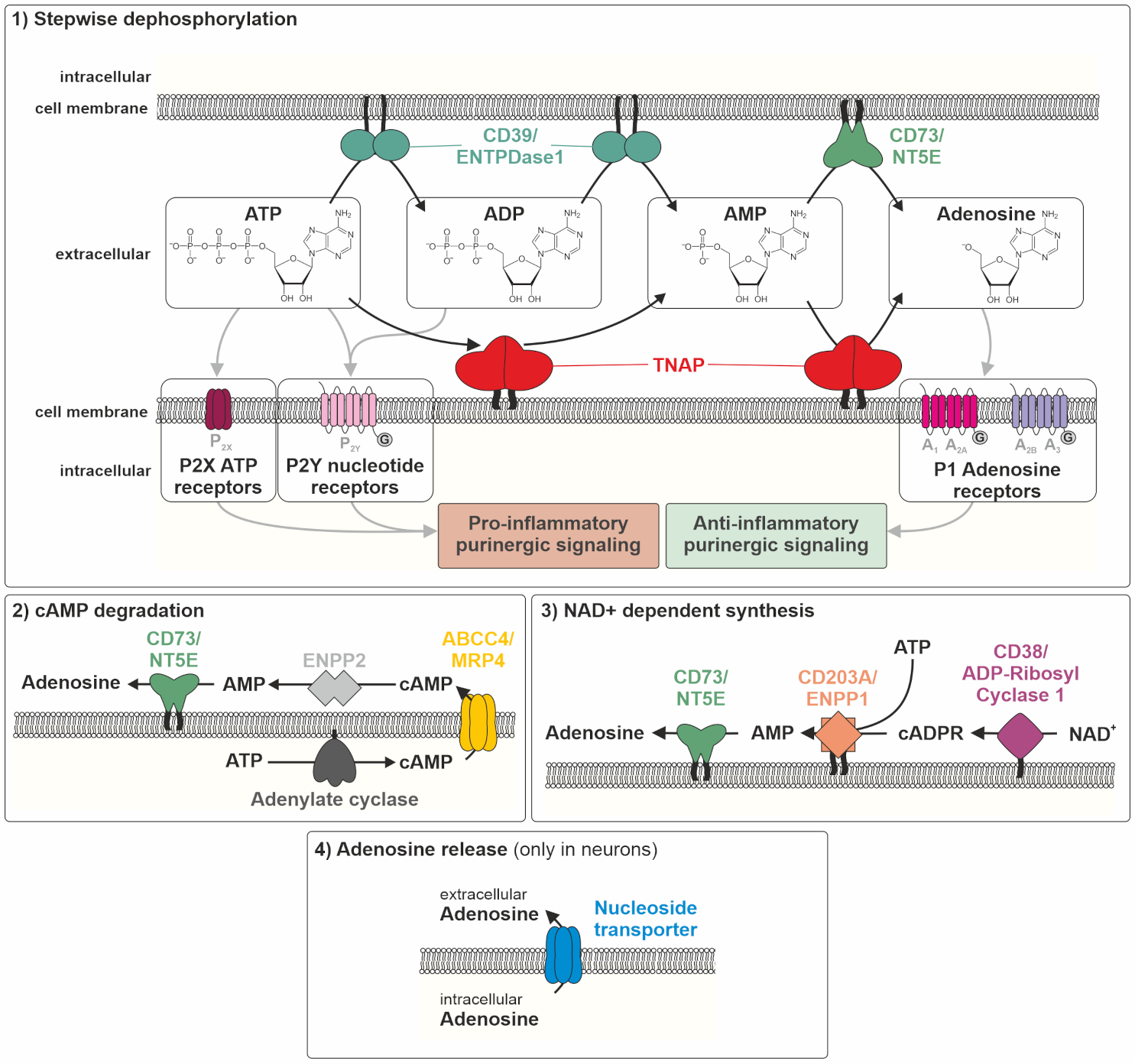
5. The Molecular Role of TNAP in Inflammation Processes
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Millan, J.L.; Whyte, M.P. Alkaline Phosphatase and Hypophosphatasia. Calcif. Tissue Int. 2016, 98, 398–416. [Google Scholar] [CrossRef] [PubMed]
- Millan, J.L. Alkaline Phosphatases: Structure, substrate specificity and functional relatedness to other members of a large superfamily of enzymes. Purinergic Signal. 2006, 2, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Harris, H. The human alkaline phosphatases: What we know and what we don’t know. Clin. Chim. Acta 1990, 186, 133–150. [Google Scholar] [CrossRef]
- Simao, A.M.; Bolean, M.; Hoylaerts, M.F.; Millan, J.L.; Ciancaglini, P. Effects of pH on the production of phosphate and pyrophosphate by matrix vesicles’ biomimetics. Calcif. Tissue Int. 2013, 93, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Fedde, K.N.; Whyte, M.P. Alkaline phosphatase (tissue-nonspecific isoenzyme) is a phosphoethanolamine and pyridoxal-5′-phosphate ectophosphatase: Normal and hypophosphatasia fibroblast study. Am. J. Hum. Genet. 1990, 47, 767–775. [Google Scholar] [PubMed]
- Narisawa, S.; Yadav, M.C.; Millan, J.L. In vivo overexpression of tissue-nonspecific alkaline phosphatase increases skeletal mineralization and affects the phosphorylation status of osteopontin. J. Bone Miner. Res. 2013, 28, 1587–1598. [Google Scholar] [CrossRef]
- Pettengill, M.; Matute, J.D.; Tresenriter, M.; Hibbert, J.; Burgner, D.; Richmond, P.; Millan, J.L.; Ozonoff, A.; Strunk, T.; Currie, A.; et al. Human alkaline phosphatase dephosphorylates microbial products and is elevated in preterm neonates with a history of late-onset sepsis. PLoS ONE 2017, 12, e0175936. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G.; Verkhratsky, A.; Zimmermann, H. Purinergic signalling in the nervous system: An overview. Trends Neurosci. 2009, 32, 19–29. [Google Scholar] [CrossRef]
- Le Du, M.H.; Stigbrand, T.; Taussig, M.J.; Menez, A.; Stura, E.A. Crystal structure of alkaline phosphatase from human placenta at 1.8 A resolution. Implication for a substrate specificity. J. Biol. Chem. 2001, 276, 9158–9165. [Google Scholar] [CrossRef]
- Hoylaerts, M.F.; Van Kerckhoven, S.; Kiffer-Moreira, T.; Sheen, C.; Narisawa, S.; Millan, J.L. Functional significance of calcium binding to tissue-nonspecific alkaline phosphatase. PLoS ONE 2015, 10, e0119874. [Google Scholar] [CrossRef]
- Weiss, M.J.; Ray, K.; Henthorn, P.S.; Lamb, B.; Kadesch, T.; Harris, H. Structure of the human liver/bone/kidney alkaline phosphatase gene. J. Biol. Chem. 1988, 263, 12002–12010. [Google Scholar] [CrossRef]
- Martins, L.; Rodrigues, T.L.; Ribeiro, M.M.; Saito, M.T.; Giorgetti, A.P.; Casati, M.Z.; Sallum, E.A.; Foster, B.L.; Somerman, M.J.; Nociti, F.H., Jr. Novel ALPL genetic alteration associated with an odontohypophosphatasia phenotype. Bone 2013, 56, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Le Du, M.H.; Millan, J.L. Structural evidence of functional divergence in human alkaline phosphatases. J. Biol. Chem. 2002, 277, 49808–49814. [Google Scholar] [CrossRef] [PubMed]
- Fauvert, D.; Brun-Heath, I.; Lia-Baldini, A.S.; Bellazi, L.; Taillandier, A.; Serre, J.L.; de Mazancourt, P.; Mornet, E. Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles. BMC Med. Genet. 2009, 10, 51. [Google Scholar] [CrossRef]
- Muller, H.L.; Yamazaki, M.; Michigami, T.; Kageyama, T.; Schonau, E.; Schneider, P.; Ozono, K. Asp361Val Mutant of alkaline phosphatase found in patients with dominantly inherited hypophosphatasia inhibits the activity of the wild-type enzyme. J. Clin. Endocrinol. Metab. 2000, 85, 743–747. [Google Scholar] [CrossRef][Green Version]
- Mornet, E.; Taillandier, A.; Domingues, C.; Dufour, A.; Benaloun, E.; Lavaud, N.; Wallon, F.; Rousseau, N.; Charle, C.; Guberto, M.; et al. Hypophosphatasia: A genetic-based nosology and new insights in genotype-phenotype correlation. Eur. J. Hum. Genet. 2020, 1–11. [Google Scholar] [CrossRef]
- Lia-Baldini, A.S.; Brun-Heath, I.; Carrion, C.; Simon-Bouy, B.; Serre, J.L.; Nunes, M.E.; Mornet, E. A new mechanism of dominance in hypophosphatasia: The mutated protein can disturb the cell localization of the wild-type protein. Hum. Genet. 2008, 123, 429–432. [Google Scholar] [CrossRef]
- Weiss, M.J.; Henthorn, P.S.; Lafferty, M.A.; Slaughter, C.; Raducha, M.; Harris, H. Isolation and characterization of a cDNA encoding a human liver/bone/kidney-type alkaline phosphatase. Proc. Natl. Acad. Sci. USA 1986, 83, 7182–7186. [Google Scholar] [CrossRef]
- Bhadada, S.K.; Pal, R.; Dhiman, V.; Alonso, N.; Ralston, S.H.; Kaur, S.; Gupta, R. Adult hypophosphatasia with a novel ALPL mutation: Report of an Indian kindred. Bone Rep. 2020, 12, 100247. [Google Scholar] [CrossRef]
- Michigami, T.; Tachikawa, K.; Yamazaki, M.; Kawai, M.; Kubota, T.; Ozono, K. Hypophosphatasia in Japan: ALPL Mutation Analysis in 98 Unrelated Patients. Calcif. Tissue Int. 2020, 106, 221–231. [Google Scholar] [CrossRef]
- Hofmann, C.; Liese, J.; Schwarz, T.; Kunzmann, S.; Wirbelauer, J.; Nowak, J.; Hamann, J.; Girschick, H.; Graser, S.; Dietz, K.; et al. Compound heterozygosity of two functional null mutations in the ALPL gene associated with deleterious neurological outcome in an infant with hypophosphatasia. Bone 2013, 55, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Girschick, H.; Mornet, E.; Schneider, D.; Jakob, F.; Mentrup, B. Unexpected high intrafamilial phenotypic variability observed in hypophosphatasia. Eur. J. Hum. Genet. 2014, 22, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Silvent, J.; Gasse, B.; Mornet, E.; Sire, J.Y. Molecular evolution of the tissue-nonspecific alkaline phosphatase allows prediction and validation of missense mutations responsible for hypophosphatasia. J. Biol. Chem. 2014, 289, 24168–24179. [Google Scholar] [CrossRef] [PubMed]
- Mornet, E. Molecular Genetics of Hypophosphatasia and Phenotype-Genotype Correlations. Subcell Biochem. 2015, 76, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Huggins, E.; Ong, R.; Rockman-Greenberg, C.; Flueckinger, L.B.; Dahir, K.M.; Kishnani, P.S. Multigenerational case examples of hypophosphatasia: Challenges in genetic counseling and disease management. Mol. Genet. Metab. Rep. 2020, 25, 100661. [Google Scholar] [CrossRef]
- Rathbun, J.C. Hypophosphatasia; A new developmental anomaly. Am. J. Dis. Child. 1948, 75, 822–831. [Google Scholar] [CrossRef]
- Hofmann, C.; Girschick, H.J.; Mentrup, B.; Graser, S.; Seefried, L.; Liese, J.; Jakob, F. Clinical Aspects of Hypophosphatasia: An Update. Clinic Rev. Bone Miner. Metab. 2013, 11, 60–70. [Google Scholar] [CrossRef]
- Mornet, E.; Yvard, A.; Taillandier, A.; Fauvert, D.; Simon-Bouy, B. A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann. Hum. Genet. 2011, 75, 439–445. [Google Scholar] [CrossRef]
- Fukushi-Irie, M.; Ito, M.; Amaya, Y.; Amizuka, N.; Ozawa, H.; Omura, S.; Ikehara, Y.; Oda, K. Possible interference between tissue-non-specific alkaline phosphatase with an Arg54-->Cys substitution and acounterpart with an Asp277-->Ala substitution found in a compound heterozygote associated with severe hypophosphatasia. Biochem. J. 2000, 348 Pt 3, 633–642. [Google Scholar] [CrossRef]
- Vogt, M.; Girschick, H.; Schweitzer, T.; Benoit, C.; Holl-Wieden, A.; Seefried, L.; Jakob, F.; Hofmann, C. Pediatric hypophosphatasia: Lessons learned from a retrospective single-center chart review of 50 children. Orphanet J. Rare Dis. 2020, 15, 212. [Google Scholar] [CrossRef]
- Whyte, M.P. Hypophosphatasia—Aetiology, nosology, pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2016, 12, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Mornet, E.; Hofmann, C.; Bloch-Zupan, A.; Girschick, H.; Le Merrer, M. Clinical utility gene card for: Hypophosphatasia—Update 2013. Eur. J. Hum. Genet. 2014, 22. [Google Scholar] [CrossRef] [PubMed]
- Eberle, F.; Hartenfels, S.; Pralle, H.; Kabisch, A. Adult hypophosphatasia without apparent skeletal disease: “odontohypophosphatasia” in four heterozygote members of a family. Klin. Wochenschr. 1984, 62, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Bloch-Zupan, A.; Vaysse, F. Hypophosphatasia: Oral cavity and dental disorders. Arch. Pediatr. 2017, 24, 5S80–5S84. [Google Scholar] [CrossRef]
- Reibel, A.; Maniere, M.C.; Clauss, F.; Droz, D.; Alembik, Y.; Mornet, E.; Bloch-Zupan, A. Orodental phenotype and genotype findings in all subtypes of hypophosphatasia. Orphanet J. Rare Dis. 2009, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.L.; Bishop, N.J.; Guanabens, N.; Hofmann, C.; Jakob, F.; Roux, C.; Zillikens, M.C.; Rare Bone Disease Action Group of the European Calcified Tissue Society. Hypophosphatasia in adolescents and adults: Overview of diagnosis and treatment. Osteoporos. Int. 2020, 31, 1445–1460. [Google Scholar] [CrossRef]
- Lopez-Delgado, L.; Riancho-Zarrabeitia, L.; Garcia-Unzueta, M.T.; Tenorio, J.A.; Garcia-Hoyos, M.; Lapunzina, P.; Valero, C.; Riancho, J.A. Abnormal bone turnover in individuals with low serum alkaline phosphatase. Osteoporos. Int. 2018, 29, 2147–2150. [Google Scholar] [CrossRef]
- Berkseth, K.E.; Tebben, P.J.; Drake, M.T.; Hefferan, T.E.; Jewison, D.E.; Wermers, R.A. Clinical spectrum of hypophosphatasia diagnosed in adults. Bone 2013, 54, 21–27. [Google Scholar] [CrossRef]
- Kramer, K.; Chavez, M.B.; Tran, A.T.; Farah, F.; Tan, M.H.; Kolli, T.N.; Dos Santos, E.J.L.; Wimer, H.F.; Millan, J.L.; Suva, L.J.; et al. Dental defects in the primary dentition associated with hypophosphatasia from biallelic ALPL mutations. Bone 2020, 143, 115732. [Google Scholar] [CrossRef]
- Seefried, L.; Dahir, K.; Petryk, A.; Hogler, W.; Linglart, A.; Martos-Moreno, G.A.; Ozono, K.; Fang, S.; Rockman-Greenberg, C.; Kishnani, P.S. Burden of Illness in Adults with Hypophosphatasia: Data From the Global Hypophosphatasia Patient Registry. J. Bone Miner. Res. 2020, 35, 2171–2178. [Google Scholar] [CrossRef]
- Kishnani, P.S.; Rush, E.T.; Arundel, P.; Bishop, N.; Dahir, K.; Fraser, W.; Harmatz, P.; Linglart, A.; Munns, C.F.; Nunes, M.E.; et al. Monitoring guidance for patients with hypophosphatasia treated with asfotase alfa. Mol. Genet. Metab. 2017, 122, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; Mahuren, J.D.; Vrabel, L.A.; Coburn, S.P. Markedly increased circulating pyridoxal-5’-phosphate levels in hypophosphatasia. Alkaline phosphatase acts in vitamin B6 metabolism. J. Clin. Investig. 1985, 76, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Michigami, T.; Ohata, Y.; Fujiwara, M.; Mochizuki, H.; Adachi, M.; Kitaoka, T.; Kubota, T.; Sawai, H.; Namba, N.; Hasegawa, K.; et al. Clinical Practice Guidelines for Hypophosphatasia. Clin. Pediatr. Endocrinol. 2020, 29, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Offiah, A.C.; Vockley, J.; Munns, C.F.; Murotsuki, J. Differential diagnosis of perinatal hypophosphatasia: Radiologic perspectives. Pediatr. Radiol. 2019, 49, 3–22. [Google Scholar] [CrossRef]
- Hogler, W.; Langman, C.; da Silva, H.G.; Fang, S.; Linglart, A.; Ozono, K.; Petryk, A.; Rockman-Greenberg, C.; Seefried, L.; Kishnani, P.S. Diagnostic delay is common among patients with hypophosphatasia: Initial findings from a longitudinal, prospective, global registry. BMC Musculoskelet Disord. 2019, 20, 80. [Google Scholar] [CrossRef]
- Girschick, H.J.; Seyberth, H.W.; Huppertz, H.I. Treatment of childhood hypophosphatasia with nonsteroidal antiinflammatory drugs. Bone 1999, 25, 603–607. [Google Scholar] [CrossRef]
- Girschick, H.J.; Schneider, P.; Haubitz, I.; Hiort, O.; Collmann, H.; Beer, M.; Shin, Y.S.; Seyberth, H.W. Effective NSAID treatment indicates that hyperprostaglandinism is affecting the clinical severity of childhood hypophosphatasia. Orphanet J. Rare Dis. 2006, 1, 24. [Google Scholar] [CrossRef][Green Version]
- Collmann, H.; Mornet, E.; Gattenlohner, S.; Beck, C.; Girschick, H. Neurosurgical aspects of childhood hypophosphatasia. Childs Nerv. Syst. 2009, 25, 217–223. [Google Scholar] [CrossRef]
- Feeney, C.; Stanford, N.; Lee, S.; Barry, S. Hypophosphatasia and the importance of the general dental practitioner—A case series and discussion of upcoming treatments. Br. Dent. J. 2018, 224, 937–943. [Google Scholar] [CrossRef]
- Weber, T.J.; Sawyer, E.K.; Moseley, S.; Odrljin, T.; Kishnani, P.S. Burden of disease in adult patients with hypophosphatasia: Results from two patient-reported surveys. Metabolism 2016, 65, 1522–1530. [Google Scholar] [CrossRef]
- Genest, F.; Seefried, L. Subtrochanteric and diaphyseal femoral fractures in hypophosphatasia—Not atypical at all. Osteoporos. Int. 2018, 29, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Klidaras, P.; Severt, J.; Aggers, D.; Payne, J.; Miller, P.D.; Ing, S.W. Fracture Healing in Two Adult Patients with Hypophosphatasia After Asfotase Alfa Therapy. JBMR Plus 2018, 2, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; Greenberg, C.R.; Salman, N.J.; Bober, M.B.; McAlister, W.H.; Wenkert, D.; Van Sickle, B.J.; Simmons, J.H.; Edgar, T.S.; Bauer, M.L.; et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N. Engl. J. Med. 2012, 366, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Seefried, L.; Jakob, F. Asfotase alfa: Enzyme replacement for the treatment of bone disease in hypophosphatasia. Drugs Today 2016, 52, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.E.; Harmatz, P.; Vockley, J.; Hogler, W.; Nakayama, H.; Bishop, N.; Martos-Moreno, G.A.; Moseley, S.; Fujita, K.P.; Liese, J.; et al. Efficacy and Safety of Asfotase Alfa in Infants and Young Children With Hypophosphatasia: A Phase 2 Open-Label Study. J. Clin. Endocrinol. Metab. 2019, 104, 2735–2747. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P.; Rockman-Greenberg, C.; Ozono, K.; Riese, R.; Moseley, S.; Melian, A.; Thompson, D.D.; Bishop, N.; Hofmann, C. Asfotase Alfa Treatment Improves Survival for Perinatal and Infantile Hypophosphatasia. J. Clin. Endocrinol. Metab. 2016, 101, 334–342. [Google Scholar] [CrossRef]
- Yadav, M.C.; de Oliveira, R.C.; Foster, B.L.; Fong, H.; Cory, E.; Narisawa, S.; Sah, R.L.; Somerman, M.; Whyte, M.P.; Millan, J.L. Enzyme replacement prevents enamel defects in hypophosphatasia mice. J. Bone Miner. Res. 2012, 27, 1722–1734. [Google Scholar] [CrossRef]
- Whyte, M.P.; Simmons, J.H.; Moseley, S.; Fujita, K.P.; Bishop, N.; Salman, N.J.; Taylor, J.; Phillips, D.; McGinn, M.; McAlister, W.H. Asfotase alfa for infants and young children with hypophosphatasia: 7 year outcomes of a single-arm, open-label, phase 2 extension trial. Lancet Diabetes Endocrinol. 2019, 7, 93–105. [Google Scholar] [CrossRef]
- Seefried, L.; Kishnani, P.S.; Moseley, S.; Denker, A.E.; Watsky, E.; Whyte, M.P.; Dahir, K.M. Pharmacodynamics of asfotase alfa in adults with pediatric-onset hypophosphatasia. Bone 2020, 142, 115664. [Google Scholar] [CrossRef]
- Magdaleno, A.L.; Singh, S.; Venkataraman, S.; Perilli, G.A.; Lee, Y.Y. Adult-Onset Hypophosphatasia: Before and after Treatment with Asfotase Alfa. AACE Clin. Case Rep. 2019, 5, e344–e348. [Google Scholar] [CrossRef]
- Remde, H.; Cooper, M.S.; Quinkler, M. Successful Asfotase Alfa Treatment in an Adult Dialysis Patient with Childhood-Onset Hypophosphatasia. J. Endocr. Soc. 2017, 1, 1188–1193. [Google Scholar] [CrossRef]
- Bowden, S.A.; Foster, B.L. Profile of asfotase alfa in the treatment of hypophosphatasia: Design, development, and place in therapy. Drug Des. Devel. Ther. 2018, 12, 3147–3161. [Google Scholar] [CrossRef]
- Seefried, L.; Baumann, J.; Hemsley, S.; Hofmann, C.; Kunstmann, E.; Kiese, B.; Huang, Y.; Chivers, S.; Valentin, M.A.; Borah, B.; et al. Efficacy of anti-sclerostin monoclonal antibody BPS804 in adult patients with hypophosphatasia. J. Clin. Investig. 2017, 127, 2148–2158. [Google Scholar] [CrossRef]
- Camacho, P.M.; Painter, S.; Kadanoff, R. Treatment of adult hypophosphatasia with teriparatide. Endocr. Pract. 2008, 14, 204–208. [Google Scholar] [CrossRef]
- Doshi, K.B.; Hamrahian, A.H.; Licata, A.A. Teriparatide treatment in adult hypophosphatasia in a patient exposed to bisphosphonate: A case report. Clin. Cases Miner. Bone Metab. 2009, 6, 266–269. [Google Scholar]
- Laroche, M. Failure of teriparatide in treatment of bone complications of adult hypophosphatasia. Calcif. Tissue Int. 2012, 90, 250. [Google Scholar] [CrossRef]
- Righetti, M.; Wach, J.; Desmarchelier, R.; Coury, F. Teriparatide treatment in an adult patient with hypophosphatasia exposed to bisphosphonate and revealed by bilateral atypical fractures. Jt. Bone Spine 2018, 85, 365–367. [Google Scholar] [CrossRef]
- Schmidt, T.; Rolvien, T.; Linke, C.; Jandl, N.M.; Oheim, R.; Amling, M.; Barvencik, F. Outcome of Teriparatide Treatment on Fracture Healing Complications and Symptomatic Bone Marrow Edema in Four Adult Patients with Hypophosphatasia. JBMR Plus 2019, 3, e10215. [Google Scholar] [CrossRef]
- Schalin-Jantti, C.; Mornet, E.; Lamminen, A.; Valimaki, M.J. Parathyroid hormone treatment improves pain and fracture healing in adult hypophosphatasia. J. Clin. Endocrinol. Metab. 2010, 95, 5174–5179. [Google Scholar] [CrossRef]
- Harmey, D.; Hessle, L.; Narisawa, S.; Johnson, K.A.; Terkeltaub, R.; Millan, J.L. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: An integrated model of the pathogenesis of mineralization disorders. Am. J. Pathol. 2004, 164, 1199–1209. [Google Scholar] [CrossRef]
- Millan, J.L. The role of phosphatases in the initiation of skeletal mineralization. Calcif. Tissue Int. 2013, 93, 299–306. [Google Scholar] [CrossRef]
- Sebastian-Serrano, A.; de Diego-Garcia, L.; Henshall, D.C.; Engel, T.; Diaz-Hernandez, M. Haploinsufficient TNAP Mice Display Decreased Extracellular ATP Levels and Expression of Pannexin-1 Channels. Front. Pharmacol. 2018, 9, 170. [Google Scholar] [CrossRef]
- Huesa, C.; Houston, D.; Kiffer-Moreira, T.; Yadav, M.M.; Millan, J.L.; Farquharson, C. The Functional co-operativity of Tissue-Nonspecific Alkaline Phosphatase (TNAP) and PHOSPHO1 during initiation of Skeletal Mineralization. Biochem. Biophys. Rep. 2015, 4, 196–201. [Google Scholar] [CrossRef]
- Colazo, J.M.; Hu, J.R.; Dahir, K.M.; Simmons, J.H. Neurological symptoms in Hypophosphatasia. Osteoporos. Int. 2019, 30, 469–480. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Bowling, F.; Carpenter, K.; Earl, J.; Chaitow, J.; Pitt, J.; Mornet, E.; Sillence, D.; Ellaway, C. Perinatal hypophosphatasia presenting as neonatal epileptic encephalopathy with abnormal neurotransmitter metabolism secondary to reduced co-factor pyridoxal-5′-phosphate availability. J. Inherit. Metab. Dis. 2010, 33 (Suppl. 3), S25–S33. [Google Scholar] [CrossRef]
- Nunes, M.L.; Mugnol, F.; Bica, I.; Fiori, R.M. Pyridoxine-dependent seizures associated with hypophosphatasia in a newborn. J. Child. Neurol. 2002, 17, 222–224. [Google Scholar] [CrossRef]
- Guzel Nur, B.; Celmeli, G.; Manguoglu, E.; Soyucen, E.; Bircan, I.; Mihci, E. Pyridoxine-Responsive Seizures in Infantile Hypophosphatasia and a Novel Homozygous Mutation in ALPL Gene. J. Clin. Res. Pediatr. Endocrinol. 2016, 8, 360–364. [Google Scholar] [CrossRef]
- Demirbilek, H.; Alanay, Y.; Alikasifoglu, A.; Topcu, M.; Mornet, E.; Gonc, N.; Ozon, A.; Kandemir, N. Hypophosphatasia presenting with pyridoxine-responsive seizures, hypercalcemia, and pseudotumor cerebri: Case report. J. Clin. Res. Pediatr. Endocrinol. 2012, 4, 34–38. [Google Scholar] [CrossRef]
- Cruz, T.; Gleizes, M.; Balayssac, S.; Mornet, E.; Marsal, G.; Millan, J.L.; Malet-Martino, M.; Nowak, L.G.; Gilard, V.; Fonta, C. Identification of altered brain metabolites associated with TNAP activity in a mouse model of hypophosphatasia using untargeted NMR-based metabolomics analysis. J. Neurochem. 2017, 140, 919–940. [Google Scholar] [CrossRef]
- Ermonval, M.; Baudry, A.; Baychelier, F.; Pradines, E.; Pietri, M.; Oda, K.; Schneider, B.; Mouillet-Richard, S.; Launay, J.M.; Kellermann, O. The cellular prion protein interacts with the tissue non-specific alkaline phosphatase in membrane microdomains of bioaminergic neuronal cells. PLoS ONE 2009, 4, e6497. [Google Scholar] [CrossRef]
- Fonta, C.; Negyessy, L.; Renaud, L.; Barone, P. Postnatal development of alkaline phosphatase activity correlates with the maturation of neurotransmission in the cerebral cortex. J. Comp. Neurol. 2005, 486, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Hanics, J.; Barna, J.; Xiao, J.; Millan, J.L.; Fonta, C.; Negyessy, L. Ablation of TNAP function compromises myelination and synaptogenesis in the mouse brain. Cell Tissue Res. 2012, 349, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Fonta, C.; Negyessy, L.; Renaud, L.; Barone, P. Areal and subcellular localization of the ubiquitous alkaline phosphatase in the primate cerebral cortex: Evidence for a role in neurotransmission. Cereb. Cortex 2004, 14, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Diez-Zaera, M.; Diaz-Hernandez, J.I.; Hernandez-Alvarez, E.; Zimmermann, H.; Diaz-Hernandez, M.; Miras-Portugal, M.T. Tissue-nonspecific alkaline phosphatase promotes axonal growth of hippocampal neurons. Mol. Biol. Cell 2011, 22, 1014–1024. [Google Scholar] [CrossRef]
- Graser, S.; Mentrup, B.; Schneider, D.; Klein-Hitpass, L.; Jakob, F.; Hofmann, C. Overexpression of tissue-nonspecific alkaline phosphatase increases the expression of neurogenic differentiation markers in the human SH-SY5Y neuroblastoma cell line. Bone 2015, 79, 150–161. [Google Scholar] [CrossRef]
- Sebastian-Serrano, A.; Engel, T.; de Diego-Garcia, L.; Olivos-Ore, L.A.; Arribas-Blazquez, M.; Martinez-Frailes, C.; Perez-Diaz, C.; Millan, J.L.; Artalejo, A.R.; Miras-Portugal, M.T.; et al. Neurodevelopmental alterations and seizures developed by mouse model of infantile hypophosphatasia are associated with purinergic signalling deregulation. Hum. Mol. Genet. 2016, 25, 4143–4156. [Google Scholar] [CrossRef]
- Theriault, O.; Poulin, H.; Thomas, G.R.; Friesen, A.D.; Al-Shaqha, W.A.; Chahine, M. Pyridoxal-5′-phosphate (MC-1), a vitamin B6 derivative, inhibits expressed P2X receptors. Can. J. Physiol. Pharmacol. 2014, 92, 189–196. [Google Scholar] [CrossRef]
- Street, S.E.; Sowa, N.A. TNAP and Pain Control. Subcell Biochem. 2015, 76, 283–305. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic receptors and pain. Curr. Pharm. Des. 2009, 15, 1717–1735. [Google Scholar] [CrossRef]
- Kermer, V.; Ritter, M.; Albuquerque, B.; Leib, C.; Stanke, M.; Zimmermann, H. Knockdown of tissue nonspecific alkaline phosphatase impairs neural stem cell proliferation and differentiation. Neurosci. Lett. 2010, 485, 208–211. [Google Scholar] [CrossRef]
- Hedrich, C.M.; Morbach, H.; Reiser, C.; Girschick, H.J. New Insights into Adult and Paediatric Chronic Non-bacterial Osteomyelitis CNO. Curr. Rheumatol. Rep. 2020, 22, 52. [Google Scholar] [CrossRef]
- Bessueille, L.; Briolay, A.; Como, J.; Mebarek, S.; Mansouri, C.; Gleizes, M.; El Jamal, A.; Buchet, R.; Dumontet, C.; Matera, E.L.; et al. Tissue-nonspecific alkaline phosphatase is an anti-inflammatory nucleotidase. Bone 2020, 133, 115262. [Google Scholar] [CrossRef]
- Jacobson, K.A.; AP, I.J.; Muller, C.E. Medicinal chemistry of P2 and adenosine receptors: Common scaffolds adapted for multiple targets. Biochem. Pharmacol. 2020, in press. [Google Scholar] [CrossRef]
- Muller, C.E.; Baqi, Y.; Namasivayam, V. Agonists and Antagonists for Purinergic Receptors. Methods Mol. Biol. 2020, 2041, 45–64. [Google Scholar] [CrossRef]
- D’Antongiovanni, V.; Fornai, M.; Pellegrini, C.; Benvenuti, L.; Blandizzi, C.; Antonioli, L. The Adenosine System at the Crossroads of Intestinal Inflammation and Neoplasia. Int. J. Mol. Sci. 2020, 21, 5089. [Google Scholar] [CrossRef]
- Gratal, P.; Lamuedra, A.; Medina, J.P.; Bermejo-Alvarez, I.; Largo, R.; Herrero-Beaumont, G.; Mediero, A. Purinergic System Signaling in Metainflammation-Associated Osteoarthritis. Front. Med. 2020, 7, 506. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of Adenosine Receptors: The State of the Art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Beck, C.; Morbach, H.; Richl, P.; Stenzel, M.; Girschick, H.J. How can calcium pyrophosphate crystals induce inflammation in hypophosphatasia or chronic inflammatory joint diseases? Rheumatol. Int. 2009, 29, 229–238. [Google Scholar] [CrossRef]
- Mulay, S.R.; Steiger, S.; Shi, C.; Anders, H.J. A guide to crystal-related and nano- or microparticle-related tissue responses. FEBS J. 2020, 287, 818–832. [Google Scholar] [CrossRef]
- Kahles, F.; Findeisen, H.M.; Bruemmer, D. Osteopontin: A novel regulator at the cross roads of inflammation, obesity and diabetes. Mol. Metab. 2014, 3, 384–393. [Google Scholar] [CrossRef]
- Castello, L.M.; Raineri, D.; Salmi, L.; Clemente, N.; Vaschetto, R.; Quaglia, M.; Garzaro, M.; Gentilli, S.; Navalesi, P.; Cantaluppi, V.; et al. Osteopontin at the Crossroads of Inflammation and Tumor Progression. Mediat. Inflamm. 2017, 2017, 4049098. [Google Scholar] [CrossRef] [PubMed]
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24. [Google Scholar] [CrossRef]
- Zhang, X.; Shu, Q.; Liu, Z.; Gao, C.; Wang, Z.; Xing, Z.; Song, J. Recombinant osteopontin provides protection for cerebral infarction by inhibiting the NLRP3 inflammasome in microglia. Brain Res. 2020, 1751, 147170. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Jeong, S.; Xia, Q.; Kong, X. Role of Osteopontin in Liver Diseases. Int. J. Biol. Sci. 2016, 12, 1121–1128. [Google Scholar] [CrossRef]
- Iida, T.; Wagatsuma, K.; Hirayama, D.; Nakase, H. Is Osteopontin a Friend or Foe of Cell Apoptosis in Inflammatory Gastrointestinal and Liver Diseases? Int. J. Mol. Sci. 2017, 19, 7. [Google Scholar] [CrossRef]
- Lei, W.; Ni, H.; Herington, J.; Reese, J.; Paria, B.C. Alkaline phosphatase protects lipopolysaccharide-induced early pregnancy defects in mice. PLoS ONE 2015, 10, e0123243. [Google Scholar] [CrossRef]
- Kellett, K.A.; Hooper, N.M. The Role of Tissue Non-specific Alkaline Phosphatase (TNAP) in Neurodegenerative Diseases: Alzheimer’s Disease in the Focus. Subcell Biochem. 2015, 76, 363–374. [Google Scholar] [CrossRef]
- Moskalev, A.; Stambler, I.; Caruso, C. Innate and Adaptive Immunity in Aging and Longevity: The Foundation of Resilience. Aging Dis. 2020, 11, 1363–1373. [Google Scholar] [CrossRef]
- Hernandez-Chirlaque, C.; Gamez-Belmonte, R.; Ocon, B.; Martinez-Moya, P.; Wirtz, S.; Sanchez de Medina, F.; Martinez-Augustin, O. Tissue Non-specific Alkaline Phosphatase Expression is Needed for the Full Stimulation of T Cells and T Cell-Dependent Colitis. J. Crohns Colitis 2017, 11, 857–870. [Google Scholar] [CrossRef][Green Version]
- Rader, B.A. Alkaline Phosphatase, an Unconventional Immune Protein. Front. Immunol. 2017, 8, 897. [Google Scholar] [CrossRef]
- Goettsch, C.; Strzelecka-Kiliszek, A.; Bessueille, L.; Quillard, T.; Mechtouff, L.; Pikula, S.; Canet-Soulas, E.; Millan, J.L.; Fonta, C.; Magne, D. TNAP as a therapeutic target for cardiovascular calcification—A discussion of its pleiotropic functions in the body. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Zebisch, M.; Strater, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H. History of ectonucleotidases and their role in purinergic signaling. Biochem. Pharmacol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Pleli, T.; Mondorf, A.; Ferreiros, N.; Thomas, D.; Dvorak, K.; Biondi, R.M.; Heringdorf, D.M.Z.; Zeuzem, S.; Geisslinger, G.; Zimmermann, H.; et al. Activation of Adenylyl Cyclase Causes Stimulation of Adenosine Receptors. Cell Physiol. Biochem. 2018, 45, 2516–2528. [Google Scholar] [CrossRef]
- Yang, D.; Ding, C.; Qi, G.; Feldmeyer, D. Cholinergic and Adenosinergic Modulation of Synaptic Release. Neuroscience 2020, in press. [Google Scholar] [CrossRef]
- Minor, M.; Alcedo, K.P.; Battaglia, R.A.; Snider, N.T. Cell type- and tissue-specific functions of ecto-5′-nucleotidase (CD73). Am. J. Physiol. Cell Physiol. 2019, 317, C1079–C1092. [Google Scholar] [CrossRef]
- Schneider, E.; Rissiek, A.; Winzer, R.; Puig, B.; Rissiek, B.; Haag, F.; Mittrucker, H.W.; Magnus, T.; Tolosa, E. Generation and Function of Non-cell-bound CD73 in Inflammation. Front. Immunol. 2019, 10, 1729. [Google Scholar] [CrossRef]
- Fredholm, B.B.; AP, I.J.; Jacobson, K.A.; Linden, J.; Muller, C.E. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—An update. Pharmacol. Rev. 2011, 63, 1–34. [Google Scholar] [CrossRef]
- Eisenstein, A.; Chitalia, S.V.; Ravid, K. Bone Marrow and Adipose Tissue Adenosine Receptors Effect on Osteogenesis and Adipogenesis. Int. J. Mol. Sci. 2020, 21, 7470. [Google Scholar] [CrossRef]
- Dwyer, K.M.; Kishore, B.K.; Robson, S.C. Conversion of extracellular ATP into adenosine: A master switch in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 509–524. [Google Scholar] [CrossRef]
- Wang, S.; Gao, S.; Zhou, D.; Qian, X.; Luan, J.; Lv, X. The role of the CD39-CD73-adenosine pathway in liver disease. J. Cell Physiol. 2021, 236, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Cekic, C. Modulation of myeloid cells by adenosine signaling. Curr. Opin. Pharmacol. 2020, 53, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; St Hilaire, C.; Huang, Y.; Yang, D.; Dmitrieva, N.I.; Negro, A.; Schwartzbeck, R.; Liu, Y.; Yu, Z.; Walts, A.; et al. Increased activity of TNAP compensates for reduced adenosine production and promotes ectopic calcification in the genetic disease ACDC. Sci. Signal. 2016, 9, ra121. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Copeland, J.; Shin, M.; Chang, Y.; Venton, B.J. CD73 or CD39 Deletion Reveals Different Mechanisms of Formation for Spontaneous and Mechanically Stimulated Adenosine and Sex Specific Compensations in ATP Degradation. ACS Chem. Neurosci. 2020, 11, 919–928. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graser, S.; Liedtke, D.; Jakob, F. TNAP as a New Player in Chronic Inflammatory Conditions and Metabolism. Int. J. Mol. Sci. 2021, 22, 919. https://doi.org/10.3390/ijms22020919
Graser S, Liedtke D, Jakob F. TNAP as a New Player in Chronic Inflammatory Conditions and Metabolism. International Journal of Molecular Sciences. 2021; 22(2):919. https://doi.org/10.3390/ijms22020919
Chicago/Turabian StyleGraser, Stephanie, Daniel Liedtke, and Franz Jakob. 2021. "TNAP as a New Player in Chronic Inflammatory Conditions and Metabolism" International Journal of Molecular Sciences 22, no. 2: 919. https://doi.org/10.3390/ijms22020919
APA StyleGraser, S., Liedtke, D., & Jakob, F. (2021). TNAP as a New Player in Chronic Inflammatory Conditions and Metabolism. International Journal of Molecular Sciences, 22(2), 919. https://doi.org/10.3390/ijms22020919