Deficiency in Tissue Non-Specific Alkaline Phosphatase Leads to Steatohepatitis in Mice Fed a High Fat Diet Similar to That Produced by a Methionine and Choline Deficient Diet

 ,
, 
 , ,
, ,  and
and
Abstract
1. Introduction
2. Results
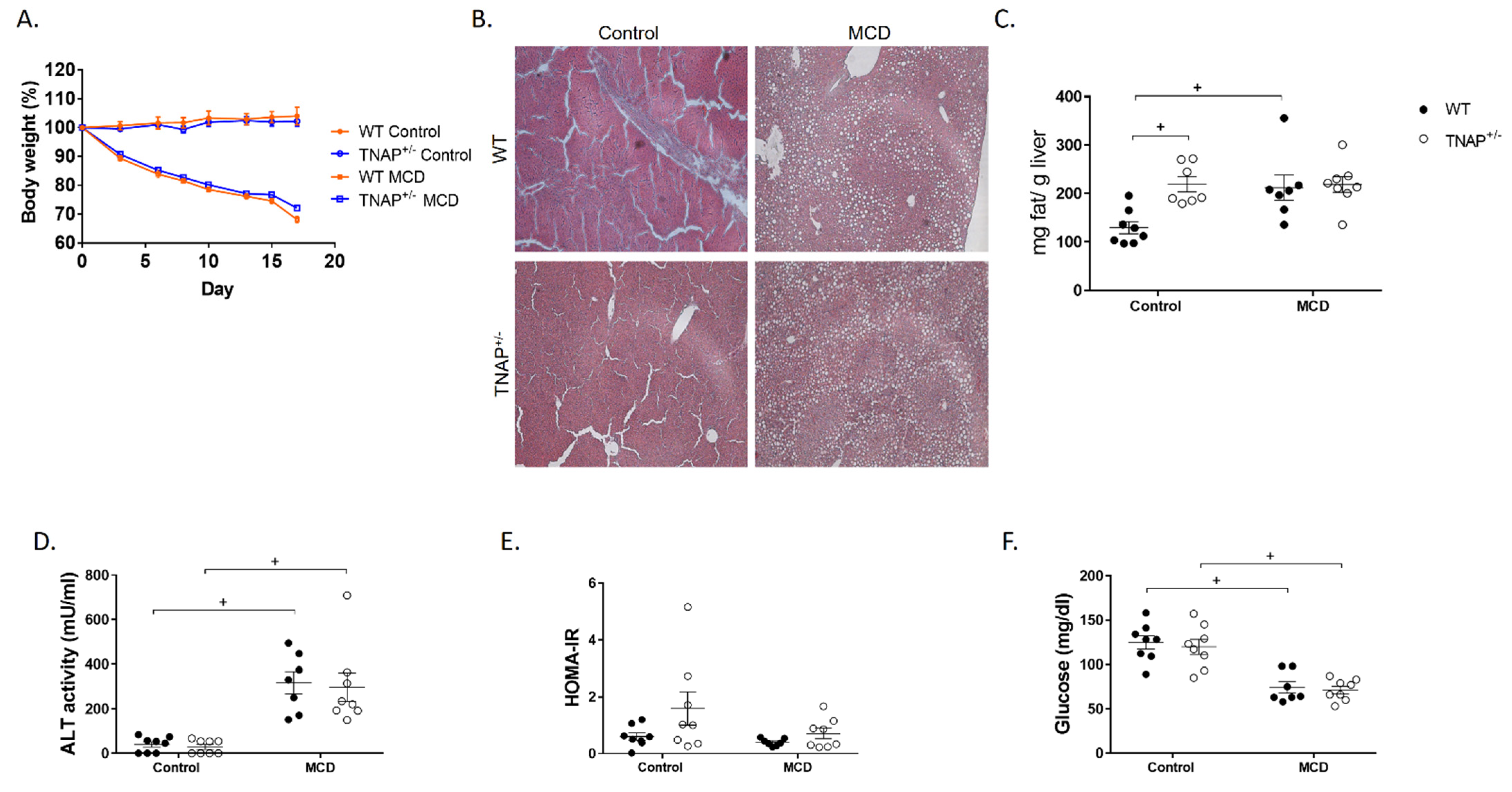
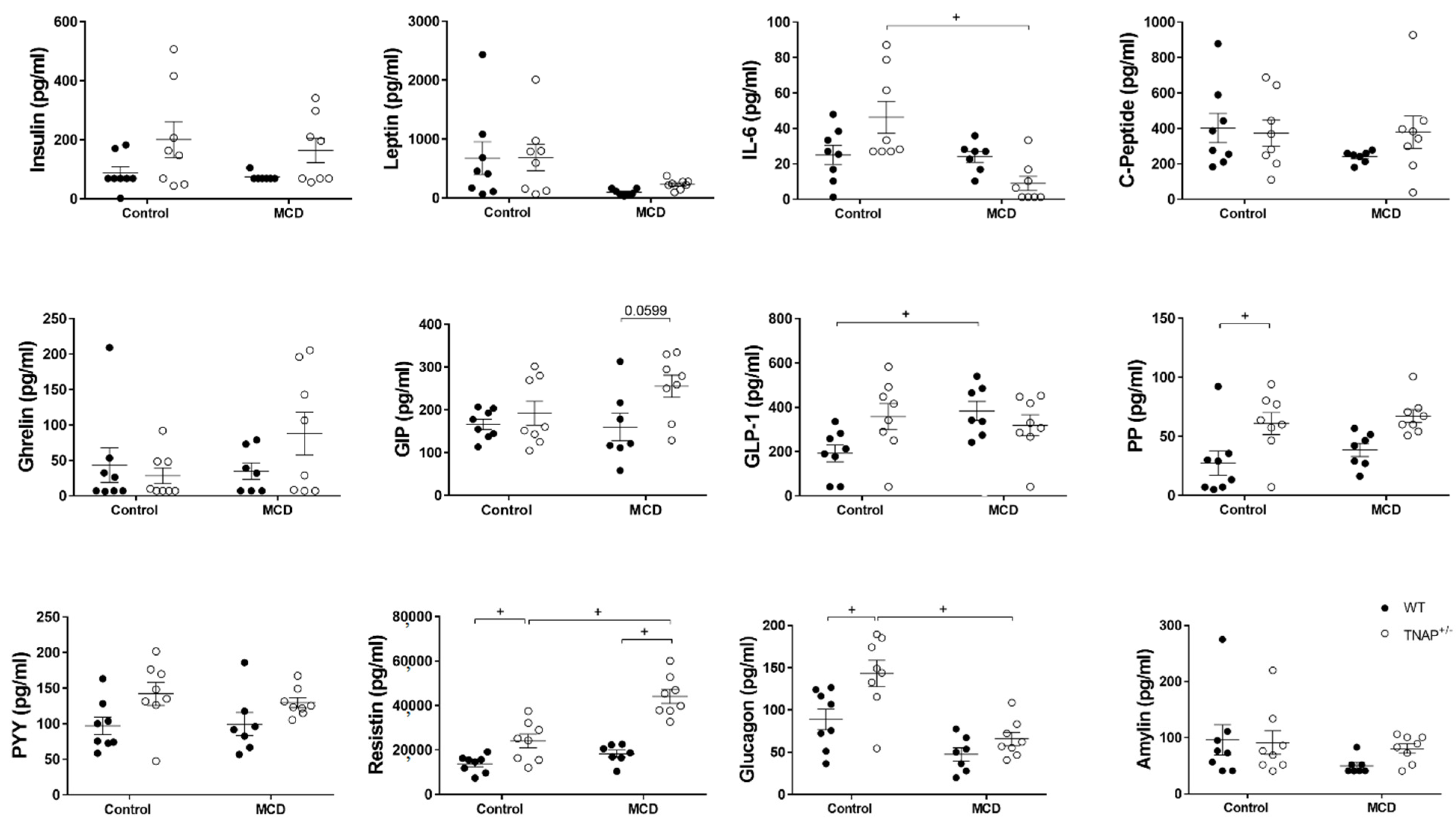
2.1. The Absence of a Single Alpl Allele Does Not Modify the Response of Mice to the MCD Diet
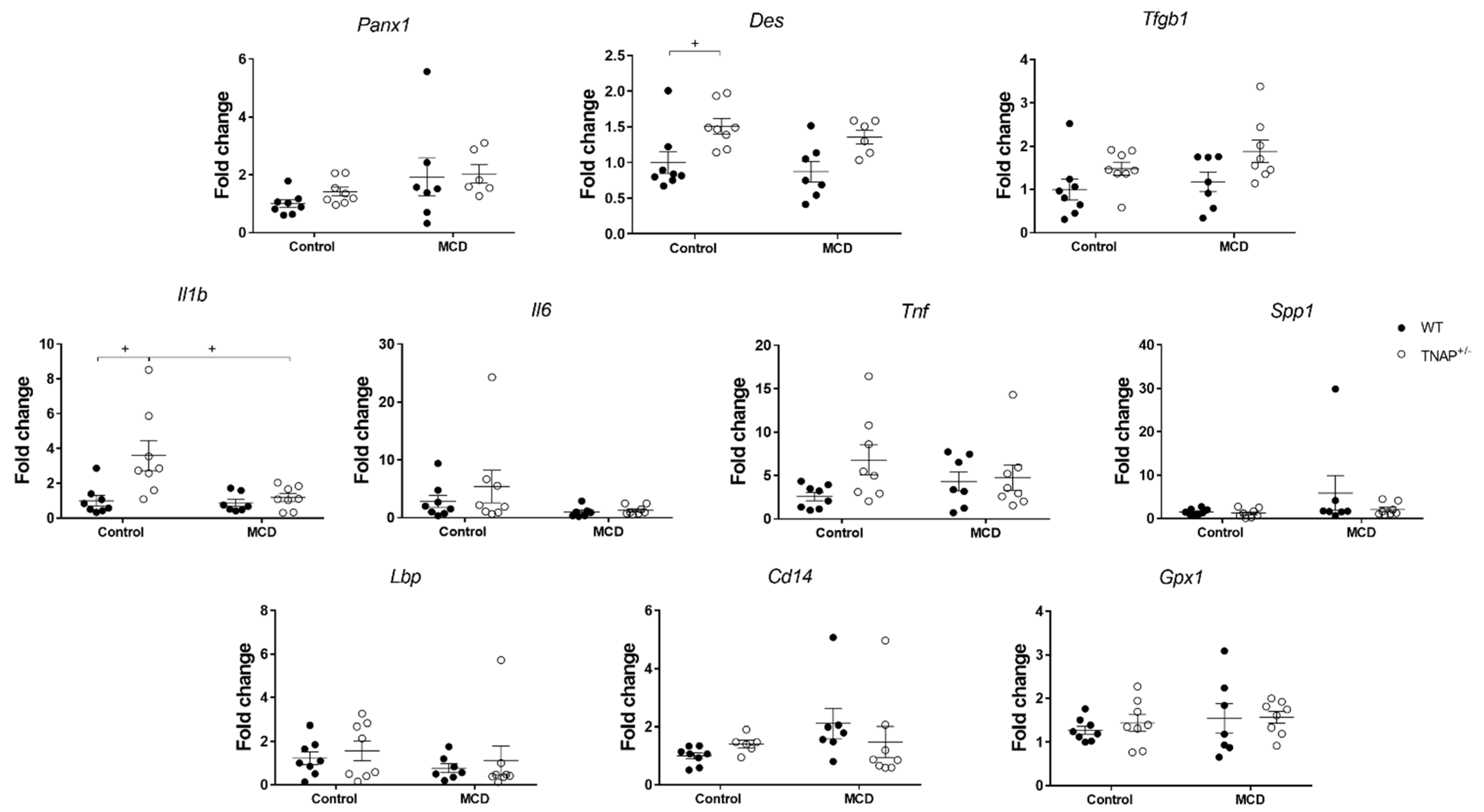
2.2. Alpl Haplodeficiency Induces Hepatic Fat Accumulation in Mice Fed a Control Diet
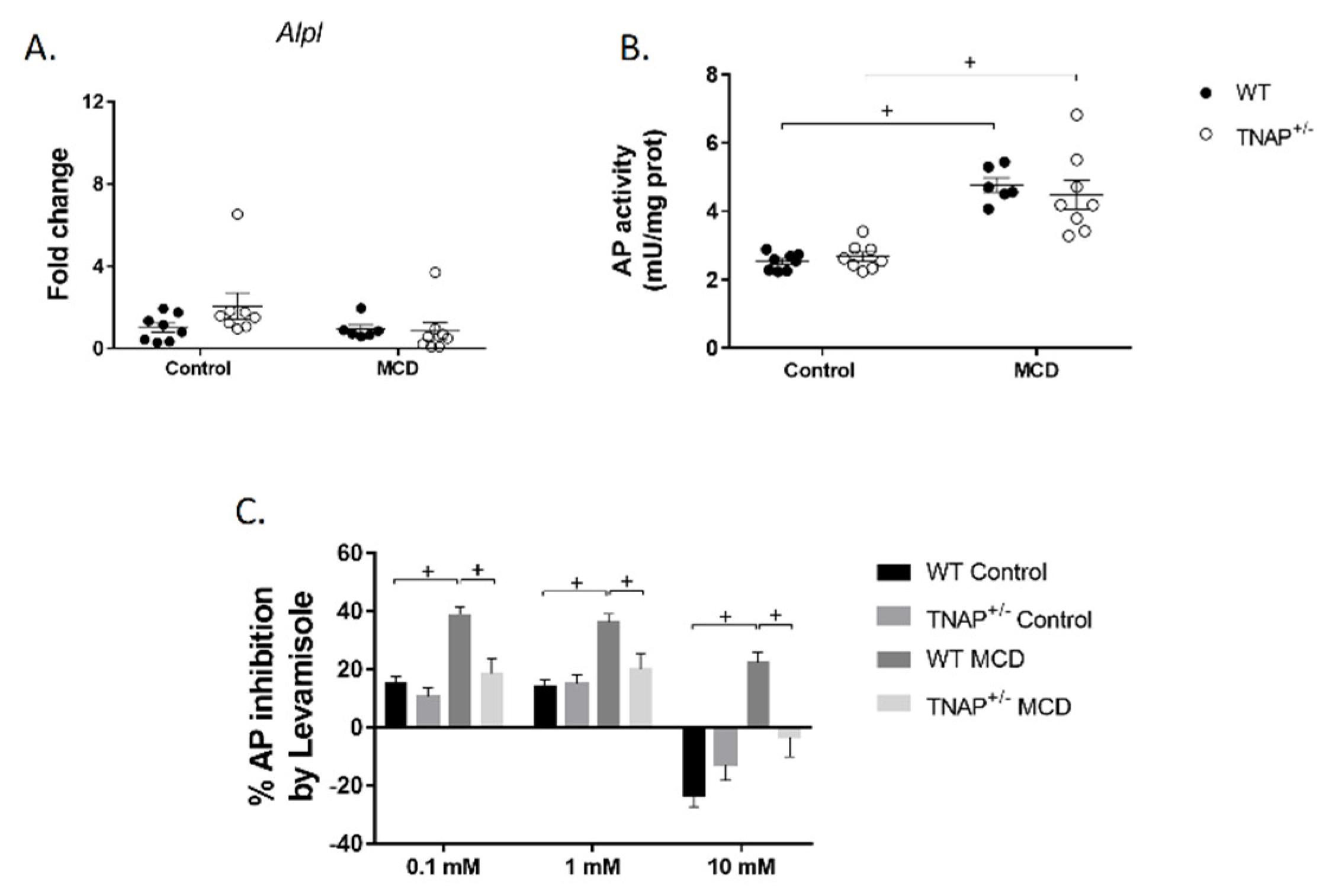
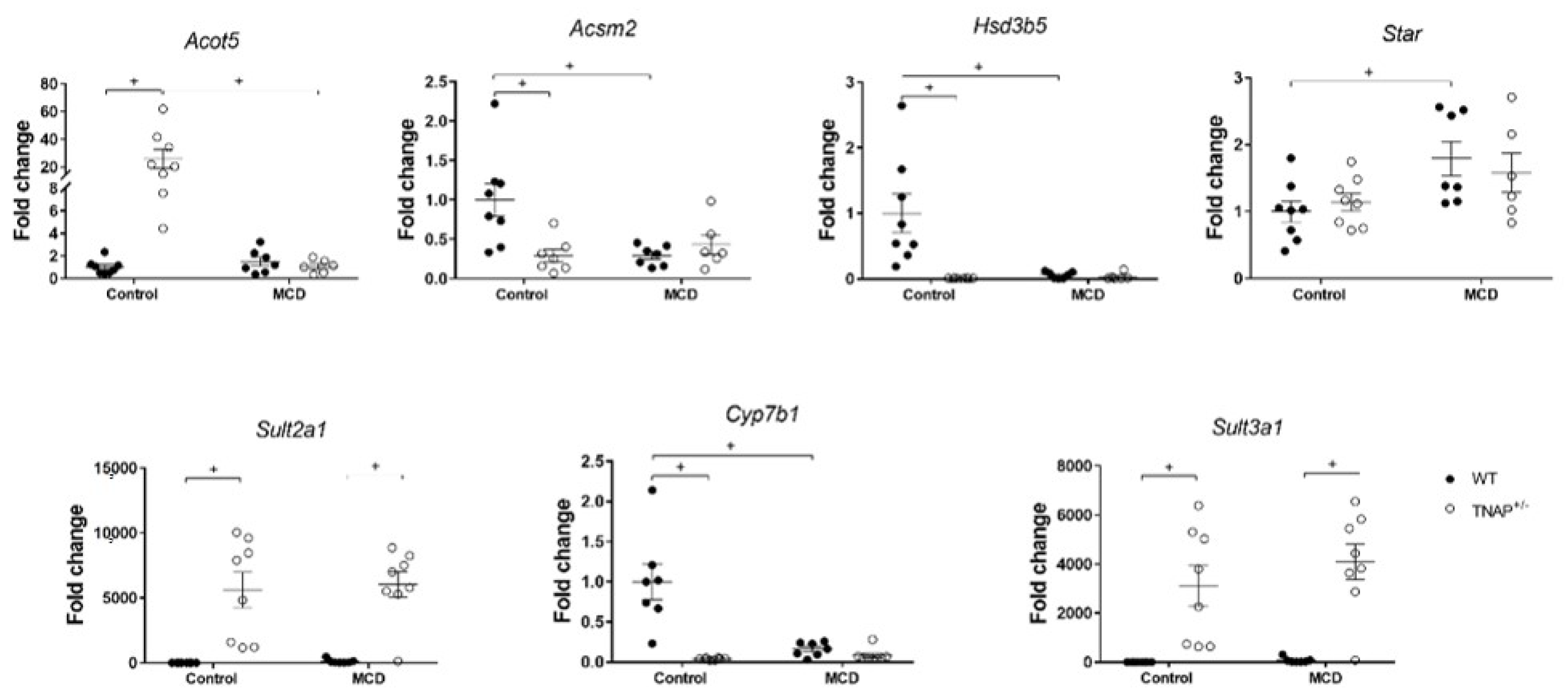
2.3. Characterization of Steatosis in Alpl Haplodeficient Mice
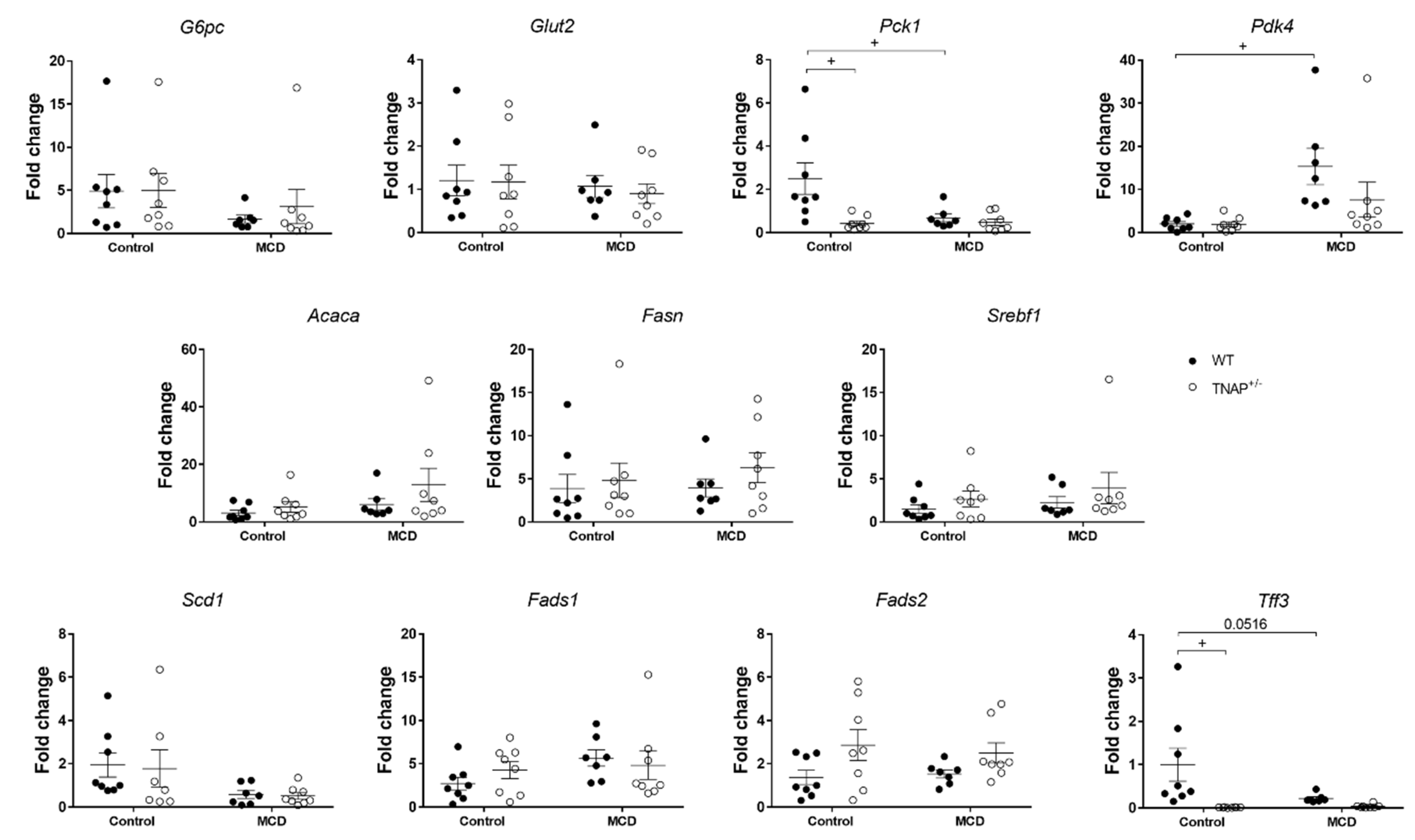
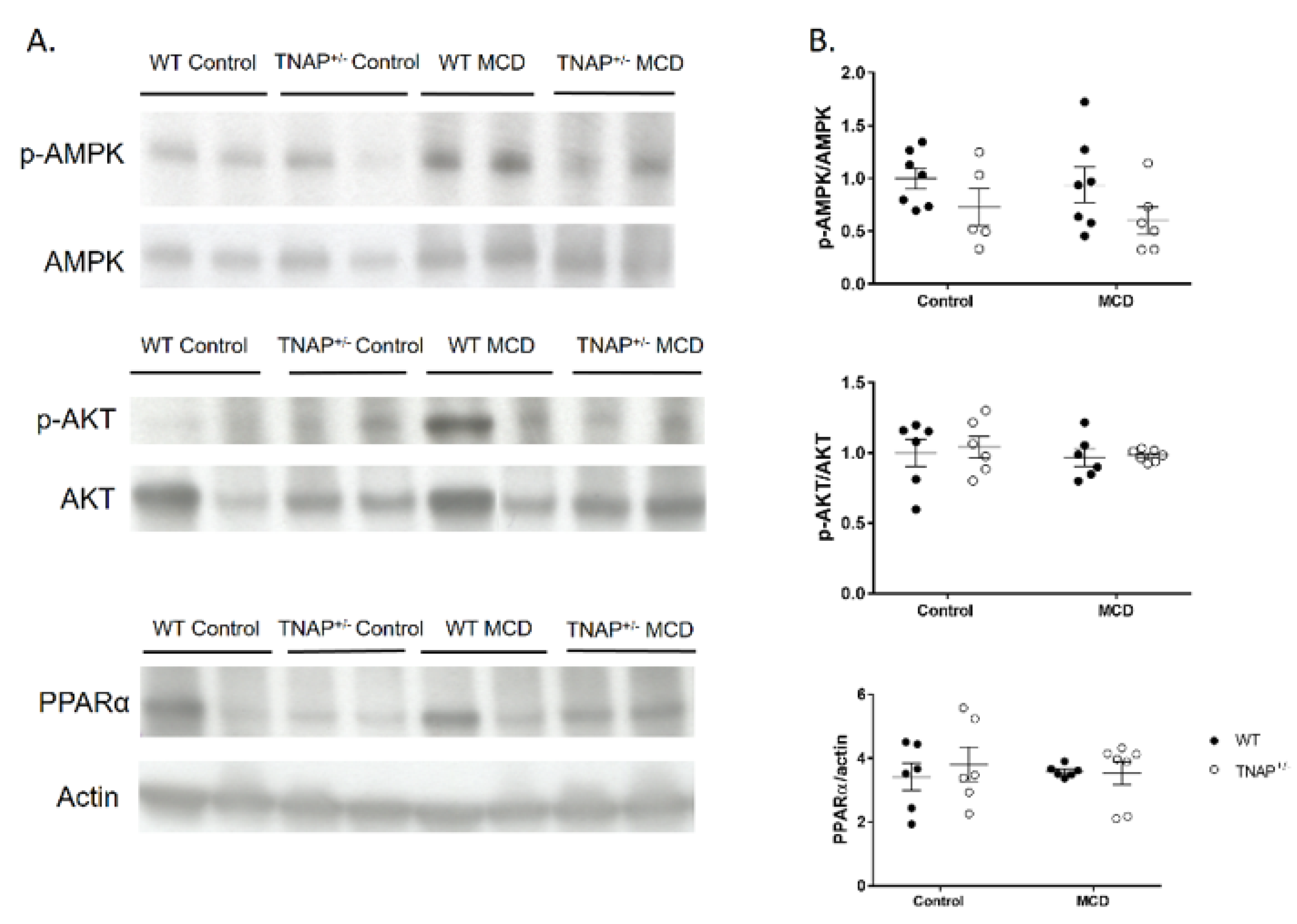
2.4. Alpl Haplodeficient Mice Exhibit a Reduced Activation of the AMPK but Not the AKT or the PPARα Pathway
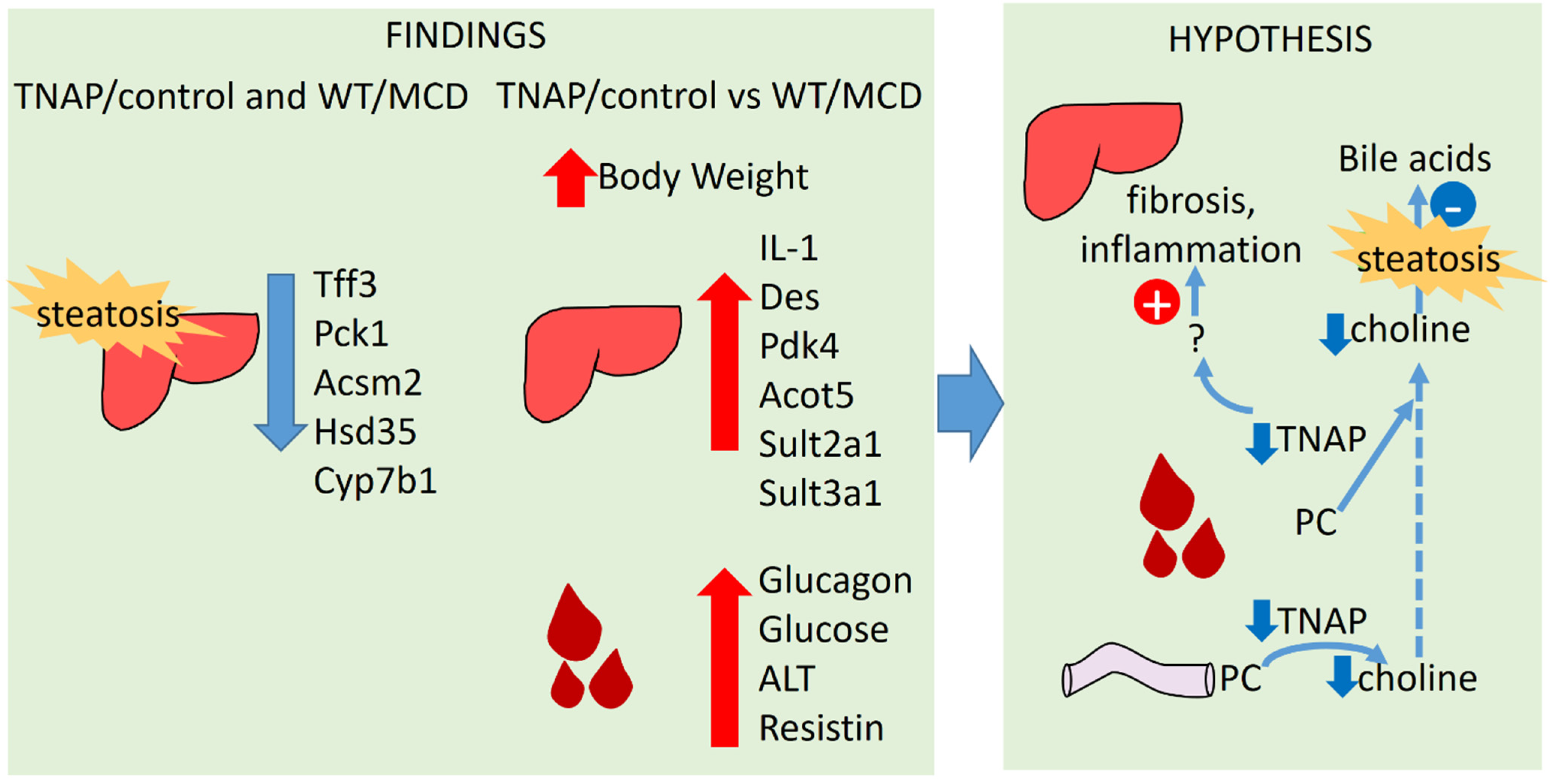
3. Discussion
4. Materials and Methods
4.1. Animals and Experimental Design
4.2. Diets
4.3. Histological Assessment
4.4. Plasmatic Parameters
4.5. Alkaline Phosphatase Activity
4.6. Hepatic Fat Content
4.7. RNA Isolation and Quantitative Reverse-Transcription Polymerase Chain Reaction (RT-qPCR) Analysis
4.8. Western Blot
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALT | alanine aminotransferase |
| BA | bile acids |
| GIP | gastric inhibitory peptide |
| GLP-1 | glucagon-like peptide 1 |
| HOMA-IR | homeostasis model assessment–insulin resistance |
| IAP | intestinal alkaline phosphatase |
| MCD | Methionine- and choline- deficient diet |
| NAFLD | nonalcoholic fatty liver disease |
| NASH | nonalcoholic steatohepatitis |
| PC | Phosphorylcholine |
| PAP | pancreatic peptide |
| PEPCK | phosphoenolpyruvate carboxykinase |
| PYY | peptide YY |
| TNAP | tissue nonspecific alkaline phosphatase |
| VLDL | very low-density lipoproteins |
References
- Raza, S.; Rajak, S.; Upadhyay, A.; Tewari, A.; Anthony Sinha, R. Current treatment paradigms and emerging therapies for NAFLD/NASH. Front. Biosci. 2021, 26, 206–237. [Google Scholar] [CrossRef]
- Pafili, K.; Roden, M. Non-alcoholic fatty liver disease (NAFLD) from pathogenesis to treatment concepts in humans. Mol. Metab. 2020, 101122. [Google Scholar] [CrossRef] [PubMed]
- Rader, B.A. Alkaline Phosphatase, an Unconventional Immune Protein. Front. Immunol. 2017, 8, 897. [Google Scholar] [CrossRef]
- Buchet, R.; Millan, J.L.; Magne, D. Multisystemic functions of alkaline phosphatases. Methods Mol. Biol. 2013, 1053, 27–51. [Google Scholar]
- Gamez-Belmonte, R.; Hernandez-Chirlaque, C.; Sanchez de Medina, F.; Martinez-Augustin, O. Experimental acute pancreatitis is enhanced in mice with tissue nonspecific alkaline phoshatase haplodeficiency due to modulation of neutrophils and acinar cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3769–3779. [Google Scholar] [CrossRef]
- Bessueille, L.; Briolay, A.; Como, J.; Mebarek, S.; Mansouri, C.; Gleizes, M.; El Jamal, A.; Buchet, R.; Dumontet, C.; Matera, E.L.; et al. Tissue-nonspecific alkaline phosphatase is an anti-inflammatory nucleotidase. Bone 2020, 133, 115262. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Chirlaque, C.; Gamez-Belmonte, R.; Ocon, B.; Martinez-Moya, P.; Wirtz, S.; Sanchez de Medina, F.; Martinez-Augustin, O. Tissue Non-specific Alkaline Phosphatase Expression is Needed for the Full Stimulation of T Cells and T Cell-Dependent Colitis. J. Crohn’s Colitis 2017, 11, 857–870. [Google Scholar]
- Kaliannan, K.; Hamarneh, S.R.; Economopoulos, K.P.; Nasrin Alam, S.; Moaven, O.; Patel, P.; Malo, N.S.; Ray, M.; Abtahi, S.M.; Muhammad, N.; et al. Intestinal alkaline phosphatase prevents metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 7003–7008. [Google Scholar] [CrossRef]
- Nakano, T.; Inoue, I.; Koyama, I.; Kanazawa, K.; Nakamura, K.-I.; Narisawa, S.; Tanaka, K.; Akita, M.; Masuyama, T.; Seo, M.; et al. Disruption of the murine intestinal alkaline phosphatase gene Akp3 impairs lipid transcytosis and induces visceral fat accumulation and hepatic steatosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1439–G1449. [Google Scholar] [CrossRef] [PubMed]
- Sherriff, J.L.; O’Sullivan, T.A.; Properzi, C.; Oddo, J.L.; Adams, L.A. Choline, Its Potential Role in Nonalcoholic Fatty Liver Disease, and the Case for Human and Bacterial Genes. Adv. Nutr. 2016, 7, 5–13. [Google Scholar] [CrossRef]
- Rinella, M.E.; Elias, M.S.; Smolak, R.R.; Fu, T.; Borensztajn, J.; Green, R.M. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 2008, 49, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Bin, P.; Huang, R.; Zhou, X. Oxidation Resistance of the Sulfur Amino Acids: Methionine and Cysteine. BioMed Res. Int. 2017, 2017, 9584932. [Google Scholar] [CrossRef] [PubMed]
- Willebrords, J.; Maes, M.; Pereira, I.V.A.; da Silva, T.C.; Govoni, V.M.; Lopes, V.V.; Crespo Yanguas, S.; Shestopalov, V.I.; Nogueira, M.S.; de Castro, I.A.; et al. Protective effect of genetic deletion of pannexin1 in experimental mouse models of acute and chronic liver disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 819–830. [Google Scholar] [CrossRef]
- Bujak, M.; Bujak, I.T.; Sobočanec, S.; Mihalj, M.; Novak, S.; Ćosić, A.; Levak, M.T.; Kopačin, V.; Mihaljević, B.; Balog, T.; et al. Trefoil Factor 3 Deficiency Affects Liver Lipid Metabolism. Cell. Physiol. Biochem. 2018, 47, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Poupon, R. Liver alkaline phosphatase: A missing link between choleresis and biliary inflammation. Hepatology 2015, 61, 2080–2090. [Google Scholar] [CrossRef] [PubMed]
- Pandak, W.M.; Kakiyama, G. The acidic pathway of bile acid synthesis: Not just an alternative pathway. Liver Res. 2019, 3, 88–98. [Google Scholar] [CrossRef]
- Lopez-Posadas, R.; Gonzalez, R.; Ballester, I.; Martinez-Moya, P.; Romero-Calvo, I.; Suarez, M.D.; Zarzuelo, A.; Martinez-Augustin, O.; Sanchez de Medina, F. Tissue-nonspecific alkaline phosphatase is activated in enterocytes by oxidative stress via changes in glycosylation. Inflamm. Bowel. Dis. 2011, 17, 543–556. [Google Scholar] [CrossRef]
- Sanchez de Medina, F.; Martinez-Augustin, O.; Gonzalez, R.; Ballester, I.; Nieto, A.; Galvez, J.; Zarzuelo, A. Induction of alkaline phosphatase in the inflamed intestine: A novel pharmacological target for inflammatory bowel disease. Biochem. Pharmacol. 2004, 68, 2317–2326. [Google Scholar] [CrossRef]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; Almeida Pereira, T.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 222–232.e2. [Google Scholar] [CrossRef]
- Burgess, S.C.; Hausler, N.; Merritt, M.; Jeffrey, F.M.; Storey, C.; Milde, A.; Koshy, S.; Lindner, J.; Magnuson, M.A.; Malloy, C.R.; et al. Impaired tricarboxylic acid cycle activity in mouse livers lacking cytosolic phosphoenolpyruvate carboxykinase. J. Biol. Chem. 2004, 279, 48941–48949. [Google Scholar] [CrossRef] [PubMed]
- Jha, P.; Knopf, A.; Koefeler, H.; Mueller, M.; Lackner, C.; Hoefler, G.; Claudel, T.; Trauner, M. Role of adipose tissue in methionine-choline-deficient model of non-alcoholic steatohepatitis (NASH). Biochim. Biophys. Acta 2014, 1842, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Boomgaarden, I.; Vock, C.; Klapper, M.; Döring, F. Comparative analyses of disease risk genes belonging to the acyl-CoA synthetase medium-chain (ACSM) family in human liver and cell lines. Biochem. Genet. 2009, 47, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Kocabayoglu, P.; Sowa, J.P.; Sydor, S.; Best, J.; Schlattjan, M.; Beilfuss, A.; Schmitt, J.; Hannivoort, R.A.; Kilicarslan, A.; et al. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology 2013, 57, 1394–1406. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Deng, W.; Wang, J.; Shao, Y.; Ou, M.; Ding, M. Primary bile acids as potential biomarkers for the clinical grading of intrahepatic cholestasis of pregnancy. Int. J. Gynaecol. Obstet. 2013, 122, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Guillén, N.; Navarro, M.A.; Arnal, C.; Noone, E.; Arbonés-Mainar, J.M.; Acín, S.; Surra, J.C.; Muniesa, P.; Roche, H.M.; Osada, J. Microarray analysis of hepatic gene expression identifies new genes involved in steatotic liver. Physiol. Genom. 2009, 37, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Setchell, K.D.R. Will the real bile acid sulfotransferase please stand up? Identification of Sult2a8 as a major hepatic bile acid sulfonating enzyme in mice. J. Lipid. Res. 2017, 58, 1033–1035. [Google Scholar] [CrossRef]
- Robben, J.; Parmentier, G.; Eyssen, H. Isolation of a rat intestinal Clostridium strain producing 5 alpha- and 5 beta-bile salt 3 alpha-sulfatase activity. Appl. Environ. Microbiol. 1986, 51, 32–38. [Google Scholar] [CrossRef]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | MCD Diet | Chow Diet | |
|---|---|---|---|
| Sucrose (g/kg) | 455 | 455 | 445 |
| Corn starch (g/kg) | 200 | 200 | 200 |
| Corn Oil (g/kg) | 100 | 100 | 40 |
| l-Methionine (g/kg) | 8.2 | - | 8.2 |
| Choline (g/kg) | 1.4 | - | 1.4 |
| g/Kg | Control Diet | MCD Diet |
|---|---|---|
| Sucrose | 443.597 | 455.294 |
| Corn starch | 198.783 | 200.0 |
| Corn Oil | 100.0 | 100.0 |
| Cellulose | 30.0 | 30.0 |
| Mineral Mix, AIN-76 (170915) | 35.0 | 35.0 |
| Calcium Phosphate, dibasic | 3.0 | 3.0 |
| L-Alanine | 3.5 | 3.5 |
| L-Arginine HCl | 12.1 | 12.1 |
| L-Asparagine | 6.0 | 6.0 |
| L-Aspartic Acid | 3.5 | 3.5 |
| L-Cysteine | 3.5 | 3.5 |
| L-Glutamic Acid | 40.0 | 40.0 |
| Glycine | 23.3 | 23.3 |
| L-Histidine HCl, monohydrate | 4.5 | 4.5 |
| L-Isoleucine | 8.2 | 8.2 |
| L-Leucine | 11.1 | 11.1 |
| L-Lysine HCl | 18.0 | 18.0 |
| L-Methionine | 8.2 | - |
| L-Phenylalanine | 7.5 | 7.5 |
| L-Proline | 3.5 | 3.5 |
| L-Serine | 3.5 | 3.5 |
| L-Threonine | 8.2 | 8.2 |
| L-Tryptophan | 1.8 | 1.8 |
| L-Tyrosine | 5.0 | 5.0 |
| L-Valine | 8.2 | 8.2 |
| Vitamin Mix, Teklad (400600) + Choline dihydrogen citrate | 10.0 (1.4) | - |
| Vitamin Mix, w/o choline, A, D, E (83171) | - | 5.0 |
| Vitamin E, DL-α tocopherol acetate (500 IU/g) | - | 0.242 |
| Vitamin A Palmitate (500,000 IU/g) | - | 0.0396 |
| Vitamin D3, cholecalciferol (500,000 IU/g) | - | 0.0044 |
| Ethoxyquin, antioxidant | 0.02 | 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gámez-Belmonte, R.; Tena-Garitaonaindia, M.; Hernández-Chirlaque, C.; Córdova, S.; Ceacero-Heras, D.; de Medina, F.S.; Martínez-Augustin, O. Deficiency in Tissue Non-Specific Alkaline Phosphatase Leads to Steatohepatitis in Mice Fed a High Fat Diet Similar to That Produced by a Methionine and Choline Deficient Diet. Int. J. Mol. Sci. 2021, 22, 51. https://doi.org/10.3390/ijms22010051
Gámez-Belmonte R, Tena-Garitaonaindia M, Hernández-Chirlaque C, Córdova S, Ceacero-Heras D, de Medina FS, Martínez-Augustin O. Deficiency in Tissue Non-Specific Alkaline Phosphatase Leads to Steatohepatitis in Mice Fed a High Fat Diet Similar to That Produced by a Methionine and Choline Deficient Diet. International Journal of Molecular Sciences. 2021; 22(1):51. https://doi.org/10.3390/ijms22010051
Chicago/Turabian StyleGámez-Belmonte, Reyes, Mireia Tena-Garitaonaindia, Cristina Hernández-Chirlaque, Samir Córdova, Diego Ceacero-Heras, Fermín Sánchez de Medina, and Olga Martínez-Augustin. 2021. "Deficiency in Tissue Non-Specific Alkaline Phosphatase Leads to Steatohepatitis in Mice Fed a High Fat Diet Similar to That Produced by a Methionine and Choline Deficient Diet" International Journal of Molecular Sciences 22, no. 1: 51. https://doi.org/10.3390/ijms22010051
APA StyleGámez-Belmonte, R., Tena-Garitaonaindia, M., Hernández-Chirlaque, C., Córdova, S., Ceacero-Heras, D., de Medina, F. S., & Martínez-Augustin, O. (2021). Deficiency in Tissue Non-Specific Alkaline Phosphatase Leads to Steatohepatitis in Mice Fed a High Fat Diet Similar to That Produced by a Methionine and Choline Deficient Diet. International Journal of Molecular Sciences, 22(1), 51. https://doi.org/10.3390/ijms22010051