PPAR Gamma: From Definition to Molecular Targets and Therapy of Lung Diseases


 and
and 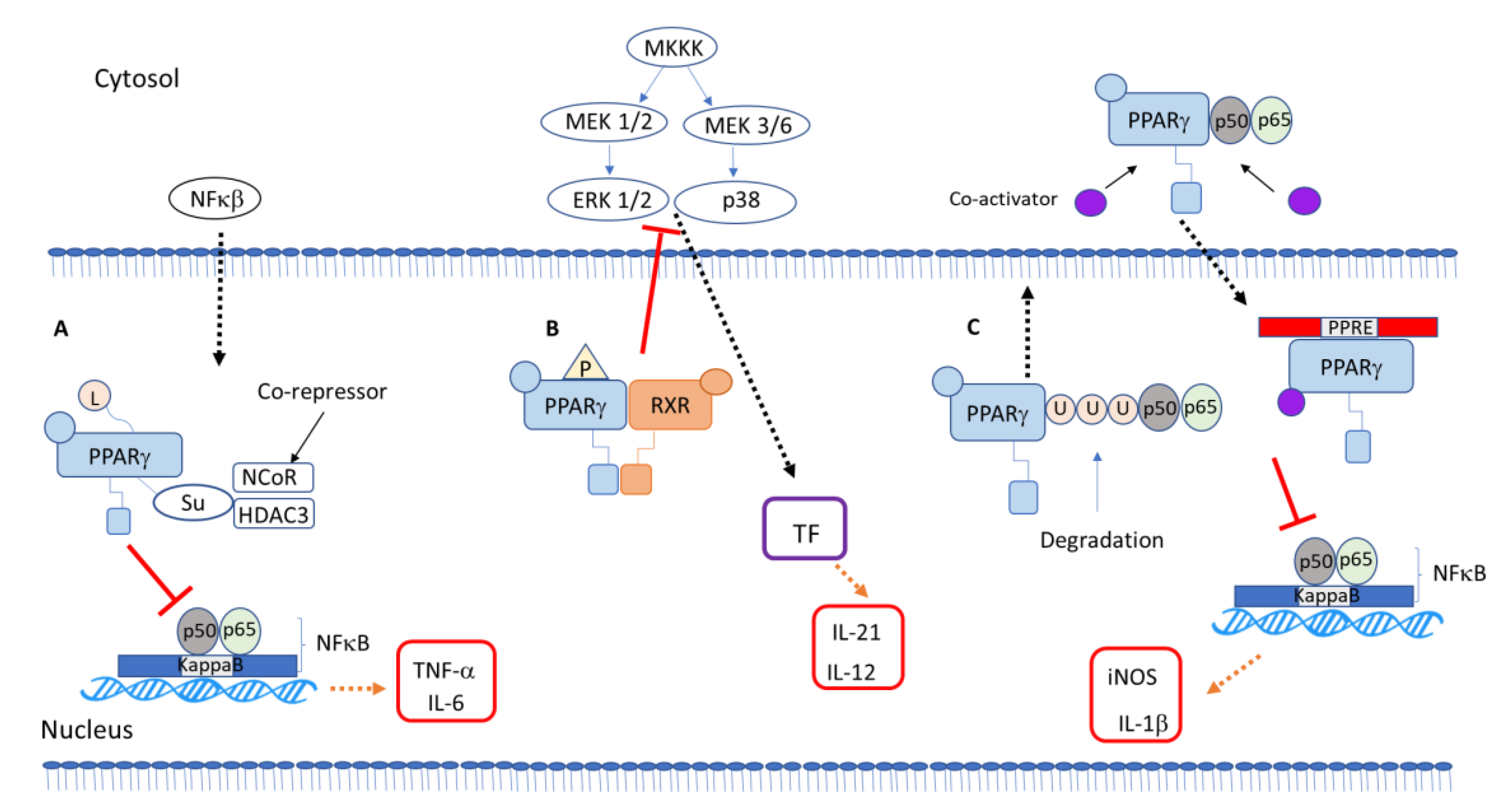{kind=link}
{kind=link}
Abstract
1. Introduction
2. Isoforms and Function of PPARγ
3. PPARγ Ligands and Overall Effects
4. Mechanisms of Regulation of PPARγ
4.1. Phosphorylation
4.2. SUMOylation
4.3. Ubiquitination
4.4. Acetylation
4.5. Glycosylation
5. PPARγ-Dependent Anti-Inflammatory Mechanisms
PPARγ Interaction with Other Transcription Factors and Intracellular Signaling Proteins
6. Pharmacologic and Therapeutic Potentials of PPARγ Ligands
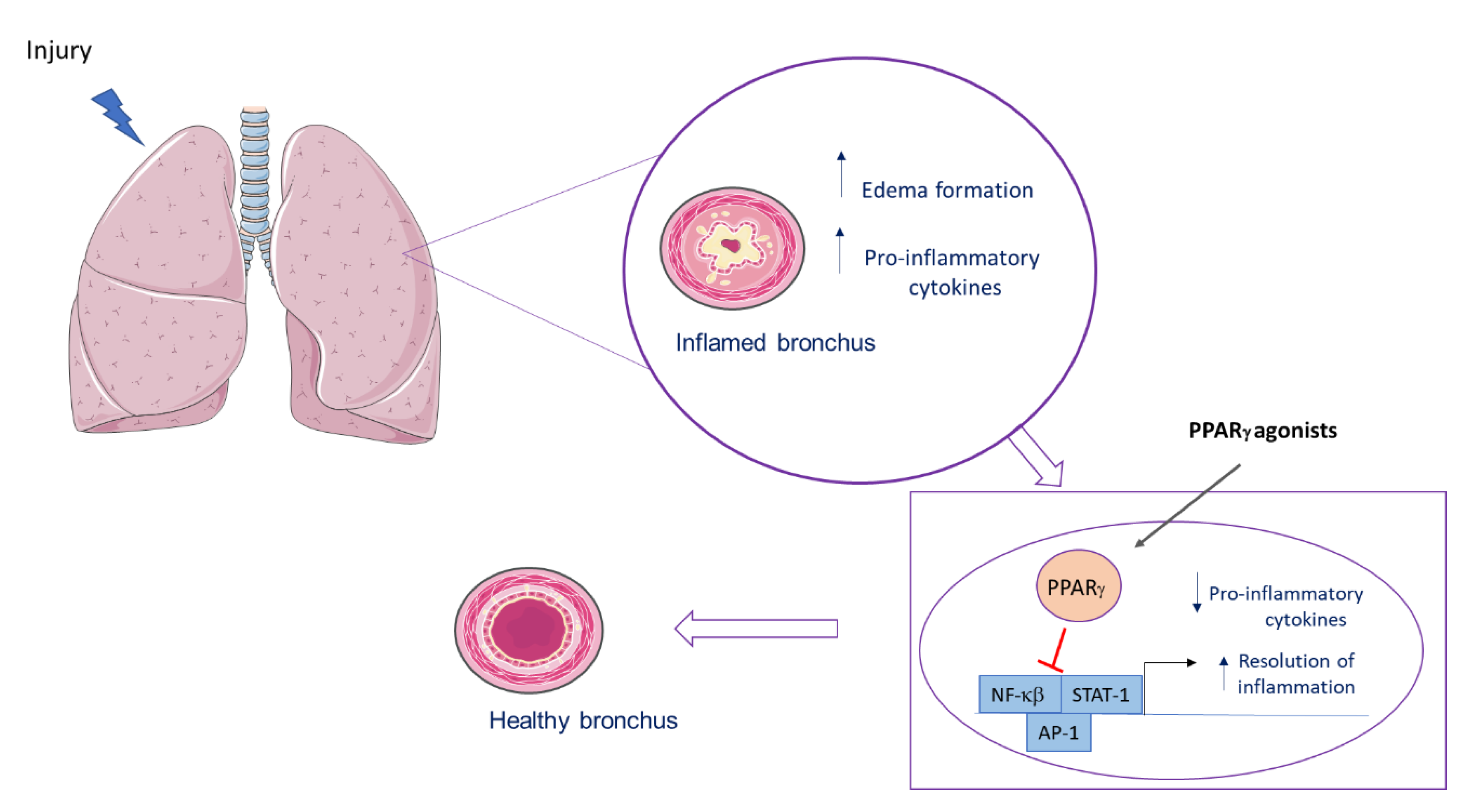
6.1. PPARγ Role in Lung Inflammatory Diseases
6.2. PPARγ’s Role in Bacterial Lung Infection
6.3. PPARγ Role in Viral Lung Infection
7. Concluding Remarks Considering Experimental Findings and Clinical Trials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin-converting enzyme 2 |
| AMPK | Cyclic adenosine monophosphate kinase |
| AKT | Protein kinase B |
| AP-1 | Activator Protein 1 |
| ARDS | Acute respiratory distress syndrome |
| BALF | Bronchoalveolar lavage flui |
| CFTR | |
| COPD | Chronic obstructive pulmonary disease |
| COVID-19 | Coronavirus disease 19 |
| COX | Cycloxygenase |
| Cq1 | Complement component 1q |
| cAMP | Cyclic adenosine monophosphate |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| CREB | cAMP-response element binding protein |
| CX3CR1 | CX3C chemokine receptor 1 |
| DHA | Docosahexaenoic acid |
| EPA | Eicosapentaenoic acid |
| ERK | Extracellular signal-regulated kinase |
| FAM3A | Family with sequence similarity 3 A |
| HDAC | Histone deacethylase |
| HO-1 | Hemoxygenase-1 |
| ICAM-1 | Intercellular adhesion molecule 1 |
| JAK | Janus kinase |
| LPS | Lipopolysaccharide |
| MAP | Mitogen-activated kinase |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| NAD | Nicotinamide adenine dinucleotide |
| NHANES | National Health and Nutrition Examination Survey |
| NEDD4 | Neural precursor cell expressed developmentally downregulated protein 4 |
| NCOR | Nuclear receptor co-repressor |
| NFκB | Nuclear factor kappa B |
| NK | Natural Killer |
| PG | Prostaglandin |
| PKA | Protein kinase A |
| PKB | Protein kinase B |
| PKC | Protein kinase C |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| PTEN | Phosphatase and tensin homolog |
| PTM | Post-translational mechanisms |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SOCS | Suppressor of cytokine signaling protein |
| STAT | Signal transducers and activators of transcription |
| SUMO | Small ubiquitin-like modifier |
| TFEB | Transcription factor EB |
| TLR | Toll-like receptor |
| TREM-2 | Triggering receptor expressed on myeloid cells |
| TRIM23 | Tripartite motif containing 23 |
| TZD | Thiazolidinedione |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| wnt | Homologous wingless and Int-1 |
References
- Han, L.; Shen, W.-J.; Bittner, S.; Kraemer, F.B.; Azhar, S. PPARs: Regulators of metabolism and as therapeutic targets in cardiovascular disease. Part II: PPAR-β/δ and PPAR-γ. Future Cardiol. 2017, 13, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, C.; Krey, G.; Keller, H.; Givel, F.; Helftenbein, G.; Wahli, W. Control of the peroxisomal β-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992, 68, 879–887. [Google Scholar] [CrossRef]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Kliewer, S.A.; Forman, B.M.; Blumberg, B.; Ong, E.S.; Borgmeyer, U.; Mangelsdorf, D.J.; Umesono, K.; Evans, R.M. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 7355–7359. [Google Scholar] [CrossRef]
- Strand, D.W.; Jiang, M.; Murphy, T.A.; Yi, Y.; Konvinse, K.C.; Franco, O.E.; Wang, Y.; Young, J.D.; Hayward, S.W. PPARγ isoforms differentially regulate metabolic networks to mediate mouse prostatic epithelial differentiation. Cell Death Dis. 2012, 3, e361. [Google Scholar] [CrossRef]
- Shao, X.; Wang, M.; Wei, X.; Deng, S.; Fu, N.; Peng, Q.; Jiang, Y.; Ye, L.; Xie, J.; Lin, Y. Peroxisome Proliferator-Activated Receptor-γ: Master Regulator of Adipogenesis and Obesity. Curr. Stem Cell Res. Ther. 2016, 11, 282–289. [Google Scholar] [CrossRef]
- Mirza, R.E.; Fang, M.M.; Novak, M.L.; Urao, N.; Sui, A.; Ennis, W.J.; Koh, T.J. Macrophage PPARγ and impaired wound healing in type 2 diabetes. J. Pathol. 2015, 236, 433–444. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Araújo, C.V.; Campbell, C.; Gonçalves-De-Albuquerque, C.F.; Molinaro, R.; Cody, M.J.; Yost, C.C.; Bozza, P.T.; Zimmerman, G.A.; Weyrich, A.S.; Castro-Faria-Neto, H.C.; et al. A Pparγ Agonist Enhances Bacterial Clearance Through Neutrophil Extracellular TRAP formation and Improves Survival in Sepsis. Shock 2016, 45, 393–403. [Google Scholar] [CrossRef]
- Araújo, C.; Estato, V.; Tibiriça, E.; Bozza, P.T.; Castro-Faria-Neto, H.; Silva, A.R. PPAR gamma activation protects the brain against microvascular dysfunction in sepsis. Microvasc. Res. 2012, 84, 218–221. [Google Scholar] [CrossRef]
- Silva, A.R.; Gonçalves-De-Albuquerque, C.F.; Pérez, A.R.; Carvalho, V.F. Immune-endocrine interactions related to a high risk of infections in chronic metabolic diseases: The role of PPAR gamma. Eur. J. Pharmacol. 2019, 854, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Villapol, S. Roles of Peroxisome Proliferator-Activated Receptor Gamma on Brain and Peripheral Inflammation. Cell. Mol. Neurobiol. 2017, 38, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Janani, C.; Kumari, B.R. PPAR gamma gene—A review. Diabetes Metab. Syndr. Clin. Res. Rev. 2015, 9, 46–50. [Google Scholar] [CrossRef]
- Moseti, D.; Regassa, A.; Kim, W.K. Molecular Regulation of Adipogenesis and Potential Anti-Adipogenic Bioactive Molecules. Int. J. Mol. Sci. 2016, 17, 124. [Google Scholar] [CrossRef] [PubMed]
- Salgia, M.M.; Elix, C.C.; Pal, S.K.; Jones, J.O. Different roles of peroxisome proliferator-activated receptor gamma isoforms in prostate cancer. Am. J. Clin. Exp. Urol. 2019, 7, 98–109. [Google Scholar]
- Chen, K.; Li, J.; Wang, J.; Xia, Y.; Dai, W.; Wang, F.; Shen, M.; Cheng, P.; Zhang, Y.; Wang, C.; et al. 15-Deoxy-γ12,14-prostaglandin J2 Reduces Liver Impairment in a Model of ConA-Induced Acute Hepatic Inflammation by Activation of PPARγand Reduction in NF-κB Activity. PPAR Res. 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Rolland, M.; Li, X.; Sellier, Y.; Martin, H.; Pérez-Berezo, T.; Rauwel, B.; Benchoua, A.; Bessières, B.; Aziza, J.; Cenac, N.; et al. PPARγ Is Activated during Congenital Cytomegalovirus Infection and Inhibits Neuronogenesis from Human Neural Stem Cells. PLoS Pathog. 2016, 12, e1005547. [Google Scholar] [CrossRef]
- Narala, V.R.; Subramani, P.A.; Narasimha, V.R.; Shaik, F.B.; Panati, K. The role of nitrated fatty acids and peroxisome proliferator-activated receptor gamma in modulating inflammation. Int. Immunopharmacol. 2014, 23, 283–287. [Google Scholar] [CrossRef]
- Elferink, R.P.O.; Bolier, R.; Beuers, U. Lysophosphatidic acid and signaling in sensory neurons. Biochim. et Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2015, 1851, 61–65. [Google Scholar] [CrossRef]
- Han, W.; Zhao, H.; Jiao, B.; Liu, F. EPA and DHA increased PPARγ expression and deceased integrin-linked kinase and integrin β1 expression in rat glomerular mesangial cells treated with lipopolysaccharide. Biosci. Trends 2014, 8, 120–125. [Google Scholar] [CrossRef]
- Takada, I.; Makishima, M. Peroxisome proliferator-activated receptor agonists and antagonists: A patent review (2014-present). Expert Opin. Ther. Patents 2019, 30, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Hu, J.-P.; Yu, S.; Li, B.-K.; Cui, Y.; Ren, L.; Zhang, L.-D. Astragaloside IV, a Natural PPARγ Agonist, Reduces Aβ Production in Alzheimer’s Disease Through Inhibition of BACE 1. Mol. Neurobiol. 2016, 54, 2939–2949. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Yuan, Q.; Meng, X.; Huo, J.; Bao, Y.; Xie, G. 6-Shogaol attenuates LPS-induced inflammation in BV2 microglia cells by activating PPAR-γ. Oncotarget 2017, 8, 42001–42006. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Lin, Q.; Li, X.; Nie, Y.; Sun, S.; Deng, X.; Wang, L.; Lu, J.; Tang, Y.; Luo, F. Alliin, a garlic organosulfur compound, ameliorates gut inflammation through MAPK-NF-κB/AP-1/STAT-1 inactivation and PPAR-γ activation. Mol. Nutr. Food Res. 2017, 61, 1601013. [Google Scholar] [CrossRef] [PubMed]
- Vallée, J.-N.; LeCarpentier, Y.; Guillevin, R.; Vallée, J.-N. Effects of cannabidiol interactions with Wnt/β-catenin pathway and PPARγ on oxidative stress and neuroinflammation in Alzheimer’s disease. Acta Biochim. Biophys. Sin. 2017, 49, 853–866. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, L.; Tang, F. The protective effects of magnolol on acute trinitrobenzene sulfonic acid-induced colitis in rats. Mol. Med. Rep. 2017, 17, 3455–3464. [Google Scholar] [CrossRef]
- Serra, D.; Almeida, L.M.; Dinis, T.C.P. Anti-inflammatory protection afforded by cyanidin-3-glucoside and resveratrol in human intestinal cells via Nrf2 and PPAR-γ: Comparison with 5-aminosalicylic acid. Chem. Interact. 2016, 260, 102–109. [Google Scholar] [CrossRef]
- Zhu, T.; Chen, Z.; Chen, G.; Wang, D.; Tang, S.; Deng, H.; Wang, J.; Li, S.; Lan, J.; Tong, J.; et al. Curcumin Attenuates Asthmatic Airway Inflammation and Mucus Hypersecretion Involving a PPARγ-Dependent NF-κB Signaling Pathway In Vivo and In Vitro. Mediat. Inflamm. 2019, 2019, 4927430-15. [Google Scholar] [CrossRef]
- Medeiros-De-Moraes, I.M.; Gonçalves-De-Albuquerque, C.F.; Kurz, A.R.M.; Oliveira, F.M.D.J.; De Abreu, V.H.P.; Torres, R.; Carvalho, V.F.; Estato, V.; Bozza, P.T.; Sperandio, M.; et al. Omega-9 Oleic Acid, the Main Compound of Olive Oil, Mitigates Inflammation during Experimental Sepsis. Oxidative Med. Cell. Longev. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Silva, A.R.; Moraes, B.P.T.; Gonçalves-De-Albuquerque, C.F. Mediterranean Diet: Lipids, Inflammation, and Malaria Infection. Int. J. Mol. Sci. 2020, 21, 4489. [Google Scholar] [CrossRef]
- Jia, Y.; Kim, J.-Y.; Jun, H.-J.; Kim, S.-J.; Lee, J.-H.; Hoang, M.H.; Hwang, K.-Y.; Um, S.-J.; Chang, H.-I.; Lee, S.-J. The natural carotenoid astaxanthin, a PPAR-α agonist and PPAR-γ antagonist, reduces hepatic lipid accumulation by rewiring the transcriptome in lipid-loaded hepatocytes. Mol. Nutr. Food Res. 2012, 56, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.M.; Moore, L.B.; Smith-Oliver, T.A.; Wilkison, W.O.; Willson, T.M.; Kliewer, S.A. An Antidiabetic Thiazolidinedione Is a High Affinity Ligand for Peroxisome Proliferator-activated Receptor γ (PPARγ). J. Biol. Chem. 1995, 270, 12953–12956. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L. Rosiglitazone approved for treatment of type 2 diabetes. Am. J. Heal. Pharm. 1999, 56, 1292. [Google Scholar] [CrossRef] [PubMed]
- Cariou, B.; Charbonnel, B.; Staels, B. Thiazolidinediones and PPARγ agonists: Time for a reassessment. Trends Endocrinol. Metab. 2012, 23, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Takada, I.; Makishima, M. PPARγ ligands and their therapeutic applications: A patent review (2008–2014). Expert Opin. Ther. Patents 2014, 25, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, R.; Fu, H.; Li, J.; Wang, X.; Cheng, L.; Korantzopoulos, P.; Tse, G.; Li, G.; Liu, T. Pioglitazone attenuates atrial remodeling and vulnerability to atrial fibrillation in alloxan-induced diabetic rabbits. Cardiovasc. Ther. 2017, 35, e12284. [Google Scholar] [CrossRef]
- Legchenko, E.; Chouvarine, P.; Borchert, P.; Fernandez-Gonzalez, A.; Snay, E.; Meier, M.; Maegel, L.; Mitsialis, S.A.; Rog-Zielinska, E.A.; Kourembanas, S.; et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med. 2018, 10, eaao0303. [Google Scholar] [CrossRef]
- Brunmeir, R.; Xu, F. Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 2018, 19, 1738. [Google Scholar] [CrossRef]
- Fang, T.; Di, Y.; Li, G.; Cui, X.; Shen, N.; Li, Y.; Xi, P.; Xie, Y.; Tian, F.; Li, G. Effects of telmisartan on TNFα induced PPARγ phosphorylation and insulin resistance in adipocytes. Biochem. Biophys. Res. Commun. 2018, 503, 3044–3049. [Google Scholar] [CrossRef]
- Bhatt, K.H.; Sodhi, A.; Chakraborty, R. Peptidoglycan induced expression of peroxisome proliferator-activated receptor γ in mouse peritoneal macrophages: Role of ERK and JNK MAP kinases. Cytokine 2012, 60, 778–786. [Google Scholar] [CrossRef]
- Choi, M.-J.; Lee, E.-J.; Park, J.-S.; Kim, S.-N.; Park, E.-M.; Kim, H.-S. Anti-inflammatory mechanism of galangin in lipopolysaccharide-stimulated microglia: Critical role of PPAR-γ signaling pathway. Biochem. Pharmacol. 2017, 144, 120–131. [Google Scholar] [CrossRef]
- Hu, T.; Lin, Q.; Guo, T.; Yang, T.; Zhou, W.; Deng, X.; Yan, J.-K.; Luo, Y.; Ju, M.; Luo, F. Polysaccharide isolated from Phellinus linteus mycelia exerts anti-inflammatory effects via MAPK and PPAR signaling pathways. Carbohydr. Polym. 2018, 200, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yang, Y.; Li, W.-X.; Wu, X.-Q.; Li, X.-F.; Ma, T.-T.; Zhang, L.; Meng, X.-M.; Li, J. Hyperin attenuates inflammation by activating PPAR-γ in mice with acute liver injury (ALI) and LPS-induced RAW264.7 cells. Int. Immunopharmacol. 2015, 29, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhou, Q.; Shi, Y.; Liu, J.; Zhong, F.; Hao, X.; Li, C.; Chen, N.; Wang, W. SUMOylation of PPARγ by Rosiglitazone Prevents LPS-Induced NCoR Degradation Mediating Down Regulation of Chemokines Expression in Renal Proximal Tubular Cells. PLoS ONE 2013, 8, e79815. [Google Scholar] [CrossRef] [PubMed]
- Eifler, K.; Vertegaal, A.C. SUMOylation-Mediated Regulation of Cell Cycle Progression and Cancer. Trends Biochem. Sci. 2015, 40, 779–793. [Google Scholar] [CrossRef]
- Zelcer, N.; Tontonoz, P. SUMOylation and PPARγ: Wrestling with inflammatory signaling. Cell Metab. 2005, 2, 273–275. [Google Scholar] [CrossRef][Green Version]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.T.; Ghosh, S. ’PPAR’ting ways with inflammation. Nat. Immunol. 2005, 6, 966–967. [Google Scholar] [CrossRef]
- Jennewein, C.; Kuhn, A.-M.; Schmidt, M.V.; Meilladec-Jullig, V.; Von Knethen, A.; Gonzalez, F.J.; Brüne, B. Sumoylation of PPARγ by apoptotic cells prevents LPS-induced NCoR removal from κB binding sites mediating transrepression of pro-inflammatory cytokines. J. Immunol. 2008, 181, 5646–5652. [Google Scholar] [CrossRef]
- Li, J.-J.; Wang, R.; Lama, R.; Wang, X.; Floyd, Z.E.; Park, E.A.; Liao, F.-F. Ubiquitin Ligase NEDD4 Regulates PPARγ Stability and Adipocyte Differentiation in 3T3-L1 Cells. Sci. Rep. 2016, 6, 38550. [Google Scholar] [CrossRef]
- Watanabe, M.; Takahashi, H.; Saeki, Y.; Ozaki, T.; Itoh, S.; Suzuki, M.; Mizushima, W.; Tanaka, K.; Hatakeyama, S. The E3 ubiquitin ligase TRIM23 regulates adipocyte differentiation via stabilization of the adipogenic activator PPARγ. eLife 2015, 4, e05615. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R.; et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell 2012, 150, 620–632. [Google Scholar] [CrossRef]
- Ji, S.; Park, S.Y.; Roth, J.; Kim, H.S.; Cho, J.W. O-GlcNAc modification of PPARγ reduces its transcriptional activity. Biochem. Biophys. Res. Commun. 2012, 417, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, U.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Al-Soudi, A.; Kaaij, M.; Tas, S.W. Endothelial cells: From innocent bystanders to active participants in immune responses. Autoimmun. Rev. 2017, 16, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, M. Dendritic cell-epithelial cell crosstalk in the gut. Immunol. Rev. 2014, 260, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Ushiki, T.; Huntington, N.D.; Glaser, S.P.; Kiu, H.; Georgiou, A.; Zhang, J.-G.; Metcalf, D.; Nicola, N.A.; Roberts, A.W.; Alexander, W.S. Rapid Inflammation in Mice Lacking Both SOCS1 and SOCS3 in Hematopoietic Cells. PLoS ONE 2016, 11, e0162111. [Google Scholar] [CrossRef]
- O’Connor, P.W. Natalizumab and the role of α4-integrin antagonism in the treatment of multiple sclerosis. Expert Opin. Biol. Ther. 2006, 7, 123–136. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Reis, E.S.; Mastellos, D.C.; Ricklin, D.; Lambris, J.D. Novel mechanisms and functions of complement. Nat. Immunol. 2017, 18, 1288–1298. [Google Scholar] [CrossRef]
- Croasdell, A.; Duffney, P.F.; Kim, N.; Lacy, S.H.; Sime, P.J.; Phipps, R.P. PPARγand the Innate Immune System Mediate the Resolution of Inflammation. PPAR Res. 2015, 2015, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Smale, S.T. Selective Transcription in Response to an Inflammatory Stimulus. Cell 2010, 140, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Koues, O.I.; Collins, P.L.; Cella, M.; Robinette, M.L.; Porter, S.I.; Pyfrom, S.C.; Payton, J.E.; Colonna, M.; Oltz, E.M. Distinct Gene Regulatory Pathways for Human Innate versus Adaptive Lymphoid Cells. Cell 2016, 165, 1134–1146. [Google Scholar] [CrossRef] [PubMed]
- Hawiger, J.; Zienkiewicz, J. Decoding inflammation, its causes, genomic responses, and emerging countermeasures. Scand. J. Immunol. 2019, 90, e12812. [Google Scholar] [CrossRef]
- Ran, L.; Yu, Q.; Zhang, S.; Xiong, F.; Cheng, J.; Yang, P.; Xu, J.-F.; Nie, H.; Zhong, Q.; Yang, X.; et al. Cx3cr1 deficiency in mice attenuates hepatic granuloma formation during acute schistosomiasis by enhancing the M2-type polarization of macrophages. Dis. Model. Mech. 2015, 8, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.-M.; Manughian-Peter, A.O.; Spivia, W.R.; Taylor, A.; Fraser, D.A. Macrophage molecular signaling and inflammatory responses during ingestion of atherogenic lipoproteins are modulated by complement protein C1q. Atherosclerosis 2016, 253, 38–46. [Google Scholar] [CrossRef]
- Geng, L.; Zhang, T.; Liu, W.; Chen, Y. Inhibition of miR-128 Abates Aβ-Mediated Cytotoxicity by Targeting PPAR-γ via NF-κB Inactivation in Primary Mouse Cortical Neurons and Neuro2a Cells. Yonsei Med. J. 2018, 59, 1096–1106. [Google Scholar] [CrossRef]
- Khosravi, F.; Kharazmi, F.; Kamran, M.; Malekzadeh, K.; Talebi, A.; Soltani, N. The role of PPAR-γ and NFKB genes expression in muscle to improve hyperglycemia in STZ-induced diabetic rat following magnesium sulfate administration. Int. J. Physiol. Pathophysiol. Pharmacol. 2018, 10, 124–131. [Google Scholar]
- Liu, Y.; Chen, S.; Liu, J.; Jin, Y.; Yu, S.; An, R. Telmisartan inhibits oxalate and calcium oxalate crystal-induced epithelial-mesenchymal transformation via PPAR-γ-AKT/STAT3/p38 MAPK-Snail pathway. Life Sci. 2020, 241, 117108. [Google Scholar] [CrossRef]
- Zhao, M.; Bian, Y.; Yang, L.-L.; Chen, Y.-Q.; Wang, Y.-J.; Ma, Y.-T.; Pei, Y.-Q.; Li, W.-L.; Zeng, L. HuoXueTongFu Formula Alleviates Intraperitoneal Adhesion by Regulating Macrophage Polarization and the SOCS/JAK2/STAT/PPAR-γ Signalling Pathway. Mediat. Inflamm. 2019, 2019, 1769374-17. [Google Scholar] [CrossRef]
- De Jong, E.; Winkel, P.; Poelstra, K.; Prakash, J. Anticancer Effects of 15d-Prostaglandin-J2 in Wild-Type and Doxorubicin-Resistant Ovarian Cancer Cells: Novel Actions on SIRT1 and HDAC. PLoS ONE 2011, 6, e25192. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.; Luo, P.; Zhou, J.; Tan, M. 15-Deoxy-Δ12,14-prostaglandin J2 as a potential regulator of bone metabolism via PPARγ-dependent and independent pathways: A review. Drug Des. Dev. Ther. 2019, 13, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhou, X.E.; Shi, J.; Zhou, Z.; Zhao, G.; Zhang, X.; Sun, Y.; Suino-Powell, K.; Ma, L.; Gao, H.; et al. Identification and structural insight of an effective PPARγ modulator with improved therapeutic index for anti-diabetic drug discovery. Chem. Sci. 2020, 11, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, S.P.; Reddy, A.T.; Reddy, R.C. Emerging pharmaceutical therapies for COPD. Int. J. Chronic Obstr. Pulm. Dis. 2017, 12, 2141–2156. [Google Scholar] [CrossRef]
- Mazidi, M.; Karimi, E.; Meydani, M.; Avan, A.; Ferns, G.A. Potential effects of curcumin on peroxisome proliferator-activated receptor-γin vitroandin vivo. World J. Methodol. 2016, 6, 112–117. [Google Scholar] [CrossRef]
- Jacob, A.; Wu, R.; Zhou, M.; Wang, P. Mechanism of the Anti-inflammatory Effect of Curcumin: PPAR-γ Activation. PPAR Res. 2008, 2007, 1–5. [Google Scholar] [CrossRef]
- Dreier, D.; Latkolik, S.; Rycek, L.; Schnürch, M.; Dymáková, A.; Atanasov, A.G.; Ladurner, A.; Heiss, E.H.; Stuppner, H.; Schuster, D.; et al. Linked magnolol dimer as a selective PPARγ agonist - Structure-based rational design, synthesis, and bioactivity evaluation. Sci. Rep. 2017, 7, 13002. [Google Scholar] [CrossRef]
- Aldridge, J.R.; Moseley, C.E.; Boltz, D.A.; Negovetich, N.J.; Reynolds, C.; Franks, J.; Brown, S.A.; Doherty, P.C.; Webster, R.G.; Thomas, P.G. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc. Natl. Acad. Sci. USA 2009, 106, 5306–5311. [Google Scholar] [CrossRef]
- Venkataraman, B.; Ojha, S.; Belur, P.D.; Bhongade, B.; Raj, V.; Collin, P.D.; Adrian, T.E.; Subramanya, S.B. Phytochemical drug candidates for the modulation of peroxisome proliferator-activated receptor γ in inflammatory bowel diseases. Phytotherapy Res. 2020, 34, 1530–1549. [Google Scholar] [CrossRef]
- Linares-Cervantes, I.; Farrokhi, K.; Echeverri, J.; Kaths, J.M.; Kollmann, D.; Hamar, M.; Urbanellis, P.; Ganesh, S.; Adeyi, O.; Yip, P.; et al. PPAR-gamma activation is associated with reduced liver ischemia-reperfusion injury and altered tissue-resident macrophages polarization in a mouse model. PLoS ONE 2018, 13, e0195212. [Google Scholar] [CrossRef]
- Yang, W.; Chen, J.; Meng, Y.; Chen, Z.; Yang, J. Novel Targets for Treating Ischemia-Reperfusion Injury in the Liver. Int. J. Mol. Sci. 2018, 19, 1302. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, S.; Chen, G. Aggravation of Cerebral Ischemia/Reperfusion Injury by Peroxisome Proliferator-Activated Receptor-Gamma Deficiency via Endoplasmic Reticulum Stress. Med. Sci. Monit. 2019, 25, 7518–7526. [Google Scholar] [CrossRef] [PubMed]
- Elshazly, S.; Soliman, E. PPAR gamma agonist, pioglitazone, rescues liver damage induced by renal ischemia/reperfusion injury. Toxicol. Appl. Pharmacol. 2019, 362, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Ciavarella, C.; Motta, I.; Valente, S.; Pasquinelli, G. Pharmacological (or Synthetic) and Nutritional Agonists of PPAR-γ as Candidates for Cytokine Storm Modulation in COVID-19 Disease. Molecules 2020, 25, 2076. [Google Scholar] [CrossRef] [PubMed]
- Chichger, H.; Rounds, S.; Harrington, E.O. Endosomes and Autophagy: Regulators of Pulmonary Endothelial Cell Homeostasis in Health and Disease. Antioxidants Redox Signal. 2019, 31, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Giaginis, C.; Tsourouflis, G.; Theocharis, S. Peroxisome Proliferator-Activated Receptor-γ (PPAR-γ) Ligands: Novel Pharmacological Agents in the Treatment of Ischemia Reperfusion Injury. Curr. Mol. Med. 2008, 8, 562–579. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, S.P.; Reddy, A.T.; Banno, A.; Reddy, R.C. PPAR Agonists for the Prevention and Treatment of Lung Cancer. PPAR Res. 2017, 2017, 1–8. [Google Scholar] [CrossRef]
- Huang, G.; Yin, L.-M.; Lan, J.; Tong, R.; Li, M.; Na, F.; Mo, X.; Chen, C.; Xue, J.-X.; Lu, Y. Synergy between peroxisome proliferator-activated receptor γ agonist and radiotherapy in cancer. Cancer Sci. 2018, 109, 2243–2255. [Google Scholar] [CrossRef]
- New, M.; White, C.M.; Mcgonigle, P.; McArthur, D.G.; Dwyer-Nield, L.D.; Merrick, D.T.; Keith, R.L.; Tennis, M.A. Prostacyclin and EMT Pathway Markers for Monitoring Response to Lung Cancer Chemoprevention. Cancer Prev. Res. 2018, 11, 643–654. [Google Scholar] [CrossRef]
- Ammu, V.R.K.; Garikapati, K.K.; Krishnamurthy, P.T.; Chintamaneni, P.K.; Pindiprolu, S.K.S. Possible role of PPAR-γ and COX-2 receptor modulators in the treatment of Non-Small Cell lung carcinoma. Med. Hypotheses 2019, 124, 98–100. [Google Scholar] [CrossRef]
- Reddy, A.T.; Lakshmi, S.P.; Reddy, R.C. PPARγas a Novel Therapeutic Target in Lung Cancer. PPAR Res. 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Banno, A.; Reddy, A.T.; Lakshmi, S.P.; Reddy, A.R.C. PPARs: Key Regulators of Airway Inflammation and Potential Therapeutic Targets in Asthma. Nucl. Recept. Res. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, S.P.; Reddy, A.T.; Banno, A.; Reddy, R.C. Airway Epithelial Cell Peroxisome Proliferator–Activated Receptor γ Regulates Inflammation and Mucin Expression in Allergic Airway Disease. J. Immunol. 2018, 201, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Yang, W.-K.; Yee, S.-M.; Kim, S.-M.; Park, Y.-C.; Shin, H.J.; Han, C.K.; Kang, H.-S.; Kim, S.-H.; Lee, Y.C. KGC3P attenuates ovalbumin-induced airway inflammation through downregulation of p-PTEN in asthmatic mice. Phytomedicine 2019, 62, 152942. [Google Scholar] [CrossRef] [PubMed]
- Rosanna, D.P.; Salvatore, C. Reactive oxygen species, inflammation, and lung diseases. Curr. Pharm. Des. 2012, 18, 3889–3900. [Google Scholar] [CrossRef]
- Butt, Y.; Kurdowska, A.; Allen, T.C. Acute Lung Injury: A Clinical and Molecular Review. Arch. Pathol. Lab. Med. 2016, 140, 345–350. [Google Scholar] [CrossRef]
- Meng, F.; Mambetsariev, I.; Tian, Y.; Beckham, Y.; Meliton, A.; Leff, A.; Gardel, M.L.; Allen, M.J.; Birukov, K.G.; Birukova, A.A. Attenuation of Lipopolysaccharide-Induced Lung Vascular Stiffening by Lipoxin Reduces Lung Inflammation. Am. J. Respir. Cell Mol. Biol. 2015, 52, 152–161. [Google Scholar] [CrossRef]
- Li, Q.; Sun, J.; Mohammadtursun, N.; Wu, J.; Dong, J.; Li, L. Curcumin inhibits cigarette smoke-induced inflammation via modulating the PPARγ-NF-κB signaling pathway. Food Funct. 2019, 10, 7983–7994. [Google Scholar] [CrossRef]
- Morissette, M.C.; Shen, P.; Thayaparan, D.; Stampfli, M.R. Impacts of peroxisome proliferator-activated receptor-γ activation on cigarette smoke-induced exacerbated response to bacteria. Eur. Respir. J. 2014, 45, 191–200. [Google Scholar] [CrossRef]
- Elborn, J. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Rab, A.; Jurkuvenaite, A.; Mazur, M.; Wakefield, J.; Collawn, J.F.; Bebok, Z. Activation of the Unfolded Protein Response by ΔF508 CFTR. Am. J. Respir. Cell Mol. Biol. 2008, 39, 448–457. [Google Scholar] [CrossRef]
- Luciani, A.; Villella, V.R.; Esposito, S.; Brunetti-Pierri, N.; Medina, D.L.; Settembre, C.; Gavina, M.; Pulze, L.; Giardino, I.; Pettoello-Mantovani, M.; et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nature 2010, 12, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Smerieri, A.; Montanini, L.; Maiuri, L.; Bernasconi, S.; Street, M.E. FOXO1 Content Is Reduced in Cystic Fibrosis and Increases with IGF-I Treatment. Int. J. Mol. Sci. 2014, 15, 18000–18022. [Google Scholar] [CrossRef] [PubMed]
- Andersson, C.; Zaman, M.M.; Jones, A.B.; Freedman, S.D. Alterations in immune response and PPAR/LXR regulation in cystic fibrosis macrophages. J. Cyst. Fibros. 2008, 7, 68–78. [Google Scholar] [CrossRef]
- Caretti, A.; Vasso, M.; Bonezzi, F.T.; Gallina, A.; Trinchera, M.; Rossi, A.; Adami, R.; Casas, J.; Falleni, M.; Tosi, D.; et al. Myriocin treatment of CF lung infection and inflammation: Complex analyses for enigmatic lipids. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2017, 390, 775–790. [Google Scholar] [CrossRef] [PubMed]
- Caretti, A.; Torelli, R.; Perdoni, F.; Falleni, M.; Tosi, D.; Zulueta, A.; Casas, J.; Sanguinetti, M.; Ghidoni, R.; Borghi, E.; et al. Inhibition of ceramide de novo synthesis by myriocin produces the double effect of reducing pathological inflammation and exerting antifungal activity against A. fumigatus airways infection. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2016, 1860, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Lee, H.-M.; Kim, J.K.; Yang, C.-S.; Kim, T.S.; Jung, M.; Jin, H.S.; Kim, S.; Jang, J.; Oh, G.T.; et al. PPAR-α Activation Mediates Innate Host Defense through Induction of TFEB and Lipid Catabolism. J. Immunol. 2017, 198, 3283–3295. [Google Scholar] [CrossRef]
- Mingione, A.; Cas, M.D.; Bonezzi, F.; Caretti, A.; Piccoli, M.; Anastasia, L.; Ghidoni, R.; Paroni, R.; Signorelli, P. Inhibition of Sphingolipid Synthesis as a Phenotype-Modifying Therapy in Cystic Fibrosis. Cell. Physiol. Biochem. 2020, 54, 110–125. [Google Scholar] [CrossRef]
- Reddy, A.T.; Lakshmi, S.P.; Kleinhenz, J.M.; Sutliff, R.L.; Hart, C.M.; Reddy, R.C. Endothelial Cell Peroxisome Proliferator–Activated Receptor γ Reduces Endotoxemic Pulmonary Inflammation and Injury. J. Immunol. 2012, 189, 5411–5420. [Google Scholar] [CrossRef]
- Belvisi, M.G.; Hele, D.J. Peroxisome Proliferator-Activated Receptors as Novel Targets in Lung Disease*. Chest 2008, 134, 152–157. [Google Scholar] [CrossRef]
- Belvisi, M.G.; Mitchell, J.A. Targeting PPAR receptors in the airway for the treatment of inflammatory lung disease. Br. J. Pharmacol. 2009, 158, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, H.E.; Thatcher, T.H.; Olsen, K.C.; Garcia-Bates, T.M.; Baglole, C.J.; Kottmann, R.M.; Strong, E.R.; Phipps, R.P.; Sime, P.J. Peroxisome proliferator-activated receptor-gamma ligands induce heme oxygenase-1 in lung fibroblasts by a PPARgamma-independent, glutathione-dependent mechanism. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L912–L919. [Google Scholar] [CrossRef]
- Krönke, G.; Kadl, A.; Ikonomu, E.; Bluml, S.; Furnkranz, A.; Sarembock, I.J.; Bochkov, V.N.; Exner, M.; Binder, B.R.; Leitinger, N. Expression of Heme Oxygenase-1 in Human Vascular Cells Is Regulated by Peroxisome Proliferator-Activated Receptors. Arter. Thromb. Vasc. Biol. 2007, 27, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, G.; Han, N.; Zhang, Y.; Xu, J.; Lu, J.; Li, S.; Xie, X.; Liu, L.; Dong, L.; et al. Activation of PPAR-γ ameliorates pulmonary arterial hypertension via inducing heme oxygenase-1 and p21WAF1: An in vivo study in rats. Life Sci. 2014, 98, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.-L.; Yang, C.-C.; Tseng, H.-C.; Hsiao, L.-D.; Lin, C.-C.; Yang, C.-M. Haem oxygenase-1 up-regulation by rosiglitazone via ROS-dependent Nrf2-antioxidant response elements axis or PPARγ attenuates LPS-mediated lung inflammation. Br. J. Pharmacol. 2018, 175, 3928–3946. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.-L.; Lin, W.-N.; Wang, C.-Y.; Yang, C.-C.; Hsiao, L.-D.; Lin, C.-C.; Yang, C.-M. Heme oxygenase-1 induction by rosiglitazone via PKCα/AMPKα/p38 MAPKα/SIRT1/PPARγ pathway suppresses lipopolysaccharide-mediated pulmonary inflammation. Biochem. Pharmacol. 2018, 148, 222–237. [Google Scholar] [CrossRef]
- Yao, J.; Pan, D.; Zhao, Y.; Zhao, L.; Sun, J.; Wang, Y.; You, Q.-D.; Xi, T.; Guo, Q.-L.; Lu, N. Wogonin prevents lipopolysaccharide-induced acute lung injury and inflammation in mice via peroxisome proliferator-activated receptor gamma-mediated attenuation of the nuclear factor-kappaB pathway. Immunology 2014, 143, 241–257. [Google Scholar] [CrossRef]
- Huang, H.; Cheng, Z.; Shi, H.; Xin, W.; Wang, T.T.Y.; Yu, L. (Lucy) Isolation and Characterization of Two Flavonoids, Engeletin and Astilbin, from the Leaves of Engelhardia roxburghiana and Their Potential Anti-inflammatory Properties. J. Agric. Food Chem. 2011, 59, 4562–4569. [Google Scholar] [CrossRef]
- Jiang, X.; Chen, L.; Zhang, Z.; Sun, Y.; Wang, X.; Wei, J. Protective and Therapeutic Effects of Engeletin on LPS-Induced Acute Lung Injury. Inflammation 2018, 41, 1259–1265. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Zhang, P.; Ruan, W.; Zhang, L.; Yuan, S.; Pang, T.; Jia, A.-Q. Smiglaside A ameliorates LPS-induced acute lung injury by modulating macrophage polarization via AMPK-PPARγ pathway. Biochem. Pharmacol. 2018, 156, 385–395. [Google Scholar] [CrossRef]
- Liao, Z.; Dong, J.; Wu, W.; Yang, T.; Wang, T.; Guo, L.; Chen, L.; Xu, D.; Wen, F.-Q. Resolvin D1 attenuates inflammation in lipopolysaccharide-induced acute lung injury through a process involving the PPARγ/NF-κB pathway. Respir. Res. 2012, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Volckaert, T.; Dill, E.; Campbell, A.; Tiozzo, C.; Majka, S.; Bellusci, S.; De Langhe, S. Parabronchial smooth muscle constitutes an airway epithelial stem cell niche in the mouse lung after injury. J. Clin. Investig. 2011, 121, 4409–4419. [Google Scholar] [CrossRef] [PubMed]
- Zulueta, A.; Colombo, M.; Peli, V.; Falleni, M.; Tosi, D.; Ricciardi, M.; Baisi, A.; Bulfamante, G.; Chiaramonte, R.; Caretti, A. Lung mesenchymal stem cells-derived extracellular vesicles attenuate the inflammatory profile of Cystic Fibrosis epithelial cells. Cell. Signal. 2018, 51, 110–118. [Google Scholar] [CrossRef]
- Heming, M.; Gran, S.; Jauch, S.-L.; Fischer-Riepe, L.; Russo, A.; Klotz, L.; Hermann, S.; Schafers, M.; Roth, J.; Barczyk-Kahlert, K. Peroxisome Proliferator-Activated Receptor-γ Modulates the Response of Macrophages to Lipopolysaccharide and Glucocorticoids. Front. Immunol. 2018, 9, 893. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, R.; Cuzzocrea, S. Peroxisome Proliferator-Activated Receptors and Acute Lung Injury. PPAR Res. 2007, 2007, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Maeno, T.; Aoyagi, K.; Ueno, M.; Aoki, F.; Aoki, N.; Nakagawa, J.; Sando, Y.; Shimizu, Y.; Suga, T.; et al. Pioglitazone, a Peroxisome Proliferator-Activated Receptor Gamma Ligand, Suppresses Bleomycin-Induced Acute Lung Injury and Fibrosis. Respiration 2008, 77, 311–319. [Google Scholar] [CrossRef]
- Ohta, A.; Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916–920. [Google Scholar] [CrossRef]
- Sharma, A.K.; Linden, J.; Kron, I.L.; Laubach, V.E. Protection from pulmonary ischemia-reperfusion injury by adenosine A2A receptor activation. Respir. Res. 2009, 10, 1–9. [Google Scholar] [CrossRef]
- He, X.; Hu, J.-L.; Li, J.; Zhao, L.; Zhang, Y.; Zeng, Y.-J.; Dai, S.-S.; He, F.-T. A feedback loop in PPARγ–adenosine A2A receptor signaling inhibits inflammation and attenuates lung damages in a mouse model of LPS-induced acute lung injury. Cell. Signal. 2013, 25, 1913–1923. [Google Scholar] [CrossRef]
- Shen, J.; Sakaida, I.; Uchida, K.; Terai, S.; Okita, K. Leptin enhances TNF-α production via p38 and JNK MAPK in LPS-stimulated Kupffer cells. Life Sci. 2005, 77, 1502–1515. [Google Scholar] [CrossRef]
- Iikuni, N.; Lam, Q.L.K.; Lu, L.; Matarese, G.; La Cava, A. Leptin and Inflammation. Curr. Immunol. Rev. 2008, 4, 70–79. [Google Scholar] [CrossRef] [PubMed]
- La Cava, A. Leptin in inflammation and autoimmunity. Cytokine 2017, 98, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Tschöp, J.; Nogueiras, R.; Lockie, S.H.; Kasten, K.R.; Castañeda, T.R.; Huber, N.; Guanciale, K.; Perez-Tilve, D.; Habegger, K.; Ottaway, N.; et al. CNS leptin action modulates immune response and survival in sepsis. J. Neurosci. 2010, 30, 6036–6047. [Google Scholar] [CrossRef]
- Paz-Filho, G.; Mastronardi, C.A.; Franco, C.B.; Wang, K.B.; Wong, M.-L.; Licinio, J. Leptin: Molecular mechanisms, systemic pro-inflammatory effects, and clinical implications. Arq. Bras. Endocrinol. Metabol. 2012, 56, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Radford, K.; Fanat, A.; Janssen, L.J.; Peters-Golden, M.; Cox, P.G. The Effects of Leptin on Airway Smooth Muscle Responses. Am. J. Respir. Cell Mol. Biol. 2008, 39, 475–481. [Google Scholar] [CrossRef]
- Vernooy, J.H.; Ubags, N.D.; Brusselle, G.G.; Tavernier, J.; Suratt, B.T.; Joos, G.F.; Wouters, E.F.; Bracke, K. Leptin as regulator of pulmonary immune responses: Involvement in respiratory diseases. Pulm. Pharmacol. Ther. 2013, 26, 464–472. [Google Scholar] [CrossRef]
- Assad, N.A.; Sood, A. Leptin, adiponectin and pulmonary diseases. Biochimie 2012, 94, 2180–2189. [Google Scholar] [CrossRef]
- Abbasi, A.; Moghadam, A.A.; Kahrarian, Z.; Abbsavaran, R.; Yari, K.; Alizadeh, E. Molecular effects of leptin on peroxisome proliferator activated receptor gamma (PPAR-γ) mRNA expression in rat’s adipose and liver tissue. Cell. Mol. Biol. 2017, 63, 89–93. [Google Scholar] [CrossRef]
- Ajuwon, K.M.; Spurlock, M.E. Adiponectin inhibits LPS-induced NF-κB activation and IL-6 production and increases PPARγ2 expression in adipocytes. Am. J. Physiol. Integr. Comp. Physiol. 2005, 288, R1220–R1225. [Google Scholar] [CrossRef]
- Wulster-Radcliffe, M.C.; Ajuwon, K.M.; Wang, J.; Christian, J.A.; Spurlock, M.E. Adiponectin differentially regulates cytokines in porcine macrophages. Biochem. Biophys. Res. Commun. 2004, 316, 924–929. [Google Scholar] [CrossRef]
- Wolf, A.M.; Wolf, D.; Rumpold, H.; Enrich, B.; Tilg, H. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem. Biophys. Res. Commun. 2004, 323, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Ishtiaq, S.M.; Rashid, H.; Hussain, Z.; Arshad, M.I.; Khan, J.A. Adiponectin and PPAR: A setup for intricate crosstalk between obesity and non-alcoholic fatty liver disease. Rev. Endocr. Metab. Disord. 2019, 20, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Yang, J.; Lin, X.; Shin, J.; Gao, J.; Zhong, X.-P. Essential Role of mTORC1 in Self-Renewal of Murine Alveolar Macrophages. J. Immunol. 2016, 198, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Hussell, T.; Bell, T.J. Alveolar macrophages: Plasticity in a tissue-specific context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef]
- Kawasaki, T.; Ito, K.; Miyata, H.; Akira, S.; Kawai, T. Deletion of PIK fyve alters alveolar macrophage populations and exacerbates allergic inflammation in mice. EMBO J. 2017, 36, 1707–1718. [Google Scholar] [CrossRef]
- Kopf, M.; Schneider, C.; Nobs, S.P. The development and function of lung-resident macrophages and dendritic cells. Nat. Immunol. 2014, 16, 36–44. [Google Scholar] [CrossRef]
- Cardani, A.; Boulton, A.; Kim, T.S.; Braciale, T.J. Alveolar Macrophages Prevent Lethal Influenza Pneumonia By Inhibiting Infection Of Type-1 Alveolar Epithelial Cells. PLoS Pathog. 2017, 13, e1006140. [Google Scholar] [CrossRef]
- Kim, H.M.; Lee, Y.-W.; Lee, K.-J.; Kim, H.S.; Cho, S.W.; Van Rooijen, N.; Guan, Y.; Seo, S.H. Alveolar Macrophages Are Indispensable for Controlling Influenza Viruses in Lungs of Pigs. J. Virol. 2008, 82, 4265–4274. [Google Scholar] [CrossRef]
- Laidlaw, B.J.; Decman, V.; Ali, M.-A.A.; Abt, M.C.; Wolf, A.I.; Monticelli, L.A.; Mozdzanowska, K.; Angelosanto, J.M.; Artis, D.; Erikson, J.; et al. Cooperativity Between CD8+ T Cells, Non-Neutralizing Antibodies, and Alveolar Macrophages Is Important for Heterosubtypic Influenza Virus Immunity. PLoS Pathog. 2013, 9, e1003207. [Google Scholar] [CrossRef]
- Purnama, C.; Ng, S.L.; Tetlak, P.; Setiagani, Y.A.; Kandasamy, M.; Baalasubramanian, S.; Karjalainen, K.; Ruedl, C. Transient ablation of alveolar macrophages leads to massive pathology of influenza infection without affecting cellular adaptive immunity. Eur. J. Immunol. 2014, 44, 2003–2012. [Google Scholar] [CrossRef]
- Gautier, E.L.; Chow, A.; Spanbroek, R.; Marcelin, G.; Greter, M.; Jakubzick, C.; Bogunovic, M.; Leboeuf, M.; Van Rooijen, N.; Habenicht, A.J.R.; et al. Systemic analysis of PPARγ in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. J. Immunol. 2012, 189, 2614–2624. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Mizgerd, J.P. Acute Lower Respiratory Tract Infection. New Engl. J. Med. 2008, 358, 716–727. [Google Scholar] [CrossRef]
- Soehnlein, O.; Lindbom, L. Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 2010, 10, 427–439. [Google Scholar] [CrossRef]
- Ray, N.B.; Durairaj, L.; Chen, B.B.; McVerry, B.J.; Ryan, A.J.; Donahoe, M.; Waltenbaugh, A.K.; O’Donnell, C.P.; Henderson, F.C.; Etscheidt, C.A.; et al. Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat. Med. 2010, 16, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, K.; Raundhal, M.; Chen, B.B.; Morse, C.; Tyurina, Y.Y.; Khare, A.; Oriss, T.B.; Huff, R.; Lee, J.S.; Croix, C.M.S.; et al. The mito-DAMP cardiolipin blocks IL-10 production causing persistent inflammation during bacterial pneumonia. Nat. Commun. 2017, 8, 13944. [Google Scholar] [CrossRef]
- Bedi, B.; Maurice, N.M.; Ciavatta, V.T.; Lynn, K.S.; Yuan, Z.; Molina, S.A.; Joo, M.; Tyor, W.R.; Goldberg, J.B.; Koval, M.; et al. Peroxisome proliferator-activated receptor-γ agonists attenuate biofilm formation by Pseudomonas aeruginosa. FASEB J. 2017, 31, 3608–3621. [Google Scholar] [CrossRef]
- Gonçalves-De-Albuquerque, C.F.; Silva, A.R.; Burth, P.; Rocco, P.R.M.; Castro-Faria, M.V.; Castro-Faria-Neto, H.C. Possible mechanisms of Pseudomonas aeruginosa-associated lung disease. Int. J. Med. Microbiol. 2016, 306, 20–28. [Google Scholar] [CrossRef]
- Guirado, E.; Rajaram, M.V.S.; Chawla, A.; Daigle, J.; La Perle, K.M.D.; Arnett, E.; Turner, J.; Schlesinger, L.S. Deletion of PPARγ in lung macrophages provides an immunoprotective response against M. tuberculosis infection in mice. Tuberculosis 2018, 111, 170–177. [Google Scholar] [CrossRef]
- Malur, A.; Mohan, A.; Barrington, R.A.; Leffler, N.; Malur, A.; Muller-Borer, B.; Murray, G.; Kew, K.; Zhou, C.; Russell, J.; et al. Peroxisome Proliferator–activated Receptor-γ Deficiency Exacerbates Fibrotic Response to Mycobacteria Peptide in Murine Sarcoidosis Model. Am. J. Respir. Cell Mol. Biol. 2019, 61, 198–208. [Google Scholar] [CrossRef]
- Gonçalves-De-Albuquerque, C.F.; Rohwedder, I.; Silva, A.R.; Ferreira, A.S.; Kurz, A.R.M.; Cougoule, C.; Klapproth, S.; Eggersmann, T.; Silva, J.D.; De Oliveira, G.P.; et al. The Yin and Yang of Tyrosine Kinase Inhibition During Experimental Polymicrobial Sepsis. Front. Immunol. 2018, 9, 901. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.M.; Nowell, M.; Chima, R.; Zingarelli, B. Pioglitazone reduces inflammation through inhibition of NF-κB in polymicrobial sepsis. Innate Immun. 2013, 20, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.M.; Hake, P.W.; Denenberg, A.; Nowell, M.; Piraino, G.; Zingarelli, B. Phosphorylation of Extracellular Signal-Regulated Kinase (ERK)-1/2 Is Associated with the Downregulation of Peroxisome Proliferator–Activated Receptor (PPAR)-γ during Polymicrobial Sepsis. Mol. Med. 2010, 16, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pritchard, D.K.; Wang, X.; Park, D.R.; Bumgarner, R.E.; Schwartz, S.M.; Liles, W.C. cDNA microarray analysis reveals fundamental differences in the expression profiles of primary human monocytes, monocyte-derived macrophages, and alveolar macrophages. J. Leukoc. Biol. 2006, 81, 328–335. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, K.; Jin, Y.; Zhu, T.; Cheng, B.; Shu, Q.; Fang, X.-M. Triggering Receptor Expressed on Myeloid Cells-2 Protects against Polymicrobial Sepsis by Enhancing Bacterial Clearance. Am. J. Respir. Crit. Care Med. 2013, 188, 201–212. [Google Scholar] [CrossRef]
- Sharif, O.; Gawish, R.; Warszawska, J.M.; Martins, R.; Lakovits, K.; Hladik, A.; Doninger, B.; Brunner, J.S.; Korosec, A.; Schwarzenbacher, R.E.; et al. The Triggering Receptor Expressed on Myeloid Cells 2 Inhibits Complement Component 1q Effector Mechanisms and Exerts Detrimental Effects during Pneumococcal Pneumonia. PLoS Pathog. 2014, 10, e1004167. [Google Scholar] [CrossRef]
- Moseley, C.E.; Webster, R.G.; Aldridge, J.R. Original Article: Peroxisome proliferator-activated receptor and AMP-activated protein kinase agonists protect against lethal influenza virus challenge in mice. Influ. Other Respir. Viruses 2010, 4, 307–311. [Google Scholar] [CrossRef]
- Darwish, I.; Mubareka, S.; Liles, W.C. Immunomodulatory therapy for severe influenza. Expert Rev. Anti-Infect. Ther. 2011, 9, 807–822. [Google Scholar] [CrossRef]
- Cloutier, A.; Marois, I.; Cloutier, D.; Verreault, C.; Cantin, A.M.; Richter, M.V. The Prostanoid 15-Deoxy-Δ12,14-Prostaglandin-J2 Reduces Lung Inflammation and Protects Mice Against Lethal Influenza Infection. J. Infect. Dis. 2012, 205, 621–630. [Google Scholar] [CrossRef]
- Fedson, D.S. Treating influenza with statins and other immunomodulatory agents. Antivir. Res. 2013, 99, 417–435. [Google Scholar] [CrossRef]
- Bassaganya-Riera, J.; Guri, A.J.; Lu, P.; Climent, M.; Carbo, A.; Sobral, B.W.; Horne, W.T.; Lewis, S.N.; Bevan, D.; Hontecillas, R. Abscisic Acid Regulates Inflammation via Ligand-binding Domain-independent Activation of Peroxisome Proliferator-activated Receptor γ. J. Biol. Chem. 2010, 286, 2504–2516. [Google Scholar] [CrossRef] [PubMed]
- Guri, A.J.; Misyak, S.A.; Hontecillas, R.; Hasty, A.H.; Liu, D.; Si, H.; Bassaganya-Riera, J. Abscisic acid ameliorates atherosclerosis by suppressing macrophage and CD4+ T cell recruitment into the aortic wall. J. Nutr. Biochem. 2010, 21, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Hontecillas, R.; Roberts, P.C.; Carbo, A.; Vives, C.; Horne, W.T.; Genis, S.; Velayudhan, B.; Bassaganya-Riera, J. Dietary abscisic acid ameliorates influenza-virus-associated disease and pulmonary immunopathology through a PPARγ-dependent mechanism. J. Nutr. Biochem. 2012, 24, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhu, B.; Cheon, I.S.; Goplen, N.P.; Jiang, L.; Zhang, R.; Peebles, R.S.; Mack, M.; Kaplan, M.H.; Limper, A.H.; et al. PPAR-γ in Macrophages Limits Pulmonary Inflammation and Promotes Host Recovery following Respiratory Viral Infection. J. Virol. 2019, 93, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Jiang, L.; Cheon, I.S.; Sun, J. Targeting Peroxisome Proliferator-Activated Receptor-Gamma Decreases Host Mortality After Influenza Infection in Obese Mice. Viral Immunol. 2019, 32, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xu, Y.-Z.; Liu, B.; Wu, R.; Yang, Y.-Y.; Xiao, X.-Q.; Zhang, X. Pioglitazone Upregulates Angiotensin Converting Enzyme 2 Expression in Insulin-Sensitive Tissues in Rats with High-Fat Diet-Induced Nonalcoholic Steatohepatitis. Sci. World J. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Aguilar, M.; Ibarra-Lara, L.; Mondragón, L.D.V.; Rubio-Ruiz, M.E.; Aguilar-Navarro, A.G.; Zamorano-Carrillo, A.; Ramírez-Ortega, M.D.C.; Pastelín-Hernández, G.; Sánchez-Mendoza, A. Rosiglitazone, a Ligand to PPARγ, Improves Blood Pressure and Vascular Function through Renin-Angiotensin System Regulation. PPAR Res. 2019, 2019, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Filardi, T.; Morano, S. COVID-19: Is there a link between the course of infection and pharmacological agents in diabetes? J. Endocrinol. Investig. 2020, 43, 1053–1060. [Google Scholar] [CrossRef]
- Ghoneim, H.E.; Thomas, P.G.; McCullers, J.A. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J. Immunol. 2013, 191, 1250–1259. [Google Scholar] [CrossRef]
- Nakamura, S.; Davis, K.M.; Weiser, J.N. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J. Clin. Investig. 2011, 121, 3657–3665. [Google Scholar] [CrossRef]
- Moorthy, A.N.; Narasaraju, T.; Rai, P.; Perumalsamy, R.; Tan, K.B.; Wang, S.; Engelward, B.P.; Chow, V.T.K. In vivo and in vitro studies on the roles of neutrophil extracellular traps during secondary pneumococcal pneumonia after primary pulmonary influenza infection. Front. Immunol. 2013, 4, 56. [Google Scholar] [CrossRef]
- Small, C.-L.; Shaler, C.R.; McCormick, S.; Jeyanathan, M.; Damjanovic, D.; Brown, E.G.; Arck, P.C.; Jordana, M.; Kaushic, C.; Ashkar, A.A.; et al. Influenza Infection Leads to Increased Susceptibility to Subsequent Bacterial Superinfection by Impairing NK Cell Responses in the Lung. J. Immunol. 2010, 184, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Finelli, L.; Fiore, A.; Dhara, R.; Brammer, L.; Shay, D.K.; Kamimoto, L.; Fry, A.; Hageman, J.; Gorwitz, R.; Bresee, J.; et al. Influenza-Associated Pediatric Mortality in the United States: Increase of Staphylococcus aureus Coinfection. Pediatrics 2008, 122, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, E.; Kollef, M.H.; Nathwani, D. Pneumonia Caused by Methicillin-ResistantStaphylococcus aureus. Clin. Infect. Dis. 2008, 46, S378–S385. [Google Scholar] [CrossRef]
- Sharma, R.; Kaundal, R.; Sharma, S.S. Amelioration of pulmonary dysfunction and neutrophilic inflammation by PPARγ agonist in LPS-exposed guinea pigs. Pulm. Pharmacol. Ther. 2009, 22, 183–189. [Google Scholar] [CrossRef]
- Gopal, R.; Mendy, A.; Marinelli, M.A.; Richwalls, L.J.; Seger, P.J.; Patel, S.; McHugh, K.J.; Rich, H.E.; Grousd, J.A.; Forno, E.; et al. Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Suppresses Inflammation and Bacterial Clearance during Influenza-Bacterial Super-Infection. Viruses 2019, 11, 505. [Google Scholar] [CrossRef]
- Botta, M.; Audano, M.; Yaribeygi, H.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19, 1197. [Google Scholar] [CrossRef]
- Rousselot, P.; Prost, S.; Guilhot, J.; Roy, L.; Etienne, G.; Legros, L.; Charbonnier, A.; Coiteux, V.; Cony-Makhoul, P.; Huguet, F.; et al. Pioglitazone together with imatinib in chronic myeloid leukemia: A proof of concept study. Cancer 2016, 123, 1791–1799. [Google Scholar] [CrossRef]
- Kumar, B.R.P.; Kumar, A.P.; Jose, J.A.; Prabitha, P.; Yuvaraj, S.; Sandhya, C.; Jeyarani, V.; Manisha, C.; Banerjee, S.; Jeyabalan, J.B.; et al. Minutes of PPAR-γ Agonism and Neuroprotection. Neurochem. Int. 2020, 140, 104814. [Google Scholar] [CrossRef]
- Anderson, J.R.; Mortimer, K.; Pang, L.; Smith, K.M.; Bailey, H.; Hodgson, D.B.; Shaw, D.E.; Knox, A.; Harrison, T.W. Evaluation of the PPAR-γ Agonist Pioglitazone in Mild Asthma: A Double-Blind Randomized Controlled Trial. PLoS ONE 2016, 11, e0160257. [Google Scholar] [CrossRef]
- Reddy, A.T.; Lakshmi, S.P.; Reddy, R.C. PPARγ in Bacterial Infections: A Friend or Foe? PPAR Res. 2016, 2016, 1–7. [Google Scholar] [CrossRef]
- Devchand, P.R.; Liu, T.; Altman, R.B.; Fitzgerald, G.A.; Schadt, E.E. The Pioglitazone Trek via Human PPAR Gamma: From Discovery to a Medicine at the FDA and Beyond. Front. Pharmacol. 2018, 9, 1093. [Google Scholar] [CrossRef] [PubMed]


© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carvalho, M.V.d.; Gonçalves-de-Albuquerque, C.F.; Silva, A.R. PPAR Gamma: From Definition to Molecular Targets and Therapy of Lung Diseases. Int. J. Mol. Sci. 2021, 22, 805. https://doi.org/10.3390/ijms22020805
Carvalho MVd, Gonçalves-de-Albuquerque CF, Silva AR. PPAR Gamma: From Definition to Molecular Targets and Therapy of Lung Diseases. International Journal of Molecular Sciences. 2021; 22(2):805. https://doi.org/10.3390/ijms22020805
Chicago/Turabian StyleCarvalho, Márcia V. de, Cassiano F. Gonçalves-de-Albuquerque, and Adriana R. Silva. 2021. "PPAR Gamma: From Definition to Molecular Targets and Therapy of Lung Diseases" International Journal of Molecular Sciences 22, no. 2: 805. https://doi.org/10.3390/ijms22020805
APA StyleCarvalho, M. V. d., Gonçalves-de-Albuquerque, C. F., & Silva, A. R. (2021). PPAR Gamma: From Definition to Molecular Targets and Therapy of Lung Diseases. International Journal of Molecular Sciences, 22(2), 805. https://doi.org/10.3390/ijms22020805