Mapping the SARS-CoV-2–Host Protein–Protein Interactome by Affinity Purification Mass Spectrometry and Proximity-Dependent Biotin Labeling: A Rational and Straightforward Route to Discover Host-Directed Anti-SARS-CoV-2 Therapeutics



 ,
,  and
and
Abstract
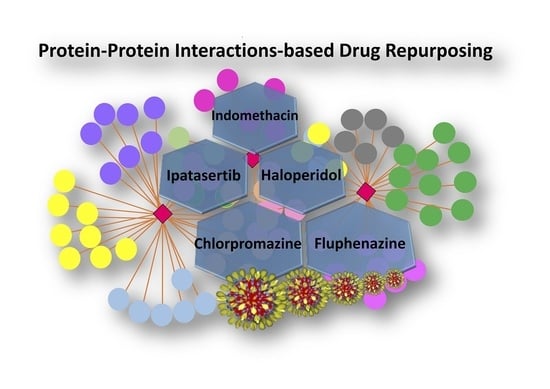
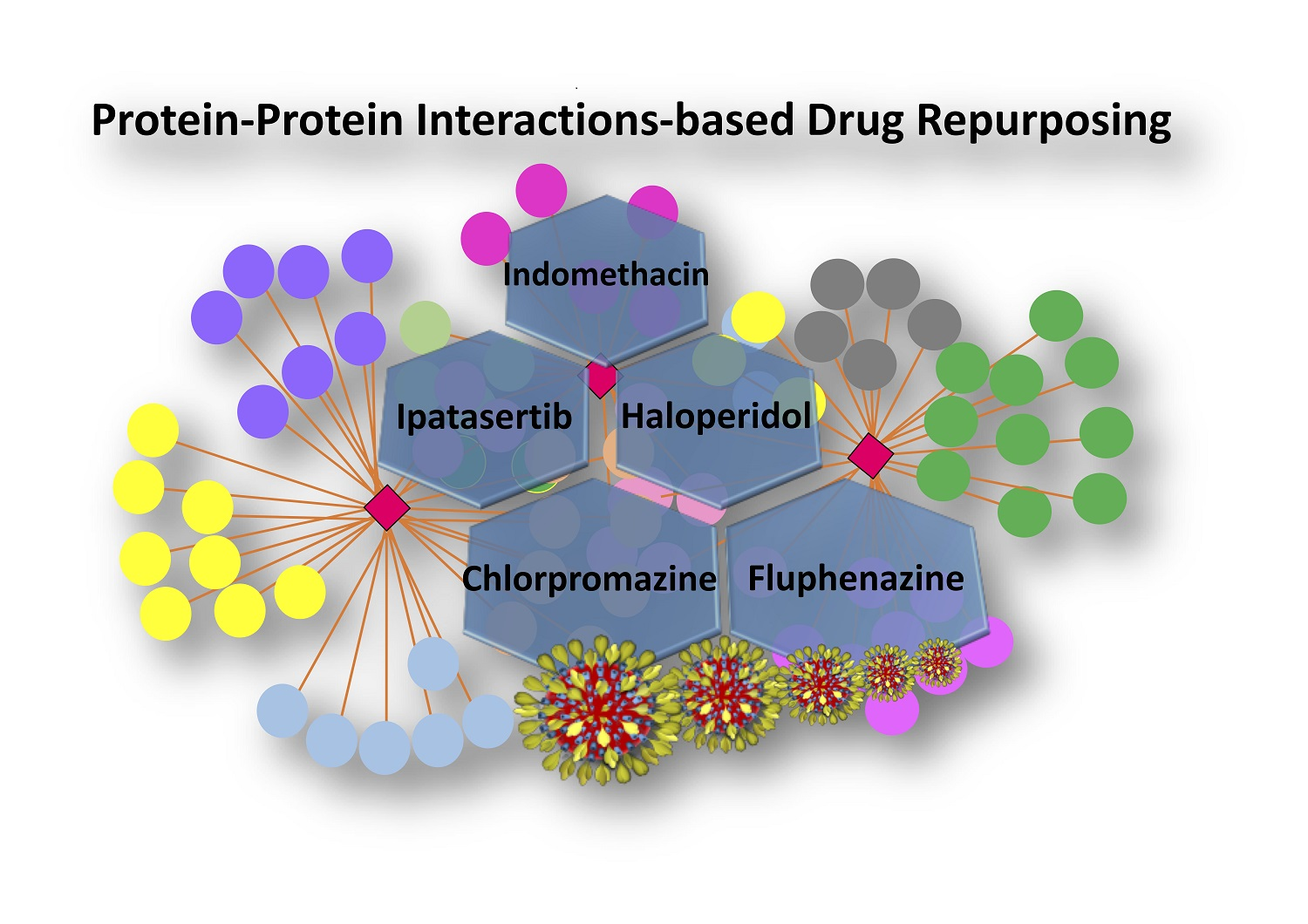
1. Introduction
1.1. Structural Features of SARS-CoV-2
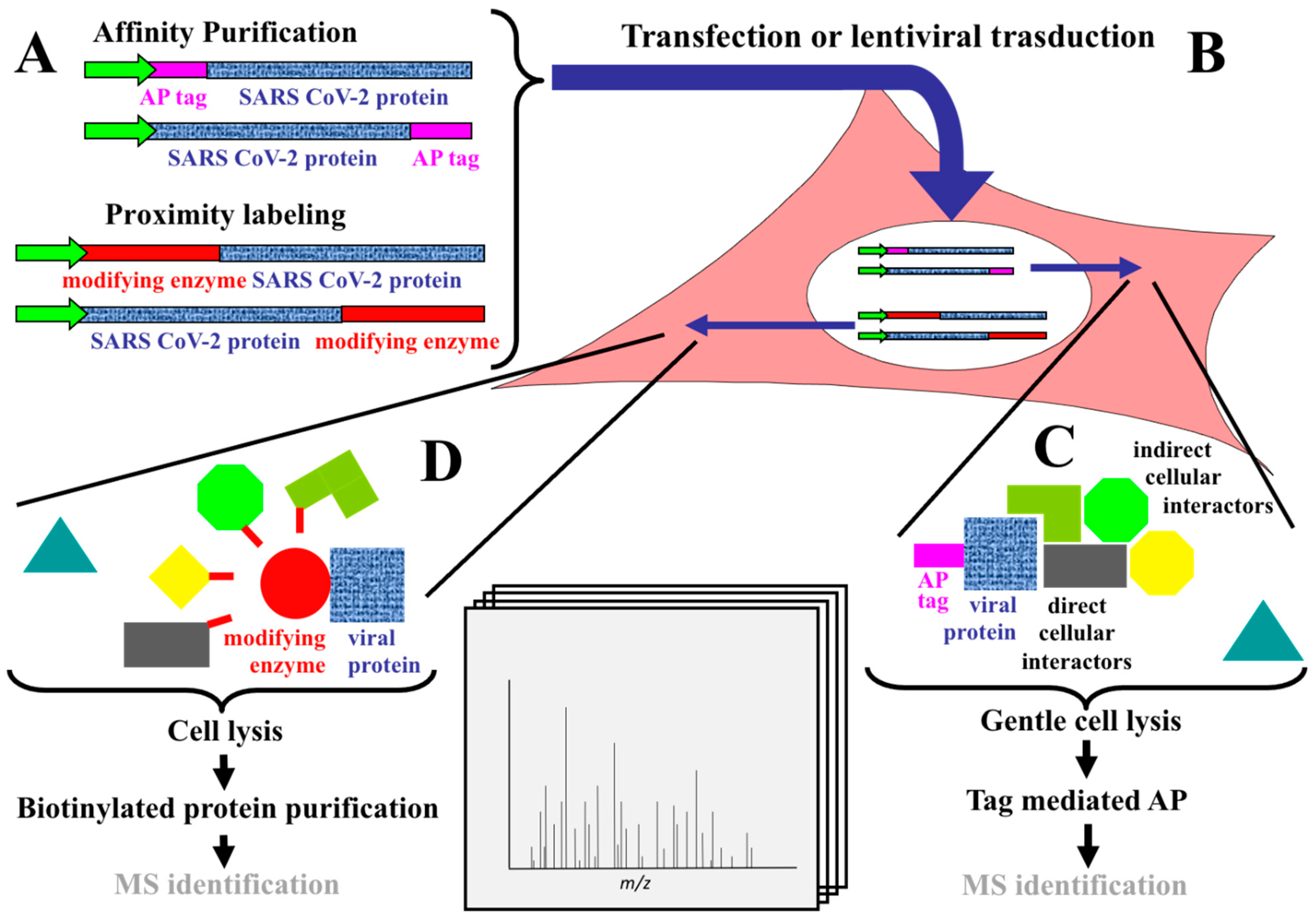
1.2. AP-MS and BioID
1.3. Data Filtering and Graphical Network Representation
1.4. Validation Experiments
1.5. Mapping PTMs Profiles
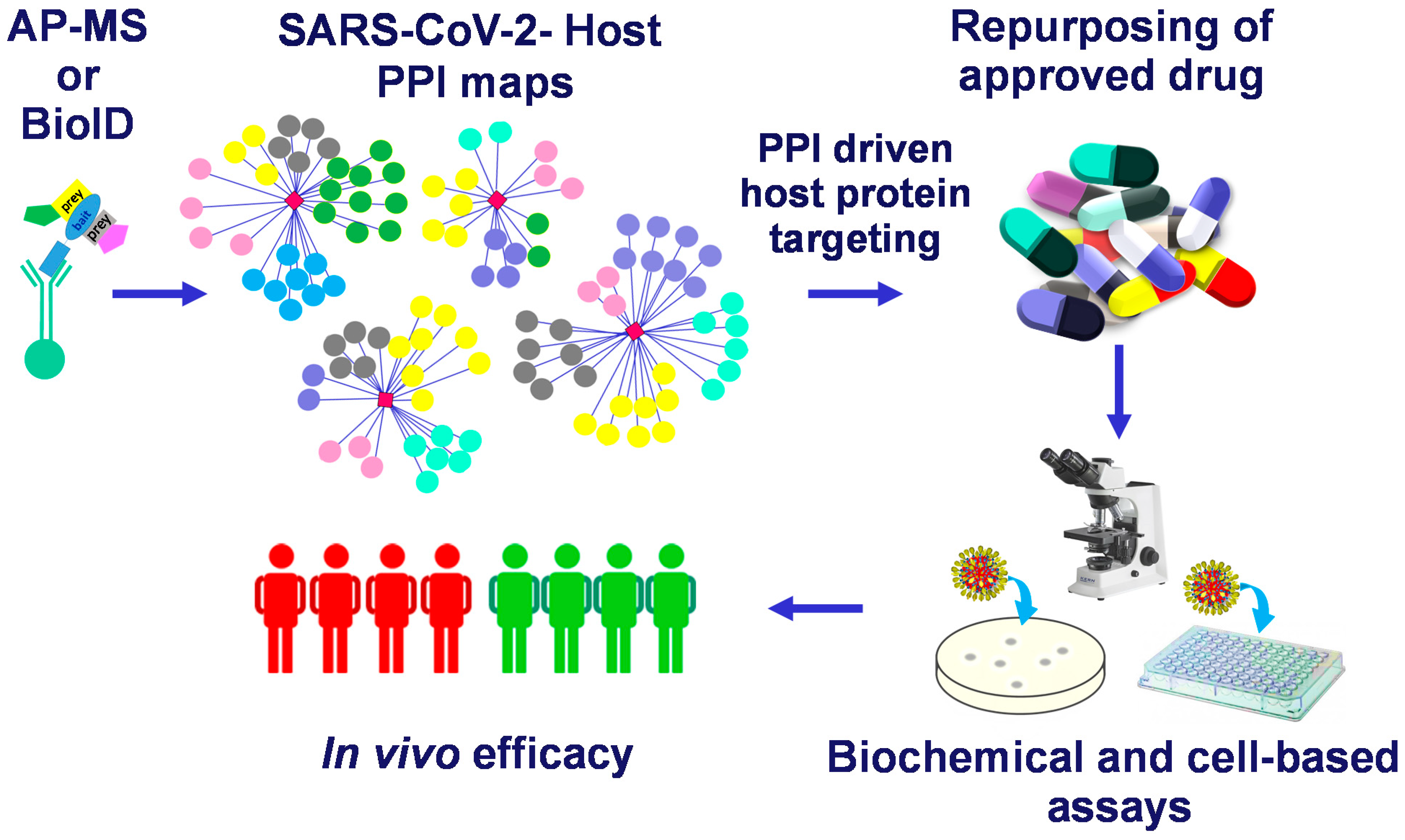
2. Mapping the SARS-CoV-2 Interactome
2.1. Mapping the SARS-CoV-2 Interactome Generated in HEK-293 Cells by AP-MS

{kind=link}
{kind=link}
{kind=link}
| References | Bait | Prey (Gene Name) or Process | Compound | In Vitro Evidence (1) | Compound Approval Status |
|---|---|---|---|---|---|
| Gordon et al. [17] | (2) | mRNA translation | PS3061 | IC50 = 20–500 nM | Preclinical compound |
| Ternatin-4 | IC50 = 71 nM | Preclinical compound | |||
| Zotatifin | IC50 = 1.5 nM | Drug in clinical trial | |||
| E | BRD2/4 | dBET6 | IC50 = 14 nM | Preclinical compound | |
| MZ1 | Kd = 120–228 nM | Preclinical compound | |||
| M | ATP6AP1/ATP6V1A | Bafilomycin A1 | IC50 = 100 nM | Preclinical compound | |
| N | CSNK2A2 | Silmitasertib | IC50 = 1 nM | Drug in clinical trial | |
| Nsp6 | SIGMAR1 | Clemastine | Ki = 67 nM | FDA-approved | |
| Haloperidol | Ki = 2.91 nM | FDA-approved | |||
| Hydroxychloroquine | Ki = 85 nM | FDA-approved | |||
| PB28 | Ki = 13 nM | Preclinical compound | |||
| Siramesine | Ki = 17 nM | Drug in clinical trial | |||
| Cloperastine | Ki = 20 nM | FDA-approved | |||
| ATP6AP1/ATP6V1A | Bafilomycin A1 | IC50 = 100 nM | Preclinical compound | ||
| Nsp12 | RIPK1 | Ponatinib | IC50 = 12 nM | FDA-approved | |
| Nsp14 | IMPDH2 | Mycophenolic acid | IC50 = 20 nM | FDA-approved | |
| ORF9c | SIGMAR2 | Clemastine | Ki = 15 nM | FDA-approved | |
| Haloperidol | Ki = 54.1 nM | FDA-approved | |||
| Hydroxychloroquine | Ki = 772 nM | FDA-approved | |||
| PB28 | Ki = 13 nM | Preclinical compound | |||
| Siramesine | Ki = 0.12 nM | Drug in clinical trial | |||
| Cloperastine | Ki = 900 nM | FDA-approved | |||
| F2RL1 | AZ3451 (PAR2 negative allosteric modulator) | pKd = 15 | Preclinical compound | ||
| ORF10 | VCP | ML240 | IC50 = 100 nM | Preclinical compound | |
| Bouhaddou et al. [63] | (3) | AXL | Gilteritinib | IC50 = 0.807 μM | FDA-approved |
| N/A | MAPK11, MAPK14 | Ralimetinib | IC50 = 0.873 μM | Drug in clinical trial | |
| (4) | MAPK13 | MAPK13-IN-1 | IC50 = 4.63 μM | Preclinical compound | |
| N/A | MAPK14 | ARRY-797 | IC50= 0.913μM | Drug in clinical trial | |
| (4) | MAPK14, MAPK11, MAPK12, MAPK13 | SB203580 | IC50 = 4.76μM | Preclinical compound | |
| N | CSNK2A1, CSNK2A2 | Silmitasertib | IC50 = 2.34 μM | Drug in clinical trial | |
| (5) | PIKFYVE | Apilimod | IC50 = 0.08 μM IC50 = 0.007 μM | Drug in clinical trial | |
| (6) | CDK | Dinaciclib | IC50 = 0.127 μM IC50 = 0.032 μM | Drug in clinical trial | |
| Gordon et al. [18] | Nsp6 | SIGMAR1 | Fluphenazine | pIC50 = 6.46 | FDA approved |
| Chlorpromazine | pIC50 = 6.05 | FDA approved | |||
| Haloperidol | pIC50 = 5.684 | FDA approved | |||
| Clemastine | pIC50 = 6.264 | FDA approved | |||
| Meclizine | pIC50 = 5177 | FDA approved | |||
| Amodiaquine | pIC50 = 6.428 | FDA approved | |||
| Hydroxychloroquine | pIC50 = 6.062 | FDA approved | |||
| Chloroquine | pIC50 = 6.036 | FDA approved | |||
| Amiodarone | pIC50 = 6.779 | FDA approved | |||
| Tamoxifen | pIC50 = 6.563 | FDA approved | |||
| Triparanol | pIC50 = 6.439 | FDA approved | |||
| Clomiphene | pIC50 = 6.257 | FDA approved | |||
| Propranolol | pIC50 = 5.435 | FDA approved | |||
| Nsp7 | PTGES2 | Indomethacin | pIC50 = 4.258 | FDA approved | |
| ORF9c | SIGMAR2 | Fluphenazine | pIC50 = 6.46 | FDA approved | |
| Chlorpromazine | pIC50 = 6.05 | FDA approved | |||
| Haloperidol | pIC50 = 5.684 | FDA approved | |||
| Clemastine | pIC50 = 6.264 | FDA approved | |||
| Meclizine | pIC50 = 5177 | FDA approved | |||
| Amodiaquine | pIC50 = 6.428 | FDA approved | |||
| Hydroxychloroquine | pIC50 = 6.062 | FDA approved | |||
| Chloroquine | pIC50 = 6.036 | FDA approved | |||
| Amiodarone | pIC50 = 6.779 | FDA approved | |||
| Tamoxifen | pIC50 = 6.563 | FDA approved | |||
| Triparanol | pIC50 = 6.439 | FDA approved | |||
| Clomiphene | pIC50 = 6.257 | FDA approved | |||
| Propranolol | pIC50 = 5.435 | FDA approved | |||
| Stukalov et al. [21] | N/A | Inducers of DNA damage | Tirapazamine | 2 µM (7) | Drug in clinical trial |
| Rabusertib | 1 µM (7) | Drug in clinical trial | |||
| N/A | mTOR inhibitor | Rapamycin | 1 µM (7) | FDA-approved | |
| ORF3 | FLT3/AXL | Gilteritinib | 0.5 µM (7) | FDA-approved | |
| (8) | AKT | Ipatasertib | 5 µM (7) | Drug in clinical trial | |
| N/A | Matrix metalloproteinase inhibitors | Prinomastat | 2 µM (7) | Drug in clinical trial | |
| Marimastat | 2 µM (7) | Drug in clinical trial |
2.2. Mapping the SARS-CoV-2 Interactome Generated in A549 Lung Carcinoma Cells by AP-MS
2.3. Mapping the SARS-CoV-2 Interactome Generated in HEK293 Cells by BioID
2.4. Mapping the SARS-CoV-2 Interactome Generated in A549 Cells by BioID
3. Discussion
3.1. Common Host Interactors Across Core PPI Datasets
3.2. Challenges and Limitations of These Approaches
4. Conclusions
5. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Teichmann, S.; Regev, A. The network effect: Studying COVID-19 pathology with the Human Cell Atlas. Nat. Rev. Mol. Cell. Biol. 2020, 21, 415–416. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Stefanelli, I.; Cerchia, C.; Beccari, A.R.; Pelliccia, S.; Summa, V. SARS-CoV-2 Entry Inhibitors: Small Molecules and Peptides Targeting Virus or Host Cells. Int. J. Mol. Sci. 2020, 21, 5707. [Google Scholar] [CrossRef] [PubMed]
- Cannalire, R.; Cerchia, C.; Beccari, A.R.; Di Leva, F.S.; Summa, V. Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J. Med. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Meyniel-Schicklin, L.; De Chassey, B.; André, P.; Lotteau, V. Viruses and interactomes in translation. Mol. Cell. Proteomics 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.L.; Ganji, G.; Paeper, B.; Proll, S.; Katze, M.G. System biology and the host response to viral infection. Nat. Biotechnol. 2007, 25, 1383–1389. [Google Scholar] [CrossRef]
- Goodacre, N.; Devkota, P.; Bae, E.; Wuchty, S.; Uetz, P. Protein-protein interactions of human viruses. Semin. Cell. Dev. Biol. 2020, 99, 31–39. [Google Scholar] [CrossRef]
- Keskin, O.; Tuncbag, N.; Gursoy, A. Predicting Protein-Protein Interactions from Molecular to the Proteome Level. Chem. Rev. 2016, 116, 4884–4909. [Google Scholar] [CrossRef]
- Perrin-Cocon, L.; Diaz, O.; Jacquemin, C.; Barthel, V.; Ogire, E.; Ramière, C.; André, P.; Lotteau, V.; Vidalain, P.O. The current landscape of coronavirus-host protein-protein interactions. J. Transl. Med. 2020, 18, 319. [Google Scholar] [CrossRef]
- Parrish, J.R.; Gulyas, K.D.; Finley, R.L. Yeast two-hybrid contributions to interactome mapping. Curr. Opin. Biotechnol. 2006, 17, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 2020, 184, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Heaton, B.E.; Trimarco, J.D.; Hamele, C.E.; Harding, A.T.; Tata, A.; Zhu, X.; Tata, P.R.; Smith, C.M.; Heaton, N.S. SRSF protein kinases 1 and 2 are essential host factors for human coronaviruses including SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Lum, K.L.; Cristea, I.M. Proteomic approaches to uncovering virus–host protein interactions during the progression of viral infection. Expert Rev. Proteom. 2016, 13, 325–340. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Gerber, M.; Kelly, J.; Pfaender, S.; Ebert, N.; Lagache, S.B.; Simillion, C.; Portmann, J.; Stalder, H.; Gaschen, V.; et al. Determination of host proteins composing the microenvironment of coronavirus replicase complexes proximity-labeling. Elife 2019, 8, e42037. [Google Scholar] [CrossRef] [PubMed]
- Cornillez-Ty, C.T.; Liao, L.; Yates, J.R., 3rd; Kuhn, P.; Buchmeier, M.J. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J. Virol. 2009, 83, 10314–10318. [Google Scholar]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J.; et al. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 2020, 370, 6521. [Google Scholar] [CrossRef]
- Davies, J.P.; Almasy, K.M.; McDonald, E.F.; Plate, L. Comparative multiplexed interactomics of SARS-CoV-2 and homologous coronavirus non-structural proteins identifies unique and shared host-cell dependencies. ACS Infect. Dis. 2020, 6, 3174–3189. [Google Scholar] [CrossRef]
- Li, J.; Guo, M.; Tian, X.; Wang, X.; Yang, X.; Wu, P.; Liu, C.; Xiao, Z.; Qu, Y.; Yin, Y.; et al. Virus-Host Interactome and Proteomic Survey Reveal Potential Virulence Factors Influencing SARS-CoV-2 Pathogenesis. Med. (N.Y.) 2020. [Google Scholar] [CrossRef]
- Stukalov, A.; Girault, V.; Grass, V.; Bergant, V.; Karayel, O.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multi-level proteomics reveals host-perturbation strategies of SARS-CoV-2 and SARS-CoV. bioRxiv 2020. [Google Scholar] [CrossRef]
- Samavarchi-Tehrani, P.; Abdouni, H.; Knight, J.D.R.; Astori, A.; Samson, R.; Lin, Z.; Kim, D.; Knapp, J.J.; St-Germain, J.; Go, C.D.; et al. A SARS-CoV-2—Host proximity interactome. bioRxiv 2020. [Google Scholar] [CrossRef]
- St-Germain, J.; Astori, A.; Samavarchi-Tehrani, P.; Abdouni, H.; Macwan, V.; Kim, D.; Knapp, J.J.; Roth, F.P.; Gingras, A.; Raught, B. A SARS-CoV-2 BioID-based virus-host membrane protein interactome and virus peptide compendium: New proteomics resources for COVID-19 research. bioRxiv 2020. [Google Scholar] [CrossRef]
- Laurent, E.M.N.; Yorgos Sofianatos, Y.; Komarova, A.; Gimeno, J.; Payman Samavarchi-Tehrani, P.; Kim, D.; Abdouni, H.; Duhamel, M.; Cassonnet, P.; Knapp, J.J.; et al. Global BioID-based SARS-CoV-2 proteins proximal interactome unveils novel ties between viral polypeptides and host factors involved in multiple COVID19-associated mechanisms. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 580, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar]
- Walls, A.C.; Park, Y.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef]
- Weekes, M.P.; Tomasec, P.; Huttlin, E.L.; Fielding, C.A.; Nusinow, D.; Stanton, R.J.; Wang, E.; Aicheler, R.; Murrell, I.; Wilkinson, G.; et al. Quantitative temporal viromics: An approach to investigate host-pathogen interaction. Cell 2014, 157, 1460–1472. [Google Scholar] [CrossRef]
- Cristea, I.M.; Moorman, N.J.; Terhune, S.S.; Cuevas, C.D.; O’Keefe, E.S.; Rout, M.P.; Chait, B.T.; Shenk, T. Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J. Virol. 2010, 84, 7803–7814. [Google Scholar] [CrossRef]
- Jäger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global landscape of HIV-human protein complexes. Nature 2011, 481, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.S.; Wojcechowskyj, J.A.; Eckhardt, M.; Krogan, N.J. Comparative mapping of host-pathogen protein-protein interactions. Curr. Opin. Microbiol. 2015, 27, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Berggård, T.; Linse, S.; James, P. Methods for the detection and analysis of protein-protein interactions. Proteomics 2007, 7, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Mann, M. Mass spectrometry–based proteomics in cell biology. J. Cell Biol. 2010, 190, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Nicod, C.; Banaei-Esfahani, A.; Collins, B.C. Elucidation of Host–Pathogen Protein–Protein Interactions to Uncover Mechanisms of Host Cell Rewiring. Curr. Opin. Microbiol. 2017, 39, 7–15. [Google Scholar] [CrossRef]
- Beltran, P.M.J.; Cook, K.C.; Cristea, I.M. Exploring and Exploiting Proteome Organization during Viral Infection. J. Virol. 2017, 91, 18. [Google Scholar] [CrossRef]
- Hesketh, G.G.; Youn, J.Y.; Samavarchi-Tehrani, P.; Raught, B.; Gingras, A.C. Parallel Exploration of Interaction Spacy BioID and Affinity Purification Coupled to Mass Spectrometry. Methodol. Biol. 2017, 1550, 115–136. [Google Scholar]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous bioiotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar]
- Streaker, E.D.; Beckett, D. Nonenzymatic biotinylation of a biotin carboxyl carrier protein: Unusual reactivity of the physiological target lysine. Protein Sci. 2006, 15, 1928–1935. [Google Scholar] [CrossRef]
- Choi-Rhee, E.; Schulman, H.; Cronan, J.E. Promiscuous protein biotinylation by Escherichia coli biotin protein ligase. Protein Sci. 2004, 13, 3043–3050. [Google Scholar]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Samavarchi-Tehrani, P.; Samson, R.; Gingras, A. Proximity Dependent Biotinylation: Key Enzymes and Adaptation to Proteomics Approaches. Mol. Cell. Proteom. 2020, 19, 757–773. [Google Scholar] [CrossRef] [PubMed]
- Lalonde, S.; Ehrhardt, D.W.; Loqué, D.; Chen, J.; Rhee, S.Y.; Frommer, W.B. Molecular and cellular approaches for the detection of protein-protein interactions: Latest techniques and current limitations. Plant. J. 2008, 53, 610–635. [Google Scholar] [CrossRef] [PubMed]
- Armean, I.M.; Lilley, K.S.; Trotter, M.W.B. Popular Computational Methods to Asses Multiprotein Complexes Derived From Label-Free Affinity Purification and Mass Spectrometry (AP-MS) Experiments. Mol. Cell. Proteom. 2012, 12, 1–13. [Google Scholar] [CrossRef]
- Teo, G.; Liu, G.; Zhang, J.; Nesvizhskii, A.I.; Gingras, A.; Choi, H. SAINTexpress: Improvements and additional features in Significance Analysis of INTeractome software. J. Proteom. 2014, 100, 37–43. [Google Scholar]
- Verschueren, E.; Von Dollen, J.; Cimermancic, P.; Gulbahce, N.; Sali, A.; Krogan, N.J. Scoring large-scale affinity purification mass spectrometry datasets with MiST. Curr. Protoc. Bioinform. 2015, 49, 8.19.1–8.19.16. [Google Scholar]
- Morris, J.H.; Knudsen, G.M.; Verschueren, E.; Johnson, J.R.; Cimermancic, P.; Greninger, A.L.; Pico, A.R. Affinity purification-mass spectrometry and network analysis to understand protein-protein interactions. Nat. Protoc. 2014, 9, 2539–2554. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar]
- Jacob, N.T. Drug promotion practices: A review. Br. J. Clin. Pharmacol. 2018, 84, 1659–1667. [Google Scholar] [CrossRef] [PubMed]
- Reineke, L.C.; Tsai, W.; Jain, A.; Kaelber, J.T.; Jung, S.Y.; Lloyd, R.E. Casein kinase 2 is linked to stress granules dynamics through phosphorylation of the stress granule nucleating protein G3BP1. Mol. Cell. Biol. 2017, 37, e00596-16. [Google Scholar] [CrossRef] [PubMed]
- Slaine, P.D.; Kleer, M.; Smith, N.K.; Khaperskyy, D.A.; McCormick, C. Stress granule-inducing eukaryotic translation initiation factor 4A inhibitors block influenza A virus replication. Viruses 2017, 9, 388. [Google Scholar] [CrossRef] [PubMed]
- Timms, R.T.; Zhang, Z.; Rhee, D.Y.; Harper, J.W.; Koren, I.; Elledge, S.J. A glycine-specific N-degron pathway mediates the quality control of protein N-myristoylation. Science 2019, 365, eaaw4912. [Google Scholar] [CrossRef] [PubMed]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Quan, B.; Seo, H.S.; Blobel, G.; Ren, Y. Vesiculoviral matrix (M) protein occupies nucleic acid binding site at nucleoporin pair (Rae1•Nup98). Proc. Natl. Acad. Sci. USA 2014, 111, 9127–9132. [Google Scholar] [CrossRef] [PubMed]
- Frieman, M.; Yount, B.; Heise, M.; Kopecky-Bromberg, S.A.; Palese, P.; Baric, R.S. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 2007, 81, 9812–9824. [Google Scholar] [CrossRef]
- Wang, D.; Eraslan, B.; Wieland, T.; Hallström, B.; Hopf, T.; Zolg, D.P.; Zecha, J.; Asplund, A.; Li, L.; Meng, C.; et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 2019, 15, e8503. [Google Scholar] [CrossRef]
- WHO Solidarity Trial Consortium; Pan, H.; Peto, R.; Henao-Restrepo, A.M.; Preziosi, M.P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M.M.; Hernández García, C.; Kieny, M.P.; et al. Repurposed Antiviral Drugs for Covid-19—Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Zecha, J.; Lee, C.Y.; Bayer, F.P.; Meng, C.; Grass, V.; Zerweck, J.; Schnatbaum, K.; Michler, T.; Pichlmair, A.; Ludwig, C.; et al. Data, Reagents, Assays and Merits of Proteomics for SARS-CoV-2 Research and Testing. Mol. Cell. Proteom. 2020, 19, 1503–1522. [Google Scholar] [CrossRef]
- Rahaman, M.M.; Reinders, F.G.; Koes, D.; Nguyen, A.T.; Mutchler, S.M.; Sparacino-Watkins, C.; Alvarez, R.A.; Miller, M.P.; Cheng, D.; Chen, B.B.; et al. Structure Guided Chemical Modifications of Propylthiouracil Reveal Novel Small Molecule Inhibitors of Cytochrom5 Reductase 3 That Increase Nitric Oxide Bioavailability. J. Biol. Chem. 2015, 290, 16861–16872. [Google Scholar] [CrossRef] [PubMed]
- Sadegh, S.; Matschinske, J.; Blumenthal, D.B.; Galindez, G.; Kacprowski, T.; List, M.; Nasirigerdeh, R.; Oubounyt, M.; Pichlmair, A.; Rose, T.D.; et al. Exploring the SARS-CoV-2 virus-host-drug interactome for drug repurposing. Nat. Commun. 2020, 11, 3518. [Google Scholar]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Correa Marrero, M.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712. [Google Scholar] [CrossRef] [PubMed]
- Kuss, S.K.; Mata, M.A.; Zhang, L.; Fontoura, B.M.A. Nuclear imprisonment: Viral strategies to arrest host mRNA nuclear export. Viruses 2013, 5, 1824–1849. [Google Scholar] [CrossRef]
- Walsh, D.; Mohr, I. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 2011, 9, 860–875. [Google Scholar] [CrossRef]
- Han, H.; Yang, L.; Liu, R.; Liu, F.; Wu, K.L.; Li, J.; Liu, X.H.; Zhu, C.L. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin. Chem. Lab. Med. 2020, 58, 1116–1120. [Google Scholar] [CrossRef]
- Davidson, A.D.; Williamson, M.K.; Lewis, S.; Shoemark, D.; Carroll, M.W.; Heesom, K.J.; Zambon, M.; Ellis, J.; Lewis, P.A.; Hiscox, J.A.; et al. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 2020, 12, 68. [Google Scholar] [CrossRef]
- Hong, S.; Freeberg, M.A.; Han, T.; Kamath, A.; Yao, Y.; Fukuda, T.; Suzuki, T.; Kim, J.K.; Inoki, K. LARP1 functions as a molecular switch for mTORC1-mediated translation of an essential class of mRNAs. Elife 2017, 6, e25237. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The role of the transcription factor CREB in immune function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef]
- Pedersen, S.F.; Ho, Y.C. SARS-CoV-2: A storm is raging. J. Clin. Invest. 2020, 130, 2202–2205. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Paz, S.; Hiscott, J. Tom70 imports antiviral immunity to the mitochondria. Cell. Res. 2010, 20, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Cui, Y.; Huang, Y.; Liu, H.; Li, L.; Li, M.; Ruan, K.C.; Zhou, Q.; Wang, C. Tom70 mediates Sendai virus-induced apoptosis on mitochondria. J. Virol. 2015, 89, 3804–3818. [Google Scholar] [CrossRef]
- Jiang, H.W.; Zhang, H.N.; Meng, Q.F.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.; Wang, X.; Qi, H.; Zhang, J.; et al. SARS-CoV-2 Orf9b suppresses type I interferon response by targeting TOM70. Cell. Mol. Immunol. 2020, 17, 998–1000. [Google Scholar] [CrossRef]
- Liu, X.Y.; Wei, B.; Shi, H.X.; Shan, Y.F.; Wang, C. Tom70 mediates activation of interferon regulatory factor 3 on mitochondria. Cell. Res. 2010, 20, 994–1011. [Google Scholar] [CrossRef]
- Amici, C.; Di Caro, A.; Ciucci, A.; Chiappa, L.; Castilletti, C.; Martella, V.; Decaro, N.; Buonavoglia, C.; Capobianchi, M.R.; Santoro, M.G. Indomethacin has a potent antiviral activity against SARS coronavirus. Antivir. Ther. 2006, 11, 1021–1030. [Google Scholar]
- Christie, D.A.; Lemke, C.D.; Elias, I.M.; Chau, L.A.; Kirchhof, M.G.; Li, B.; Ball, E.H.; Dunn, S.D.; Hatch, G.M.; Madrenas, J. Stomatin-Likrotein 2 Binds Cardiolipin and Regulates Mitochondrial Biogenesis and Function. Mol. Cell. Biol. 2011, 31, 3845–3856. [Google Scholar] [CrossRef]
- Kim, J.H.; Rhee, J.K.; Ahn, D.G.; Kim, K.P.; Oh, J.W. Interaction of Stomatin with Hepatitis C Virus RNA Polymerase Stabilizes the Viral RNA Replicase Complexes on Detergent-Resistant Membranes. J. Microbiol. Biotechnol. 2014, 24, 1744–1754. [Google Scholar] [CrossRef]
- Wintachai, P.; Wikan, N.; Kuadkitkan, A.; Jaimipuk, T.; Ubol, S.; Pulmanausahakul, R.; Auewarakul, P.; Kasinrerk, W.; Weng, W.; Panyasrivanit, M.; et al. Identification of Prohibitin as a Chikungunya Virus Receptor Protein. J. Med. Virol. 2012, 84, 1757–1770. [Google Scholar] [CrossRef]
- Too, I.H.K.; Bonne, I.; Tan, E.L.; Chu, J.J.H.; Alonso, S. Prohibitin Plays a Critical Role in Enterovirus 71 Neuropathogenesis. PLoS Pathog. 2018, 14, e1006778. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chan, J.F.; Wang, Y.; Yuen, T.T.; Chai, Y.; Hou, Y.; Shuai, H.; Yang, D.; Hu, B.; Huang, X.; et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: An ex vivo study with implications for the pathogenesis of COVID-19. Clin. Infect. Dis. 2020, 71, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Boopathy, G.T.K.; Wanjin Hong, W. Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Front. Cell Dev. Biol. 2019, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Weiss, C.M.; Trobaugh, D.W.; Sun, C.; Lucas, T.M.; Diamond, M.S.; Ryman, K.D.; Klimstra, W.B. The Interferon-Induced Exonuclease ISG20 Exerts Antiviral Activity through Upregulation of Type I Interferon Response Proteins. mSphere 2018, 3, e00209-18. [Google Scholar] [CrossRef] [PubMed]
- Kopecky-Bromberg, S.A.; Martínez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Vaira, L.A.; Salzano, G.; Deiana, G.; De Riu, G. Anosmia and Ageusia: Common Findings in COVID-19 Patients. Laryngoscope 2020, 130. [Google Scholar] [CrossRef]
- Abuin, L.; Bargeton, B.; Ulbrich, M.H.; Isacoff, E.Y.; Kellenberger, S.; Benton, R. Functional architecture of olfactory ionotropic glutamate receptors. Neuron 2011, 69, 44–60. [Google Scholar] [CrossRef]
- Fouquet, C.; Di Meglio, T.; Ma, L.; Kawasaki, T.; Long, H.; Hirata, T.; Tessier-Lavigne, M.; Chédotal, A.; Nguyen-Ba-Charvet, K.T. Robo1 and robo2 control the development of the lateral olfactory tract. J. Neurosci. 2007, 27, 3037–3045. [Google Scholar] [CrossRef]
- Roulin, P.S.; Lotzerich, M.; Torta, F.; Tanner, L.B.; van Kuppeveld, F.J.; Wenk, M.R.; Greber, U.F. Rhinoviruses a phosphatidylinositol 4-phosphate/cholesterol countercurrent for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe 2014, 16, 677–690. [Google Scholar] [CrossRef]
- van der Schaar, H.M.; Dorobantu, C.M.; Albulescu, L.; Strating, J.; van Kuppeveld, F.J.M. Fat(al) attraction: Picornaviruses Usurp Lipid Transfer at Membrane Contact Sites to Create Replication Organelles. Trendicrobiology 2016, 24, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; He, G.; Filipowicz, N.A.; Randall, G.; Belov, G.A.; Kopek, B.G.; Wang, X. Host Lipids in Positive-Strand RNA Virus Genome Replication. Front. Microbiol. 2019, 10, 286. [Google Scholar] [CrossRef] [PubMed]
- Antonny, B.; Bigay, J.; Mesmin, B. The Oxysterol-Binding Protein Cycle: Burning Off PI(4)P to Transport Cholesterol. Annu. Rev. Biochem. 2018, 87, 809–837. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; van Kuppeveld, F.J. (+)RNA viruses rewire cellular pathways to build replication organelles. Curr. Opin. Virol. 2012, 2, 740–747. [Google Scholar] [CrossRef]
- Ren, Z.; Ding, T.; Zuo, Z.; Xu, Z.; Deng, J.; Wei, Z. Regulation of MAVS Expression and Signaling Function in the Antiviral Innate Immune Response. Front. Immunol. 2020, 11, 1030. [Google Scholar] [CrossRef] [PubMed]
- Jaafar, Z.A.; Kieft, J.S. Viral RNA structure-based strategies to manipulate translation. Nat. Rev. Microbiol. 2019, 17, 110–123. [Google Scholar] [CrossRef]
- Sicari, D.; Chatziioannou, A.; Koutsandreas, T.; Sitia, R.; Chevet, E. Role of the early secretory pathway in SARS-CoV-2 infection. J. Cell Biol. 2020, 219, e202006005. [Google Scholar] [CrossRef]
- Ludwig, A.; Nguyen, T.H.; Leong, D.; Ravi, L.I.; Tan, B.H.; Sandin, S.; Sugrue, R.J. Caveolae provide a specialized membrane environment for respiratory syncytial virus assembly. J. Cell Sci. 2017, 130, 1037–1050. [Google Scholar] [CrossRef]
- Kaakinen, M.; Reichelt, M.E.; Ma, Z.; Ferguson, C.; Martel, N.; Porrello, E.R.; Hudson, J.E.; Thomas, W.G.; Parton, R.G.; Headrick, J.P. Cavin-1 deficiency modifies myocardial and coronary function, stretch responses and ischaemic tolerance: Roles of NOS over-activity. Basic Res. Cardiol. 2017, 112, 24. [Google Scholar] [CrossRef]
- Chen, L.Q.; Burdowski, J.; Marfatia, R.; Weber, J.; Gliganic, K.; Diaz, N.; Ramjattan, N.; Zheng, H.; Mihalatos, D.; Wang, L.; et al. Reduced cardiac function is associated with cardiac injury and mortality risk in hospitalized COVID-19 Patients. Clin. Cardiol. 2020, 43, 1547–1554. [Google Scholar] [CrossRef]
- Agarwal, S.; Al Hashimi, H.; Agarwal, S.K.; Albastaki, U. Possible association between myocardial infarction with nonobstructed coronary arteries and SARS-CoV-2 infection. CMAJ 2020, 192, E1633–E1636. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Thomas, T.; Dzieciatkowska, M.; Hill, R.C.; Francis, R.O.; Hudson, K.E.; Zimring, J.C.; Hod, E.A.; Spitalnik, S.L.; Hansen, K.C. Serum Proteomics in COVID-19 Patients: Altered Coagulation and Complement Status as a Function of IL-6 Level. J. Proteome Res. 2020, 19, 4417–4427. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.; Nukala, S.B.; Srivastava, S.; Miyamoto, H.; Ismail, N.I.; Rehman, J.; Ong, S.B.; Lee, W.H.; Ong, S.G. Detection of Viral RNA Fragments in Human iPSC-Cardiomyocytes following Treatment with Extracellular Vesicles from SARS-CoV-2 Coding-Sequence-Overexpressing Lung Epithelial Cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Casas, A.I.; Hassan, A.A.; Larsen, S.J.; Gomez-Rangel, V.; Elbatreek, M.; Kleikers, P.W.M.; Guney, E.; Egea, J.; López, M.G.; Baumbach, J.; et al. From single drug targets to synergistic network pharmacology in ischemic stroke. Proc. Natl. Acad. Sci. USA 2019, 116, 7129–7136. [Google Scholar] [CrossRef]
- Baumbach, J.; Schmidt, H.H.H.W. The end of medicine as we know it: Introduction to the new journal, systems medicine. Netw. Syst. Med. 2018, 1, 1–2. [Google Scholar] [CrossRef]
- Halu, A.; De Domenico, M.; Arenas, A.; Sharma, A. The multiplex network of human diseases. NPJ Syst. Biol. Appl. 2019, 5, 15. [Google Scholar] [PubMed]
- Nadeau, R.; Fard, S.S.; Scheer, A.; Hashimoto-Roth, E.; Nygard, D.; Abramchuk, I.; Chung, Y.; Bennett, S.A.L.; Lavallée-Adam, M. Computational Identification of Human Biological Processes and Protein Sequence Motifs Putatively Targeted by SARS-CoV-2 Proteins Using Protein–Protein Interaction Networks. J. Proteome Res. 2020, 19, 4553–4566. [Google Scholar] [PubMed]
- Verstraete, N.; Jurman, G.; Bertagnolli, G.; Ghavasieh, A.; Pancaldi, V.; De Domenico, M. CovMulNet19, Integrating Proteins, Diseases, Drugs, and Symptoms: A Network Medicine Approach to COVID-19. Netw. Syst. Med. 2020, 3, 130–141. [Google Scholar] [PubMed]
- Srinivasan, S.; Cui, H.; Gao, Z.; Liu, M.; Lu, S.; Mkandawire, W.; Narykov, O.; Sun, M.; Korkin, D. Structural Genomics of SARS-CoV-2 Indicates Evolutionary Conserved Functional Regions of Viral Proteins. Viruses 2020, 12, 360. [Google Scholar]
- Vandelli, A.; Monti, M.; Milanetti, E.; Armaos, A.; Rupert, J.; Zacco, E.; Bechara, E.; Delli Ponti, R.; Tartaglia, G.G. Structural analysis of SARS-CoV-2 genome and predictions of the human interactome. Nucleic Acids Res. 2020, 48, 11270–11283. [Google Scholar] [CrossRef]
- European Center for Disease Prevention and Control. Risk Related to Spread of New SARS-CoV-2 Variants of Concern in the EU/EEA. Available online: https://www.ecdc.europa.eu/sites/default/files/documents/COVID-19-risk-related-to-spread-of-new-SARS-CoV-2-variants-EU-EEA.pdf (accessed on 2 January 2021).
- Gisaid.org. Available online: https://www.gisaid.org/ (accessed on 2 January 2021).
- GOV.UK—Public Health England (PHE). Investigation of Novel SARS-COV-2 Variant: Variant of Concern 202012/01. Available online: https://www.gov.uk/government/publications/investigation-of-novel-sars-cov-2-variant-variant-of-concern-20201201 (accessed on 2 January 2021).
- Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 2 January 2021).
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310. [Google Scholar] [CrossRef] [PubMed]


| References | Biological Systems | Interactors Identification Methods | PPI Analysis | SARS-CoV-2-Human Interaction Network and Enrichment Analysis (Main Pathways) | Data Availability and Web Resources |
|---|---|---|---|---|---|
| Gordon et al. [17] | Transient transfection in HEK-293 cells for PPI studies; Vero E6 cells for drug repurposing. | AP-MS: either N- or C- terminus 2xStrep tagging followed by AP-MS. | SAINTexpress (1); MiST (2); Cytoscape; GO (3) enrichment analysis. | DNA replication, epigenetic and gene-expression regulators, vesicle trafficking, lipid modification, RNA processing and regulation, ubiquitin ligases, signaling, nuclear transport machinery, cytoskeleton, mitochondria and the extracellular matrix. | MS raw data deposited to the PX (4) Consortium (www.ebi.ac.uk/pride/archive/projects/PXD018117). PPI networks uploaded to NDEx (5) (https://public.ndexbio.org/#/network/43803262-6d69-11ea-bfdc-0ac135e8bacf). |
| Gordon et al. [18] | Transient transfection in HEK-293 cells for PPI studies; HeLa cells for IF5 experiments; A549-ACE2 and Caco2 cells for validation on viral life cycle; Vero E6 and A549-ACE2 cells for drug repurposing. | AP-MS: either N- or C- terminus 2xStrep tagging followed by AP-MS. | SAINTexpress; MiST; Cytoscape; GO (3) enrichment analysis. | Regulation of RNA metabolism and ribosome biogenesis, endosomal and Golgi vesicle transport, proteasomal catabolism, cellular response to heat and regulation of intracellular protein transport. | MS-proteomics data deposited to the PX (6) Consortium (https://www.ebi.ac.uk/pride/archive/projects/PXD021588). PPI networks can be found either in NDEx (5) and at https://kroganlab.ucsf.edu/network-maps. |
| Davies et al. [19] | Transient transfection in HEK-293 cells. | AP-MS: either N- (nsp2) or C- (nsp4) terminus FLAG tagging followed by AP-MS. | R statistics software. Cytoscape; GO (3) enrichment analysis. | Nsp2 interactors are involved in a number of host cell processes, including metabolic processing and transport. Nsp4 interactors showed multiple enriched biological processes, such as cell organization and biogenesis, transport, and metabolic processes. | N/A |
| Li et al. [20] | Transient transfection in HEK-293 cells. PBMC for proteomic perturbation in COVID-19 patients primary cells. | AP-MS: N- terminus 3xFlag-tagging followed by AP-MS. | MiST; Cytoscape; GO (3) enrichment analysis. | Inflammation and immune responses, ATP biosynthesis and metabolic processes, nucleotide-excision repair, protein methylation and alkylation, translation initiation, reactive oxygen species metabolic process, ER stress, and mRNA transport. | Datasets deposited to the PX (4) Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository (dataset identifier IPX0002285000). |
| Stukalov et al. [21] | Lentiviral mediated transduction of A549 cells for PPI identification. A549-ACE2 for both OMICS perturbation and drug repurposing. | AP-MS: C- terminus HA tagging followed by AP-MS. | R statistics software; GO (3) enrichment analysis. | Stress and DNA damage response, regulation of transcription, cell junction organization, cell survival, motility and innate immune responses. | N/A |
| Samavarchi-Tehrani et al. [22] | A549 cells transduced by lentiviral constructs (except for nsp1 and nsp3, whose constructs where transfected). | BioID: miniTurbo enzyme fused separately at both N- and C- terminus of each bait, modified proteins purification followed by MS. | SAINTexpress; Cytoscape; GO (3) enrichment analysis; Humancellmap.org. | Regulation of cell cycle processes, antigen processing, viral genome replication, transcription, regulation of innate immunity, DNA damage checkpoint, histone binding, proteasomal degradation. | Virus–host proximity interactome dataset is available at https://covid19interactome.org/ |
| St-Germain et al. [23] | Stably transfected HEK-293 cells | BioID: BirA* enzyme fused at the N- terminus of 14 viral baits, modified proteins purification followed by MS. | SAINTexpress; Cytoscape; GO (3) enrichment analysis. | Vesicle-mediated transport, Golgi vesicle transport, ER to Golgi vesicle-mediated transport, response to ER stress, retrograde transport endosome to Golgi, lipid biosynthetic process, ER organization, retrograde vesicle-mediated transport, COPII-coated vesicle budding. | All virus MS data available at https://massive.ucsd.edu |
| Laurent et al. [24] | Stably transfected HEK-293 cells | BioID: BirA* enzyme fused separately at both N- and C- terminus of each bait, modified proteins purification followed by MS. | ToppCluster; Metascape; GO (3) enrichment analysis. | Innate immune response, autophagy, apoptosis, lipid metabolism, vesicular transport, chromatin remodeling, mRNA processing, inflammation, viral signal transduction, nucleic acid processing, cell adhesion and migration, platelet activation, coagulation regulation, olfactory receptors homeostasis and olfactory cell signal transmission. | Data exploitation available at http://www.sars-cov-2-interactome.org/ |
| Viral Bait | AP-MS Article | Cellular Preys Identified by AP-MS in at Least Two Different Reports | Preys Found Also by Proximity Labeling | AP-MS/BioID | ||
|---|---|---|---|---|---|---|
| Laurent et al. [24] | Samavarchi-Tehrani et al. [22] | St-Germain et al. [23] | ||||
| E | Gordon et al. [17] | No common interactors | N/A | N/A | N/A | N/A |
| Stukalov et al. [21] | ||||||
| M | Gordon et al. [17] | ATP1B1, COQ8B, INTS4, PITRM1, PMPCB, REEP5, RTN4 | REEP5, RTN4 | ATG9A, ATP1B1, REEP5, RTN4 | PMPCB | ATG9AATP1B1, PMPCB, REEP5, RTN4 |
| Stukalov et al. [21] | ARFGEF2, ATG9A, COQ8B, INTS4, PITRM1, PMPCB, RTN4, | |||||
| Li et al. [20] | ARFGEF2, ATG9A, ATP1B1, REEP5 | |||||
| N | Gordon et al. [17] | G3BP1, G3BP2 | CAVIN1, G3BP1, G3BP2, | CAVIN1, G3BP1, G3BP2, | Viral bait not tested | CAVIN1 G3BP1, G3BP2, |
| Stukalov et al. [21] | CAVIN1, G3BP1, G3BP2 | |||||
| Li et al. [20] | CAVIN1, G3BP1, G3BP2 | |||||
| S | Gordon et al. [17] | GOLGA7, ZDHHC5 | ZDHHC5 | ZDHHC5 | No common interactors | ZDHHC5 |
| Stukalov et al. [21] | GOLGA7, ZDHHC5 | |||||
| Li et al. [20] | No common interactors | |||||
| ORF3a | Gordon et al. [17] | ALG5, ARL6IP6, CLCC1, HMOX1, TRIM59, VPS11; VPS-39 | CLCC1, CPD, HMOX2, VPS39 | CLCC1, CPD, RAB13, RAB14, TBL2, VPS39 | CLCC1, VPS-39, | CLCC1 CPD, HMOX2, RAB13, RAB14, TBL2VPS39 |
| Stukalov et al. [21] | CLCC1, CPD, HMOX2, PROCR, RAB13, RAB14, SUMF2, TBL2, VPS11; VPS-39 | |||||
| Li et al. [20] | ALG5, ARL6IP6, CLCC1, CPD, HMOX1, HMOX2, PROCR, RAB13, RAB14, SUMF2, TBL2, TRIM59, VPS-39 | |||||
| ORF6 | Gordon et al. [17] | RAE1 | RAE1 | RAE1 | RAE1 | RAE1 |
| Stukalov et al. [21] | No interactors identified for this bait | |||||
| Li et al. [20] | RAE1 | |||||
| ORF7a | Gordon et al. [17] | MDN1 | No common interactors | No common interactors | No common interactors | N/A |
| Stukalov et al. [21] | ATR, MDN1 | |||||
| Li et al. [20] | ATR | |||||
| ORF8 | Gordon et al. [17] | GGH, NPTX1, UGGT2 | CNNM3 | CNNM3, GGH, NPTX1, UGGT2, | CNNM3 | GGH, NPTX1, UGGT2, CNNM3 |
| Stukalov et al. [21] | CNNM3, GGH, NPTX1, UGGT2 | |||||
| Li et al. [20] | CNNM3 | |||||
| ORF9b | Gordon et al. [17] | TOMM70 | TOMM70 | TOMM70 | TOMM70 | TOMM70 |
| Stukalov et al. [21] | TOMM70 | |||||
| Nsp1 | Gordon et al. [17] | No common interactors | N/A | N/A | Viral bait not tested | N/A |
| Stukalov et al. [21] | ||||||
| Li et al. [20] | ||||||
| Nsp2 | Gordon et al. [17] | GIGYF2, RAP1GDS1 | RAP1GDS1 | RAP1GDS1, | No common interactors | RAP1GDS1 |
| Stukalov et al. [21] | RAP1GDS1 | |||||
| Li et al. [20] | FOXK1, GIGYF2, RAP1GDS1 | |||||
| Davies et al. [19] | FOXK1 | |||||
| Nsp3 | Stukalov et al. [21] | No common interactors | N/A | N/A | N/A | N/A |
| Li et al. [20] | ||||||
| Nsp4 | Gordon et al. [17] | No common interactors | HSPA5 | No common interactors | HSPA5 | HSPA5 |
| Stukalov et al. [21] | HSPA5 | |||||
| Li et al. [20] | No common interactors | |||||
| Davies et al. [19] | HSPA5 | |||||
| Nsp5 | Gordon et al. [17] | No common interactors | N/A | N/A | Viral bait not tested | N/A |
| Li et al. [20] | ||||||
| Nsp6 | Gordon et al. [17] | ATP6AP1, ATP13A3, SIGMAR1 | ATP6AP1, SIGMAR1, | ATP6AP1 | ATP6AP1 | ATP6AP1, SIGMAR1 |
| Stukalov et al. [21] | ATP6AP1, ATP13A3, SIGMAR1 | |||||
| Nsp7 | Gordon et al. [17] | No common interactors | N/A | N/A | Viral bait not tested | N/A |
| Stukalov et al. [21] | ||||||
| Nsp8 | Gordon et al. [17] | ATE1, HECTD1 | HECTD1, HERC1 | HECTD1, HERC1 | Viral bait not tested | HECTD1 HERC1 |
| Stukalov et al. [21] | ATE1, HERC1 | |||||
| Li et al. [20] | HECTD1, HERC1 | |||||
| Nsp9 | Gordon et al. [17] | EIF4H, GTF2F2, SPART | GTF2F2 | No common interactors | Viral bait not tested | GTF2F2 |
| Stukalov et al. [21] | GTF2F2 | |||||
| Li et al. [20] | EIF4H, GTF2F2, SPART | |||||
| Nsp10 | Gordon et al. [17] | No common interactors | N/A | N/A | Viral bait not tested | N/A |
| Stukalov et al. [21] | ||||||
| Li et al. [20] | ||||||
| Nsp12 | Gordon et al. [17] | No common interactors | N/A | N/A | Viral bait not tested | N/A |
| Stukalov et al. [21] | ||||||
| Nsp13 | Gordon et al. [17] | No common interactors | N/A | N/A | Viral bait not tested | N/A |
| Stukalov et al. [21] | ||||||
| Li et al. [20] | ||||||
| Nsp14 | Gordon et al. [17] | SIRT5 | SIRT5 | No common interactors | Viral bait not tested | SIRT5 |
| Stukalov et al. [21] | No common interactors | |||||
| Li et al. [20] | SIRT5 | |||||
| Nsp15 | Gordon et al. [17] | No common interactors | N/A | N/A | Viral bait not tested | N/A |
| Stukalov et al. [21] | ||||||
| Li et al. [20] | ||||||
| Nsp16 | Gordon et al. [17] | Viral bait not tested | CCDC22 | No common interactors | Viral bait not tested | CCDC22 |
| Stukalov et al. [21] | No common interactors | |||||
| Li et al. [20] | CCDC22 | |||||
| Gordon et al. [18] | CCDC22 | |||||
| Viral Proteins | Gordon et al. [17,18] | Stukalov et al. [21] | Li et al. [20] | Davies et al. [19] | Laurent et al. [24] | Samavarchi-Tehrani et al. [22] | St-Germain et al. [23] | |
|---|---|---|---|---|---|---|---|---|
| 1 | M | ✓ | ✓ | ✓ | - | ✓ | ✓ | ✓ |
| 2 | N | ✓ | ✓ | ✓ | - | ✓ | ✓ | - |
| 3 | E | ✓ | ✓ | - | - | ✓ | ✓ | ✓ |
| 4 | S | ✓ | ✓ | ✓ | - | ✓ | ✓ | ✓ |
| 5 | ORF3a | ✓ | ✓ (referred as ORF3) | ✓ | - | ✓ | ✓ | ✓ |
| 6 | ORF3b | ✓ | - | - | - | ✓ | ✓ | ✓ |
| 7 | ORF6 | ✓ | ✓ | ✓ | - | ✓ | ✓ | ✓ |
| 8 | ORF7a | ✓ | ✓ | ✓ | - | ✓ | ✓ | ✓ |
| 9 | ORF7b | - | ✓ | - | - | ✓ | ✓ | ✓ |
| 10 | ORF8 | ✓ | ✓ | ✓ | - | ✓ | ✓ | ✓ |
| 11 | ORF9b | ✓ | ✓ | - | - | ✓ | ✓ | ✓ |
| 12 | ORF10 | ✓ | - | - | - | ✓ | - | - |
| 13 | ORF14 | ✓(referred as ORF9c) | - | - | - | ✓ | ✓ | - |
| 14 | Nsp1 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - |
| 15 | Nsp2 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| 16 | Nsp3 | - | ✓ | ✓ | - | ✓ | ✓ | ✓ |
| 17 | Nsp4 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| 18 | Nsp5 | ✓ | - | ✓ | - | ✓ | ✓ | - |
| 19 | Nsp6 | ✓ | ✓ | - | - | ✓ | ✓ | ✓ |
| 20 | Nsp7 | ✓ | ✓ | - | - | ✓ | ✓ | - |
| 21 | Nsp8 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - |
| 22 | Nsp9 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - |
| 23 | Nsp10 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - |
| 24 | Nsp11 | ✓ | - | - | - | - | - | - |
| 25 | Nsp12 | ✓ | ✓ | - | - | ✓ | ✓ | - |
| 26 | Nsp13 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - |
| 27 | Nsp14 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - |
| 28 | Nsp15 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - |
| 29 | Nsp16 | ✓ | ✓ | ✓ | - | ✓ | ✓ | - |
| TOT | 27 | 24 | 19 | 2 | 28 | 27 | 14 | |
| Drug | Chemical Structure | Mechanism of Action | SARS-CoV-2 PPI | Studies Which Detected Virus–Host PPI | In Vitro SARS-CoV-2 Antiviral Effects | In Vivo SARS-CoV-2 Activity |
|---|---|---|---|---|---|---|
| Chlorpromazine FDA approved |  | Ligand of Sigma-1 receptor | Nsp6-Sigma-1 (1) | Gordon et al. [17] Stukalov et al. [21] Laurent et al. [24] | A549-ACE2 pIC50 = 6.05 | Reduced requirement to mechanical ventilation (2) |
| Fluphenazine FDA approved |  | Ligand of Sigma-1 receptor | Nsp6-Sigma-1 (1) | Gordon et al. [17] Stukalov et al. [21] Laurent et al. [24] | A549-ACE2 pIC50 = 6.46 | Reduced requirement to mechanical ventilation (2) |
| Haloperidol FDA approved |  | Ligand of Sigma-1 receptor | Nsp6-Sigma-1 (1) | Gordon et al. [17] Stukalov et al. [21] Laurent et al. [24] | A549-ACE2 pIC50 = 5.68 | Reduced requirement to mechanical ventilation (2) |
| Indomethacin FDA approved |  | PGES-2 inhibitor | Nsp7-PGES-2 (3) | Gordon et al. [17] Gordon et al. [18] | A549-ACE2 pIC50 = 4.25 | Reduced hospitalization (4) |
| Ipatasertib compound in clinical trial |  | AKT inhibitor a potential kinase phosphorylating SARS-CoV-2 protein N | N-M (5) and M-AKT (6) kinase | (5) Li et al. [20] (6) Laurent et al. [24] | A549-ACE2 5 μM | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terracciano, R.; Preianò, M.; Fregola, A.; Pelaia, C.; Montalcini, T.; Savino, R. Mapping the SARS-CoV-2–Host Protein–Protein Interactome by Affinity Purification Mass Spectrometry and Proximity-Dependent Biotin Labeling: A Rational and Straightforward Route to Discover Host-Directed Anti-SARS-CoV-2 Therapeutics. Int. J. Mol. Sci. 2021, 22, 532. https://doi.org/10.3390/ijms22020532
Terracciano R, Preianò M, Fregola A, Pelaia C, Montalcini T, Savino R. Mapping the SARS-CoV-2–Host Protein–Protein Interactome by Affinity Purification Mass Spectrometry and Proximity-Dependent Biotin Labeling: A Rational and Straightforward Route to Discover Host-Directed Anti-SARS-CoV-2 Therapeutics. International Journal of Molecular Sciences. 2021; 22(2):532. https://doi.org/10.3390/ijms22020532
Chicago/Turabian StyleTerracciano, Rosa, Mariaimmacolata Preianò, Annalisa Fregola, Corrado Pelaia, Tiziana Montalcini, and Rocco Savino. 2021. "Mapping the SARS-CoV-2–Host Protein–Protein Interactome by Affinity Purification Mass Spectrometry and Proximity-Dependent Biotin Labeling: A Rational and Straightforward Route to Discover Host-Directed Anti-SARS-CoV-2 Therapeutics" International Journal of Molecular Sciences 22, no. 2: 532. https://doi.org/10.3390/ijms22020532
APA StyleTerracciano, R., Preianò, M., Fregola, A., Pelaia, C., Montalcini, T., & Savino, R. (2021). Mapping the SARS-CoV-2–Host Protein–Protein Interactome by Affinity Purification Mass Spectrometry and Proximity-Dependent Biotin Labeling: A Rational and Straightforward Route to Discover Host-Directed Anti-SARS-CoV-2 Therapeutics. International Journal of Molecular Sciences, 22(2), 532. https://doi.org/10.3390/ijms22020532