Can SARS-CoV-2 Virus Use Multiple Receptors to Enter Host Cells?


 ,
, 
Abstract
1. Introduction
2. Non-Immune Receptors Involved in Coronavirus Disease 2019
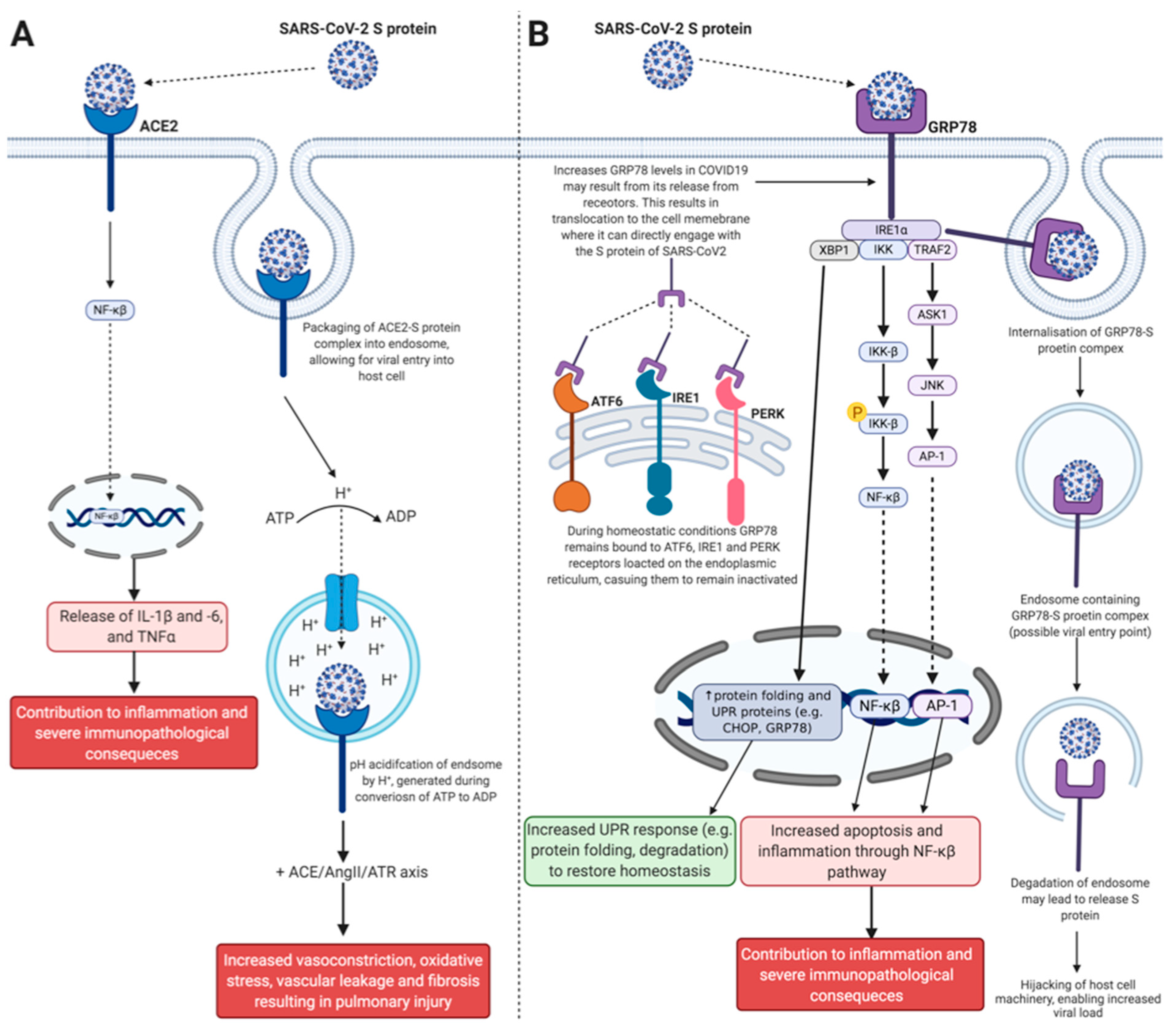
2.1. Angiotensin-Converting Enzyme 2
2.2. Glucose-Regulated Protein 78
2.3. Ezrin
3. Toll-Like Receptors Participating in Coronavirus Disease 2019 Pathogenesis and Progression
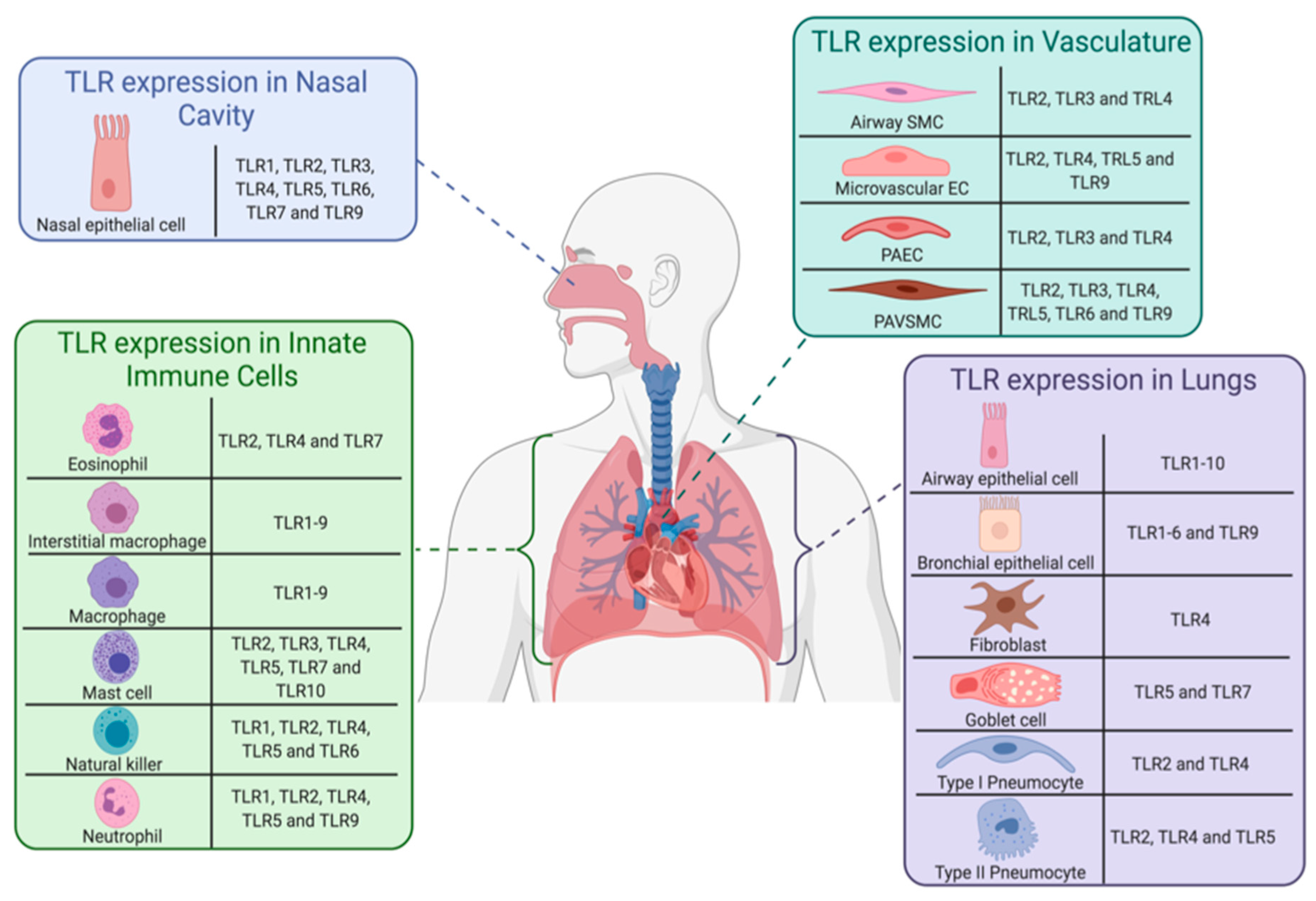
3.1. Introduction to Toll-Like Receptors
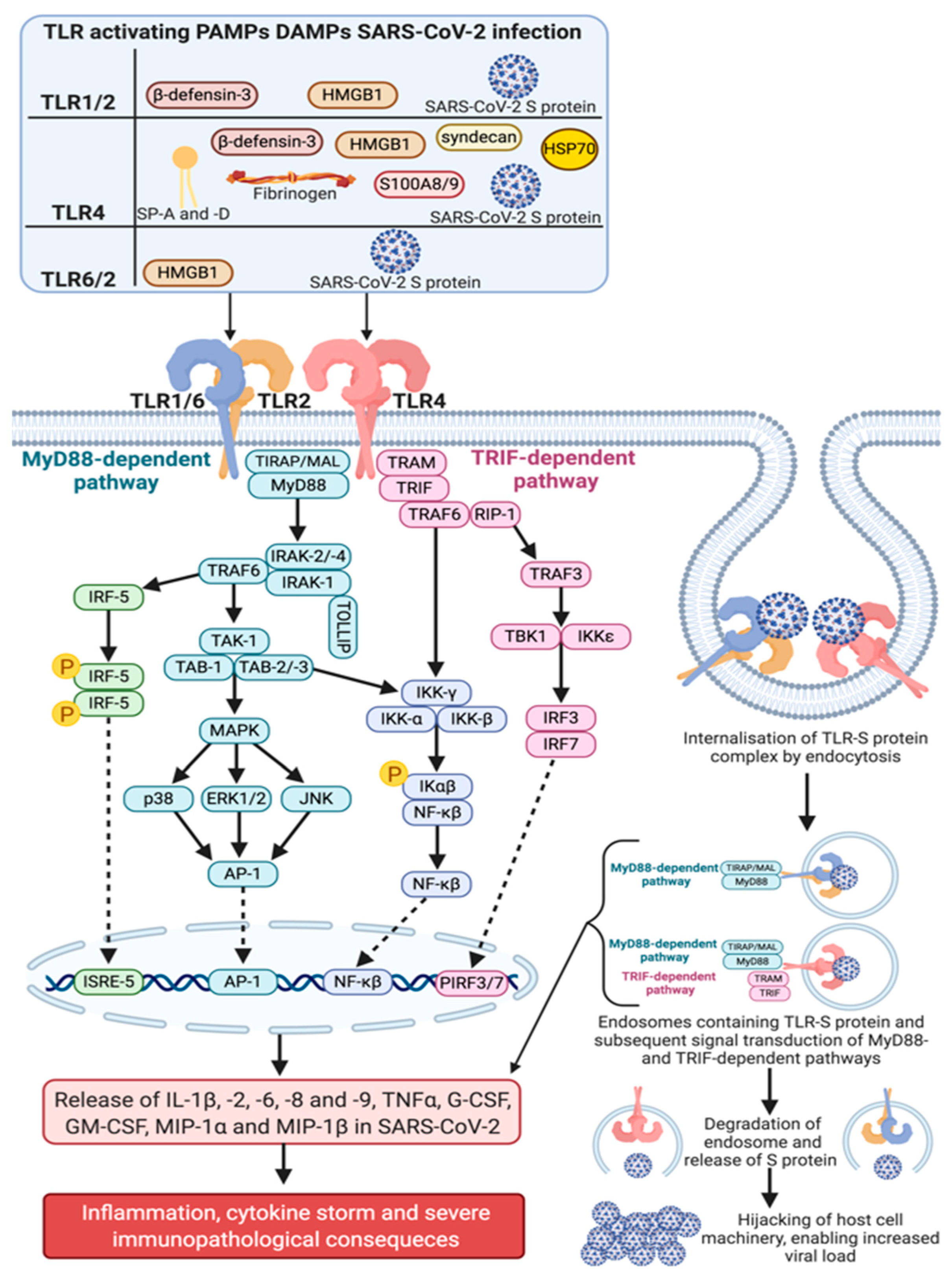
3.2. TLR1/2/6 as Potential Therapeutic Targets and Alternative Viral Entry Points for SARS-CoV-2
3.3. TLR3 as a Potential Therapeutic Target in SARS-CoV-2 Infection
3.4. TLR4 as a Potential Therapeutic Target and Alternative Viral Entry Point in SARS-CoV-2 Infection
3.5. TLR 5 as a Potential Vaccine Target in Coronavirus 2019
3.6. TLR7/8 as Potential Therapeutic Targets for SARS-CoV-2 Infection
4. C-Lectin Type Receptors Involved in COVID-19
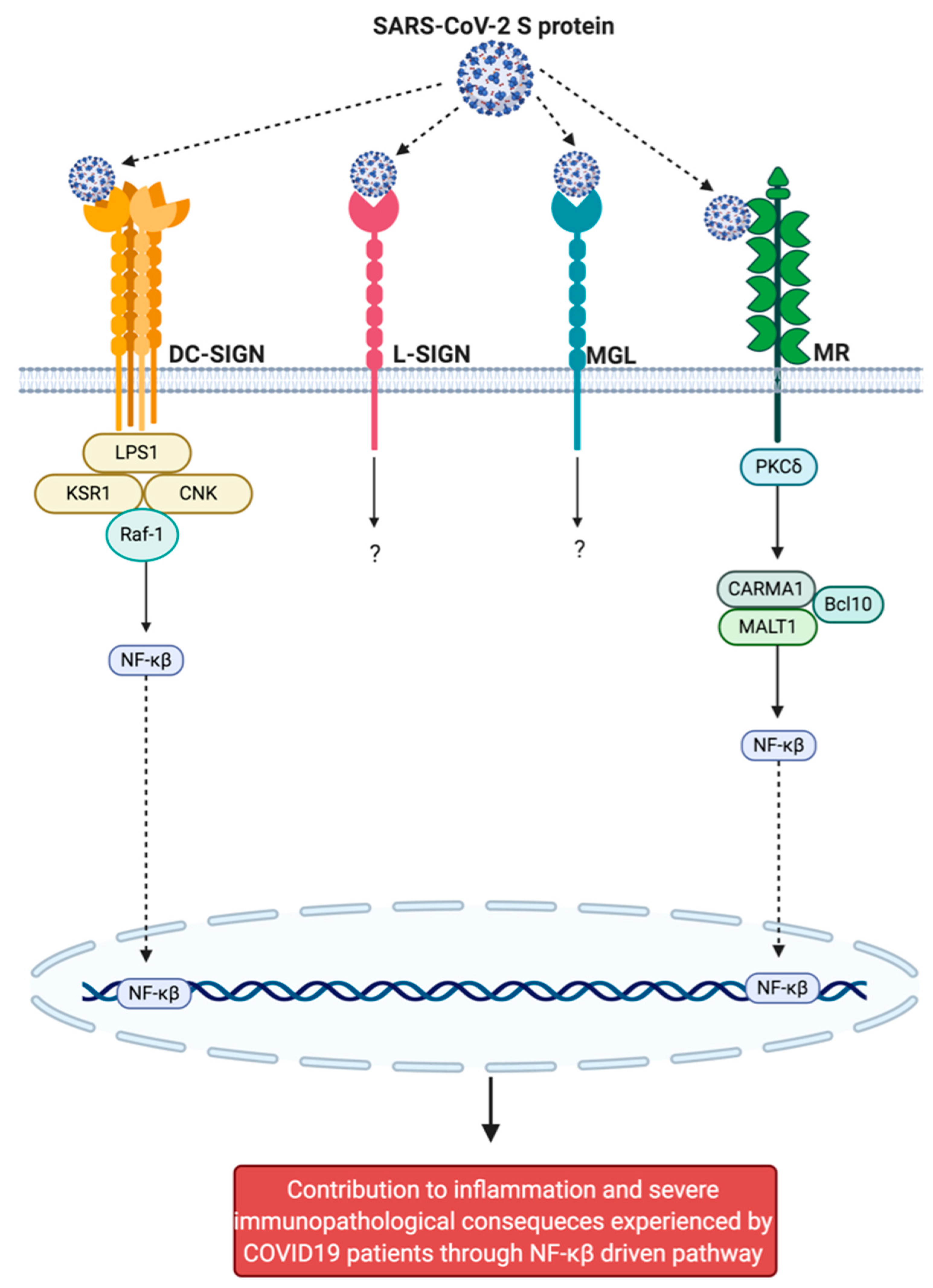
4.1. Introduction to the C-Lectin Type Receptors
4.2. Blood Dendritic Cell Antigen-2
4.3. C-Type Lectin-Like Receptor 2
4.4. Dendritic Cell-Associated C-Type Lectin-1
4.5. Dendritic Cell-Associated C-Type Lectin-2
4.6. Dendritic Cell Immunoreceptor
4.7. Dendritic Cell Natural Killer Lectin Group Receptor-1
4.8. Dendritic Cell-Specific Intracellular Adhesion Molecule-3-Grabbing Non-Integrins and Homologue Dendritic Cell-Specific Intercellular Adhesion Molecule-3-Grabbing Nonintegrin Related
4.9. Lectin-Like Oxidized Low-Density Lipoprotein Receptor-1
4.10. Liver and Lymph Node Sinusoidal Endothelial Cell C-Type Lectin
4.11. Macrophage Galactose Type C-Type Lectin
4.12. Mannose Receptor
5. Other Immune Receptors That May Participate in SARS-CoV-2 Infection
5.1. Dipeptidyl Peptidase-4
5.2. Neuropilin-1
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACE2 | Angiotensin-converting enzyme 2 |
| Angiotensin II | AngII |
| AngII receptor blocker | ARB |
| aPL | Antiphosphilipid antibody |
| BDCA-2 | Blood dendritic cell antigen-2 |
| CRD | Carbohydrate recognition domain |
| CLR | C-lectin type receptors |
| COVID-19 | Coronavirus disease 2019 |
| CLEC2 | C-type lectin-like receptor 2 |
| CVD | Cardiovascular disease |
| DAMP | Danger associated molecular patterns |
| DC | Dendritic cell |
| DCIR | Dendritic cell immunoreceptor |
| DC-SIGN | Dendritic cell-specific intracellular adhesion molecule-3-grabbing non-integrin |
| Dectin-1 | Dendritic cell-associated C-type lectin-1 |
| Dectin-2 | Dendritic cell-associated C-type lectin-2 |
| DPP4 | Dipeptidyl peptidase-4 |
| EPOV GP | Ebola virus G protein |
| GRP78 | Glucose regulated protein 78 |
| HIV-1 | Human immunodeficiency virus-1 |
| LSECtin | Liver/lymph node sinusoidal endothelial cell C-type lectin |
| L-SIGN | Liver/lymph node-SIGN |
| LOX-1 | Lectin-like oxidized low-density lipoprotein receptor-1 |
| MGL | Macrophage galactose-type lectin |
| DIZE | Diminazene aceturate |
| MR | Mannose receptor |
| Myd88 | Myeloid differentiation protein 88 |
| NF-kB | Nuclear factor kappa light chain enhancer of activated B cells |
| PAMP | Pathogen-associated molecular pattern |
| Poly(I:C) | Polyinosoinic-polycytidylic acid |
| PRR | Pattern recognition receptors |
| S | Spike protein |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus-2 |
| Subunit 1 | S1 |
| Subunit 2 | S2 |
| TLR | Toll-like receptor |
References
- Casalino, L.; Gaieb, Z.; Goldsmith, J.; Hjorth, C.; Dommer, A.; Harbison, A.; Fogarty, C.; Barros, E.; Taylor, B.; Taylor, B. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, D.; Vanelli, M. WHO Declares COVID-19 a Pandemic. Acta BioMed. 2020, 91, 157–160. [Google Scholar] [PubMed]
- Yuan, B.; Liu, H.-Q.; Yang, Z.-R.; Chen, Y.-X.; Liu, Z.-Y.; Zhang, K.; Wang, C.; Li, W.-X.; An, Y.-W.; Wang, J.-C. Recurrence of Positive SARS-CoV-2 Viral RNA in Recovered COVID-19 Patients During Medical Isolation Observation. Nat. Res. 2020. Under Review. [Google Scholar]
- WHO. Coronavirus Disease (COVID-19): Weekly Epidemiological Update; WHO: Geneva, The Switzerland, 2020. [Google Scholar]
- McKee, M.; Stuckler, D. If the world fails to protect the economy, COVID-19 will damage health not just now but also in the future. Nat. Med. 2020, 26, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.-U.; Kim, M.-J.; Ra, S.H.; Lee, J.; Bae, S.; Jung, J.; Kim, S.-H. Clinical characteristics of asymptomatic and symptomatic patients with mild COVID-19. Clin. Microbiol. Infect. 2020, 26, 948.e1–948.e3. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Chrysant, S.G. Effectiveness of the fixed-dose combination of olmesartan/amlodipine/hydrochlorothiazide for the treatment of hypertension in patients stratified by age, race and diabetes, CKD and chronic CVD. Expert Rev. Cardiovasc. Ther. 2013, 11, 1115–1124. [Google Scholar] [CrossRef]
- Rein, J.; Bader, M. Renin-Angiotensin System in Diabetes. Protein Pept. Lett. 2017, 24, 833–840. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, R.; Wang, G.; Wang, A.; Zhong, C.; Zhang, M.; Li, H.; Xu, T.; Zhang, Y. Coexistence effect of hypertension and angiotensin II on the risk of coronary heart disease: A population-based prospective cohort study among Inner Mongolians in China. Curr. Med. Res. Opin. 2019, 35, 1473–1478. [Google Scholar] [CrossRef] [PubMed]
- Barnett, A.H. The role of angiotensin II receptor antagonists in the management of diabetes. Blood Press. Suppl. 2001, 1, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Wu, C.; Ye, D.; Mullick, A.; Li, Z.; Danser, J.; Daugherty, A.; Lu, H. Effects of renin-angiotensin inhibition on ACE2 (angiotensin-converting enzyme 2) and TMPRSS2 (transmembrane protease serine 2) expression: Insights into COVID-19. Hypertension 2020, 76, e29–e30. [Google Scholar] [CrossRef] [PubMed]
- Lopes, R.D.; Macedo, A.V.S.; de Barros E Silva, P.G.M.; Moll-Bernardes, R.J.; Feldman, A.; D’Andréa Saba Arruda, G.; de Souza, A.S.; de Albuquerque, D.C.; Mazza, L.; Santos, M.F.; et al. Continuing versus suspending angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: Impact on adverse outcomes in hospitalized patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)—The BRACE CORONA Trial. Am. Heart J. 2020, 226, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Diaz, J.H. Hypothesis: Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers may increase the risk of severe COVID-19. J. Travel Med. 2020, 27. [Google Scholar] [CrossRef]
- Sommerstein, R.; Kochen, M.M.; Messerli, F.H.; Gräni, C. Coronavirus Disease 2019 (COVID-19): Do Angiotensin-Converting Enzyme Inhibitors/Angiotensin Receptor Blockers Have a Biphasic Effect? J. Am. Heart Assoc. 2020, 9, e016509. [Google Scholar] [CrossRef]
- Lopes, R. BRACE CORONA: Continuing vs. Suspending ACE Inhibitors and ARBs in COVID-19. In Proceedings of the European Society of Cardiology Virtual Congress, Digital Congress, 29 August–1 September 2020; Available online: https://www.pcronline.com/News/Whats-new-on-PCRonline/2020/ESC-2020-BRACE-CORONA-Continuing-vs.-suspending-ACE-inhibitors-and-ARBs-COVID-19 (accessed on 19 January 2021).
- Zaman, S.; MacIsaac, A.I.; Jennings, G.L.; Schlaich, M.P.; Inglis, S.C.; Arnold, R.; Kumar, S.; Thomas, L.; Wahi, S.; Lo, S.; et al. Cardiovascular disease and COVID-19: Australian and New Zealand consensus statement. Med. J. Aust. 2020, 213, 182–187. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Shajahan, A.; Supekar, N.T.; Gleinich, A.S.; Azadi, P. Deducing the N-and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. BioRxiv 2020. [Google Scholar]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.D.; Williamson, M.K.; Lewis, S.; Shoemark, D.; Carroll, M.W.; Heesom, K.J.; Zambon, M.; Ellis, J.; Lewis, P.A.; Hiscox, J.A. Characterisation of the transcriptome and proteome of SARS-CoV-2 reveals a cell passage induced in-frame deletion of the furin-like cleavage site from the spike glycoprotein. Genome Med. 2020, 12, 1–15. [Google Scholar] [CrossRef]
- Watanabe, Y.; Bowden, T.A.; Wilson, I.A.; Crispin, M. Exploitation of glycosylation in enveloped virus pathobiology. Biochim. Biophys. Acta (BBA) Gen. Subj. 2019, 1863, 1480–1497. [Google Scholar] [CrossRef]
- Vigerust, D.J.; Shepherd, V.L. Virus glycosylation: Role in virulence and immune interactions. Trends Microbiol. 2007, 15, 211–218. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific analysis of the SARS-CoV-2 glycan shield. BioRxiv 2020. [Google Scholar] [CrossRef]
- Bunyavanich, S.; Do, A.; Vicencio, A. Nasal gene expression of angiotensin-converting enzyme 2 in children and adults. JAMA 2020, 323, 2427–2429. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-cell RNA expression profiling of ACE2, thereceptor of SARS-CoV-2. BioRxiv 2020. [Google Scholar] [CrossRef]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Amraei, R.; Napoleon, M.; Yin, W.; Berrigan, J.; Suder, E.; Zhao, G.; Olejnik, J.; Gummuluru, S.; Muhlberger, E.; Chitalia, V. CD209L/L-SIGN and CD209/DC-SIGN act as receptors for SARS-CoV-2 and are differentially expressed in lung and kidney epithelial and endothelial cells. BioRxiv 2020. [Google Scholar] [CrossRef]
- Gao, C.; Zeng, J.; Jia, N.; Stavenhagen, K.; Matsumoto, Y.; Zhang, H.; Li, J.; Hume, A.J.; Mühlberger, E.; van Die, I. SARS-CoV-2 Spike Protein Interacts with Multiple Innate Immune Receptors. BioRxiv 2020. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elshahat, M.E.; Elfiky, A.A. COVID-19 spike-host cell receptor GRP78 binding site prediction. J. Infect. 2020, 80, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Bessler, W.; Holms, R.; Konopleva, M.; Ataullakhanov, R. Review of Russian ezrin peptide treatment of acute viral respiratory disease and virus induced pneumonia; A potential treatment for covid-19. Ezrin Peptide Ther. 2020. [Google Scholar] [CrossRef]
- Solerte, S.B.; Di Sabatino, A.; Galli, M.; Fiorina, P. Dipeptidyl peptidase-4 (DPP4) inhibition in COVID-19. Acta Diabetol. 2020, 57, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Nemoto, E.; Shimauchi, H.; Watanabe, T.; Mikami, T.; Matsumoto, T.; Ohno, N.; Tamura, H.; Shibata, K.I.; Akashi, S. Saccharomyces cerevisiae-and Candida albicans-derived mannan induced production of tumor necrosis factor alpha by human monocytes in a CD14-and Toll-like receptor 4-dependent manner. Microbiol. Immunol. 2002, 46, 503–512. [Google Scholar] [CrossRef]
- Sheng, K.C.; Pouniotis, D.S.; Wright, M.D.; Tang, C.K.; Lazoura, E.; Pietersz, G.A.; Apostolopoulos, V. Mannan derivatives induce phenotypic and functional maturation of mouse dendritic cells. Immunology 2006, 118, 372–383. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Penninger, J.M. The renin–angiotensin system in acute respiratory distress syndrome. Drug Discov. Today: Dis. Mech. 2006, 3, 225–229. [Google Scholar] [CrossRef]
- Lautner, R.Q.; Villela, D.C.; Fraga-Silva, R.A.; Silva, N.; Verano-Braga, T.; Costa-Fraga, F.; Jankowski, J.; Jankowski, V.; Sousa, F.; Alzamora, A. Discovery and characterization of alamandine: A novel component of the renin-angiotensin system. Circ. Res. 2013, 112, 1104–1111. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor recognition by the novel coronavirus from Wuhan: An analysis based on decade-long structural studies of SARS coronavirus. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Neufeldt, C.J.; Cerikan, B.; Cortese, M.; Frankish, J.; Lee, J.-Y.; Plociennikowska, A.; Heigwer, F.; Joecks, S.; Burkart, S.S.; Zander, D.Y. SARS-CoV-2 infection induces a pro-inflammatory cytokine response through cGAS-STING and NF-κB. BioRxiv 2020. [Google Scholar] [CrossRef]
- Codo, A.C.; DaVanzo, G.G.; Monteiro, L.B.; Souza, G.; Muraro, S.; Carregari, V.; Biagi, C.; Crunfli, F.; Restrepo, J.; Vendramini, P.; et al. Elevated Glucose Levels Favor Sars-Cov-2 Infection and Monocyte Response Through a Hif-1α/Glycolysis Dependent Axis. Cell Metab. 2020, 32, 437–446.e5. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Sousa, E.; Köseler, A.; Sabirli, R.; Gören, T.; Türkçüer, İ.; Kurt, Ö.; Pinto, M.M.; Vasconcelos, M.H. Preliminary virtual screening studies to identify GRP78 inhibitors which may interfere with SARS-CoV-2 infection. Pharmaceuticals 2020, 13, 132. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, J.; Tremblay, B.; Mansfield, M.; Woody, O.; Lobb, B.; Banerjee, A.; Chandiramohan, A.; Tiessen, N.; Dvorkin-Gheva, A.; Revill, S. Gene expression and in situ protein profiling of candidate SARS-CoV-2 receptors in human airway epithelial cells and lung tissue. BioRxiv 2020. [Google Scholar] [CrossRef]
- Triantafilou, K.; Fradelizi, D.; Wilson, K.; Triantafilou, M. GRP78, a Coreceptor for Coxsackievirus A9, Interacts with Major Histocompatibility Complex Class I Molecules Which Mediate Virus Internalization. J. Virol. 2002, 76, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the Unfolded Protein Response Regulator GRP78/BiP in Development, Cancer, and Neurological Disorders. Antioxid. Redox Signal. 2009, 11, 2307–2316. [Google Scholar] [CrossRef]
- Shimasaki, S.; Koga, T.; Shuto, T.; Suico, M.A.; Sato, T.; Watanabe, K.; Morino-Koga, S.; Taura, M.; Okada, S.; Mori, K.; et al. Endoplasmic reticulum stress increases the expression and function of toll-like receptor-2 in epithelial cells. Biochem. Biophys. Res. Commun. 2010, 402, 235–240. [Google Scholar] [CrossRef]
- Wei, D.; Li, N.L.; Zeng, Y.; Liu, B.; Kumthip, K.; Wang, T.T.; Huo, D.; Ingels, J.F.; Lu, L.; Shang, J.; et al. The Molecular Chaperone GRP78 Contributes to Toll-like Receptor 3-mediated Innate Immune Response to Hepatitis C Virus in Hepatocytes. J. Biol. Chem. 2016, 291, 12294–12309. [Google Scholar] [CrossRef]
- Parameswaran, N.; Enyindah-Asonye, G.; Bagheri, N.; Shah, N.B.; Gupta, N. Spatial coupling of JNK activation to the B cell antigen receptor by tyrosine-phosphorylated ezrin. J. Immunol. 2013, 190, 2017–2026. [Google Scholar] [CrossRef] [PubMed]
- Pore, D.; Matsui, K.; Parameswaran, N.; Gupta, N. Cutting Edge: Ezrin Regulates Inflammation by Limiting B Cell IL-10 Production. J. Immunol. 2015, 196, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.H.; Lambelé, M.; Chan, J.; Symeonides, M.; Thali, M. Ezrin is a Component of the HIV-1 Virological Presynapse and Contributes to the Inhibition of Cell-Cell Fusion. J. Virol. 2014, 88, 7645–7658. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Kien, F.; Cheung, C.-Y.; Siu, Y.-L.; Chan, W.-L.; Li, H.; Leung, N.H.L.; Jaume, M.; Bruzzone, R.; Peiris, J.S.M.; et al. Ezrin Interacts with the SARS Coronavirus Spike Protein and Restrains Infection at the Entry Stage. PLoS ONE 2012, 7, e49566. [Google Scholar] [CrossRef] [PubMed]
- Zarember, K.A.; Godowski, P.J. Tissue Expression of Human Toll-Like Receptors and Differential Regulation of Toll-Like Receptor mRNAs in Leukocytes in Response to Microbes, Their Products, and Cytokines. J. Immunol. 2002, 168, 554–561. [Google Scholar] [CrossRef]
- Kimbrell, D.A.; Beutler, B. The evolution and genetics of innate immunity. Nat. Rev. Genet. 2001, 2, 256–267. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Godfroy, J.I.; Roostan, M.; Moroz, Y.S.; Korendovych, I.V.; Yin, H. Isolated Toll-like Receptor Transmembrane Domains Are Capable of Oligomerization. PLoS ONE 2012, 7, e48875. [Google Scholar] [CrossRef]
- Patel, S. Danger-Associated Molecular Patterns (DAMPs): The Derivatives and Triggers of Inflammation. Curr. Allergy Asthma Rep. 2018, 18, 63. [Google Scholar] [CrossRef]
- Komai, K.; Shichita, T.; Ito, M.; Kanamori, M.; Chikuma, S.; Yoshimura, A. Role of scavenger receptors as damage-associated molecular pattern receptors in Toll-like receptor activation. Int. Immunol. 2017, 29, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, A.; Herrup, E.A.; Warren, H.S.; Trigilio, J.; Shin, H.-S.; Valentine, C.; Hellman, J. MyD88-Dependent and MyD88-Independent Pathways in Synergy, Priming, and Tolerance between TLR Agonists. J. Immunol. 2007, 178, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of Adaptor TRIF in the MyD88-Independent Toll-Like Receptor Signaling Pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Van Tongeren, J.; Röschmann, K.I.L.; Reinartz, S.M.; Luiten, S.; Fokkens, W.; De Jong, E.C.; Van Drunen, C.M. Expression profiling and functional analysis of Toll-like receptors in primary healthy human nasal epithelial cells shows no correlation and a refractory LPS response. Clin. Transl. Allergy 2015, 5, 1–9. [Google Scholar] [CrossRef]
- Tengroth, L.; Millrud, C.R.; Kvarnhammar, A.M.; Georén, S.K.; Latif, L.; Cardell, L.O. Functional Effects of Toll-Like Receptor (TLR)3, 7, 9, RIG-I and MDA-5 Stimulation in Nasal Epithelial Cells. PLoS ONE 2014, 9, e98239. [Google Scholar] [CrossRef]
- Porsbjerg, C.; Baines, K.; Sverrild, A.; Backer, V.; Gibson, P. Eosinophilic airway inflammation is associated with increased TLR2 and TLR4 expression in adult asthmatics. Eur. Respirat. J. 2014, 44, 58. [Google Scholar]
- Shikhagaie, M.; Andersson, C.; Mori, M.; Kortekaas Krohn, I.; Bergqvist, A.; Dahl, R.; Ekblad, E.; Hoffmann, H.J.; Bjermer, L.; Erjefält, J.S. Mapping of TLR5 and TLR7 in central and distal human airways and identification of reduced TLR expression in severe asthma. Clin. Exp. Allergy 2014, 44, 184–196. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Diesel, B.; Zarbock, R.; Breinig, T.; Monz, D.; Koch, M.; Meyerhans, A.; Gortner, L.; Lehr, C.-M.; Huwer, H.; et al. Differential cell reaction upon Toll-like receptor 4 and 9 activation in human alveolar and lung interstitial macrophages. Respir. Res. 2010, 11, 124. [Google Scholar] [CrossRef]
- Grassin-Delyle, S.; Abrial, C.; Salvator, H.; Brollo, M.; Naline, E.; DeVillier, P. The Role of Toll-Like Receptors in the Production of Cytokines by Human Lung Macrophages. J. Innate Immun. 2020, 12, 63–73. [Google Scholar] [CrossRef]
- Kulka, M.; Metcalfe, D.D. TLR3 activation inhibits human mast cell attachment to fibronectin and vitronectin. Mol. Immunol. 2006, 43, 1579–1586. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Freeman, C.M.; Martinez, F.J.; Han, M.K.; Washko, G.R.; McCubbrey, A.L.; Chensue, S.W.; Arenberg, D.; Meldrum, C.A.; McCloskey, L.; Curtis, J.L. Lung CD8+ T cells in COPD have increased expression of bacterial TLRs. Respir. Res. 2013, 14, 13. [Google Scholar] [CrossRef] [PubMed]
- Koller, B.; Kappler, M.; Latzin, P.; Gaggar, A.; Schreiner, M.; Takyar, S.; Kormann, M.; Kabesch, M.; Roos, D.; Griese, M.; et al. TLR Expression on Neutrophils at the Pulmonary Site of Infection: TLR1/TLR2-Mediated Up-Regulation of TLR5 Expression in Cystic Fibrosis Lung Disease. J. Immunol. 2008, 181, 2753–2763. [Google Scholar] [CrossRef] [PubMed]
- Sukkar, M.B.; Xie, S.; Khorasani, N.M.; Kon, O.M.; Stanbridge, R.; Issa, R.; Chung, K.F. Toll-like receptor 2, 3, and 4 expression and function in human airway smooth muscle. J. Allergy Clin. Immunol. 2006, 118, 641–648. [Google Scholar] [CrossRef]
- Hilbert, T.; Dornbusch, K.; Baumgarten, G.; Hoeft, A.; Frede, S.; Klaschik, S. Pulmonary vascular inflammation: Effect of TLR signalling on angiopoietin/TIE regulation. Clin. Exp. Pharmacol. Physiol. 2017, 44, 123–131. [Google Scholar] [CrossRef]
- Chen, S.; Wong, M.H.; Schulte, D.J.; Arditi, M.; Michelsen, K.S. Differential expression of Toll-like receptor 2 (TLR2) and responses to TLR2 ligands between human and murine vascular endothelial cells. J. Endotoxin Res. 2007, 13, 281–296. [Google Scholar] [CrossRef]
- Farkas, D.; Thompson, A.R.; Bhagwani, A.R.; Hultman, S.; Ji, H.; Kotha, N.; Farr, G.; Arnold, N.D.; Braithwaite, A.T.; Casbolt, H.; et al. Toll-like Receptor 3 is a Therapeutic Target for Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2019, 199, 199–210. [Google Scholar] [CrossRef]
- Bauer, E.M.; Shapiro, R.; Billiar, T.R.; Bauer, P.M. High Mobility Group Box 1 Inhibits Human Pulmonary Artery Endothelial Cell Migration via a Toll-like Receptor 4- and Interferon Response Factor 3-dependent Mechanism(s). J. Biol. Chem. 2013, 288, 1365–1373. [Google Scholar] [CrossRef]
- George, P.M.; Badiger, R.; Shao, D.; Edwards, M.R.; Wort, S.J.; Paul-Clark, M.J.; Mitchell, J.A. Viral Toll Like Receptor activation of pulmonary vascular smooth muscle cells results in endothelin-1 generation; relevance to pathogenesis of pulmonary arterial hypertension. Biochem. Biophys. Res. Commun. 2012, 426, 486–491. [Google Scholar] [CrossRef]
- Muir, A.; Soong, G.; Sokol, S.; Reddy, B.; Gómez, M.I.; Van Heeckeren, A.; Prince, A. Toll-Like Receptors in Normal and Cystic Fibrosis Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2004, 30, 777–783. [Google Scholar] [CrossRef]
- Mayer, A.K.; Muehmer, M.; Mages, J.; Gueinzius, K.; Hess, C.; Heeg, K.; Bals, R.; Lang, R.; Dalpke, A.H. Differential Recognition of TLR-Dependent Microbial Ligands in Human Bronchial Epithelial Cells. J. Immunol. 2007, 178, 3134–3142. [Google Scholar] [CrossRef] [PubMed]
- Esnault, S.; Bernau, K.; Torr, E.E.; Bochkov, Y.A.; Jarjour, N.N.; Sandbo, N. RNA-sequencing analysis of lung primary fibroblast response to eosinophil-degranulation products predicts downstream effects on inflammation, tissue remodeling and lipid metabolism. Respir. Res. 2017, 18, 188. [Google Scholar] [CrossRef] [PubMed]
- Thorley, A.J.; Grandolfo, D.; Lim, E.; Goldstraw, P.; Young, A.; Tetley, T.D. Innate Immune Responses to Bacterial Ligands in the Peripheral Human Lung—Role of Alveolar Epithelial TLR Expression and Signalling. PLoS ONE 2011, 6, e21827. [Google Scholar] [CrossRef] [PubMed]
- Motoi, Y.; Shibata, T.; Takahashi, K.; Kanno, A.; Murakami, Y.; Li, X.; Kasahara, T.; Miyake, K. Lipopeptides are signaled by Toll-like receptor 1, 2 and 6 in endolysosomes. Int. Immunol. 2014, 26, 563–573. [Google Scholar] [CrossRef][Green Version]
- Takeuchi, O.; Sato, S.; Horiuchi, T.; Hoshino, K.; Takeda, K.; Dong, Z.; Modlin, R.L.; Akira, S. Cutting Edge: Role of Toll-Like Receptor 1 in Mediating Immune Response to Microbial Lipoproteins. J. Immunol. 2002, 169, 10–14. [Google Scholar] [CrossRef]
- Buwitt-Beckmann, U.; Heine, H.; Wiesmüller, K.-H.; Jung, G.; Brock, R.; Akira, S.; Ulmer, A.J. TLR1- and TLR6-independent Recognition of Bacterial Lipopeptides. J. Biol. Chem. 2006, 281, 9049–9057. [Google Scholar] [CrossRef]
- Misch, E.A.; Macdonald, M.; Ranjit, C.; Sapkota, B.R.; Wells, R.D.; Siddiqui, M.R.; Kaplan, G.; Hawn, T.R. Human TLR1 deficiency is associated with impaired mycobacterial signaling and protection from leprosy reversal reaction. PLoS Negl. Trop. Dis. 2008, 2, e231. [Google Scholar] [CrossRef]
- Fuchs, K.; Gloria, Y.C.; Wolz, O.; Herster, F.; Sharma, L.; A Dillen, C.; Täumer, C.; Dickhöfer, S.; Bittner, Z.; Dang, T.; et al. The fungal ligand chitin directly binds TLR 2 and triggers inflammation dependent on oligomer size. EMBO Rep. 2018, 19, e201846065. [Google Scholar] [CrossRef]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human Cytomegalovirus Envelope Glycoproteins B and H Are Necessary for TLR2 Activation in Permissive Cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar] [CrossRef]
- Cuevas, C.D.; Ross, S.R. Toll-Like Receptor 2-Mediated Innate Immune Responses against Junín Virus in Mice Lead to Antiviral Adaptive Immune Responses during Systemic Infection and Do Not Affect Viral Replication in the Brain. J. Virol. 2014, 88, 7703–7714. [Google Scholar] [CrossRef]
- Murawski, M.R.; Bowen, G.N.; Cerny, A.M.; Anderson, L.J.; Haynes, L.M.; Tripp, R.A.; Kurt-Jones, E.A.; Finberg, R.W. Respiratory Syncytial Virus Activates Innate Immunity through Toll-Like Receptor 2. J. Virol. 2008, 83, 1492–1500. [Google Scholar] [CrossRef] [PubMed]
- Panfili, F.M.; Roversi, M.; D’Argenio, P.; Rossi, P.; Cappa, M.; Fintini, D. Possible role of vitamin D in Covid-19 infection in pediatric population. J. Endocrinol. Investig. 2021, 44, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Funderburg, N.; Lederman, M.M.; Feng, Z.; Drage, M.G.; Jadlowsky, J.; Harding, C.V.; Weinberg, A.; Sieg, S.F. Human β-defensin-3 activates professional antigen-presenting cells via Toll-like receptors 1 and 2. Proc. Natl. Acad. Sci. USA 2007, 104, 18631–18635. [Google Scholar] [CrossRef]
- Chen, L.; Long, X.; Xu, Q.; Tan, J.; Wang, G.; Cao, Y.; Wei, J.; Luo, H.; Zhu, H.; Huang, L. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 992–994. [Google Scholar] [CrossRef]
- Matsumiya, M.; Stylianou, E.; Griffiths, K.; Lang, Z.; Meyer, J.; Harris, S.A.; Rowland, R.; Minassian, A.M.; Pathan, A.A.; Fletcher, H.A.; et al. Roles for Treg Expansion and HMGB1 Signaling through the TLR1-2-6 Axis in Determining the Magnitude of the Antigen-Specific Immune Response to MVA85A. PLoS ONE 2013, 8, e67922. [Google Scholar] [CrossRef] [PubMed]
- Tapping, R.I.; Tobias, P.S. Mycobacterial lipoarabinomannan mediates physical interactions between TLR1 and TLR2 to induce signaling. J. Endotoxin Res. 2003, 9, 264–268. [Google Scholar] [CrossRef]
- Yuan, C.; Qu, Z.-L.; Tang, X.-L.; Liu, Q.; Luo, W.; Huang, C.; Pan, Q.; Zhang, X. Mycobacterium tuberculosis Mannose-Capped Lipoarabinomannan Induces IL-10-Producing B Cells and Hinders CD4+Th1 Immunity. iScience 2019, 11, 13–30. [Google Scholar] [CrossRef]
- Sohn, K.M.; Lee, S.-G.; Kim, H.J.; Cheon, S.; Jeong, H.; Lee, J.; Kim, I.S.; Silwal, P.; Jo, E.-K.; Paik, S.; et al. COVID-19 Patients Upregulate Toll-like Receptor 4-mediated Inflammatory Signaling That Mimics Bacterial Sepsis. J. Korean Med Sci. 2020, 35. [Google Scholar] [CrossRef]
- Brandt, K.J.; Fickentscher, C.; Kruithof, E.K.; De Moerloose, P. TLR2 ligands induce NF-κB activation from endosomal compartments of human monocytes. PLoS ONE 2013, 8, e80743. [Google Scholar] [CrossRef]
- Aguilar-Briseño, J.A.; Upasani, V.; Ter Ellen, B.M.; Moser, J.; Pauzuolis, M.; Ruiz-Silva, M.; Heng, S.; Laurent, D.; Choeung, R.; Dussart, P.; et al. TLR2 on blood monocytes senses dengue virus infection and its expression correlates with disease pathogenesis. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Grabowski, M.; Murgueitio, M.S.; Bermudez, M.; Wolber, G.; Weindl, G. The novel small-molecule antagonist MMG-11 preferentially inhibits TLR2/1 signaling. Biochem. Pharmacol. 2020, 171, 113687. [Google Scholar] [CrossRef] [PubMed]
- AlQasrawi, D.; Naser, S.A. Nicotine Modulates MyD88-Dependent Signaling Pathway in Macrophages during Mycobacterial Infection. Microorganisms 2020, 8, 1804. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, K.; Rahman, M.A.; Mitra, S.; Knoell, D.L.; Woodiga, S.A.; King, S.J.; Wewers, M.D. IκBζ Regulates Human Monocyte Pro-Inflammatory Responses Induced by Streptococcus pneumoniae. PLoS ONE 2016, 11, e0161931. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, J.; Zahlten, J.; Pollok, I.; Lippmann, J.; Scharf, S.; N’Guessan, P.D.; Opitz, B.; Flieger, A.; Suttorp, N.; Hippenstiel, S. Legionella pneumophila-induced IκBζ-dependent expression of interleukin-6 in lung epithelium. Eur. Respirat. J. 2011, 37, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, K.; Mitra, S.; Gavrilin, M.A.; Wewers, M.D. House dust mite allergens and the induction of monocyte interleukin 1β production that triggers an IκBζ-dependent granulocyte macrophage colony-stimulating factor release from human lung epithelial cells. Am. J. Respirat. Cell Mol. Biol. 2015, 53, 400–411. [Google Scholar] [CrossRef]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-Stranded RNA Is Produced by Positive-Strand RNA Viruses and DNA Viruses but Not in Detectable Amounts by Negative-Strand RNA Viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Karikó, K.; Bhuyan, P.; Capodici, J.; Weissman, D. Small interfering RNAs mediate sequence-independent gene suppression and induce immune activation by signaling through toll-like receptor 3. J. Immunol. 2004, 172, 6545–6549. [Google Scholar] [CrossRef]
- Tatematsu, M.; Nishikawa, F.; Seya, T.; Matsumoto, M. Toll-like receptor 3 recognizes incomplete stem structures in single-stranded viral RNA. Nat. Commun. 2013, 4, 1833. [Google Scholar] [CrossRef]
- Karikó, K.; Ni, H.; Capodici, J.; Lamphier, M.; Weissman, D. mRNA Is an Endogenous Ligand for Toll-like Receptor 3. J. Biol. Chem. 2004, 279, 12542–12550. [Google Scholar] [CrossRef]
- Suresh, M.V.; Thomas, B.; Machado-Aranda, D.; Dolgachev, V.A.; Ramakrishnan, S.K.; Talarico, N.; Cavassani, K.; Sherman, M.A.; Hemmila, M.R.; Kunkel, S.L.; et al. Double-Stranded RNA Interacts with Toll-Like Receptor 3 in Driving the Acute Inflammatory Response Following Lung Contusion. Crit. Care Med. 2016, 44, e1054–e1066. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wohlford-Lenane, C.; Fleming, E.; Lane, T.E.; McCray, P.B.; Perlman, S. Intranasal Treatment with Poly(I:C) Protects Aged Mice from Lethal Respiratory Virus Infections. J. Virol. 2012, 86, 11416–11424. [Google Scholar] [CrossRef] [PubMed]
- Barnard, D.L.; Day, C.W.; Bailey, K.; Heiner, M.; Montgomery, R.; Lauridsen, L.; Chan, P.K.; Sidwell, R.W. Evaluation of immunomodulators, interferons and known in vitro SARS-coV inhibitors for inhibition of SARS-coV replication in BALB/c mice. Antivir. Chem. Chemother. 2006, 17, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Kumaki, Y.; Salazar, A.M.; Wandersee, M.K.; Barnard, D.L. Prophylactic and therapeutic intranasal administration with an immunomodulator, Hiltonol® (Poly IC:LC), in a lethal SARS-CoV-infected BALB/c mouse model. Antivir. Res. 2017, 139, 1–12. [Google Scholar] [CrossRef]
- Totura, A.L.; Whitmore, A.C.; Agnihothram, S.; Schäfer, A.; Katze, M.G.; Heise, M.T.; Baric, R.S. Toll-Like Receptor 3 Signaling via TRIF Contributes to a Protective Innate Immune Response to Severe Acute Respiratory Syndrome Coronavirus Infection. mBio 2015, 6, e00638-15. [Google Scholar] [CrossRef]
- Lokugamage, K.G.; Hage, A.; de Vries, M.; Valero-Jimenez, A.M.; Schindewolf, C.; Dittmann, M.; Rajsbaum, R.; Menachery, V.D. Type I interferon susceptibility distinguishes SARS-CoV-2 from SARS-CoV. J. Virol. 2020. [Google Scholar] [CrossRef]
- Mantlo, E.; Bukreyeva, N.; Maruyama, J.; Paessler, S.; Huang, C. Antiviral activities of type I interferons to SARS-CoV-2 infection. Antivir. Res. 2020, 179, 104811. [Google Scholar] [CrossRef]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef]
- Stockinger, S.; Kastner, R.; Kernbauer, E.; Pilz, A.; Westermayer, S.; Reutterer, B.; Soulat, D.; Stengl, G.; Vogl, C.; Frenz, T.; et al. Characterization of the Interferon-Producing Cell in Mice Infected with Listeria monocytogenes. PLOS Pathog. 2009, 5, e1000355. [Google Scholar] [CrossRef]
- Park, B.S.; Lee, J.-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef]
- Park, B.S.; Song, D.H.; Kim, H.M.; Choi, B.-S.; Lee, H.; Lee, J.-O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nat. Cell Biol. 2009, 458, 1191–1195. [Google Scholar] [CrossRef] [PubMed]
- Marr, N.; Turvey, S.E. Role of human TLR4 in respiratory syncytial virus-induced NF-κB activation, viral entry and replication. Innate Immun. 2012, 18, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Jones, E.A.; Popova, L.; A Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Gao, W.; Xiong, Y.; Li, Q.; Yang, H. Inhibition of Toll-Like Receptor Signaling as a Promising Therapy for Inflammatory Diseases: A Journey from Molecular to Nano Therapeutics. Front. Physiol. 2017, 8, 508. [Google Scholar] [CrossRef]
- Bi, X.; Su, Z.; Yan, H.; Du, J.; Wang, J.; Chen, L.; Peng, M.; Chen, S.; Shen, B.; Li, J. Prediction of severe illness due to COVID-19 based on an analysis of initial Fibrinogen to Albumin Ratio and Platelet count. Platelets 2020, 31, 674–679. [Google Scholar] [CrossRef]
- Spiezia, L.; Boscolo, A.; Poletto, F.; Cerruti, L.; Tiberio, I.; Campello, E.; Navalesi, P.; Simioni, P. COVID-19-Related Severe Hypercoagulability in Patients Admitted to Intensive Care Unit for Acute Respiratory Failure. Thromb. Haemost. 2020, 120, 998–1000. [Google Scholar] [CrossRef] [PubMed]
- Fraser, D.D.; Cepinskas, G.; Slessarev, M.; Martin, C.; Daley, M.; Miller, M.R.; O’Gorman, D.B.; Gill, S.E.; Patterson, E.K.; Dos Santos, C.C. Inflammation profiling of critically ill coronavirus disease 2019 patients. Crit. Care Explor. 2020, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, X.; Zhou, Y.; Sun, J.; Liu, X.; Zhang, J.; Mei, X.; Zhong, J.; Zhao, J.; Ran, P. COVID-19 Severity Correlates with Weaker T-Cell Immunity, Hypercytokinemia, and Lung Epithelium Injury. Am. J. Respir. Crit. Care Med. 2020, 202, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A.-G.; Dubuisson, A.; DeRosa, L.; Almire, C.; Hénon, C.; Kosmider, O.; Droin, N.; et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, 1401–1418.e18. [Google Scholar] [CrossRef]
- Saito, A.; Kuronuma, K.; Moniwa, K.; Kodama, K.; Takahashi, S.; Takahashi, H.; Chiba, H. Serum surfactant protein A and D may be novel biomarkers of COVID-19 pneumonia severity. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Jiang, Q.; Yi, M.; Guo, Q.; Wang, C.; Wang, H.; Meng, S.; Liu, C.; Fu, Y.; Ji, H.; Chen, T. Protective effects of polydatin on lipopolysaccharide-induced acute lung injury through TLR4-MyD88-NF-κB pathway. Int. Immunopharmacol. 2015, 29, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Debierre-Grockiego, F.; Campos, M.A.; Azzouz, N.; Schmidt, J.; Bieker, U.; Resende, M.G.; Mansur, D.S.; Weingart, R.; Schmidt, R.R.; Golenbock, D.T.; et al. Activation of TLR2 and TLR4 by Glycosylphosphatidylinositols Derived from Toxoplasma gondii. J. Immunol. 2007, 179, 1129–1137. [Google Scholar] [CrossRef]
- Adanitsch, F.; Shi, J.; Shao, F.; Beyaert, R.; Heine, H.; Zamyatina, A. Synthetic glycan-based TLR4 agonists targeting caspase-4/11 for the development of adjuvants and immunotherapeutics. Chem. Sci. 2018, 9, 3957–3963. [Google Scholar] [CrossRef] [PubMed]
- Sheng, K.-C.; Kalkanidis, M.; Pouniotis, D.S.; Wright, M.D.; Pietersz, G.A.; Apostolopoulos, V. The Adjuvanticity of a Mannosylated Antigen Reveals TLR4 Functionality Essential for Subset Specialization and Functional Maturation of Mouse Dendritic Cells. J. Immunol. 2008, 181, 2455–2464. [Google Scholar] [CrossRef] [PubMed]
- Ness, T.; Abdallah, M.; Adams, J.; Alvarado, C.; Gunn, E.; House, B.; Lamb, J.; Macguire, J.; Norris, E.; Robinson, R. Candida albicans-derived mannoproteins activate NF-κB in reporter cells expressing TLR4, MD2 and CD14. PLoS ONE 2017, 12, e0189939. [Google Scholar] [CrossRef]
- Haynes, L.M.; Moore, D.D.; Kurt-Jones, E.A.; Finberg, R.W.; Anderson, L.J.; Tripp, R.A. Involvement of Toll-Like Receptor 4 in Innate Immunity to Respiratory Syncytial Virus. J. Virol. 2001, 75, 10730–10737. [Google Scholar] [CrossRef]
- Okumura, A.; Pitha, P.M.; Yoshimura, A.; Harty, R.N. Interaction between Ebola Virus Glycoprotein and Host Toll-Like Receptor 4 Leads to Induction of Proinflammatory Cytokines and SOCS1. J. Virol. 2009, 84, 27–33. [Google Scholar] [CrossRef]
- Husebye, H.; Halaas, Ø.; Stenmark, H.; Tunheim, G.; Sandanger, Ø.; Bogen, B.; Brech, A.; Latz, E.; Espevik, T. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006, 25, 683–692. [Google Scholar] [CrossRef]
- Zanoni, I.; Ostuni, R.; Marek, L.R.; Barresi, S.; Barbalat, R.; Barton, G.M.; Granucci, F.; Kagan, J.C. CD14 Controls the LPS-Induced Endocytosis of Toll-like Receptor 4. Cell 2011, 147, 868–880. [Google Scholar] [CrossRef]
- Rajaiah, R.; Perkins, D.J.; Ireland, D.D.C.; Vogel, S.N. CD14 dependence of TLR4 endocytosis and TRIF signaling displays ligand specificity and is dissociable in endotoxin tolerance. Proc. Natl. Acad. Sci. USA 2015, 112, 8391–8396. [Google Scholar] [CrossRef] [PubMed]
- Birra, D.; Benucci, M.; Landolfi, L.; Merchionda, A.; Loi, G.; Amato, P.; Licata, G.; Quartuccio, L.; Triggiani, M.; Moscato, P. COVID 19: A clue from innate immunity. Immunol. Res. 2020, 68, 161–168. [Google Scholar] [CrossRef]
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- Duthie, M.S.; Windish, H.P.; Fox, C.B.; Reed, S.G. Use of defined TLR ligands as adjuvants within human vaccines. Immunol. Rev. 2010, 239, 178–196. [Google Scholar] [CrossRef] [PubMed]
- A Hajam, I.; A Dar, P.; Shahnawaz, I.; Jaume, J.C.; Lee, J.H. Bacterial flagellin—A potent immunomodulatory agent. Exp. Mol. Med. 2017, 49, e373. [Google Scholar] [CrossRef] [PubMed]
- Felgner, S.; Spöring, I.; Pawar, V.; Kocijancic, D.; Preusse, M.; Falk, C.; Rohde, M.; Häussler, S.; Weiss, S.; Erhardt, M. The immunogenic potential of bacterial flagella for Salmonella -mediated tumor therapy. Int. J. Cancer 2020, 147, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Georgel, A.-F.; Cayet, D.; Pizzorno, A.; Rosa-Calatrava, M.; Paget, C.; Sencio, V.; Dubuisson, J.; Trottein, F.; Sirard, J.-C.; Carnoy, C. Toll-like receptor 5 agonist flagellin reduces influenza A virus replication independently of type I interferon and interleukin 22 and improves antiviral efficacy of oseltamivir. Antivir. Res. 2019, 168, 28–35. [Google Scholar] [CrossRef]
- Ramos, H.C.; Rumbo, M.; Sirard, J.-C. Bacterial flagellins: Mediators of pathogenicity and host immune responses in mucosa. Trends Microbiol. 2004, 12, 509–517. [Google Scholar] [CrossRef]
- Song, W.S.; Jeon, Y.J.; Namgung, B.; Hong, M.; Yoon, S.-I. A conserved TLR5 binding and activation hot spot on flagellin. Sci. Rep. 2017, 7, srep40878. [Google Scholar] [CrossRef]
- Kim, J.R.; Holbrook, B.C.; Hayward, S.L.; Blevins, L.K.; Jorgensen, M.J.; Kock, N.D.; De Paris, K.; D’Agostino, R.B.; Aycock, S.T.; Mizel, S.B.; et al. Inclusion of Flagellin during Vaccination against Influenza Enhances Recall Responses in Nonhuman Primate Neonates. J. Virol. 2015, 89, 7291–7303. [Google Scholar] [CrossRef]
- Delaney, K.N.; Phipps, J.P.; Johnson, J.B.; Mizel, S.B. A Recombinant Flagellin-Poxvirus Fusion Protein Vaccine Elicits Complement-Dependent Protection Against Respiratory Challenge with Vaccinia Virus in Mice. Viral Immunol. 2010, 23, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, L.A.; Mardanova, E.S.; Shuklina, M.A.; Blokhina, E.A.; Kotlyarov, R.Y.; Potapchuk, M.V.; Kovaleva, A.A.; Vidyaeva, I.G.; Korotkov, A.V.; Eletskaya, E.I. Flagellin-fused protein targeting M2e and HA2 induces potent humoral and T-cell responses and protects mice against various influenza viruses a subtypes. J. Biomed. Sci. 2018, 25, 33. [Google Scholar] [CrossRef] [PubMed]
- Huleatt, J.W.; Foellmer, H.G.; Hewitt, D.; Tang, J.; Desai, P.; Price, A.; Jacobs, A.; Takahashi, V.N.; Huang, Y.; Nakaar, V. A West Nile virus recombinant protein vaccine that coactivates innate and adaptive immunity. J. Infect. Dis. 2007, 195, 1607–1617. [Google Scholar]
- Bhattacharya, M.; Sharma, A.R.; Patra, P.; Ghosh, P.; Sharma, G.; Patra, B.C.; Lee, S.S.; Chakraborty, C. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): Immunoinformatics approach. J. Med. Virol. 2020, 92, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Erdos, G.; Huang, S.; Kenniston, T.W.; Balmert, S.C.; Carey, C.D.; Raj, V.S.; Epperly, M.W.; Klimstra, W.B.; Haagmans, B.L. Microneedle array delivered recombinant coronavirus vaccines: Immunogenicity and rapid translational development. EBioMedicine 2020, 55, 102743. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Park, A.; Huh, J.-W.; You, G.; Jung, D.-J.; Song, M.; Lee, H.K.; Kim, Y.-M. Flagellin-Stimulated Production of Interferon-β Promotes Anti-Flagellin IgG2c and IgA Responses. Mol. Cells 2020, 43, 251–263. [Google Scholar]
- Fougeron, D.; Van Maele, L.; Songhet, P.; Cayet, D.; Hot, D.; Van Rooijen, N.; Mollenkopf, H.-J.; Hardt, W.-D.; Benecke, A.G.; Sirard, J.-C. Indirect Toll-like receptor 5-mediated activation of conventional dendritic cells promotes the mucosal adjuvant activity of flagellin in the respiratory tract. Vaccine 2015, 33, 3331–3341. [Google Scholar] [CrossRef]
- Thomas, P.G.; Carter, M.R.; Atochina, O.; Da’Dara, A.A.; Piskorska, D.; McGuire, E.; Harn, D.A. Maturation of dendritic cell 2 phenotype by a helminth glycan uses a Toll-like receptor 4-dependent mechanism. J. Immunol. 2003, 171, 5837–5841. [Google Scholar] [CrossRef]
- Mancuso, G.; Gambuzza, M.; Midiri, A.; Biondo, C.; Papasergi, S.; Akira, S.; Teti, G.; Beninati, C. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat. Immunol. 2009, 10, 587–594. [Google Scholar] [CrossRef]
- Zhang, S.; Shuting, Z.; Tanji, H.; Jiang, S.; Das, N.; Li, J.; Sakaniwa, K.; Jin, J.; Bian, Y.; Ohto, U.; et al. Small-molecule inhibition of TLR8 through stabilization of its resting state. Nat. Chem. Biol. 2018, 14, 58–64. [Google Scholar] [CrossRef]
- Chen, D.-Y.; Lin, C.-C.; Chen, Y.-M.; Lan, J.-L.; Hung, W.-T.; Chen, H.-H.; Lai, K.-L.; Hsieh, C.-W. Involvement of TLR7 MyD88-dependent signaling pathway in the pathogenesis of adult-onset Still’s disease. Arthr. Res. Ther. 2013, 15, R39. [Google Scholar] [CrossRef] [PubMed]
- Melchjorsen, J.; Jensen, S.B.; Malmgaard, L.; Rasmussen, S.B.; Weber, F.; Bowie, A.G.; Matikainen, S.; Paludan, S.R. Activation of Innate Defense against a Paramyxovirus is Mediated by RIG-I and TLR7 and TLR8 in a Cell-Type-Specific Manner. J. Virol. 2005, 79, 12944–12951. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Gong, J.; Jamitzky, F.; Heckl, W.M.; Stark, R.W.; Rössle, S.C. Homology modeling of human Toll-like receptors TLR7, 8, and 9 ligand-binding domains. Protein Sci. 2009, 18, 1684–1691. [Google Scholar] [CrossRef] [PubMed]
- Mikula, I.; Bhide, M.; Pastorekova, S. Characterization of ovine TLR7 and TLR8 protein coding regions, detection of mutations and Maedi Visna virus infection. Veter. Immunol. Immunopathol. 2010, 138, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.I.; Yhee, J.Y.; Na, J.H.; Lee, S.; Lee, H.; Kang, S.-W.; Chang, H.; Ryu, J.H.; Lee, S.; Kwon, I.C.; et al. Bioorthogonal Copper Free Click Chemistry for Labeling and Tracking of Chondrocytes In Vivo. Bioconjugate Chem. 2016, 27, 927–936. [Google Scholar] [CrossRef]
- Channappanavar, R.; Fehr, A.R.; Zheng, J.; Wohlford-Lenane, C.; Abrahante, J.E.; Mack, M.; Sompallae, R.; McCray, P.B.; Meyerholz, D.K.; Perlman, S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Investig. 2019, 129, 3625–3639. [Google Scholar] [CrossRef]
- Pawar, R.D.; Ramanjaneyulu, A.; Kulkarni, O.P.; Lech, M.; Segerer, S.; Anders, H.-J. Inhibition of Toll-Like Receptor-7 (TLR-7) or TLR-7 plus TLR-9 Attenuates Glomerulonephritis and Lung Injury in Experimental Lupus. J. Am. Soc. Nephrol. 2007, 18, 1721–1731. [Google Scholar] [CrossRef]
- Ignatz-Hoover, J.J.; Wang, H.; A Moreton, S.; Chakrabarti, A.; Agarwal, M.K.; Sun, K.; Gupta, K.; Wald, D.N. The role of TLR8 signaling in acute myeloid leukemia differentiation. Leukemia 2015, 29, 918–926. [Google Scholar] [CrossRef]
- Yang, K.; Puel, A.; Zhang, S.; Eidenschenk, C.; Ku, C.-L.; Casrouge, A.; Picard, C.; Von Bernuth, H.; Senechal, B.; Plancoulaine, S. Human TLR-7-,-8-, and-9-mediated induction of IFN-α/β and-λ is IRAK-4 dependent and redundant for protective immunity to viruses. Immunity 2005, 23, 465–478. [Google Scholar] [CrossRef]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef]
- Zhang, Z.; Ohto, U.; Shibata, T.; Taoka, M.; Yamauchi, Y.; Sato, R.; Shukla, N.M.; David, S.A.; Isobe, T.; Miyake, K.; et al. Structural Analyses of Toll-like Receptor 7 Reveal Detailed RNA Sequence Specificity and Recognition Mechanism of Agonistic Ligands. Cell Rep. 2018, 25, 3371–3381. [Google Scholar] [CrossRef] [PubMed]
- Tanji, H.; Ohto, U.; Shibata, T.; Taoka, M.; Yamauchi, Y.; Isobe, T.; Miyake, K.; Shimizu, T. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat. Struct. Mol. Biol. 2015, 22, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Eutimio, M.A.; Lopez-Macias, C.; Pastelin-Palacios, R. Bioinformatic analysis and identification of single-stranded RNA sequences recognized by TLR7/8 in the SARS-CoV-2, SARS-CoV, and MERS-CoV genomes. Microbes Infect. 2020, 22, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Van Der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; Van Den Heuvel, G.; Mantere, T.; Kersten, S.; Van Deuren, R.C.; Steehouwer, M.; Van Reijmersdal, S.V.; Jaeger, M. Presence of genetic variants among young men with severe COVID-19. JAMA 2020, 324, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Hurst, J.; Prinz, N.; Lorenz, M.; Bauer, S.; Chapman, J.; Lackner, K.J.; Von Landenberg, P. TLR7 and TLR8 ligands and antiphospholipid antibodies show synergistic effects on the induction of IL-1β and caspase-1 in monocytes and dendritic cells. Immunobiology 2009, 214, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Prinz, N.; Clemens, N.; Strand, D.; Pütz, I.; Lorenz, M.; Daiber, A.; Stein, P.; Degreif, A.; Radsak, M.; Schild, H.; et al. Antiphospholipid antibodies induce translocation of TLR7 and TLR8 to the endosome in human monocytes and plasmacytoid dendritic cells. Blood 2011, 118, 2322–2332. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Hurst, J.; Lorenz, M.; Prinz, N.; Clemens, N.; Drechsler, M.D.; Bauer, S.; Chapman, J.; Shoenfeld, Y.; Blank, M. Human antiphospholipid antibodies induce TNFα in monocytes via Toll-like receptor 8. Immunobiol. 2010, 215, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Amezcua-Guerra, L.M.; Rojas-Velasco, G.; Brianza-Padilla, M.; Vázquez-Rangel, A.; Márquez-Velasco, R.; Baranda-Tovar, F.; Springall, R.; Gonzalez-Pacheco, H.; Juárez-Vicuña, Y.; Tavera-Alonso, C.; et al. Presence of antiphospholipid antibodies in COVID-19: Case series study. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef]
- Borghi, M.O.; Beltagy, A.; Garrafa, E.; Curreli, D.; Cecchini, G.; Bodio, C.; Grossi, C.; Blengino, S.; Tincani, A.; Franceschini, F. Anti-phospholipid antibodies in COVID-19 are different from those detectable in the anti-phospholipid syndrome. Front. Immunol. 2020, 11, 2692. [Google Scholar] [CrossRef]
- Angelopoulou, A.; Alexandris, N.; Konstantinou, E.; Mesiakaris, K.; Zanidis, C.; Farsalinos, K.; Poulas, K. Imiquimod—A toll like receptor 7 agonist—Is an ideal option for management of COVID 19. Environ. Res. 2020, 188, 109858. [Google Scholar] [CrossRef]
- To, E.E.; Erlich, J.; Liong, F.; Luong, R.; Liong, S.; Bozinovski, S.; Seow, H.J.; O’Leary, J.J.; Brooks, D.A.; Vlahos, R.; et al. Intranasal and epicutaneous administration of Toll-like receptor 7 (TLR7) agonists provides protection against influenza A virus-induced morbidity in mice. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; To, K.K.; Zhang, A.J.; Lee, A.C.; Zhu, H.; Mak, W.W.; Hung, I.F.; Yuen, K.-Y. Co-stimulation with TLR7 agonist imiquimod and inactivated influenza virus particles promotes mouse B cell activation, differentiation, and accelerated antigen specific antibody production. Front. Immunol. 2018, 9, 2370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.J.X.; Li, C.; To, K.K.W.; Zhu, H.-S.; Lee, A.C.Y.; Li, C.-G.; Chan, J.F.W.; Hung, I.F.N.; Yuen, K.Y. Toll-Like Receptor 7 Agonist Imiquimod in Combination with Influenza Vaccine Expedites and Augments Humoral Immune Responses against Influenza A(H1N1)pdm09 Virus Infection in BALB/c Mice. Clin. Vaccine Immunol. 2014, 21, 570–579. [Google Scholar] [CrossRef]
- Egea, S.C.; Dickerson, I.M. Direct Interactions between Calcitonin-Like Receptor (CLR) and CGRP-Receptor Component Protein (RCP) Regulate CGRP Receptor Signaling. Endocrinology 2012, 153, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.H.; Gringhuis, S.I. C-type lectin receptors in the control of T helper cell differentiation. Nat. Rev. Immunol. 2016, 16, 433–448. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; Gringhuis, S.I. Signalling through C-type lectin receptors: Shaping immune responses. Nat. Rev. Immunol. 2009, 9, 465–479. [Google Scholar] [CrossRef]
- Lee, R.T.; Hsu, T.-L.; Huang, S.K.; Hsieh, S.-L.; Wong, C.-H.; Lee, Y.C. Survey of immune-related, mannose/fucose-binding C-type lectin receptors reveals widely divergent sugar-binding specificities. Glycobiology 2011, 21, 512–520. [Google Scholar] [CrossRef]
- Palomino-Segura, M.; Perez, L.; Farsakoglu, Y.; Virgilio, T.; Latino, I.; D’antuono, R.; Chatziandreou, N.; Pizzagalli, D.U.; Wang, G.; García-Sastre, A. Protection against influenza infection requires early recognition by inflammatory dendritic cells through C-type lectin receptor SIGN-R1. Nat. Microbiol. 2019, 4, 1930–1940. [Google Scholar] [CrossRef]
- Iliev, I.D.; Funari, V.A.; Taylor, K.D.; Nguyen, Q.; Reyes, C.N.; Strom, S.P.; Brown, J.; Becker, C.A.; Fleshner, P.R.; Dubinsky, M. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 2012, 336, 1314–1317. [Google Scholar] [CrossRef]
- Rothfuchs, A.G.; Bafica, A.; Feng, C.G.; Egen, J.G.; Williams, D.L.; Brown, G.D.; Sher, A. Dectin-1 interaction with Mycobacterium tuberculosis leads to enhanced IL-12p40 production by splenic dendritic cells. J. Immunol. 2007, 179, 3463–3471. [Google Scholar] [CrossRef]
- Schreibelt, G.; Klinkenberg, L.J.; Cruz, L.J.; Tacken, P.J.; Tel, J.; Kreutz, M.; Adema, G.J.; Brown, G.D.; Figdor, C.G.; de Vries, I.J.M. The C-type lectin receptor CLEC9A mediates antigen uptake and (cross-) presentation by human blood BDCA3+ myeloid dendritic cells. Blood 2012, 119, 2284–2292. [Google Scholar] [CrossRef] [PubMed]
- Gorjestani, S.; Darnay, B.G.; Lin, X. TRAF6 and TAK1 play essential roles in C-type lectin receptor signaling in response to Candida albicans infection. J. Biol. Chem. 2012, M112, 414276. [Google Scholar]
- Gantner, B.N.; Simmons, R.M.; Canavera, S.J.; Akira, S.; Underhill, D.M. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J. Exp. Med. 2003, 197, 1107–1117. [Google Scholar] [CrossRef]
- Kanitakis, J.; Lesort, C.; Danset, M.; Jullien, D. Chilblain-like acral lesions during the COVID-19 pandemic (“COVID toes”): Histologic, immunofluorescence, and immunohistochemical study of 17 cases. J. Am. Acad. Dermatol. 2020, 83, 870–875. [Google Scholar] [CrossRef]
- Hsieh, S.-L.; Sung, P.-S. CLEC2 and CLEC5A Pathogenic Host Factors in Acute Viral Infections. Front. Immunol. 2019, 10, 2867. [Google Scholar]
- Chiodo, F.; Bruijns, S.C.; Rodriguez, E.; Li, R.E.; Molinaro, A.; Silipo, A.; Di Lorenzo, F.; Garcia-Rivera, D.; Valdes-Balbin, Y.; Verez-Bencomo, V. Novel ACE2-Independent Carbohydrate-Binding of SARS-CoV-2 Spike Protein to Host Lectins and Lung Microbiota. BioRxiv 2020. [Google Scholar] [CrossRef]
- Zhao, X.; Chu, H.; Wong, B.H.-Y.; Chiu, M.C.; Wang, D.; Li, C.; Liu, X.; Yang, D.; Poon, V.K.-M.; Cai, J.; et al. Activation of C-Type Lectin Receptor and (RIG)-I-Like Receptors Contributes to Proinflammatory Response in Middle East Respiratory Syndrome Coronavirus-Infected Macrophages. J. Infect. Dis. 2019, 221, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Onodi, F.; Bonnet-Madin, L.; Karpf, L.; Meertens, L.; Poirot, J.; Legoff, J.; Delaugerre, C.; Amara, A.; Soumelis, V. SARS-CoV-2 induces activation and diversification of human plasmacytoid pre-dendritic cells. BioRxiv 2020. [Google Scholar] [CrossRef]
- Zhou, R.; To, K.K.-W.; Wong, Y.-C.; Liu, L.; Zhou, B.; Li, X.; Huang, H.; Mo, Y.; Luk, T.-Y.; Lau, T.T.-K. Acute SARS-CoV-2 infection impairs dendritic cell and T cell responses. Immunity 2020, 53, 864–877. [Google Scholar] [CrossRef]
- Combadiere, B.; Adam, L.; Quentric, P.; Rosenbaum, P.; Dorgham, K.; Bonduelle, O.; Parizot, C.; Sauce, D.; Mayaux, J.; Luyt, C.-E. LOX-1+ immature neutrophils predict severe COVID-19 patients at risk of thrombotic complications. BioRxiv 2020. [Google Scholar] [CrossRef]
- Damas, J.; Hughes, G.M.; Keough, K.C.; Painter, C.A.; Persky, N.S.; Corbo, M.; Hiller, M.; Koepfli, K.-P.; Pfenning, A.R.; Zhao, H.; et al. Broad Host Range of SARS-CoV-2 Predicted by Comparative and Structural Analysis of ACE2 in Vertebrates. BioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Dzionek, A.; Sohma, Y.; Nagafune, J.; Cella, M.; Colonna, M.; Facchetti, F.; Guünther, G.; Johnston, I.C.D.; Lanzavecchia, A.; Nagasaka, T.; et al. BDCA-2, a Novel Plasmacytoid Dendritic Cell–specific Type II C-type Lectin, Mediates Antigen Capture and Is a Potent Inhibitor of Interferon α/β Induction. J. Exp. Med. 2001, 194, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, A.; Otero, K.; Czerkowicz, J.M.; Kerns, H.M.; Shapiro, R.I.; Ranger, A.M.; Otipoby, K.L.; Taylor, F.R.; Cameron, T.O.; Viney, J.L.; et al. Anti-BDCA 2 monoclonal antibody inhibits plasmacytoid dendritic cell activation through Fc-dependent and Fc-independent mechanisms. EMBO Mol. Med. 2015, 7, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Röck, J.; Schneider, E.; Grün, J.R.; Grützkau, A.; Küppers, R.; Schmitz, J.; Winkels, G. CD303 (BDCA-2) signals in plasmacytoid dendritic cells via a BCR-like signalosome involving Syk, Slp65 and PLCγ2. Eur. J. Immunol. 2007, 37, 3564–3575. [Google Scholar] [CrossRef]
- Florentin, J.; Aouar, B.; Dental, C.; Thumann, C.; Firaguay, G.; Gondois-Rey, F.; Soumelis, V.; Baumert, T.F.; Nunès, J.A.; Olive, D.; et al. HCV glycoprotein E2 is a novel BDCA-2 ligand and acts as an inhibitor of IFN production by plasmacytoid dendritic cells. Blood 2012, 120, 4544–4551. [Google Scholar] [CrossRef]
- May, F.; Hagedorn, I.; Pleines, I.; Bender, M.; Vögtle, T.; Eble, J.; Elvers, M.; Nieswandt, B. CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood 2009, 114, 3464–3472. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Thalhammer, T.; Tzakos, A.G.; Stojanovska, L. Targeting Antigens to Dendritic Cell Receptors for Vaccine Development. J. Drug Deliv. 2013, 2013, 1–22. [Google Scholar] [CrossRef]
- Chaipan, C.; Soilleux, E.J.; Simpson, P.; Hofmann, H.; Gramberg, T.; Marzi, A.; Geier, M.; Stewart, E.A.; Eisemann, J.; Steinkasserer, A.; et al. DC-SIGN and CLEC-2 Mediate Human Immunodeficiency Virus Type 1 Capture by Platelets. J. Virol. 2006, 80, 8951–8960. [Google Scholar] [CrossRef]
- Cummings, R.D.; McEver, R.P. C-type lectins. In Essentials of Glycobiology, 2nd ed.; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 2009. [Google Scholar]
- Adachi, Y.; Ishii, T.; Ikeda, Y.; Hoshino, A.; Tamura, H.; Aketagawa, J.; Tanaka, S.; Ohno, N. Characterization of β-Glucan Recognition Site on C-Type Lectin, Dectin 1. Infect. Immun. 2004, 72, 4159–4171. [Google Scholar] [CrossRef]
- Esteban, A.; Popp, M.W.; Vyas, V.K.; Strijbis, K.; Ploegh, H.L.; Fink, G.R. Fungal recognition is mediated by the association of dectin-1 and galectin-3 in macrophages. Proc. Natl. Acad. Sci. USA 2011, 108, 14270–14275. [Google Scholar] [CrossRef]
- Hanashima, S.; Ikeda, A.; Tanaka, H.; Adachi, Y.; Ohno, N.; Takahashi, T.; Yamaguchi, Y. NMR study of short β(1-3)-glucans provides insights into the structure and interaction with Dectin-1. Glycoconj. J. 2014, 31, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Palma, A.S.; Feizi, T.; Zhang, Y.; Stoll, M.S.; Lawson, A.M.; Díaz-Rodríguez, E.; Campanero-Rhodes, M.A.; Costa, J.; Gordon, S.; Brown, G.D.; et al. Ligands for the β-Glucan Receptor, Dectin-1, Assigned Using “Designer” Microarrays of Oligosaccharide Probes (Neoglycolipids) Generated from Glucan Polysaccharides. J. Biol. Chem. 2006, 281, 5771–5779. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.; Agrawal, S.; Banerjee, K.; Letterio, J.; Denning, T.L.; Oswald-Richter, K.; Kasprowicz, D.J.; Kellar, K.; Pare, J.; Van Dyke, T.; et al. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J. Clin. Investig. 2006, 116, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabudhe, N.M.; Dokter-Fokkens, J.; De Vos, P. Particulate β-glucans synergistically activate TLR4 and Dectin-1 in human dendritic cells. Mol. Nutr. Food Res. 2016, 60, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, H.; Jégouzo, S.A.F.; Rex, M.J.; Drickamer, K.; Weis, W.I.; Taylor, M.E. Mechanism of pathogen recognition by human dectin-2. J. Biol. Chem. 2017, 292, 13402–13414. [Google Scholar] [CrossRef]
- Ritter, M.; Gross, O.; Kays, S.; Ruland, J.; Nimmerjahn, F.; Saijo, S.; Tschopp, J.; Layland, L.E.; Da Costa, C.P. Schistosoma mansoni triggers Dectin-2, which activates the Nlrp3 inflammasome and alters adaptive immune responses. Proc. Natl. Acad. Sci. USA 2010, 107, 20459–20464. [Google Scholar] [CrossRef]
- McGreal, E.P.; Rosas, M.; Brown, G.D.; Zamze, S.; Wong, S.Y.; Gordon, S.; Martinez-Pomares, L.; Taylor, P.R. The carbohydrate-recognition domain of Dectin-2 is a C-type lectin with specificity for high mannose. Glycobiology 2006, 16, 422–430. [Google Scholar] [CrossRef]
- Taylor, P.R.; Reid, D.M.; Heinsbroek, S.E.; Brown, G.D.; Gordon, S.; Wong, S.Y. Dectin-2 is predominantly myeloid restricted and exhibits unique activation-dependent expression on maturing inflammatory monocytes elicited in vivo. Eur. J. Immunol. 2005, 35, 2163–2174. [Google Scholar] [CrossRef]
- Bermejo-Jambrina, M.; Eder, J.; Helgers, L.C.; Hertoghs, N.; Nijmeijer, B.M.; Stunnenberg, M.; Geijtenbeek, T.B. C-Type Lectin Receptors in Antiviral Immunity and Viral Escape. Front. Immunol. 2018, 9, 590. [Google Scholar] [CrossRef]
- Meyer-Wentrup, F.; Cambi, A.; Joosten, B.; Looman, M.W.; De Vries, I.J.M.; Figdor, C.G.; Adema, G.; Gosse, J. DCIR is endocytosed into human dendritic cells and inhibits TLR8-mediated cytokine production. J. Leukoc. Biol. 2008, 85, 518–525. [Google Scholar] [CrossRef]
- Meyer-Wentrup, F.; Benitez-Ribas, D.; Tacken, P.J.; Punt, C.J.A.; Figdor, C.G.; De Vries, I.J.M.; Adema, G.; Gosse, J. Targeting DCIR on human plasmacytoid dendritic cells results in antigen presentation and inhibits IFN-α production. Blood 2008, 111, 4245–4253. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.A.; Gilbert, C.; Richard, M.; Beaulieu, A.D.; Tremblay, M.J. The C-type lectin surface receptor DCIR acts as a new attachment factor for HIV-1 in dendritic cells and contributes to trans- and cis-infection pathways. Blood 2008, 112, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Bloem, K.; Vuist, I.M.; Van Der Plas, A.-J.; Knippels, L.M.J.; Garssen, J.; García-Vallejo, J.J.; Van Vliet, S.J.; Van Kooyk, Y. Ligand Binding and Signaling of Dendritic Cell Immunoreceptor (DCIR) Is Modulated by the Glycosylation of the Carbohydrate Recognition Domain. PLoS ONE 2013, 8, e66266. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Barragan, L.; Züst, R.; Weber, F.; Spiegel, M.; Lang, K.S.; Akira, S.; Thiel, V.; Ludewig, B. Control of coronavirus infection through plasmacytoid dendritic-cell–derived type I interferon. Blood 2006, 109, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Cueto, F.J.; Del Fresno, C.; Sancho, D. DNGR-1, a Dendritic Cell-Specific Sensor of Tissue Damage That Dually Modulates Immunity and Inflammation. Front. Immunol. 2020, 10, 3146. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Fan, C.-Y.; Wang, A.-l.; Zou, Y.-l.; Yu, Y.-H.; He, C.; Xia, W.-G.; Zhang, J.-X.; Miao, Q. Suppressed T cell-mediated immunity in patients with COVID-19: A clinical retrospective study in Wuhan, China. J. Infect. 2020, 81, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.-R.; Li, D.; Yu, L.; Wang, F.-J.; Xing, H.; Yang, G.-B. The levels of DNGR-1 and its ligand-bearing cells were altered after human and simian immunodeficiency virus infection. Immunol. Res. 2017, 65, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Marzi, A.; Gramberg, T.; Simmons, G.; Möller, P.; Rennekamp, A.J.; Krumbiegel, M.; Geier, M.; Eisemann, J.; Turza, N.; Saunier, B.; et al. DC-SIGN and DC-SIGNR Interact with the Glycoprotein of Marburg Virus and the S Protein of Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2004, 78, 12090–12095. [Google Scholar] [CrossRef]
- Geurtsen, J.; Driessen, N.N.; Appelmelk, B.J. Mannose–fucose recognition by DC-SIGN. In Microbial Glycobiology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 673–695. [Google Scholar]
- Chan, V.S.F.; Chan, K.Y.K.; Chen, Y.; Poon, L.L.M.; Cheung, A.N.Y.; Zheng, B.; Chan, K.-H.; Mak, W.; Ngan, H.Y.S.; Xu, X.; et al. Homozygous L-SIGN (CLEC4M) plays a protective role in SARS coronavirus infection. Nat. Genet. 2005, 38, 38–46. [Google Scholar] [CrossRef]
- Lee, B.; Leslie, G.; Soilleux, E.J.; O’Doherty, U.; Baik, S.; Levroney, E.; Flummerfelt, K.; Swiggard, W.; Coleman, N.; Malim, M.H.; et al. cis Expression of DC-SIGN Allows for More Efficient Entry of Human and Simian Immunodeficiency Viruses via CD4 and a Coreceptor. J. Virol. 2001, 75, 12028–12038. [Google Scholar] [CrossRef]
- Hillaire, M.L.B.; Nieuwkoop, N.J.; Boon, A.C.M.; De Mutsert, G.; Trierum, S.E.V.-V.; Fouchier, R.A.M.; Osterhaus, A.D.M.E.; Rimmelzwaan, G.F. Binding of DC-SIGN to the Hemagglutinin of Influenza a Viruses Supports Virus Replication in DC-SIGN Expressing Cells. PLoS ONE 2013, 8, e56164. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.-P.; Chen, C.-Y.; Liu, S.-J.; Chen, K.-H.; Lee, Y.-M.; Chao, Y.-C.; Chen, Y.-M.A. Identifying Epitopes Responsible for Neutralizing Antibody and DC-SIGN Binding on the Spike Glycoprotein of the Severe Acute Respiratory Syndrome Coronavirus. J. Virol. 2006, 80, 10315–10324. [Google Scholar] [CrossRef] [PubMed]
- Arrighi, J.-F.; Pion, M.; Garcia, E.; Escola, J.-M.; Van Kooyk, Y.; Geijtenbeek, T.B.; Piguet, V. DC-SIGN–mediated Infectious Synapse Formation Enhances X4 HIV-1 Transmission from Dendritic Cells to T Cells. J. Exp. Med. 2004, 200, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.Y.K.; Xu, M.S.; Ching, J.C.Y.; Chan, V.S.; Ip, Y.C.; Yam, L.; Chu, C.M.; Lai, S.T.; So, K.M.; Wong, T.Y.; et al. Association of a single nucleotide polymorphism in the CD209 (DC-SIGN) promoter with SARS severity. Hong Kong Med. J. 2010, 16, 37. [Google Scholar]
- Grubaugh, N.D.; Hanage, W.P.; Rasmussen, A.L. Making Sense of Mutation: What D614G Means for the COVID-19 Pandemic Remains Unclear. Cell 2020, 182, 794–795. [Google Scholar] [CrossRef]
- Brufsky, A.; Lotze, M.T. DC/L-SIGNs of Hope in the COVID-19 Pandemic. J. Med. Virol. 2020, 92, 1396–1398. [Google Scholar] [CrossRef]
- Jeffers, S.A.; Tusell, S.M.; Gillim-Ross, L.; Hemmila, E.M.; Achenbach, J.E.; Babcock, G.J.; Thomas, J.W.D.; Thackray, L.B.; Young, M.D.; Mason, R.J.; et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 2004, 101, 15748–15753. [Google Scholar] [CrossRef]
- Jeffers, S.A.; Hemmila, E.M.; Holmes, K.V. Human Coronavirus 229E can Use CD209L (L-Sign) to Enter Cells. In Advances in Experimental Medicine and Biology; Springer Nature: Cham, Switzerland, 2006; Volume 581, pp. 265–269. [Google Scholar]
- Joo, H.; Melissa, D.; Dullaers, M.; Kim, T.-W.; Duluc, D.; Upchurch, K.; Xue, Y.; Zurawski, S.; Le Grand, R.; Liu, Y.-J.; et al. C-Type Lectin-like Receptor LOX-1 Promotes Dendritic Cell-Mediated Class-Switched B Cell Responses. Immunology 2014, 41, 592–604. [Google Scholar] [CrossRef]
- Sorokin, A.V.; Karathanasis, S.K.; Yang, Z.H.; Freeman, L.; Kotani, K.; Remaley, A.T. COVID-19—Associated dyslipidemia: Implications for mechanism of impaired resolution and novel therapeutic approaches. FASEB J. 2020, 34, 9843–9853. [Google Scholar] [CrossRef]
- Murphy, J.E.; Vohra, R.S.; Dunn, S.; Holloway, Z.G.; Monaco, A.P.; Homer-Vanniasinkam, S.; Walker, J.H.; Ponnambalam, S. Oxidised LDL internalisation by the LOX-1 scavenger receptor is dependent on a novel cytoplasmic motif and is regulated by dynamin-2. J. Cell Sci. 2008, 121, 2136–2147. [Google Scholar] [CrossRef]
- Gramberg, T.; Hofmann, H.; Möller, P.; Lalor, P.F.; Marzi, A.; Geier, M.; Krumbiegel, M.; Winkler, T.; Kirchhoff, F.; Adams, D.H.; et al. LSECtin interacts with filovirus glycoproteins and the spike protein of SARS coronavirus. Virology 2005, 340, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Han, X.; Zheng, X.; Wang, H.; Yang, Z.; Liu, D.; Han, K.; Liu, J.; Wang, X.; Yang, W.; et al. The myeloid LSECtin is a DAP12-coupled receptor that is crucial for inflammatory response induced by Ebola virus glycoprotein. PLoS Pathog. 2016, 12, e1005487. [Google Scholar]
- Domínguez-Soto, A.; Aragoneses-Fenoll, L.; Martín-Gayo, E.; Martínez-Prats, L.; Colmenares, M.; Naranjo-Gómez, M.; Borràs, F.E.; Muñoz, P.; Zubiaur, M.; Toribio, M.L.; et al. The DC-SIGN–related lectin LSECtin mediates antigen capture and pathogen binding by human myeloid cells. Blood 2007, 109, 5337–5345. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, Q.; Wang, X.; Jiang, X.; Zhao, D.; Lin, X.; He, F.; Tang, L. C-type lectin receptor LSECtin-mediated apoptotic cell clearance by macrophages directs intestinal repair in experimental colitis. Proc. Natl. Acad. Sci. USA 2018, 115, 11054–11059. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Manick, B.; Renelt, M.; Gerassenkov, T.; Bi, M.; Kalabokis, V.; Person, A.; Wu, G. LSECtin interacts with BTN3A1 to inhibit T cell activation. Am. Assoc. Immnol. 2020, 204, 78. [Google Scholar]
- Martina, B.E.E.; Haagmans, B.L.; Kuiken, T.; Fouchier, R.A.M.; Rimmelzwaan, G.F.; Van Amerongen, G.; Peiris, J.S.M.; Lim, W.; Osterhaus, A.D.M.E. SARS virus infection of cats and ferrets. Nat. Cell Biol. 2003, 425, 915. [Google Scholar] [CrossRef]
- Richard, M.; Kok, A.; de Meulder, D.; Bestebroer, T.M.; Lamers, M.M.; Okba, N.M.; van Vlissingen, M.F.; Rockx, B.; Haagmans, B.L.; Koopmans, M.P.; et al. SARS-CoV-2 is transmitted via contact and via the air between ferrets. BioRxiv 2020. [Google Scholar] [CrossRef]
- Upham, J.P.; Pickett, D.; Irimura, T.; Anders, E.M.; Reading, P.C. Macrophage Receptors for Influenza A Virus: Role of the Macrophage Galactose-Type Lectin and Mannose Receptor in Viral Entry. J. Virol. 2010, 84, 3730–3737. [Google Scholar] [CrossRef]
- Thépaut, M.; Luczkowiak, J.; Vivès, C.; Labiod, N.; Bally, I.; Lasala, F.; Grimoire, Y.; Fenel, D.; Sattin, S.; Thielens, N. DC/L-SIGN recognition of spike glycoprotein promotes SARS-CoV-2 trans-infection and can be inhibited by a glycomimetic antagonist. BioRxiv 2020. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; McKenzie, I.F. Role of the mannose receptor in the immune response. Curr. Mol. Med. 2001, 1, 469–474. [Google Scholar] [CrossRef]
- Sheng, K.C.; Kalkanidis, M.; Pouniotis, D.S.; Esparon, S.; Tang, C.K.; Apostolopoulos, V.; Pietersz, G.A. Delivery of antigen using a novel mannosylated dendrimer potentiates immunogenicity in vitro and in vivo. Eur. J. Immunol. 2008, 38, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.K.; Rajaram, M.V.S.; Schlesinger, L.S. Exploitation of the Macrophage Mannose Receptor (CD206) in Infectious Disease Diagnostics and Therapeutics. J. Cytol. Mol. Biol. 2014, 1. [Google Scholar] [CrossRef]
- Uslupehlivan, M.; Şener, E. Glycoinformatics approach for identifying target positions to inhibit initial binding of SARS-CoV-2 S1 protein to the host cell. BioRxiv 2020. [Google Scholar] [CrossRef]
- Nakaira-Takahagi, E.; Golim, M.A.; Bannwart, C.F.; Puccia, R.; Peraçoli, M.T. Interactions between TLR2, TLR4, and mannose receptors with gp43 from Paracoccidioides brasiliensis induce cytokine production by human monocytes. Med. Mycol. 2011, 49, 694–703. [Google Scholar]
- Ramirez-Ortiz, Z.G.; Means, T.K. The role of dendritic cells in the innate recognition of pathogenic fungi (A. fumigatus, C. neoformans and C. albicans). Virulence 2012, 3, 635–646. [Google Scholar] [CrossRef]
- Pitocco, D.; Tartaglione, L.; Viti, L.; Di Leo, M.; Pontecorvi, A.; Caputo, S. SARS-CoV-2 and DPP4 inhibition: Is it time to pray for Janus Bifrons? Diabetes Res. Clin. Pract. 2020, 163, 108162. [Google Scholar] [CrossRef]
- Lu, G.; Hu, Y.; Wang, Q.; Qi, J.; Gao, F.; Li, Y.; Zhang, Y.; Zhang, W.; Yuan, Y.; Bao, J. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013, 500, 227–231. [Google Scholar] [CrossRef]
- Vankadari, N.; Wilce, J.A. Emerging COVID-19 coronavirus: Glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg. Microbes Infect. 2020, 9, 601–604. [Google Scholar] [CrossRef]
- Smelcerovic, A.; Kocic, G.; Gajic, M.; Tomovic, K.; Djordjevic, V.; Stankovic-Djordjevic, D.; Anderluh, M. DPP-4 Inhibitors in the Prevention/Treatment of Pulmonary Fibrosis, Heart and Kidney Injury Caused by COVID-19—A Therapeutic Approach of Choice in Type 2 Diabetic Patients? Front. Pharmacol. 2020, 11, 1185. [Google Scholar] [CrossRef]
- Strollo, R.; Pozzilli, P. DPP4 inhibition: Preventing SARS-CoV-2 infection and/or progression of COVID-19? Diabetes/Metab. Res. Rev. 2020, 36, e3330. [Google Scholar] [CrossRef]
- Daly, J.; Simonetti, B.; Plagaro, A.; Shoemark, D.; Simon-Gracia, L.; Klein, K.; Bauer, M.; Hollandi, R.; Greber, U.; Horvath, P. Neuropilin-1 is a host factor for SARS-CoV-2 infection. BioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Ye, Q.; Chen, M.; Li, A.; Mi, W.; Fang, Y.; Zaytseva, Y.Y.; O’Connor, K.L.; Kooi, C.W.V.; Liu, S.; et al. N-glycosylation-defective splice variants of neuropilin-1 promote metastasis by activating endosomal signals. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Körner, S.; Thau-Habermann, N.; Kefalakes, E.; Bursch, F.; Petri, S. Expression of the axon-guidance protein receptor Neuropilin 1 is increased in the spinal cord and decreased in muscle of a mouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 2019, 49, 1529–1543. [Google Scholar] [CrossRef] [PubMed]
- Maden, C.H.; Gomes, J.; Schwarz, Q.; Davidson, K.; Tinker, A.; Ruhrberg, C. NRP1 and NRP2 cooperate to regulate gangliogenesis, axon guidance and target innervation in the sympathetic nervous system. Dev. Biol. 2012, 369, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Roth, L.; Prahst, C.; Ruckdeschel, T.; Savant, S.; Weström, S.; Fantin, A.; Riedel, M.; Héroult, M.; Ruhrberg, C.; Augustin, H.G. Neuropilin-1 mediates vascular permeability independently of vascular endothelial growth factor receptor-2 activation. Sci. Signal. 2016, 9, ra42. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Örd, M.; Faustova, I.; Loog, M. The sequence at Spike S1/S2 site enables cleavage by furin and phospho-regulation in SARS-CoV2 but not in SARS-CoV1 or MERS-CoV. Sci. Rep. 2020, 10, 1–10. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | Mechanism of Action | Reference |
|---|---|---|
| ACE2 | S protein receptor binding motif binds to the N-terminal extracellular catalytic ectodomain of ACE2 | [25] |
| DC-SIGN | Receptor binding domain of SARS-CoV-2 S protein | [35] |
| GRP78 | III and IV cyclic regions of S protein | [36] |
| L-SIGN | Receptor binding domain of SARS-CoV-2 S protein | [35] |
| MGL | N- and O-glycans present on the S1 of the S protein | [35] |
| MR | Mannose at N-glycosylation positions at N-terminal domain present on the S1 of S protein | [258] |
| Internalization of SARS-CoV-2 substrate | ||
| NRP1 | [266,271] | |
| TLR1/4/6 | Hydrogen bonding and hydrophobic interactions with S1 of S protein | [2] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gadanec, L.K.; McSweeney, K.R.; Qaradakhi, T.; Ali, B.; Zulli, A.; Apostolopoulos, V. Can SARS-CoV-2 Virus Use Multiple Receptors to Enter Host Cells? Int. J. Mol. Sci. 2021, 22, 992. https://doi.org/10.3390/ijms22030992
Gadanec LK, McSweeney KR, Qaradakhi T, Ali B, Zulli A, Apostolopoulos V. Can SARS-CoV-2 Virus Use Multiple Receptors to Enter Host Cells? International Journal of Molecular Sciences. 2021; 22(3):992. https://doi.org/10.3390/ijms22030992
Chicago/Turabian StyleGadanec, Laura Kate, Kristen Renee McSweeney, Tawar Qaradakhi, Benazir Ali, Anthony Zulli, and Vasso Apostolopoulos. 2021. "Can SARS-CoV-2 Virus Use Multiple Receptors to Enter Host Cells?" International Journal of Molecular Sciences 22, no. 3: 992. https://doi.org/10.3390/ijms22030992
APA StyleGadanec, L. K., McSweeney, K. R., Qaradakhi, T., Ali, B., Zulli, A., & Apostolopoulos, V. (2021). Can SARS-CoV-2 Virus Use Multiple Receptors to Enter Host Cells? International Journal of Molecular Sciences, 22(3), 992. https://doi.org/10.3390/ijms22030992