In Silico Identification of Potential Druggable Binding Sites on CIN85 SH3 Domain

 ,
,  , , , and
, , , and
Abstract
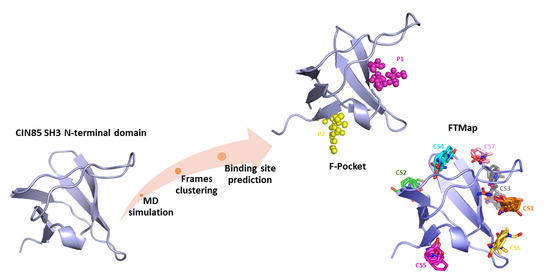
1. Introduction
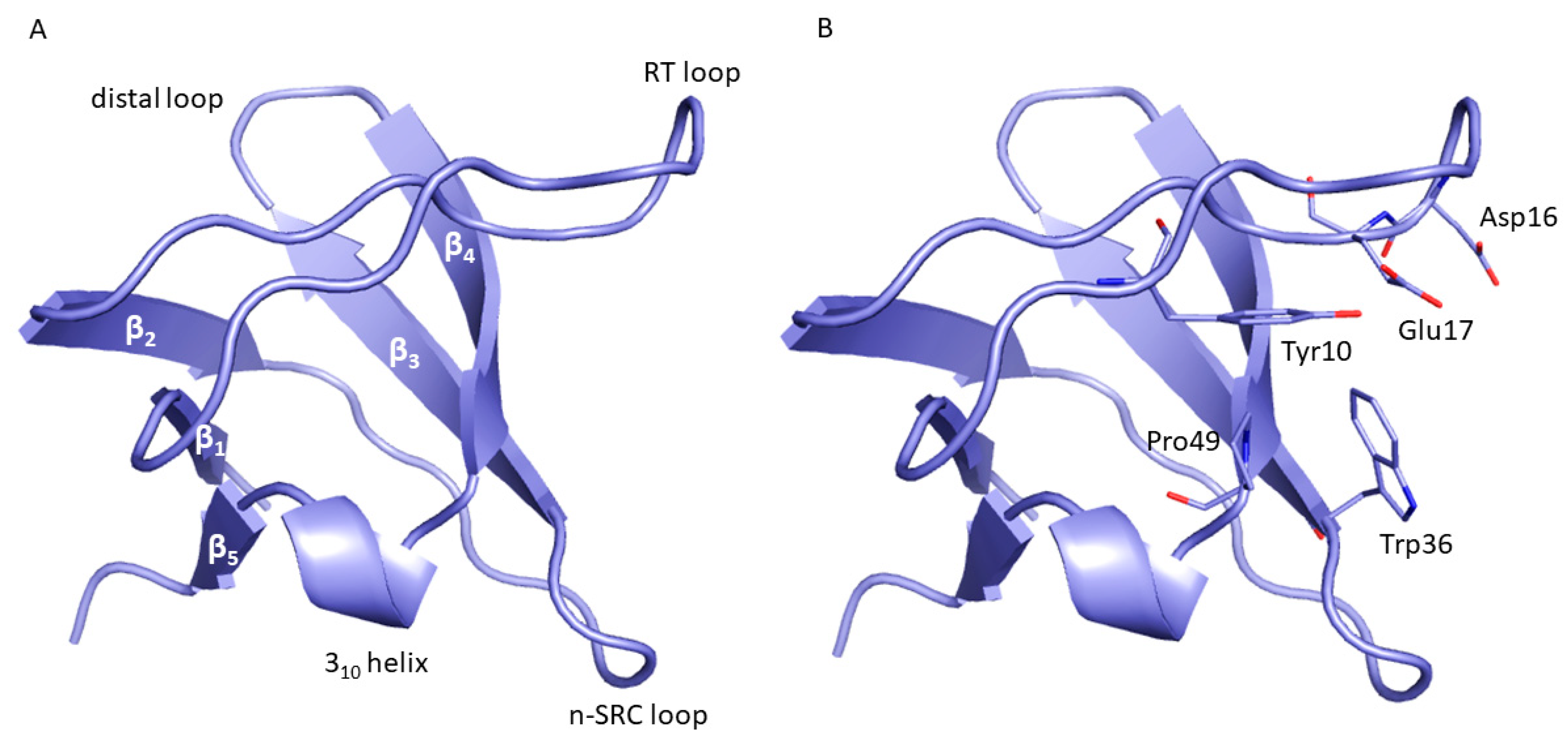
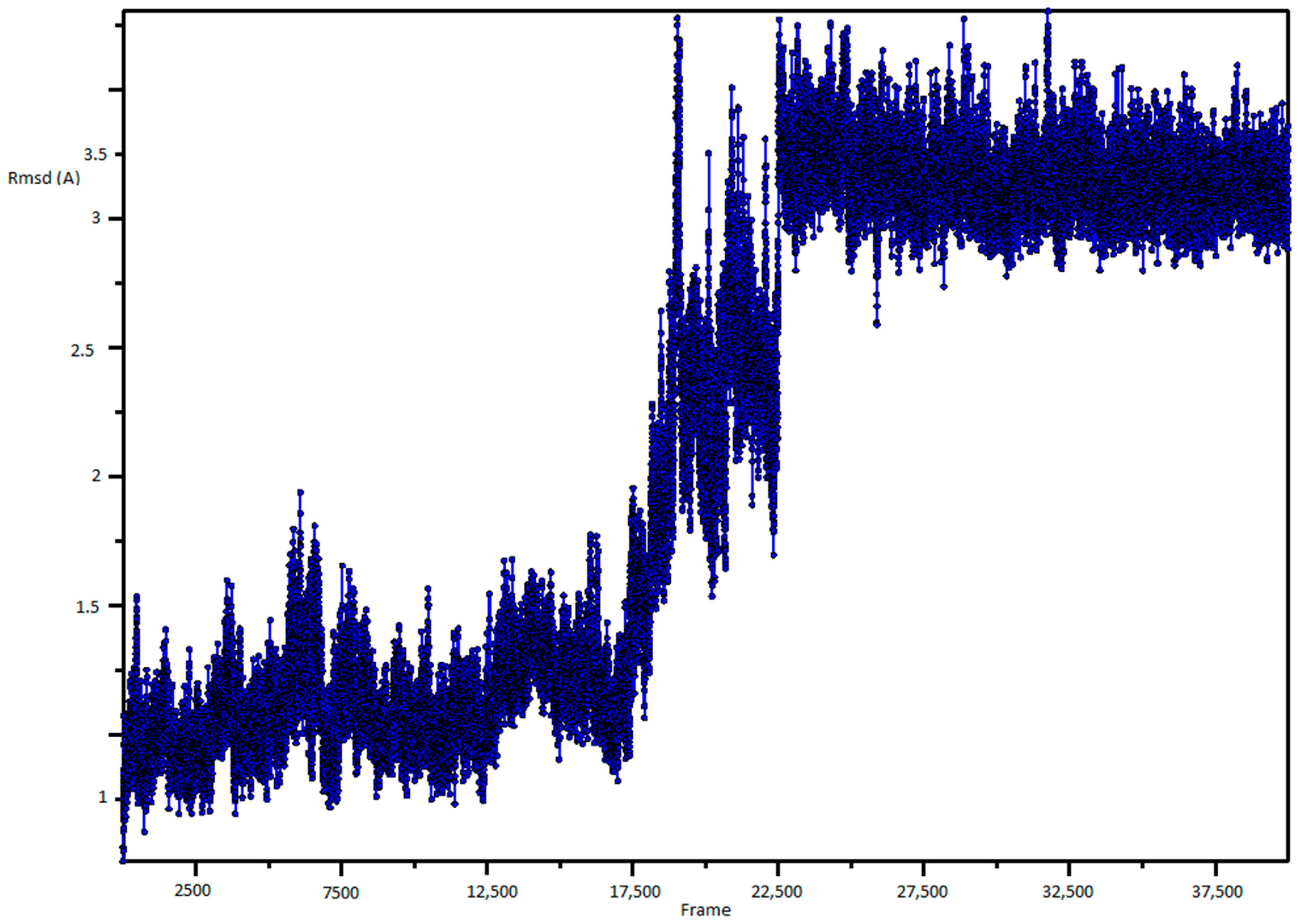
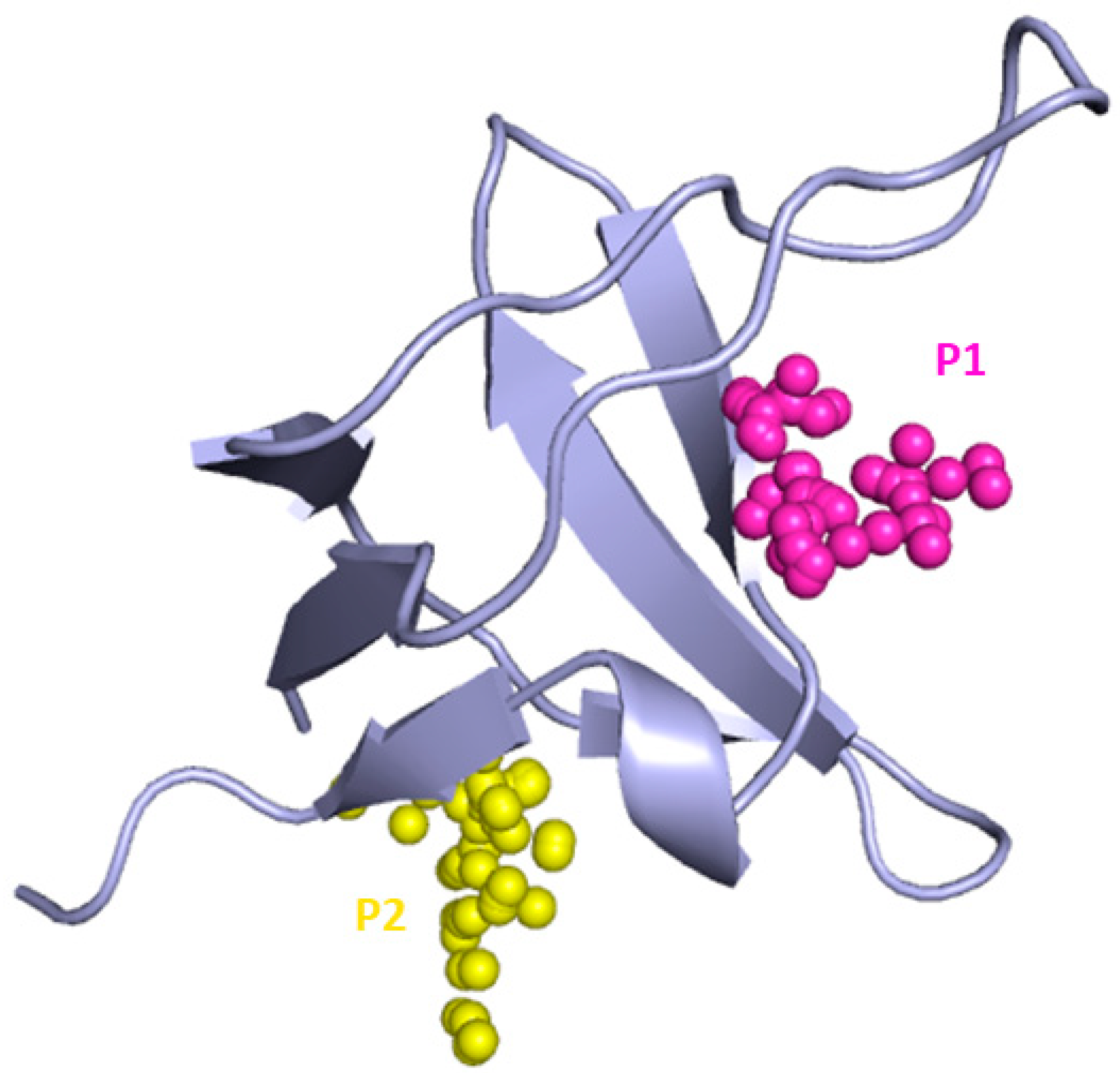
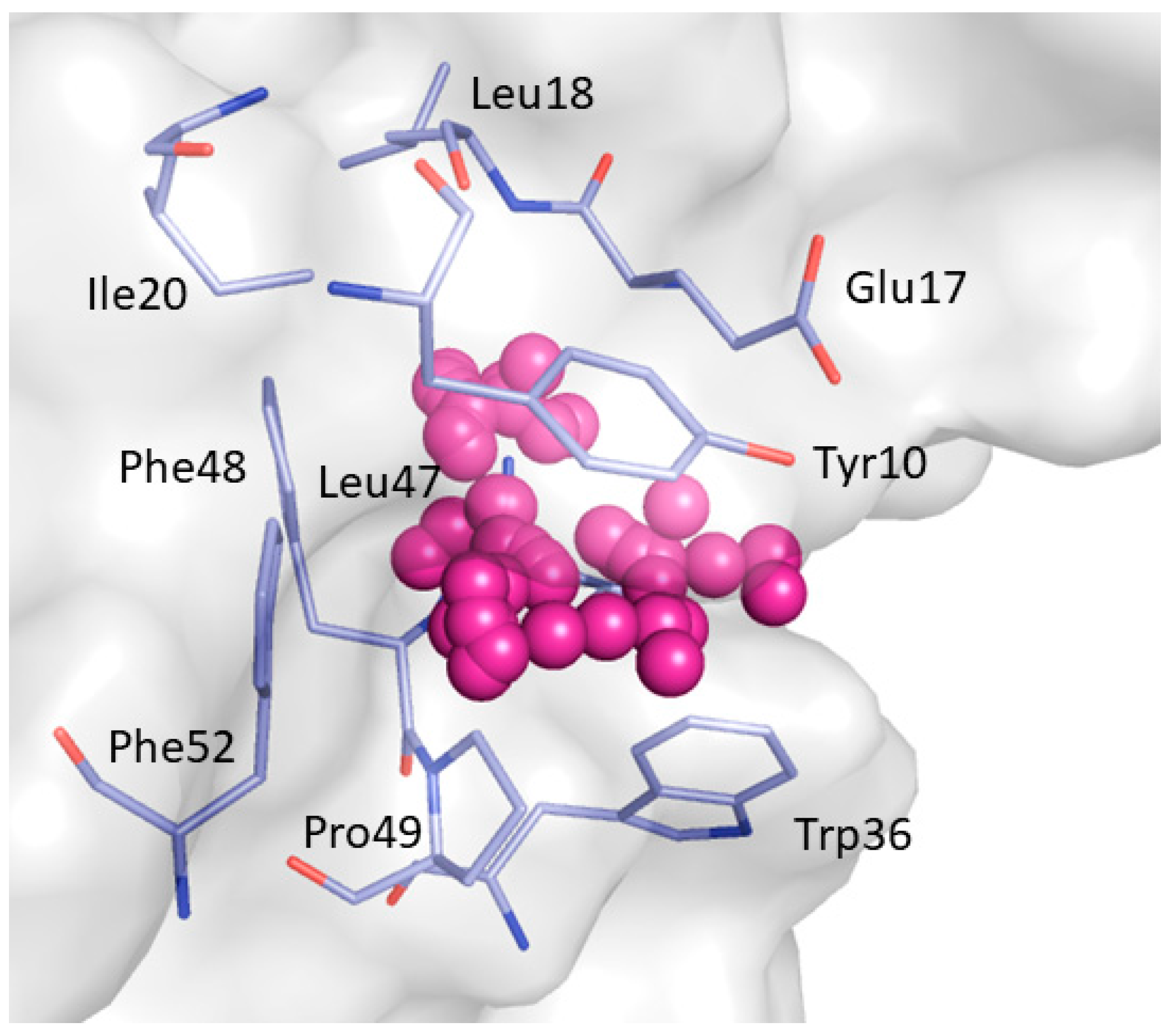
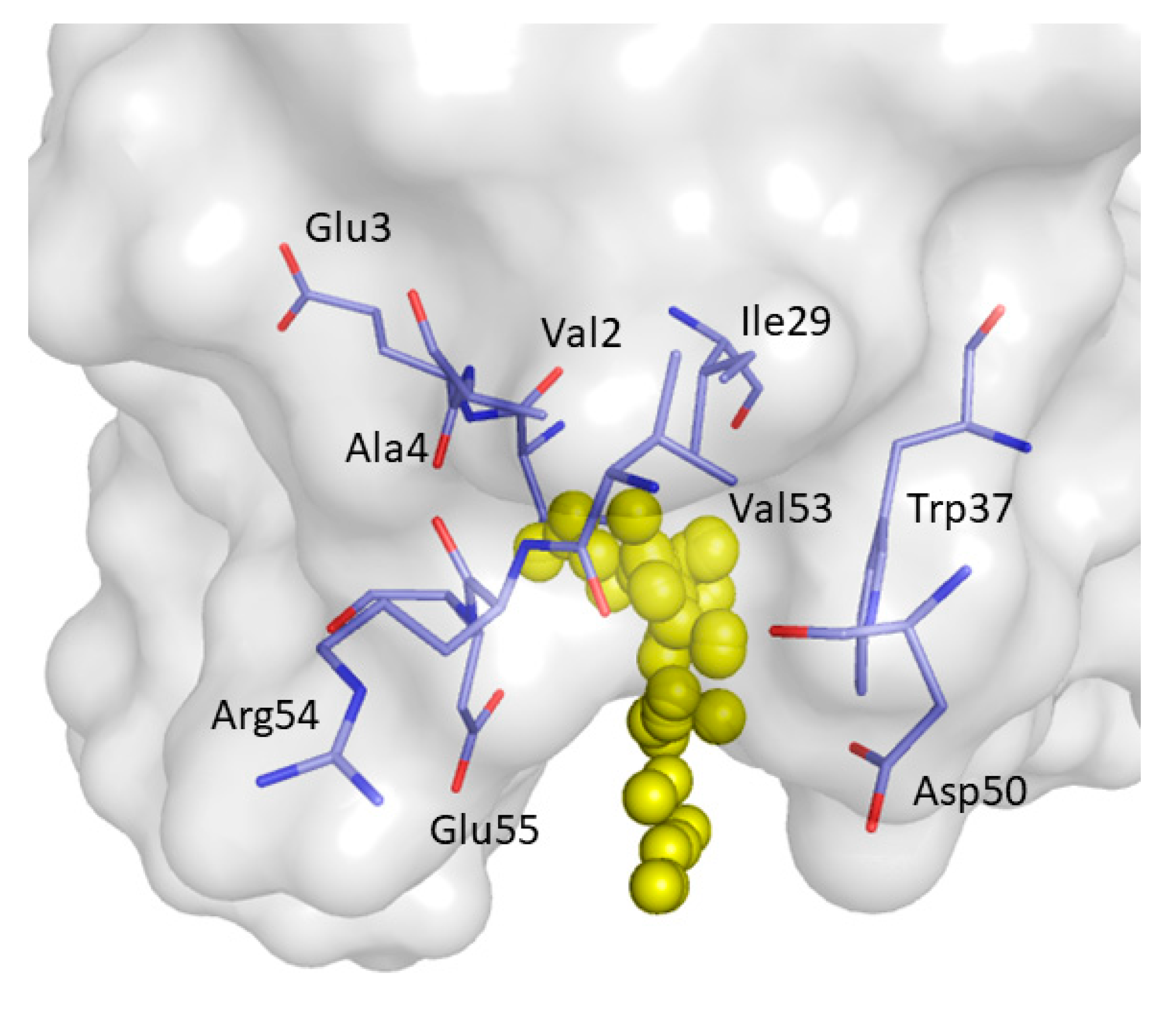
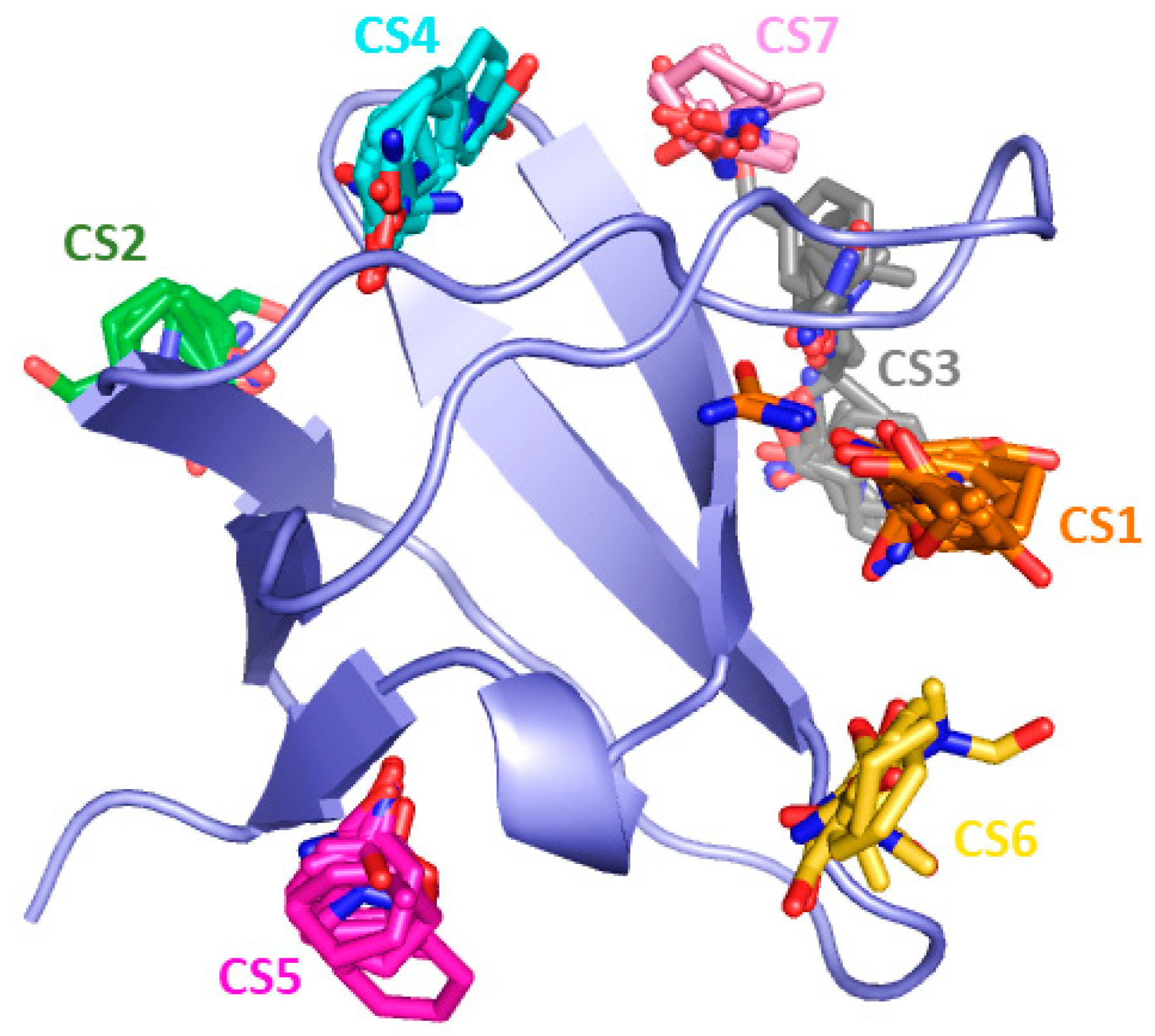
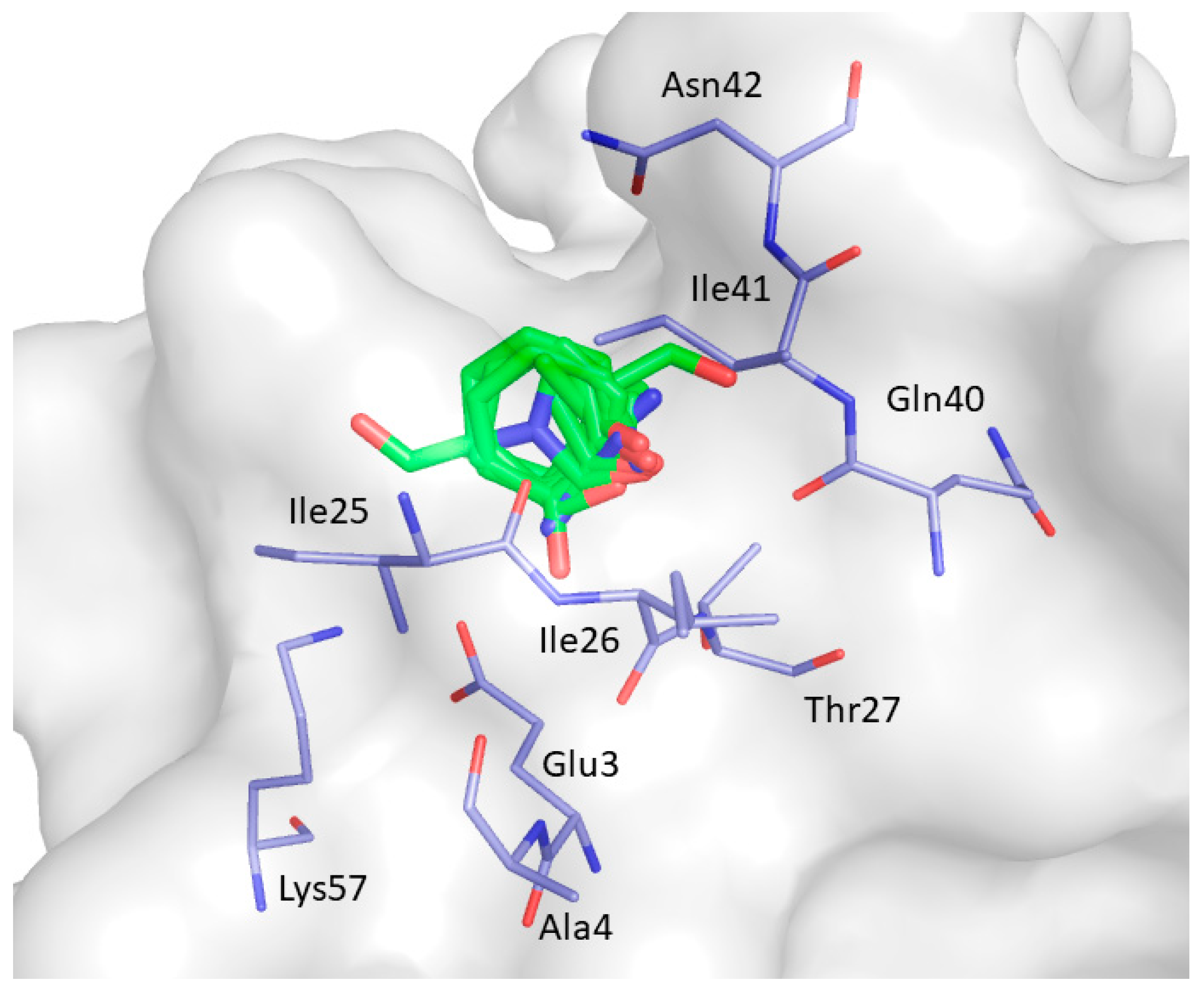
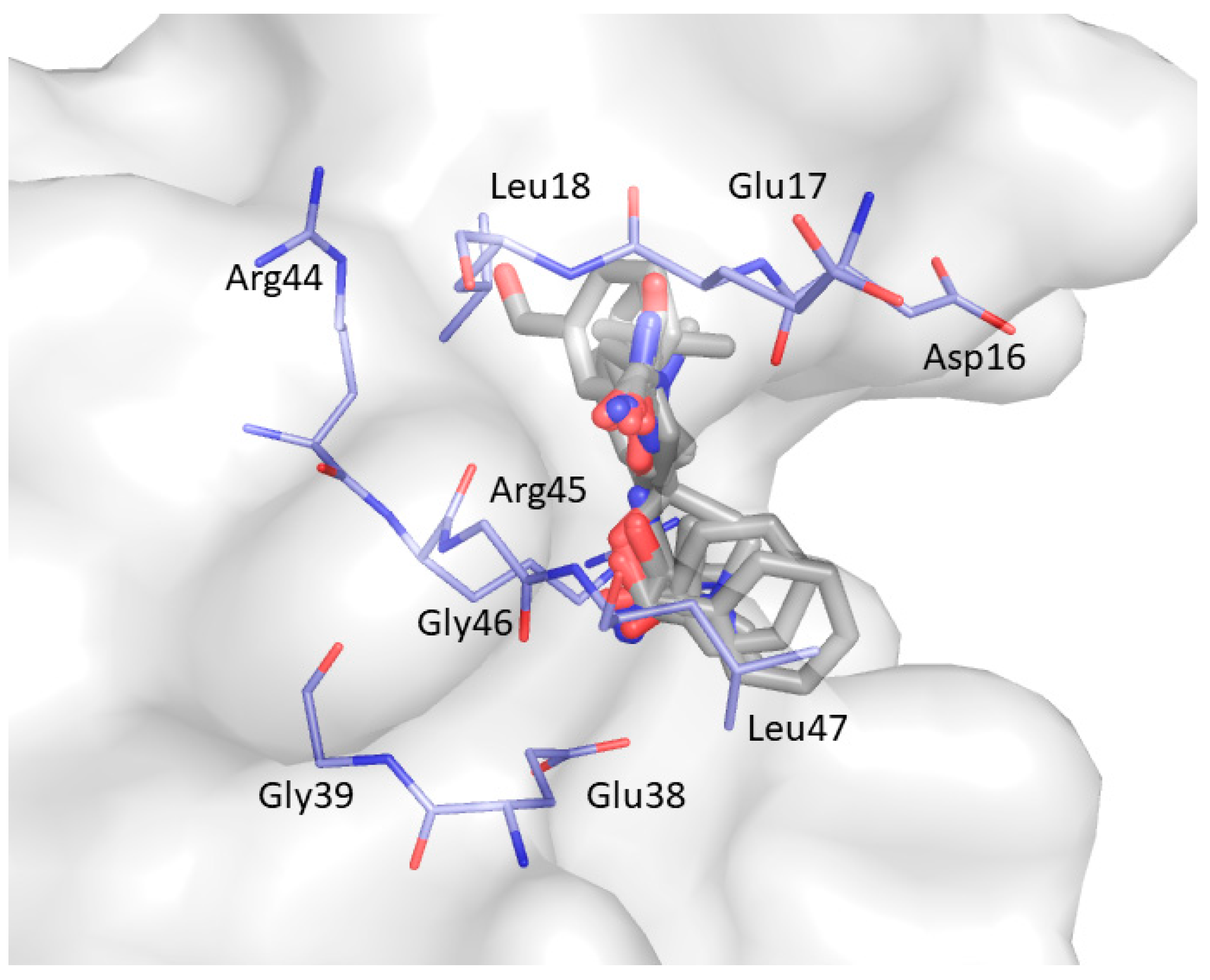
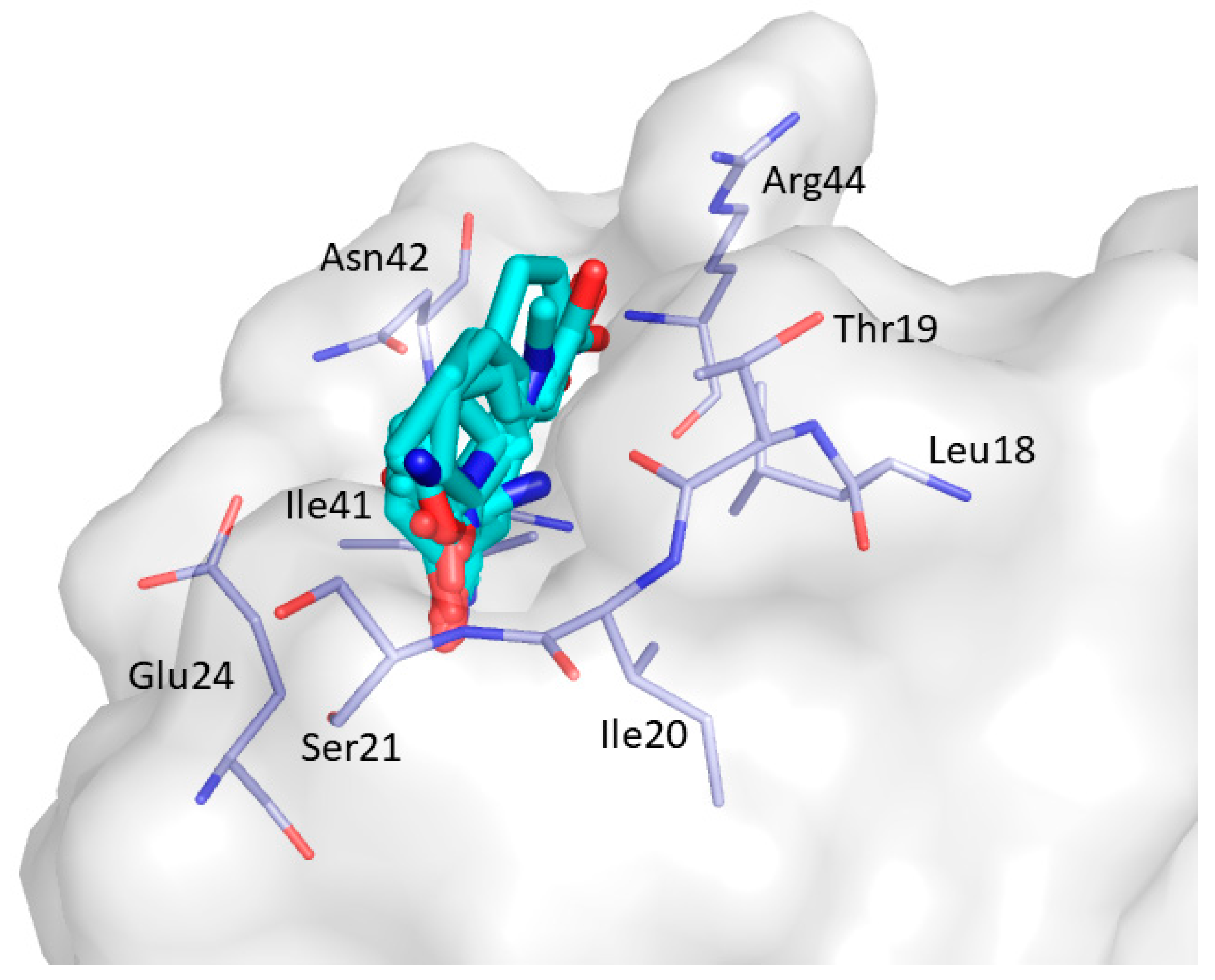
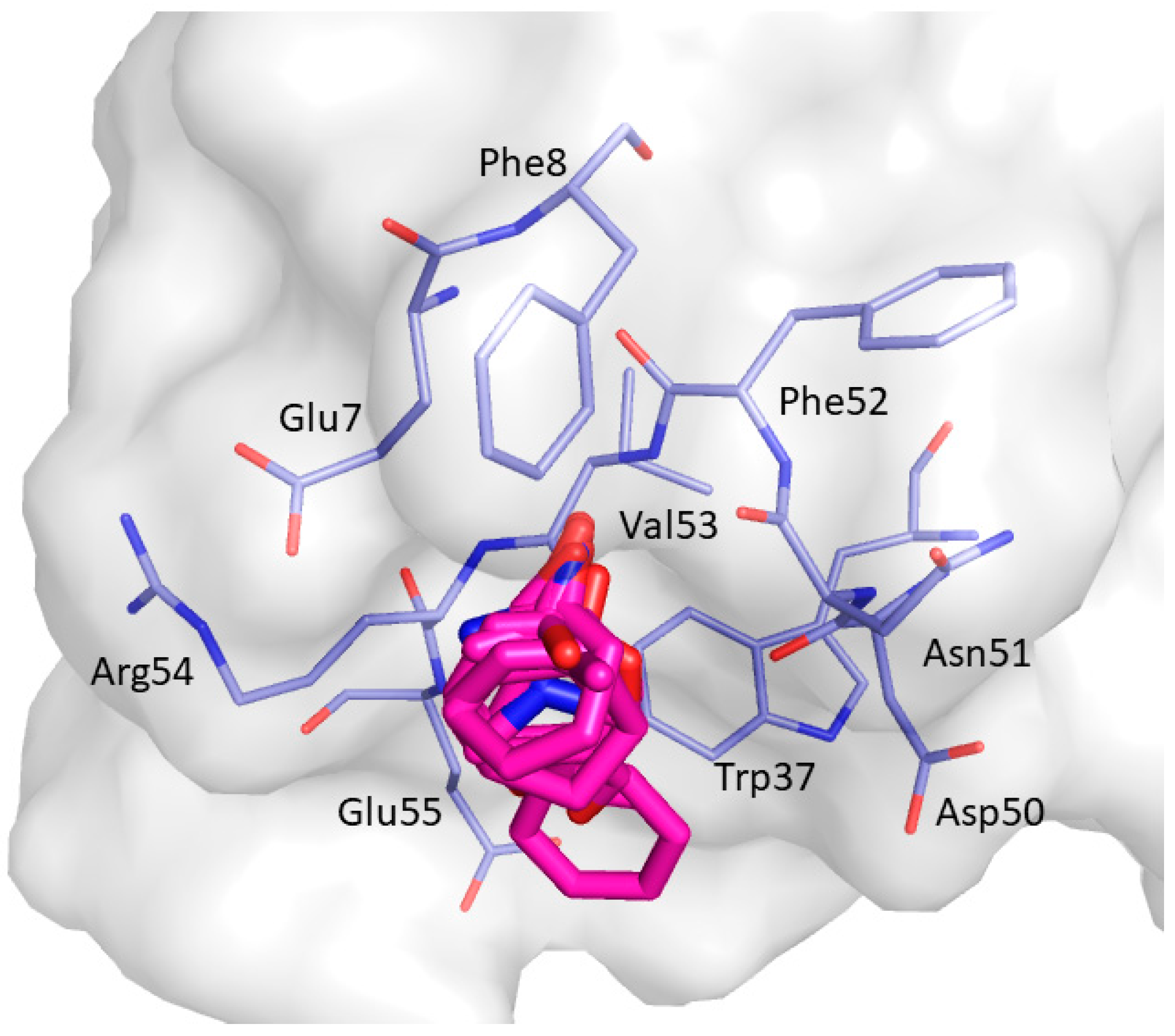
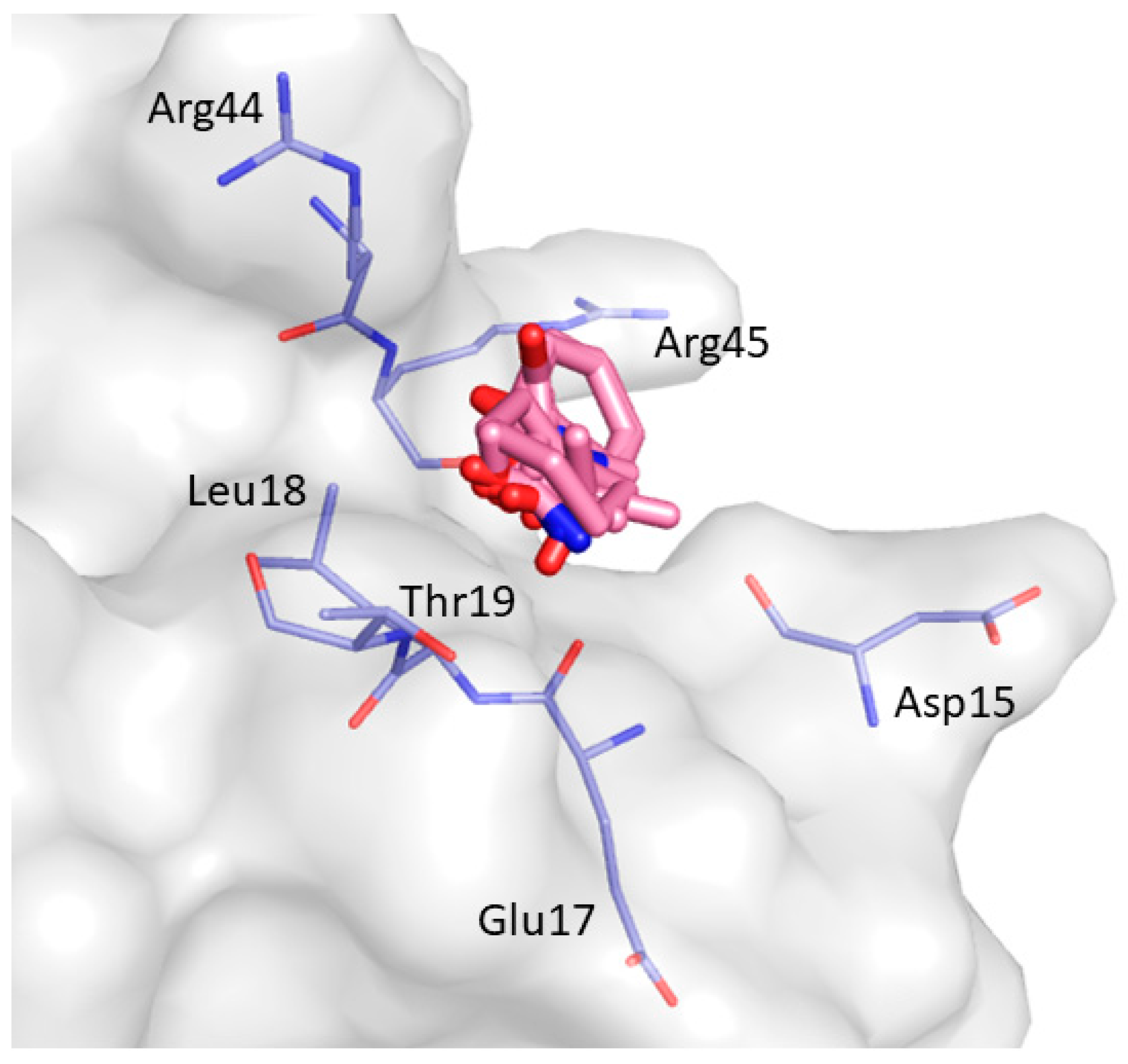
2. Results and Discussion
3. Material and Methods
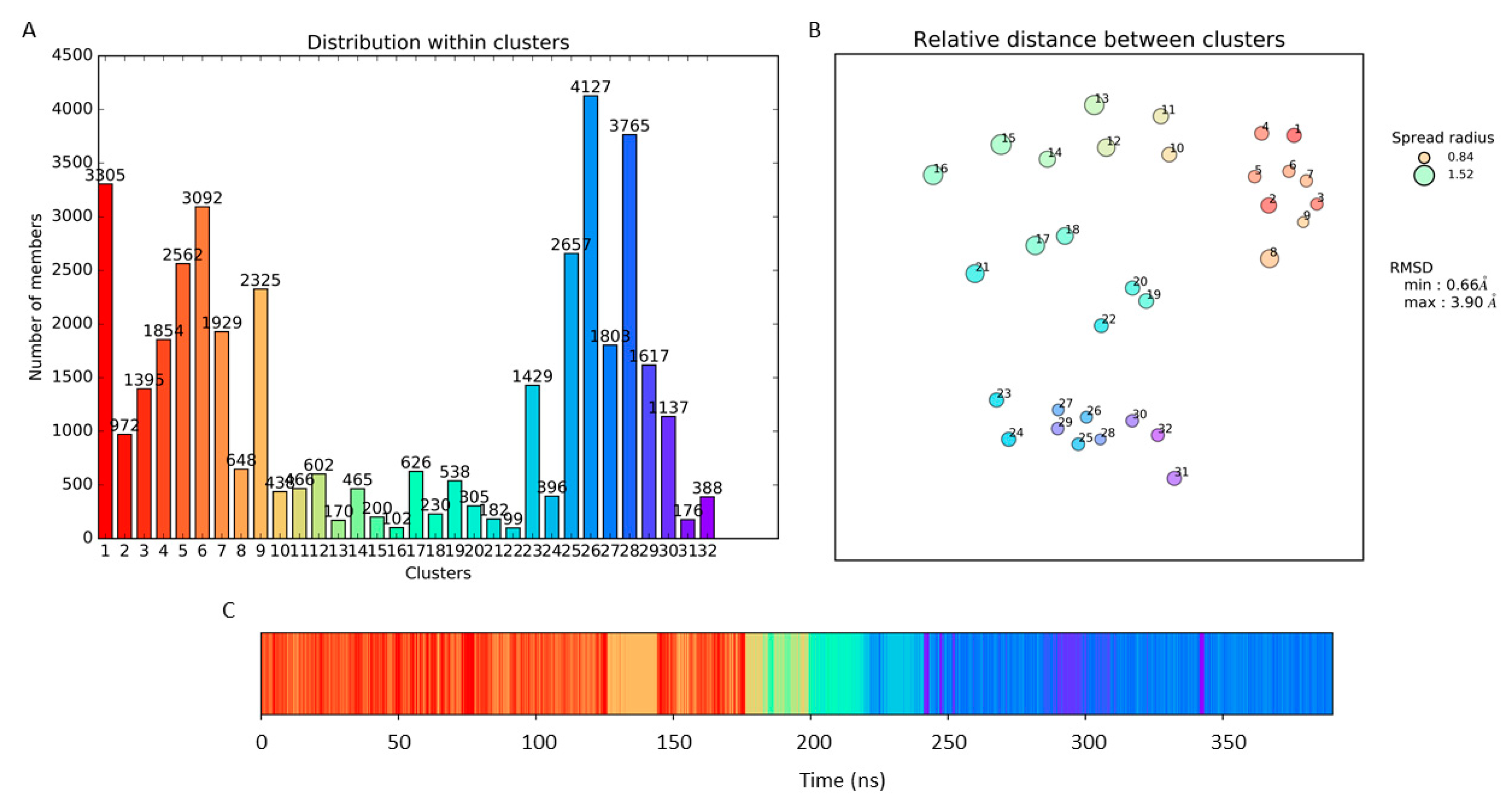
3.1. Molecular Dynamic Simulation and Frames Clustering
3.2. Binding Site Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PPI | Protein-protein interaction |
| VNTR | Variable number of tandem repeats regions |
| SH3 | Src homology 3 |
| MD | Molecular dynamics |
| RMSD | Root mean square deviation |
| C | Cluster |
| CS | Consensus site |
| VMD | Visual molecular dynamics |
References
- Bhattacharya, A.; Kim, Y.C.; Mittal, J. Protein-protein interactions in a crowded environment. Biophys. Rev. 2013, 5, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nat. Rev. Drug Discov. 2004, 3, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Bojadzic, D.; Buchwald, P. Toward Small-Molecule Inhibition of Protein-Protein Interactions: General Aspects and Recent Progress in Targeting Costimulatory and Coinhibitory (Immune Checkpoint) Interactions. Curr. Top. Med. Chem. 2018, 18, 674–699. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef]
- Mabonga, L.; Kappo, A.P. Protein-protein interaction modulators: Advances, successes and remaining challenges. Biophys. Rev. 2019, 11, 559–581. [Google Scholar] [CrossRef]
- Ran, X.; Gestwicki, J.E. Inhibitors of protein-protein interactions (PPIs): An analysis of scaffold choices and buried surface area. Curr. Opin. Chem. Biol. 2018, 44, 75–86. [Google Scholar] [CrossRef]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein-protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef]
- Cheng, S.S.; Yang, G.J.; Wang, W.; Leung, C.H.; Ma, D.L. The design and development of covalent protein-protein interaction inhibitors for cancer treatment. J. Hematol. Oncol. 2020, 13, 26. [Google Scholar] [CrossRef]
- Metz, A.; Pfleger, C.; Kopitz, H.; Pfeiffer-Marek, S.; Baringhaus, K.H.; Gohlke, H. Hot spots and transient pockets: Predicting the determinants of small-molecule binding to a protein-protein interface. J. Chem. Inf. Model. 2012, 52, 120–133. [Google Scholar] [CrossRef]
- Arkin, M.R.; Randal, M.; DeLano, W.L.; Hyde, J.; Luong, T.N.; Oslob, J.D.; Raphael, D.R.; Taylor, L.; Wang, J.; McDowell, R.S.; et al. Binding of small molecules to an adaptive protein-protein interface. Proc. Natl. Acad. Sci. USA 2003, 100, 1603–1608. [Google Scholar] [CrossRef]
- Eyrisch, S.; Helms, V. Transient pockets on protein surfaces involved in protein-protein interaction. J. Med. Chem. 2007, 50, 3457–3464. [Google Scholar] [CrossRef]
- Cascio, S.; Farkas, A.M.; Hughey, R.P.; Finn, O.J. Altered glycosylation of MUC1 influences its association with CIN85: The role of this novel complex in cancer cell invasion and migration. Oncotarget 2013, 4, 1686–1697. [Google Scholar] [CrossRef]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef]
- Dikic, I. CIN85/CMS family of adaptor molecules. FEBS Lett. 2002, 529, 110–115. [Google Scholar] [CrossRef]
- Saksela, K.; Permi, P. SH3 domain ligand binding: What’s the consensus and where’s the specificity? FEBS Lett. 2012, 586, 2609–2614. [Google Scholar] [CrossRef]
- Kurochkina, N.; Guha, U. SH3 domains: Modules of protein-protein interactions. Biophys. Rev. 2013, 5, 29–39. [Google Scholar] [CrossRef]
- Jozic, D.; Cárdenes, N.; Deribe, Y.L.; Moncalián, G.; Hoeller, D.; Groemping, Y.; Dikic, I.; Rittinger, K.; Bravo, J. CBL promotes clustering of endocytic adaptor proteins. Nat. Struct. Mol. Biol. 2005, 12, 972–979. [Google Scholar] [CrossRef]
- Kowanetz, K.; Szymkiewicz, I.; Haglund, K.; Kowanetz, M.; Husnjak, K.; Taylor, J.D.; Soubeyran, P.; Engstrom, U.; Ladbury, J.E.; Dikic, I. Identification of a novel proline-arginine motif involved in CIN85-dependent clustering of Cbl and down-regulation of epidermal growth factor receptors. J. Biol. Chem. 2003, 278, 39735–39746. [Google Scholar] [CrossRef]
- Kowanetz, K.; Husnjak, K.; Höller, D.; Kowanetz, M.; Soubeyran, P.; Hirsch, D.; Schmidt, M.H.H.; Pavelic, K.; De Camilli, P.; Randazzo, P.A.; et al. CIN85 associates with multiple effectors controlling intracellular trafficking of epidermal growth factor receptors. Mol. Biol. Cell 2004, 15, 3155–3166. [Google Scholar] [CrossRef]
- Cascio, S.; Sciurba, J.; Hughey, R.; Camacho, C.; Finn, O. Muc1/Cin85 complex is a new molecular target for control of cancer invasion and metastasis. In Proceedings of the 105th Annual Meeting of the American Association for Cancer Research AACR, San Diego, CA, USA, 5–9 April 2014; p. 3151. [Google Scholar]
- Ivetac, A.; McCammon, J.A. Mapping the druggable allosteric space of G-protein coupled receptors: A fragment-based molecular dynamics approach. Chem. Biol. Drug Des. 2010, 76, 201–217. [Google Scholar] [CrossRef]
- Landon, M.R.; Amaro, R.E.; Baron, R.; Ngan, C.H.; Ozonoff, D.; McCammon, J.A.; Vajda, S. Novel druggable hot spots in avian influenza neuraminidase H5N1 revealed by computational solvent mapping of a reduced and representative receptor ensemble. Chem. Biol. Drug Des. 2008, 71, 106–116. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- Tubiana, T.; Carvaillo, J.C.; Boulard, Y.; Bressanelli, S. TTClust: A Versatile Molecular Simulation Trajectory Clustering Program with Graphical Summaries. J. Chem. Inf. Model. 2018, 58, 2178–2182. [Google Scholar] [CrossRef]
- Philippe, D.L.; Ladbury, J.E.; Pfuhl, M. PDB ID: 2K9G. Solution Structure of the Third SH3 Domain of the Cin85 Adapter Protein. Released: 13 October 2009. Available online: https://www.rcsb.org/structure/2K9G (accessed on 10 December 2020). [CrossRef]
- Schmidtke, P.; Barril, X. Understanding and predicting druggability. A high-throughput method for detection of drug binding sites. J. Med. Chem. 2010, 53, 5858–5867. [Google Scholar] [CrossRef]
- Eastman, P.; Friedrichs, M.S.; Chodera, J.D.; Radmer, R.J.; Bruns, C.M.; Ku, J.P.; Beauchamp, K.A.; Lane, T.J.; Wang, L.P.; Shukla, D.; et al. OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J. Chem. Theory Comput. 2013, 9, 461–469. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 27–38. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Bonded Interactions | H-Bonding Interactions |
|---|---|
| Phe8, Tyr10, Gln13, Asp16, Glu17, Leu18, Glu24, Ile25, Trp36, Trp37, Glu38, Asn42 | Tyr10, Gln11, Gln13, Glu17 Thr19, Ser21, Ile25, Thr27, Asn42, Arg44, Arg45, Leu47, Asp50, Val53, Glu55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vittorio, S.; Seidel, T.; Garon, A.; Gitto, R.; Langer, T.; De Luca, L. In Silico Identification of Potential Druggable Binding Sites on CIN85 SH3 Domain. Int. J. Mol. Sci. 2021, 22, 534. https://doi.org/10.3390/ijms22020534
Vittorio S, Seidel T, Garon A, Gitto R, Langer T, De Luca L. In Silico Identification of Potential Druggable Binding Sites on CIN85 SH3 Domain. International Journal of Molecular Sciences. 2021; 22(2):534. https://doi.org/10.3390/ijms22020534
Chicago/Turabian StyleVittorio, Serena, Thomas Seidel, Arthur Garon, Rosaria Gitto, Thierry Langer, and Laura De Luca. 2021. "In Silico Identification of Potential Druggable Binding Sites on CIN85 SH3 Domain" International Journal of Molecular Sciences 22, no. 2: 534. https://doi.org/10.3390/ijms22020534
APA StyleVittorio, S., Seidel, T., Garon, A., Gitto, R., Langer, T., & De Luca, L. (2021). In Silico Identification of Potential Druggable Binding Sites on CIN85 SH3 Domain. International Journal of Molecular Sciences, 22(2), 534. https://doi.org/10.3390/ijms22020534