E3 Ubiquitin Ligases: Key Regulators of TGFβ Signaling in Cancer Progression


Abstract
1. Introduction
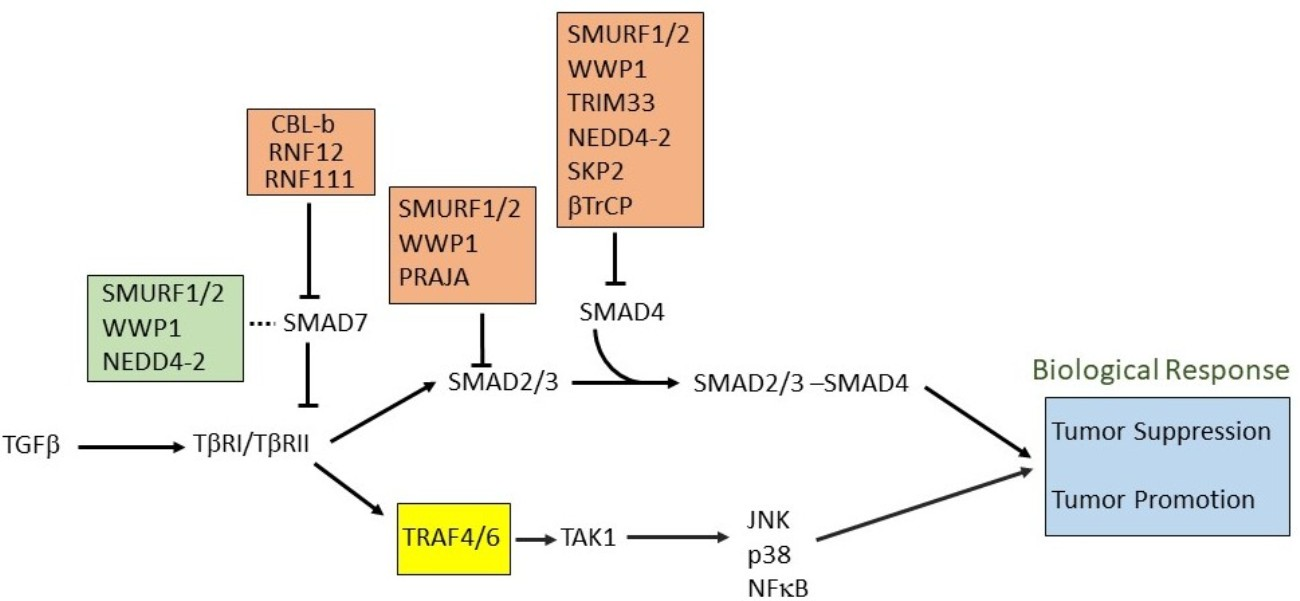
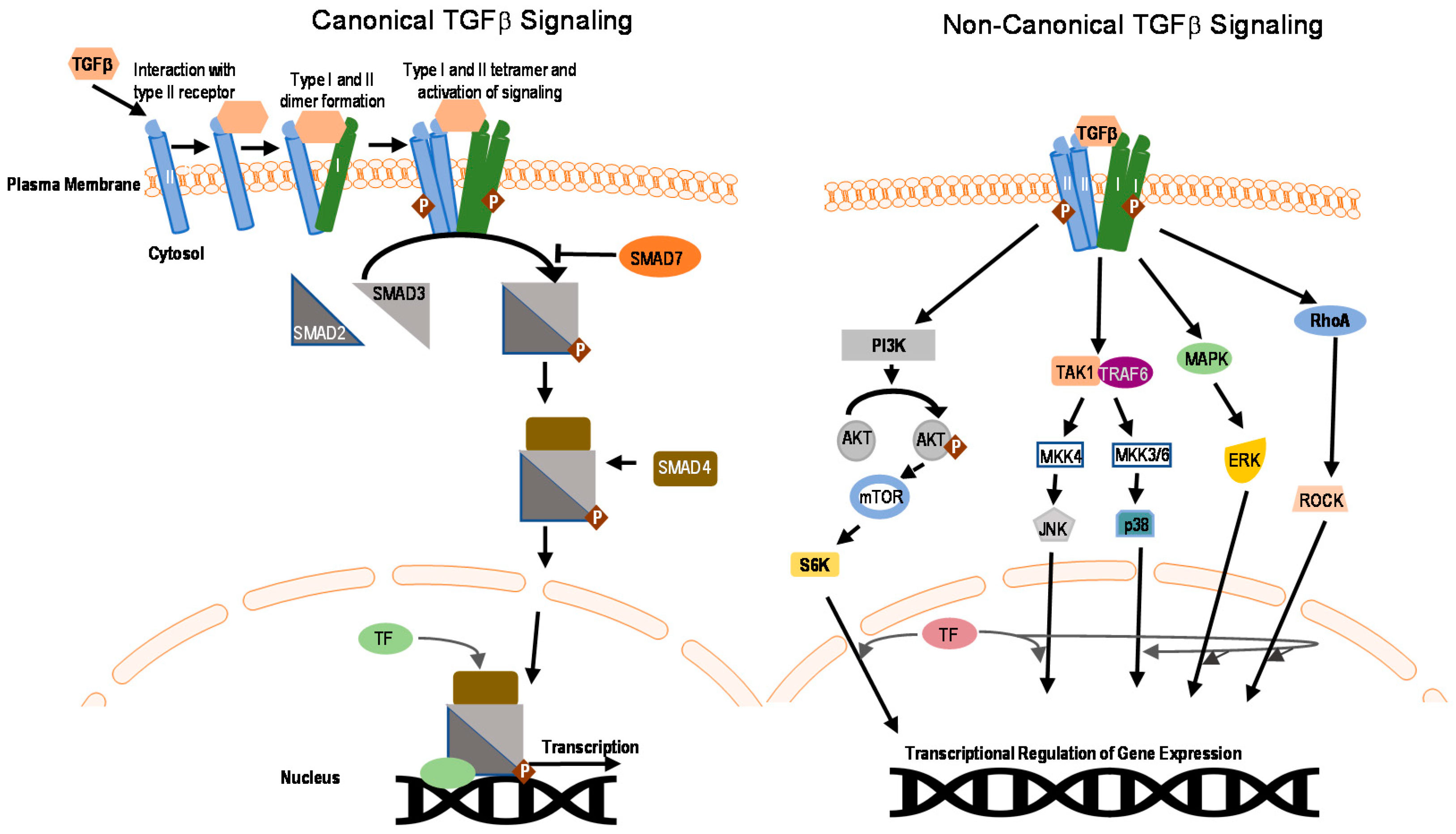
2. SMAD and Non-SMAD Signaling
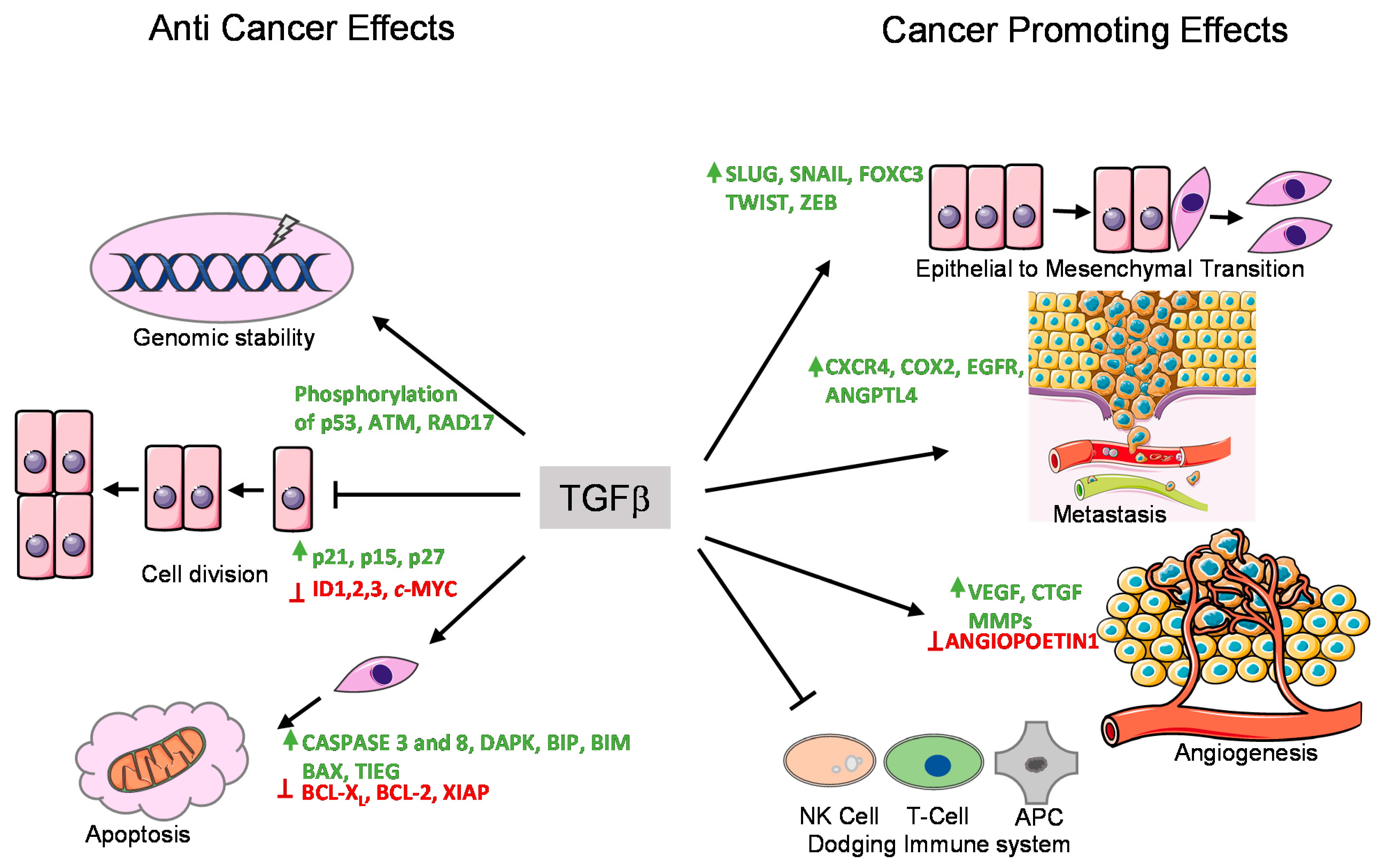
3. TGFβ as Tumor Suppressor and Tumor Promoter
3.1. Epithelial-to-Mesenchymal Transition (EMT) and Invasion
3.2. Promoting Angiogenesis
3.3. Immunomodulatory Effects
4. Controlling Protein Function by Ubiquitination
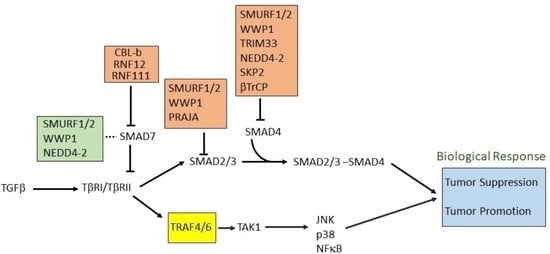
5. E3 Ligases and Their Involvement in TGFβ Signaling
5.1. Role of HECT E3 Ligases in TGFβ Signaling Pathway
5.2. Role of RING E3 Ligases in the TGFβ Signaling Pathway
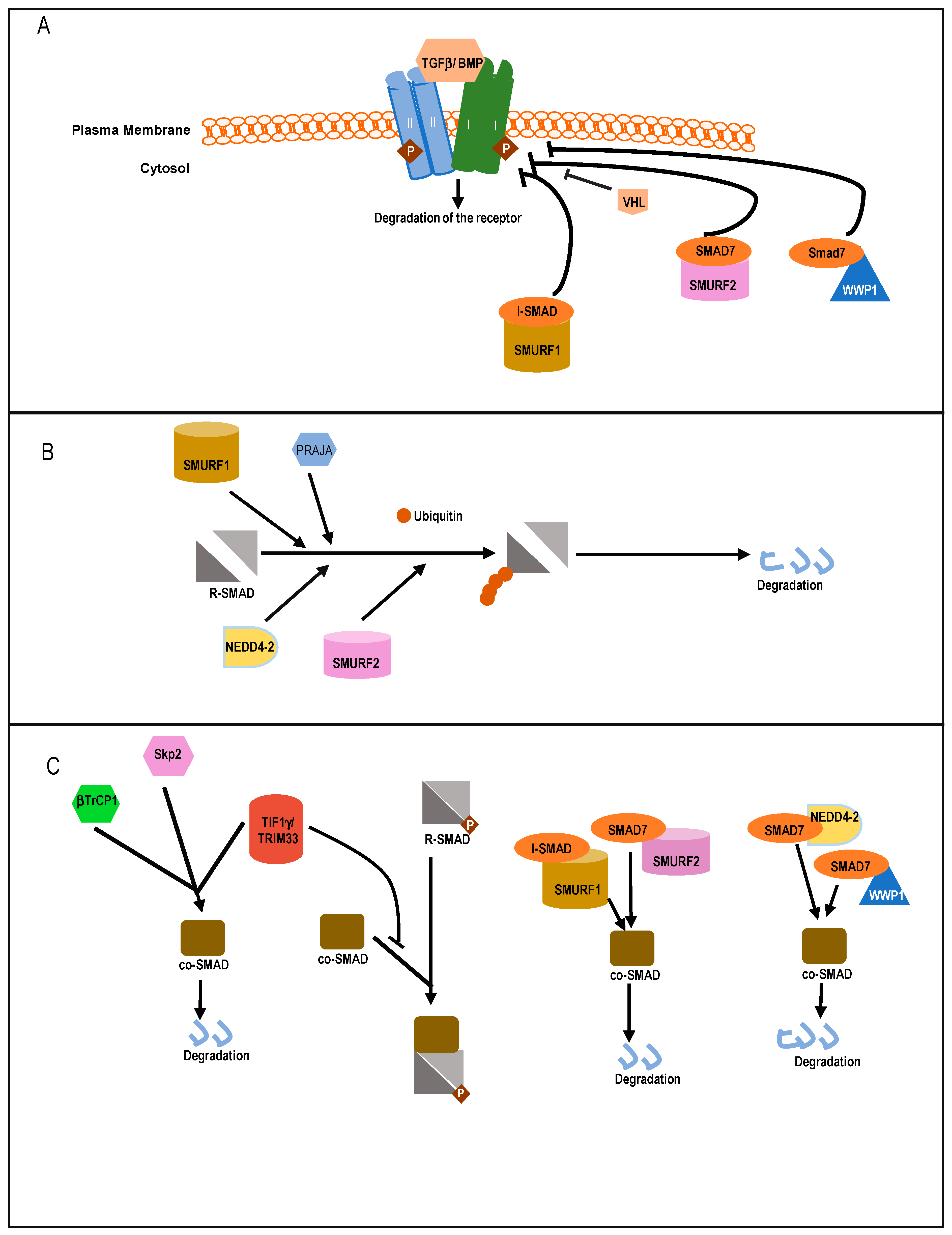
5.2.1. Targeting Receptors Directly
5.2.2. Targeting R-SMADs
5.2.3. Acting on Co-SMAD
5.2.4. Regulation of I-SMADs
5.3. Other E3 Ligases
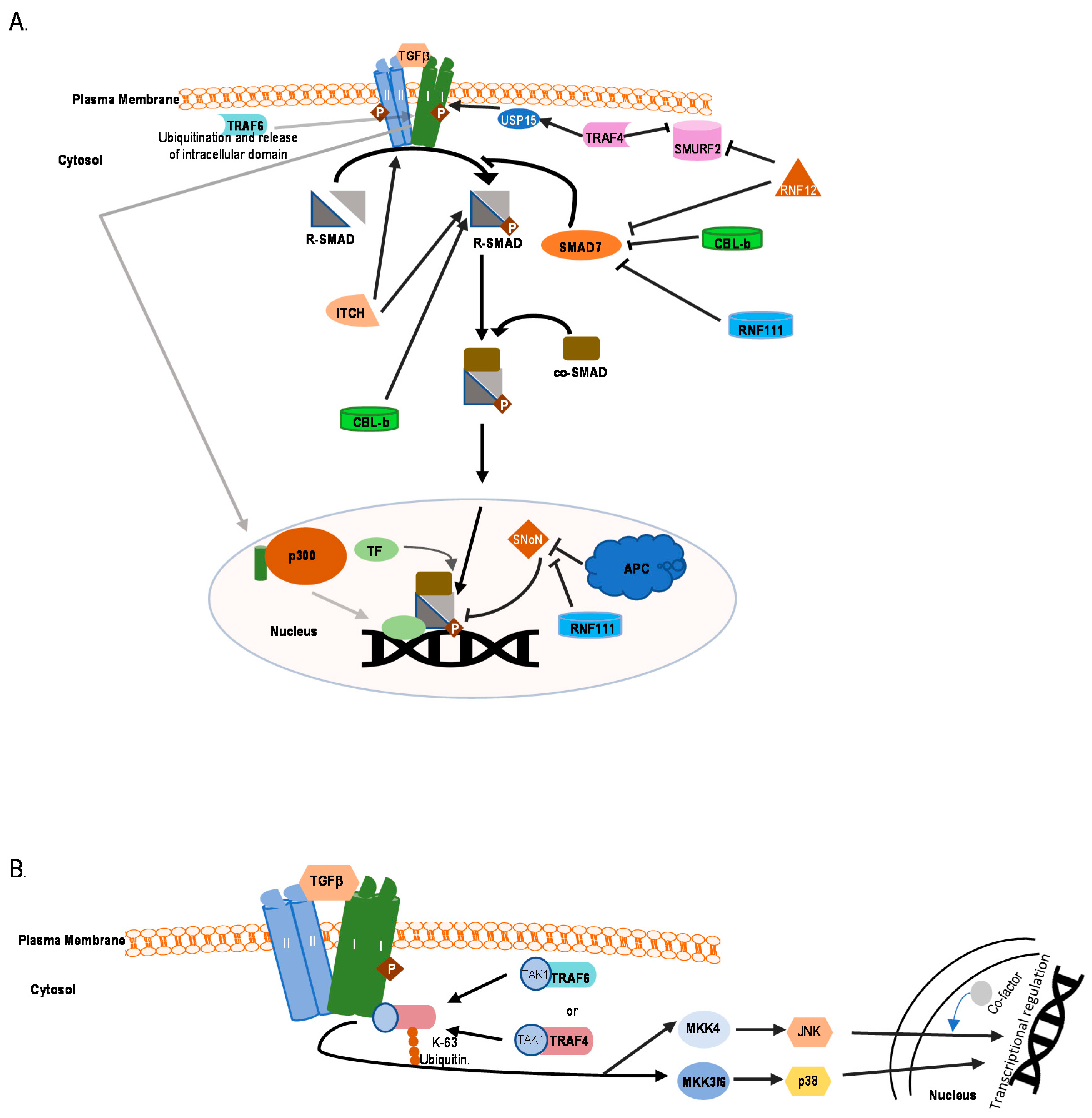
6. E3 Ubiquitin Ligases in Non-Canonical TGFβ Pathways
7. Role of E3 Ligases Associated with TGFβ Signaling in Mediating Tumorigenesis
7.1. SMURF1 and SMURF2
7.2. WWP1
7.3. NEDD4-2
7.4. TRAFs
7.5. TRIMs
7.6. RNFs
7.7. Other E3 Ligases
8. E3 Ligases as Emerging Drug Targets
9. Emerging Roles of PROTACs and Molecular Glues
10. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Twardzik, D.R.; Brown, J.P.; Ranchalis, J.E.; Todaro, G.J.; Moss, B. Vaccinia virus-infected cells release a novel polypeptide functionally related to transforming and epidermal growth factors. Proc. Natl. Acad. Sci. USA 1985, 82, 5300–5304. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Anzano, M.A.; Meyers, C.A.; Wideman, J.; Blacher, R.; Pan, Y.C.; Stein, S.; Lehrman, S.R.; Smith, J.M.; Lamb, L.C.; et al. Purification and properties of a type beta transforming growth factor from bovine kidney. Biochemistry 1983, 22, 5692–5698. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, M.; Moustakas, A.; Lin, H.Y.; Lodish, H.F.; Carr, B.I. Growth inhibition by transforming growth factor beta (TGF-β) type I is restored in TGF-β-resistant hepatoma cells after expression of TGF-beta receptor type II cDNA. Proc. Natl. Acad. Sci. USA 1993, 90, 5359–5363. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. The transforming growth factor-β family. Annu. Rev. Cell Biol. 1990, 6, 597–641. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-β family signaling. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.D.; Davies, D.R. The cystine-knot growth-factor superfamily. Annu. Rev. Biophys. Biomol. Struct. 1995, 24, 269–291. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- Levy, L.; Hill, C.S. Alterations in components of the TGF-β superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006, 17, 41–58. [Google Scholar] [CrossRef]
- Massague, J. TGF-β signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Lebrun, J.J.; Vale, W.W. Activin and inhibin have antagonistic effects on ligand-dependent heteromerization of the type I and type II activin receptors and human erythroid differentiation. Mol. Cell. Biol. 1997, 17, 1682–1691. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; ten Dijke, P.; Franzen, P.; Miyazono, K.; Heldin, C.H. Formation of hetero-oligomeric complexes of type I and type II receptors for transforming growth factor-β. J. Biol. Chem. 1994, 269, 20172–20178. [Google Scholar] [CrossRef]
- Weis-Garcia, F.; Massague, J. Complementation between kinase-defective and activation-defective TGF-β receptors reveals a novel form of receptor cooperativity essential for signaling. EMBO J. 1996, 15, 276–289. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massague, J. Mechanism of activation of the TGF-β receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef]
- Wieser, R.; Wrana, J.L.; Massague, J. GS domain mutations that constitutively activate TβR-I, the downstream signaling component in the TGF-β receptor complex. EMBO J. 1995, 14, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-beta Family. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Hata, A.; Chen, Y.G. TGF-β Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- David, C.J.; Massague, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Luo, K. Signaling Cross Talk between TGF-β/Smad and Other Signaling Pathways. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Xu, P.; Liu, J.; Derynck, R. Post-translational regulation of TGF-β receptor and Smad signaling. FEBS Lett. 2012, 586, 1871–1884. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, P.V. Regulation of Ubiquitin Enzymes in the TGF-β Pathway. Int. J. Mol. Sci. 2017, 18, 877. [Google Scholar] [CrossRef] [PubMed]
- Tu, A.W.; Luo, K. Acetylation of Smad2 by the co-activator p300 regulates activin and transforming growth factor beta response. J. Biol. Chem. 2007, 282, 21187–21196. [Google Scholar] [CrossRef] [PubMed]
- Simonsson, M.; Kanduri, M.; Gronroos, E.; Heldin, C.H.; Ericsson, J. The DNA binding activities of Smad2 and Smad3 are regulated by coactivator-mediated acetylation. J. Biol. Chem. 2006, 281, 39870–39880. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Saunier, E.F.; Akhurst, R.J.; Derynck, R. The type I TGF-β receptor is covalently modified and regulated by sumoylation. Nat. Cell Biol. 2008, 10, 654–664. [Google Scholar] [CrossRef]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGFβ receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar] [CrossRef]
- Kuratomi, G.; Komuro, A.; Goto, K.; Shinozaki, M.; Miyazawa, K.; Miyazono, K.; Imamura, T. NEDD4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates TGF-β (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and TGF-β type I receptor. Biochem. J. 2005, 386, 461–470. [Google Scholar] [CrossRef]
- Wicks, S.J.; Haros, K.; Maillard, M.; Song, L.; Cohen, R.E.; Dijke, P.T.; Chantry, A. The deubiquitinating enzyme UCH37 interacts with Smads and regulates TGF-β signalling. Oncogene 2005, 24, 8080–8084. [Google Scholar] [CrossRef]
- De Boeck, M.; Ten Dijke, P. Key role for ubiquitin protein modification in TGFβ signal transduction. Ups. J. Med. Sci. 2012, 117, 153–165. [Google Scholar] [CrossRef]
- Imamura, T.; Oshima, Y.; Hikita, A. Regulation of TGF-β family signalling by ubiquitination and deubiquitination. J. Biochem. 2013, 154, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; de Boeck, M.; van Dam, H.; Ten Dijke, P. Regulation of the TGF-β pathway by deubiquitinases in cancer. Int. J. Biochem. Cell Biol. 2016, 76, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Herhaus, L.; Sapkota, G.P. The emerging roles of deubiquitylating enzymes (DUBs) in the TGFβ and BMP pathways. Cell. Signal. 2014, 26, 2186–2192. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Baek, K.H. TGF-β signaling pathway mediated by deubiquitinating enzymes. Cell. Mol. Life Sci. 2019, 76, 653–665. [Google Scholar] [CrossRef]
- Abdollah, S.; Macias-Silva, M.; Tsukazaki, T.; Hayashi, H.; Attisano, L.; Wrana, J.L. TβRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J. Biol. Chem. 1997, 272, 27678–27685. [Google Scholar] [CrossRef]
- Souchelnytskyi, S.; Tamaki, K.; Engstrom, U.; Wernstedt, C.; ten Dijke, P.; Heldin, C.H. Phosphorylation of Ser465 and Ser467 in the C terminus of Smad2 mediates interaction with Smad4 and is required for transforming growth factor-β signaling. J. Biol. Chem. 1997, 272, 28107–28115. [Google Scholar] [CrossRef]
- Sanchez-Duffhues, G.; Williams, E.; Goumans, M.J.; Heldin, C.H.; Ten Dijke, P. Bone morphogenetic protein receptors: Structure, function and targeting by selective small molecule kinase inhibitors. Bone 2020, 138, 115472. [Google Scholar] [CrossRef]
- Lagna, G.; Hata, A.; Hemmati-Brivanlou, A.; Massague, J. Partnership between DPC4 and SMAD proteins in TGF-β signalling pathways. Nature 1996, 383, 832–836. [Google Scholar] [CrossRef]
- Wu, R.Y.; Zhang, Y.; Feng, X.H.; Derynck, R. Heteromeric and homomeric interactions correlate with signaling activity and functional cooperativity of Smad3 and Smad4/DPC4. Mol. Cell. Biol. 1997, 17, 2521–2528. [Google Scholar] [CrossRef]
- Nakao, A.; Imamura, T.; Souchelnytskyi, S.; Kawabata, M.; Ishisaki, A.; Oeda, E.; Tamaki, K.; Hanai, J.; Heldin, C.H.; Miyazono, K.; et al. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997, 16, 5353–5362. [Google Scholar] [CrossRef]
- Hill, C.S. Transcriptional Control by the SMADs. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Itoh, S.; Vivien, D.; ten Dijke, P.; Huet, S.; Gauthier, J.M. Direct binding of Smad3 and Smad4 to critical TGFβ-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998, 17, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; ten Dijke, P. Negative regulation of TGF-β receptor/Smad signal transduction. Curr. Opin. Cell Biol. 2007, 19, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- de Ceuninck van Capelle, C.; Spit, M.; Ten Dijke, P. Current perspectives on inhibitory SMAD7 in health and disease. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 691–715. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.K.; Moses, H.L. STRAP and Smad7 synergize in the inhibition of transforming growth factor β signaling. Mol. Cell. Biol. 2000, 20, 3157–3167. [Google Scholar] [CrossRef] [PubMed]
- Ferrigno, O.; Lallemand, F.; Verrecchia, F.; L’Hoste, S.; Camonis, J.; Atfi, A.; Mauviel, A. Yes-associated protein (YAP65) interacts with Smad7 and potentiates its inhibitory activity against TGF-β/Smad signaling. Oncogene 2002, 21, 4879–4884. [Google Scholar] [CrossRef]
- Lallemand, F.; Seo, S.R.; Ferrand, N.; Pessah, M.; L’Hoste, S.; Rawadi, G.; Roman-Roman, S.; Camonis, J.; Atfi, A. AIP4 restricts transforming growth factor-β signaling through a ubiquitination-independent mechanism. J. Biol. Chem. 2005, 280, 27645–27653. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-β signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef]
- Hanafusa, H.; Ninomiya-Tsuji, J.; Masuyama, N.; Nishita, M.; Fujisawa, J.; Shibuya, H.; Matsumoto, K.; Nishida, E. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-β-induced gene expression. J. Biol. Chem. 1999, 274, 27161–27167. [Google Scholar] [CrossRef] [PubMed]
- Bakin, A.V.; Rinehart, C.; Tomlinson, A.K.; Arteaga, C.L. p38 mitogen-activated protein kinase is required for TGFβ-mediated fibroblastic transdifferentiation and cell migration. J. Cell. Sci. 2002, 115, 3193–3206. [Google Scholar] [PubMed]
- Yu, L.; Hebert, M.C.; Zhang, Y.E. TGF-β receptor-activated p38 MAP kinase mediates Smad-independent TGF-β responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Harada, J.; Tashiro, S.; Gotoh-Mandeville, R.; Maekawa, T.; Ishii, S. ATF-2 is a common nuclear target of Smad and TAK1 pathways in transforming growth factor-β signaling. J. Biol. Chem. 1999, 274, 8949–8957. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-β activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, F.; ten Dijke, P. Signaling interplay between transforming growth factor-β receptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 2013, 38, 612–620. [Google Scholar] [CrossRef]
- Edlund, S.; Landstrom, M.; Heldin, C.H.; Aspenstrom, P. Transforming growth factor-β-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell 2002, 13, 902–914. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-β1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef]
- Shen, X.; Li, J.; Hu, P.P.; Waddell, D.; Zhang, J.; Wang, X.F. The activity of guanine exchange factor NET1 is essential for transforming growth factor-β-mediated stress fiber formation. J. Biol. Chem. 2001, 276, 15362–15368. [Google Scholar] [CrossRef]
- Papadimitriou, E.; Kardassis, D.; Moustakas, A.; Stournaras, C. TGFβ-induced early activation of the small GTPase RhoA is Smad2/3-independent and involves Src and the guanine nucleotide exchange factor Vav2. Cell. Physiol. Biochem. 2011, 28, 229–238. [Google Scholar] [CrossRef]
- Kim, S.J.; Im, Y.H.; Markowitz, S.D.; Bang, Y.J. Molecular mechanisms of inactivation of TGF-β receptors during carcinogenesis. Cytokine Growth Factor Rev. 2000, 11, 159–168. [Google Scholar] [CrossRef]
- Li, J.M.; Nichols, M.A.; Chandrasekharan, S.; Xiong, Y.; Wang, X.F. Transforming growth factor β activates the promoter of cyclin-dependent kinase inhibitor p15INK4B through an Sp1 consensus site. J. Biol. Chem. 1995, 270, 26750–26753. [Google Scholar] [CrossRef] [PubMed]
- Datto, M.B.; Li, Y.; Panus, J.F.; Howe, D.J.; Xiong, Y.; Wang, X.F. Transforming growth factor β induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc. Natl. Acad. Sci. USA 1995, 92, 5545–5549. [Google Scholar] [CrossRef] [PubMed]
- Coffey, R.J., Jr.; Bascom, C.C.; Sipes, N.J.; Graves-Deal, R.; Weissman, B.E.; Moses, H.L. Selective inhibition of growth-related gene expression in murine keratinocytes by transforming growth factor β. Mol. Cell. Biol. 1988, 8, 3088–3093. [Google Scholar] [CrossRef]
- Ho, J.; Cocolakis, E.; Dumas, V.M.; Posner, B.I.; Laporte, S.A.; Lebrun, J.J. The G protein-coupled receptor kinase-2 is a TGFβ-inducible antagonist of TGFβ signal transduction. EMBO J. 2005, 24, 3247–3258. [Google Scholar] [CrossRef]
- Kang, Y.; Chen, C.R.; Massague, J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol. Cell 2003, 11, 915–926. [Google Scholar] [CrossRef]
- Kowanetz, M.; Valcourt, U.; Bergstrom, R.; Heldin, C.H.; Moustakas, A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor β and bone morphogenetic protein. Mol. Cell. Biol. 2004, 24, 4241–4254. [Google Scholar] [CrossRef]
- Ohgushi, M.; Kuroki, S.; Fukamachi, H.; O’Reilly, L.A.; Kuida, K.; Strasser, A.; Yonehara, S. Transforming growth factor β-dependent sequential activation of Smad, Bim, and caspase-9 mediates physiological apoptosis in gastric epithelial cells. Mol. Cell. Biol. 2005, 25, 10017–10028. [Google Scholar] [CrossRef]
- Wildey, G.M.; Patil, S.; Howe, P.H. Smad3 potentiates transforming growth factor beta (TGFβ)-induced apoptosis and expression of the BH3-only protein Bim in WEHI 231 B lymphocytes. J. Biol. Chem. 2003, 278, 18069–18077. [Google Scholar] [CrossRef]
- Francis, J.M.; Heyworth, C.M.; Spooncer, E.; Pierce, A.; Dexter, T.M.; Whetton, A.D. Transforming growth factor-β1 induces apoptosis independently of p53 and selectively reduces expression of Bcl-2 in multipotent hematopoietic cells. J. Biol. Chem. 2000, 275, 39137–39145. [Google Scholar] [CrossRef]
- Saltzman, A.; Munro, R.; Searfoss, G.; Franks, C.; Jaye, M.; Ivashchenko, Y. Transforming growth factor-β-mediated apoptosis in the Ramos B-lymphoma cell line is accompanied by caspase activation and Bcl-XL downregulation. Exp. Cell Res. 1998, 242, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Bhat, M.; Hsing, A.Y.; Ma, J.; Danielpour, D. Bcl-xL blocks transforming growth factor-β1-induced apoptosis by inhibiting cytochrome c release and not by directly antagonizing Apaf-1-dependent caspase activation in prostate epithelial cells. J. Biol. Chem. 2001, 276, 26614–26621. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.W.; Chen, C.H.; Chen, C.C.; Chen, J.Y.; Su, Y.H.; Chen, R.H. TGF-β induces apoptosis through Smad-mediated expression of DAP-kinase. Nat. Cell Biol. 2002, 4, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.C.; Freimuth, J.; Akhurst, R.J. Complexities of TGF-β targeted cancer therapy. Int. J. Biol. Sci. 2012, 8, 964–978. [Google Scholar] [CrossRef]
- Glick, A.B.; Weinberg, W.C.; Wu, I.H.; Quan, W.; Yuspa, S.H. Transforming growth factor β1 suppresses genomic instability independent of a G1 arrest, p53, and Rb. Cancer Res. 1996, 56, 3645–3650. [Google Scholar]
- Katz, L.H.; Li, Y.; Chen, J.S.; Munoz, N.M.; Majumdar, A.; Chen, J.; Mishra, L. Targeting TGF-β signaling in cancer. Expert Opin. Ther. Targets 2013, 17, 743–760. [Google Scholar] [CrossRef]
- Kirshner, J.; Jobling, M.F.; Pajares, M.J.; Ravani, S.A.; Glick, A.B.; Lavin, M.J.; Koslov, S.; Shiloh, Y.; Barcellos-Hoff, M.H. Inhibition of transforming growth factor-β1 signaling attenuates ataxia telangiectasia mutated activity in response to genotoxic stress. Cancer Res. 2006, 66, 10861–10869. [Google Scholar] [CrossRef]
- Bornstein, S.; White, R.; Malkoski, S.; Oka, M.; Han, G.; Cleaver, T.; Reh, D.; Andersen, P.; Gross, N.; Olson, S.; et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J. Clin. Investig. 2009, 119, 3408–3419. [Google Scholar] [CrossRef]
- Kubiczkova, L.; Sedlarikova, L.; Hajek, R.; Sevcikova, S. TGF-β—An excellent servant but a bad master. J. Transl. Med. 2012, 10, 183. [Google Scholar] [CrossRef]
- Valcourt, U.; Kowanetz, M.; Niimi, H.; Heldin, C.H.; Moustakas, A. TGF-β and the Smad signaling pathway support transcriptomic reprogramming during epithelial-mesenchymal cell transition. Mol. Biol. Cell 2005, 16, 1987–2002. [Google Scholar] [CrossRef]
- Meulmeester, E.; Ten Dijke, P. The dynamic roles of TGF-β in cancer. J. Pathol. 2011, 223, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007, 98, 1512–1520. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.J.; Blobe, G.C. Dichotomous roles of TGF-β in human cancer. Biochem. Soc. Trans. 2016, 44, 1441–1454. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible TGFβ signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296. [Google Scholar] [CrossRef]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massague, J.; Mundy, G.R.; Guise, T.A. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef]
- Sanchez-Elsner, T.; Botella, L.M.; Velasco, B.; Corbi, A.; Attisano, L.; Bernabeu, C. Synergistic cooperation between hypoxia and transforming growth factor-β pathways on human vascular endothelial growth factor gene expression. J. Biol. Chem. 2001, 276, 38527–38535. [Google Scholar] [CrossRef]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef]
- Rossant, J.; Howard, L. Signaling pathways in vascular development. Annu. Rev. Cell Dev. Biol. 2002, 18, 541–573. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Hurd, S.D.; Winnier, A.R.; Johnson, M.D.; Fendly, B.M.; Forbes, J.T. Anti-transforming growth factor (TGF)- β antibodies inhibit breast cancer cell tumorigenicity and increase mouse spleen natural killer cell activity. Implications for a possible role of tumor cell/host TGF-β interactions in human breast cancer progression. J. Clin. Investig. 1993, 92, 2569–2576. [Google Scholar] [CrossRef] [PubMed]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGFβ. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Jaffrey, S.R. The new landscape of protein ubiquitination. Nat. Biotechnol. 2011, 29, 1098–1100. [Google Scholar] [CrossRef] [PubMed]
- Deol, K.K.; Lorenz, S.; Strieter, E.R. Enzymatic Logic of Ubiquitin Chain Assembly. Front. Physiol. 2019, 10, 835. [Google Scholar] [CrossRef] [PubMed]
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001, 70, 503–533. [Google Scholar] [CrossRef] [PubMed]
- Husnjak, K.; Dikic, I. Ubiquitin-binding proteins: Decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef]
- Braten, O.; Livneh, I.; Ziv, T.; Admon, A.; Kehat, I.; Caspi, L.H.; Gonen, H.; Bercovich, B.; Godzik, A.; Jahandideh, S.; et al. Numerous proteins with unique characteristics are degraded by the 26S proteasome following monoubiquitination. Proc. Natl. Acad. Sci. USA 2016, 113, E4639–E4647. [Google Scholar] [CrossRef]
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180. [Google Scholar] [CrossRef]
- Li, W.; Ye, Y. Polyubiquitin chains: Functions, structures, and mechanisms. Cell. Mol. Life Sci. 2008, 65, 2397–2406. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Riezman, H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Akutsu, M.; Dikic, I.; Bremm, A. Ubiquitin chain diversity at a glance. J. Cell Sci. 2016, 129, 875–880. [Google Scholar] [CrossRef]
- Ohtake, F.; Tsuchiya, H. The emerging complexity of ubiquitin architecture. J. Biochem. 2017, 161, 125–133. [Google Scholar] [CrossRef]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.P.; Chen, J.; Tse, W.K.F. Role of Deubiquitinases in Human Cancers: Potential Targeted Therapy. Int. J. Mol. Sci. 2020, 21, 2548. [Google Scholar] [CrossRef]
- Ciechanover, A. The unravelling of the ubiquitin system. Nat. Rev. Mol. Cell Biol. 2015, 16, 322–324. [Google Scholar] [CrossRef]
- Kliza, K.; Husnjak, K. Resolving the Complexity of Ubiquitin Networks. Front. Mol. Biosci. 2020, 7, 21. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Oh, E.; Akopian, D.; Rape, M. Principles of Ubiquitin-Dependent Signaling. Annu. Rev. Cell Dev. Biol. 2018, 34, 137–162. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef] [PubMed]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome Structure and Assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [CrossRef] [PubMed]
- Satija, Y.K.; Bhardwaj, A.; Das, S. A portrayal of E3 ubiquitin ligases and deubiquitylases in cancer. Int. J. Cancer 2013, 133, 2759–2768. [Google Scholar] [CrossRef] [PubMed]
- Senft, D.; Qi, J.; Ronai, Z.A. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat. Rev. Cancer 2018, 18, 69–88. [Google Scholar] [CrossRef]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef]
- Metzger, M.B.; Hristova, V.A.; Weissman, A.M. HECT and RING finger families of E3 ubiquitin ligases at a glance. J. Cell Sci. 2012, 125, 531–537. [Google Scholar] [CrossRef]
- Fu, L.; Cui, C.P.; Zhang, X.; Zhang, L. The functions and regulation of Smurfs in cancers. Semin. Cancer Biol. 2019. [Google Scholar] [CrossRef]
- Zhu, H.; Kavsak, P.; Abdollah, S.; Wrana, J.L.; Thomsen, G.H. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature 1999, 400, 687–693. [Google Scholar] [CrossRef]
- Murakami, G.; Watabe, T.; Takaoka, K.; Miyazono, K.; Imamura, T. Cooperative inhibition of bone morphogenetic protein signaling by Smurf1 and inhibitory Smads. Mol. Biol. Cell 2003, 14, 2809–2817. [Google Scholar] [CrossRef]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-β type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef] [PubMed]
- Moren, A.; Imamura, T.; Miyazono, K.; Heldin, C.H.; Moustakas, A. Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J. Biol. Chem. 2005, 280, 22115–22123. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, E.; Mishra, L. Transforming growth factor-β signaling and ubiquitinators in cancer. Endocr. Relat. Cancer 2008, 15, 59–72. [Google Scholar] [CrossRef]
- Zhang, Y.; Chang, C.; Gehling, D.J.; Hemmati-Brivanlou, A.; Derynck, R. Regulation of Smad degradation and activity by Smurf2, an E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2001, 98, 974–979. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.Y.; Yamashita, M.; Coussens, N.P.; Tang, Y.; Wang, X.; Li, C.; Deng, C.X.; Cheng, S.Y.; Zhang, Y.E. Ablation of Smurf2 reveals an inhibition in TGF-β signalling through multiple mono-ubiquitination of Smad3. EMBO J. 2011, 30, 4777–4789. [Google Scholar] [CrossRef]
- Sim, W.J.; Iyengar, P.V.; Lama, D.; Lui, S.K.L.; Ng, H.C.; Haviv-Shapira, L.; Domany, E.; Kappei, D.; Tan, T.Z.; Saei, A.; et al. c-Met activation leads to the establishment of a TGFβ-receptor regulatory network in bladder cancer progression. Nat. Commun. 2019, 10, 4349. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, P.V.; Jaynes, P.; Rodon, L.; Lama, D.; Law, K.P.; Lim, Y.P.; Verma, C.; Seoane, J.; Eichhorn, P.J. USP15 regulates SMURF2 kinetics through C-lobe mediated deubiquitination. Sci. Rep. 2015, 5, 14733. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Fan, Y.; Xie, F.; Zhou, H.; Jin, K.; Shao, L.; Shi, W.; Fang, P.; Yang, B.; van Dam, H.; et al. Breast cancer metastasis suppressor OTUD1 deubiquitinates SMAD7. Nat. Commun. 2017, 8, 2116. [Google Scholar] [CrossRef]
- Komuro, A.; Imamura, T.; Saitoh, M.; Yoshida, Y.; Yamori, T.; Miyazono, K.; Miyazawa, K. Negative regulation of transforming growth factor-beta (TGF-β) signaling by WW domain-containing protein 1 (WWP1). Oncogene 2004, 23, 6914–6923. [Google Scholar] [CrossRef]
- Seo, S.R.; Lallemand, F.; Ferrand, N.; Pessah, M.; L’Hoste, S.; Camonis, J.; Atfi, A. The novel E3 ubiquitin ligase Tiul1 associates with TGIF to target Smad2 for degradation. EMBO J. 2004, 23, 3780–3792. [Google Scholar] [CrossRef]
- Bai, Y.; Yang, C.; Hu, K.; Elly, C.; Liu, Y.C. Itch E3 ligase-mediated regulation of TGF-β signaling by modulating smad2 phosphorylation. Mol. Cell 2004, 15, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Sundar, R.; Thakur, N.; Ekman, M.; Gudey, S.K.; Yakymovych, M.; Hermansson, A.; Dimitriou, H.; Bengoechea-Alonso, M.T.; Ericsson, J.; et al. TRAF6 ubiquitinates TGFβ type I receptor to promote its cleavage and nuclear translocation in cancer. Nat. Commun. 2011, 2, 330. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-β. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, F.; Garcia de Vinuesa, A.; de Kruijf, E.M.; Mesker, W.E.; Hui, L.; Drabsch, Y.; Li, Y.; Bauer, A.; Rousseau, A.; et al. TRAF4 promotes TGF-β receptor signaling and drives breast cancer metastasis. Mol. Cell 2013, 51, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, P.V.; Marvin, D.L.; Lama, D.; Tan, T.Z.; Suriyamurthy, S.; Xie, F.; van Dinther, M.; Mei, H.; Verma, C.S.; Zhang, L.; et al. TRAF4 inhibits bladder cancer progression by promoting BMP/SMAD signalling pathway. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mallikarjuna, P. The Role of Transforming Growth Factor-β Signaling and Hypoxia-Inducible Factors in Renal Cell Carcinoma; Umeå University: Umea, Sweden, 2019. [Google Scholar]
- Wohlfert, E.A.; Gorelik, L.; Mittler, R.; Flavell, R.A.; Clark, R.B. Cutting edge: Deficiency in the E3 ubiquitin ligase Cbl-b results in a multifunctional defect in T cell TGF-β sensitivity in vitro and in vivo. J. Immunol. 2006, 176, 1316–1320. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Balasenthil, S.; Reuther, J.; Frayna, A.; Wang, Y.; Chandler, D.S.; Abruzzo, L.V.; Rashid, A.; Rodriguez, J.; Lozano, G.; et al. DEAR1 is a chromosome 1p35 tumor suppressor and master regulator of TGF-β-driven epithelial-mesenchymal transition. Cancer Discov. 2013, 3, 1172–1189. [Google Scholar] [CrossRef]
- Mishra, L.; Katuri, V.; Evans, S. The role of PRAJA and ELF in TGF-β signaling and gastric cancer. Cancer Biol. Ther. 2005, 4, 694–699. [Google Scholar] [CrossRef][Green Version]
- Saha, T.; Vardhini, D.; Tang, Y.; Katuri, V.; Jogunoori, W.; Volpe, E.A.; Haines, D.; Sidawy, A.; Zhou, X.; Gallicano, I.; et al. RING finger-dependent ubiquitination by PRAJA is dependent on TGF-β and potentially defines the functional status of the tumor suppressor ELF. Oncogene 2006, 25, 693–705. [Google Scholar] [CrossRef][Green Version]
- Fukuchi, M.; Imamura, T.; Chiba, T.; Ebisawa, T.; Kawabata, M.; Tanaka, K.; Miyazono, K. Ligand-dependent degradation of Smad3 by a ubiquitin ligase complex of ROC1 and associated proteins. Mol. Biol. Cell 2001, 12, 1431–1443. [Google Scholar] [CrossRef]
- Wan, Y.; Liu, X.; Kirschner, M.W. The anaphase-promoting complex mediates TGF-β signaling by targeting SnoN for destruction. Mol. Cell 2001, 8, 1027–1039. [Google Scholar] [CrossRef]
- Stroschein, S.L.; Wang, W.; Zhou, S.; Zhou, Q.; Luo, K. Negative feedback regulation of TGF-β signaling by the SnoN oncoprotein. Science 1999, 286, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Nagano, Y.; Mavrakis, K.J.; Lee, K.L.; Fujii, T.; Koinuma, D.; Sase, H.; Yuki, K.; Isogaya, K.; Saitoh, M.; Imamura, T.; et al. Arkadia induces degradation of SnoN and c-Ski to enhance transforming growth factor-β signaling. J. Biol. Chem. 2007, 282, 20492–20501. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.; Howell, M.; Das, D.; Harkin, S.; Episkopou, V.; Hill, C.S. Arkadia activates Smad3/Smad4-dependent transcription by triggering signal-induced SnoN degradation. Mol. Cell. Biol. 2007, 27, 6068–6083. [Google Scholar] [CrossRef]
- Xia, T.; Levy, L.; Levillayer, F.; Jia, B.; Li, G.; Neuveut, C.; Buendia, M.A.; Lan, K.; Wei, Y. The four and a half LIM-only protein 2 (FHL2) activates transforming growth factor beta (TGF-β) signaling by regulating ubiquitination of the E3 ligase Arkadia. J. Biol. Chem. 2013, 288, 1785–1794. [Google Scholar] [CrossRef]
- Dupont, S.; Mamidi, A.; Cordenonsi, M.; Montagner, M.; Zacchigna, L.; Adorno, M.; Martello, G.; Stinchfield, M.J.; Soligo, S.; Morsut, L.; et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFβ signaling, controls Smad4 monoubiquitination. Cell 2009, 136, 123–135. [Google Scholar] [CrossRef]
- Dupont, S.; Zacchigna, L.; Cordenonsi, M.; Soligo, S.; Adorno, M.; Rugge, M.; Piccolo, S. Germ-layer specification and control of cell growth by Ectodermin, a Smad4 ubiquitin ligase. Cell 2005, 121, 87–99. [Google Scholar] [CrossRef]
- He, W.; Dorn, D.C.; Erdjument-Bromage, H.; Tempst, P.; Moore, M.A.; Massague, J. Hematopoiesis controlled by distinct TIF1γ and Smad4 branches of the TGFβ pathway. Cell 2006, 125, 929–941. [Google Scholar] [CrossRef]
- Liang, M.; Liang, Y.Y.; Wrighton, K.; Ungermannova, D.; Wang, X.P.; Brunicardi, F.C.; Liu, X.; Feng, X.H.; Lin, X. Ubiquitination and proteolysis of cancer-derived Smad4 mutants by SCFSkp2. Mol. Cell. Biol. 2004, 24, 7524–7537. [Google Scholar] [CrossRef]
- Wan, M.; Tang, Y.; Tytler, E.M.; Lu, C.; Jin, B.; Vickers, S.M.; Yang, L.; Shi, X.; Cao, X. Smad4 protein stability is regulated by ubiquitin ligase SCFβ-TrCP1. J. Biol. Chem. 2004, 279, 14484–14487. [Google Scholar] [CrossRef]
- Gruber, T.; Hinterleitner, R.; Hermann-Kleiter, N.; Meisel, M.; Kleiter, I.; Wang, C.M.; Viola, A.; Pfeifhofer-Obermair, C.; Baier, G. Cbl-b mediates TGFβ sensitivity by downregulating inhibitory SMAD7 in primary T cells. J. Mol. Cell Biol. 2013, 5, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Koinuma, D.; Shinozaki, M.; Komuro, A.; Goto, K.; Saitoh, M.; Hanyu, A.; Ebina, M.; Nukiwa, T.; Miyazawa, K.; Imamura, T.; et al. Arkadia amplifies TGF-β superfamily signalling through degradation of Smad7. EMBO J. 2003, 22, 6458–6470. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Huang, H.; Zhou, F.; Schimmel, J.; Pardo, C.G.; Zhang, T.; Barakat, T.S.; Sheppard, K.A.; Mickanin, C.; Porter, J.A.; et al. RNF12 controls embryonic stem cell fate and morphogenesis in zebrafish embryos by targeting Smad7 for degradation. Mol. Cell 2012, 46, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, Y.; Gao, R.; Yang, X.; Yan, X.; Wang, C.; Jiang, S.; Yu, L. RLIM interacts with Smurf2 and promotes TGF-β induced U2OS cell migration. Biochem. Biophys. Res. Commun. 2011, 414, 181–185. [Google Scholar] [CrossRef]
- Li, R.F.; Zhang, F.; Lu, Y.J.; Sui, S.F. Specific interaction between Smad1 and CHIP: A surface plasmon resonance study. Colloids Surf. B Biointerfaces 2005, 40, 133–136. [Google Scholar] [CrossRef]
- Li, L.; Xin, H.; Xu, X.; Huang, M.; Zhang, X.; Chen, Y.; Zhang, S.; Fu, X.Y.; Chang, Z. CHIP mediates degradation of Smad proteins and potentially regulates Smad-induced transcription. Mol. Cell. Biol. 2004, 24, 856–864. [Google Scholar] [CrossRef]
- Xin, H.; Xu, X.; Li, L.; Ning, H.; Rong, Y.; Shang, Y.; Wang, Y.; Fu, X.Y.; Chang, Z. CHIP controls the sensitivity of transforming growth factor-β signaling by modulating the basal level of Smad3 through ubiquitin-mediated degradation. J. Biol. Chem. 2005, 280, 20842–20850. [Google Scholar] [CrossRef]
- Sorrentino, A.; Thakur, N.; Grimsby, S.; Marcusson, A.; von Bulow, V.; Schuster, N.; Zhang, S.; Heldin, C.H.; Landstrom, M. The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 2008, 10, 1199–1207. [Google Scholar] [CrossRef]
- Delaney, J.R.; Mlodzik, M. TGF-β activated kinase-1: New insights into the diverse roles of TAK1 in development and immunity. Cell Cycle 2006, 5, 2852–2855. [Google Scholar] [CrossRef]
- Jung, S.M.; Lee, J.H.; Park, J.; Oh, Y.S.; Lee, S.K.; Park, J.S.; Lee, Y.S.; Kim, J.H.; Lee, J.Y.; Bae, Y.S.; et al. Smad6 inhibits non-canonical TGF-β1 signalling by recruiting the deubiquitinase A20 to TRAF6. Nat. Commun. 2013, 4, 2562. [Google Scholar] [CrossRef]
- Bonni, S.; Wang, H.R.; Causing, C.G.; Kavsak, P.; Stroschein, S.L.; Luo, K.; Wrana, J.L. TGF-β induces assembly of a Smad2-Smurf2 ubiquitin ligase complex that targets SnoN for degradation. Nat. Cell Biol. 2001, 3, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhao, K.; Miao, C.; Xu, A.; Zhang, J.; Zhu, J.; Su, S.; Wang, Z. Silencing Trim59 inhibits invasion/migration and epithelial-to-mesenchymal transition via TGF-β/Smad2/3 signaling pathway in bladder cancer cells. OncoTargets Ther. 2017, 10, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, W.B. Down-regulation of tripartite motif protein 59 inhibits proliferation, migration and invasion in breast cancer cells. Biomed. Pharmacother. 2017, 89, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, P.; Xie, Z.; Wang, S.; Cen, S.; Li, M.; Liu, W.; Tang, S.; Ye, G.; Zheng, G.; et al. TRAF4 positively regulates the osteogenic differentiation of mesenchymal stem cells by acting as an E3 ubiquitin ligase to degrade Smurf2. Cell Death Differ. 2019, 26, 2652–2666. [Google Scholar] [CrossRef] [PubMed]
- Bashyam, M.D.; Bair, R.; Kim, Y.H.; Wang, P.; Hernandez-Boussard, T.; Karikari, C.A.; Tibshirani, R.; Maitra, A.; Pollack, J.R. Array-based comparative genomic hybridization identifies localized DNA amplifications and homozygous deletions in pancreatic cancer. Neoplasia 2005, 7, 556–562. [Google Scholar] [CrossRef] [PubMed]
- van Dekken, H.; Tilanus, H.W.; Hop, W.C.; Dinjens, W.N.; Wink, J.C.; Vissers, K.J.; van Marion, R. Array comparative genomic hybridization, expression array, and protein analysis of critical regions on chromosome arms 1q, 7q, and 8p in adenocarcinomas of the gastroesophageal junction. Cancer Genet. Cytogenet. 2009, 189, 37–42. [Google Scholar] [CrossRef]
- Ke, M.; Mo, L.; Li, W.; Zhang, X.; Li, F.; Yu, H. Ubiquitin ligase SMURF1 functions as a prognostic marker and promotes growth and metastasis of clear cell renal cell carcinoma. FEBS Open Bio. 2017, 7, 577–586. [Google Scholar] [CrossRef]
- Tao, Y.; Sun, C.; Zhang, T.; Song, Y. SMURF1 promotes the proliferation, migration and invasion of gastric cancer cells. Oncol. Rep. 2017, 38, 1806–1814. [Google Scholar] [CrossRef]
- Gang, X.; Wang, G.; Huang, H. Androgens regulate SMAD ubiquitination regulatory factor-1 expression and prostate cancer cell invasion. Prostate 2015, 75, 561–572. [Google Scholar] [CrossRef]
- Wang, W.; Ren, F.; Wu, Q.; Jiang, D.; Li, H.; Peng, Z.; Wang, J.; Shi, H. MicroRNA-497 inhibition of ovarian cancer cell migration and invasion through targeting of SMAD specific E3 ubiquitin protein ligase 1. Biochem. Biophys. Res. Commun. 2014, 449, 432–437. [Google Scholar] [CrossRef]
- Khammanivong, A.; Gopalakrishnan, R.; Dickerson, E.B. SMURF1 silencing diminishes a CD44-high cancer stem cell-like population in head and neck squamous cell carcinoma. Mol. Cancer 2014, 13, 260. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Su, H.; Song, X.; Cao, H.; Kong, L.; Cui, W. Smad Ubiquitination Regulatory Factor 1 (Smurf1) Promotes Thyroid Cancer Cell Proliferation and Migration via Ubiquitin-Dependent Degradation of Kisspeptin-1. Cell. Physiol. Biochem. 2018, 49, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, E.; Inoue, Y.; Komiya, S.; Horiguchi, K.; Goto, K.; Saitoh, M.; Miyazawa, K.; Koinuma, D.; Hanyu, A.; Imamura, T. Smurf2 induces ubiquitin-dependent degradation of Smurf1 to prevent migration of breast cancer cells. J. Biol. Chem. 2008, 283, 35660–35667. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.M.; Ooi, L.L.; Hui, K.M. Identification and validation of a novel gene signature associated with the recurrence of human hepatocellular carcinoma. Clin. Cancer Res. 2007, 13, 6275–6283. [Google Scholar] [CrossRef] [PubMed]
- Klupp, F.; Giese, C.; Halama, N.; Franz, C.; Lasitschka, F.; Warth, A.; Schmidt, T.; Kloor, M.; Ulrich, A.; Schneider, M. E3 ubiquitin ligase Smurf2: A prognostic factor in microsatellite stable colorectal cancer. Cancer Manag. Res. 2019, 11, 1795–1803. [Google Scholar] [CrossRef]
- Jin, C.; Yang, Y.A.; Anver, M.R.; Morris, N.; Wang, X.; Zhang, Y.E. Smad ubiquitination regulatory factor 2 promotes metastasis of breast cancer cells by enhancing migration and invasiveness. Cancer Res. 2009, 69, 735–740. [Google Scholar] [CrossRef]
- Fukuchi, M.; Fukai, Y.; Masuda, N.; Miyazaki, T.; Nakajima, M.; Sohda, M.; Manda, R.; Tsukada, K.; Kato, H.; Kuwano, H. High-level expression of the Smad ubiquitin ligase Smurf2 correlates with poor prognosis in patients with esophageal squamous cell carcinoma. Cancer Res. 2002, 62, 7162–7165. [Google Scholar]
- Emanuelli, A.; Manikoth Ayyathan, D.; Koganti, P.; Shah, P.A.; Apel-Sarid, L.; Paolini, B.; Detroja, R.; Frenkel-Morgenstern, M.; Blank, M. Altered Expression and Localization of Tumor Suppressive E3 Ubiquitin Ligase SMURF2 in Human Prostate and Breast Cancer. Cancers 2019, 11, 556. [Google Scholar] [CrossRef]
- Wu, B.; Guo, B.; Kang, J.; Deng, X.; Fan, Y.; Zhang, X.; Ai, K. Downregulation of Smurf2 ubiquitin ligase in pancreatic cancer cells reversed TGF-β-induced tumor formation. Tumour Biol. 2016. [Google Scholar] [CrossRef]
- Chae, D.K.; Park, J.; Cho, M.; Ban, E.; Jang, M.; Yoo, Y.S.; Kim, E.E.; Baik, J.H.; Song, E.J. MiR-195 and miR-497 suppress tumorigenesis in lung cancer by inhibiting SMURF2-induced TGF-β receptor I ubiquitination. Mol. Oncol. 2019, 13, 2663–2678. [Google Scholar] [CrossRef]
- David, D.; Jagadeeshan, S.; Hariharan, R.; Nair, A.S.; Pillai, R.M. Smurf2 E3 ubiquitin ligase modulates proliferation and invasiveness of breast cancer cells in a CNKSR2 dependent manner. Cell. Div. 2014, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Sun, X.; Guo, P.; Dong, X.Y.; Sethi, P.; Zhou, W.; Zhou, Z.; Petros, J.; Frierson, H.F., Jr.; Vessella, R.L.; et al. Ubiquitin E3 ligase WWP1 as an oncogenic factor in human prostate cancer. Oncogene 2007, 26, 2386–2394. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, Z.; Ross, J.S.; Zhou, W.; Dong, J.T. The amplified WWP1 gene is a potential molecular target in breast cancer. Int. J. Cancer 2007, 121, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Huu, N.S.; Ryder, W.D.; Zeps, N.; Flasza, M.; Chiu, M.; Hanby, A.M.; Poulsom, R.; Clarke, R.B.; Baron, M. Tumour-promoting activity of altered WWP1 expression in breast cancer and its utility as a prognostic indicator. J. Pathol. 2008, 216, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Courivaud, T.; Ferrand, N.; Elkhattouti, A.; Kumar, S.; Levy, L.; Ferrigno, O.; Atfi, A.; Prunier, C. Functional Characterization of a WWP1/Tiul1 Tumor-derived Mutant Reveals a Paradigm of Its Constitutive Activation in Human Cancer. J. Biol. Chem. 2015, 290, 21007–21018. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Hsieh, S.C.; Chen, J.N.; Tsai, M.H.; Chang, C.C. WWP1 gene is a potential molecular target of human oral cancer. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2013, 116, 221–231. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, Z.; Ma, Z.; Liu, H.; Wu, Y.; Zhang, Q. WWP1 as a potential tumor oncogene regulates PTEN-Akt signaling pathway in human gastric carcinoma. Tumor Biol. 2015, 36, 787–798. [Google Scholar] [CrossRef]
- Zhang, X.F.; Chao, J.; Pan, Q.Z.; Pan, K.; Weng, D.S.; Wang, Q.J.; Zhao, J.J.; He, J.; Liu, Q.; Jiang, S.S.; et al. Overexpression of WWP1 promotes tumorigenesis and predicts unfavorable prognosis in patients with hepatocellular carcinoma. Oncotarget 2015, 6, 40920–40933. [Google Scholar] [CrossRef]
- He, H.; Huang, C.; Chen, Z.; Huang, H.; Wang, X.; Chen, J. An outlined review for the role of Nedd4-1 and Nedd4-2 in lung disorders. Biomed. Pharmacother. 2020, 125, 109983. [Google Scholar] [CrossRef]
- Singh, R.; Karri, D.; Shen, H.; Shao, J.; Dasgupta, S.; Huang, S.; Edwards, D.P.; Ittmann, M.M.; O’Malley, B.W.; Yi, P. TRAF4-mediated ubiquitination of NGF receptor TrkA regulates prostate cancer metastasis. J. Clin. Investig. 2018, 128, 3129–3143. [Google Scholar] [CrossRef]
- Song, J.; Landstrom, M. TGFβ activates PI3K-AKT signaling via TRAF6. Oncotarget 2017, 8, 99205–99206. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Xue, Y.; Dai, T.; Li, X.; Zheng, N. Tripartite motif containing 25 promotes proliferation and invasion of colorectal cancer cells through TGF-β signaling. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Wang, Y.; Zhang, C.; Yu, S.; Zhu, Q.; Hou, K.; Yan, B. TRIM25 blockade by RNA interference inhibited migration and invasion of gastric cancer cells through TGF-β signaling. Sci. Rep. 2016, 6, 19070. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Munoz-Antonia, T.; Cress, W.D. Trim28 contributes to EMT via regulation of E-cadherin and N-cadherin in lung cancer cell lines. PLoS ONE 2014, 9, e101040. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dong, Y.; Zhao, L.; Su, L.; Diao, K.; Mi, X. TRIM59 overexpression correlates with poor prognosis and contributes to breast cancer progression through AKT signaling pathway. Mol. Carcinog. 2018, 57, 1792–1802. [Google Scholar] [CrossRef]
- Zhan, W.; Han, T.; Zhang, C.; Xie, C.; Gan, M.; Deng, K.; Fu, M.; Wang, J.B. TRIM59 Promotes the Proliferation and Migration of Non-Small Cell Lung Cancer Cells by Upregulating Cell Cycle Related Proteins. PLoS ONE 2015, 10, e0142596. [Google Scholar] [CrossRef]
- Lee, H.J. The Role of Tripartite Motif Family Proteins in TGF-β Signaling Pathway and Cancer. J. Cancer Prev. 2018, 23, 162–169. [Google Scholar] [CrossRef]
- Ding, Z.Y.; Jin, G.N.; Wang, W.; Chen, W.X.; Wu, Y.H.; Ai, X.; Chen, L.; Zhang, W.G.; Liang, H.F.; Laurence, A.; et al. Reduced expression of transcriptional intermediary factor 1γ promotes metastasis and indicates poor prognosis of hepatocellular carcinoma. Hepatology 2014, 60, 1620–1636. [Google Scholar] [CrossRef]
- Xue, J.; Lin, X.; Chiu, W.T.; Chen, Y.H.; Yu, G.; Liu, M.; Feng, X.H.; Sawaya, R.; Medema, R.H.; Hung, M.C.; et al. Sustained activation of SMAD3/SMAD4 by FOXM1 promotes TGF-β-dependent cancer metastasis. J. Clin. Investig. 2014, 124, 564–579. [Google Scholar] [CrossRef]
- Ligr, M.; Wu, X.; Daniels, G.; Zhang, D.; Wang, H.; Hajdu, C.; Wang, J.; Pan, R.; Pei, Z.; Zhang, L.; et al. Imbalanced expression of Tif1γ inhibits pancreatic ductal epithelial cell growth. Am. J. Cancer Res. 2014, 4, 196–210. [Google Scholar]
- Zhou, F.; Drabsch, Y.; Dekker, T.J.; de Vinuesa, A.G.; Li, Y.; Hawinkels, L.J.; Sheppard, K.A.; Goumans, M.J.; Luwor, R.B.; de Vries, C.J.; et al. Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-β signalling. Nat. Commun. 2014, 5, 3388. [Google Scholar] [CrossRef] [PubMed]
- Briones-Orta, M.A.; Levy, L.; Madsen, C.D.; Das, D.; Erker, Y.; Sahai, E.; Hill, C.S. Arkadia regulates tumor metastasis by modulation of the TGF-β pathway. Cancer Res. 2013, 73, 1800–1810. [Google Scholar] [CrossRef]
- Peng, R.; Zhang, P.F.; Yang, X.; Wei, C.Y.; Huang, X.Y.; Cai, J.B.; Lu, J.C.; Gao, C.; Sun, H.X.; Gao, Q.; et al. Overexpression of RNF38 facilitates TGF-β signaling by Ubiquitinating and degrading AHNAK in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 113. [Google Scholar] [CrossRef]
- Ohshiro, K.; Chen, J.; Jogunoori, W.; Deng, C.X.; Mishra, B.; Li, S.; Mishra, L. Targeting E3 ligase PJA1 via TGF-β pathway in hepatocellular carcinoma. Cancer Res. 2019, 79, 4443. [Google Scholar]
- Teixeira, A.F.; Ten Dijke, P.; Zhu, H.J. On-Target Anti-TGF-β Therapies Are Not Succeeding in Clinical Cancer Treatments: What Are Remaining Challenges? Front. Cell Dev. Biol. 2020, 8, 605. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Huynh, L.K.; Hipolito, C.J.; Ten Dijke, P. A Perspective on the Development of TGF-β Inhibitors for Cancer Treatment. Biomolecules 2019, 9, 743. [Google Scholar] [CrossRef]
- Tian, M.; Zeng, T.; Liu, M.; Han, S.; Lin, H.; Lin, Q.; Li, L.; Jiang, T.; Li, G.; Lin, H.; et al. A cell-based high-throughput screening method based on a ubiquitin-reference technique for identifying modulators of E3 ligases. J. Biol. Chem. 2019, 294, 2880–2891. [Google Scholar] [CrossRef]
- Chen, D.; Gehringer, M.; Lorenz, S. Developing Small-Molecule Inhibitors of HECT-Type Ubiquitin Ligases for Therapeutic Applications: Challenges and Opportunities. Chembiochem 2018, 19, 2123–2135. [Google Scholar] [CrossRef]
- Chan, C.H.; Morrow, J.K.; Li, C.F.; Gao, Y.; Jin, G.; Moten, A.; Stagg, L.J.; Ladbury, J.E.; Cai, Z.; Xu, D.; et al. Pharmacological inactivation of Skp2 SCF ubiquitin ligase restricts cancer stem cell traits and cancer progression. Cell 2013, 154, 556–568. [Google Scholar] [CrossRef]
- Brenke, J.K.; Popowicz, G.M.; Schorpp, K.; Rothenaigner, I.; Roesner, M.; Meininger, I.; Kalinski, C.; Ringelstetter, L.; R’Kyek, O.; Jurjens, G.; et al. Targeting TRAF6 E3 ligase activity with a small-molecule inhibitor combats autoimmunity. J. Biol. Chem. 2018, 293, 13191–13203. [Google Scholar] [CrossRef]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef]
- Galdeano, C. Drugging the undruggable: Targeting challenging E3 ligases for personalized medicine. Future Med. Chem. 2017, 9, 347–350. [Google Scholar] [CrossRef]
- Skaar, J.R.; Pagan, J.K.; Pagano, M. SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov. 2014, 13, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Buckley, D.L.; Van Molle, I.; Gareiss, P.C.; Tae, H.S.; Michel, J.; Noblin, D.J.; Jorgensen, W.L.; Ciulli, A.; Crews, C.M. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1α interaction. J. Am. Chem. Soc. 2012, 134, 4465–4468. [Google Scholar] [CrossRef]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef] [PubMed]
- An, S.; Fu, L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine 2018, 36, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Sun, X.; Rao, Y. PROTAC Technology: Opportunities and Challenges. ACS Med. Chem. Lett. 2020, 11, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Maniaci, C.; Hughes, S.J.; Testa, A.; Chen, W.; Lamont, D.J.; Rocha, S.; Alessi, D.R.; Romeo, R.; Ciulli, A. Homo-PROTACs: Bivalent small-molecule dimerizers of the VHL E3 ubiquitin ligase to induce self-degradation. Nat. Commun. 2017, 8, 830. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.; Galdeano, C.; Soares, P.; Gadd, M.S.; Grzes, K.M.; Ellis, L.; Epemolu, O.; Shimamura, S.; Bantscheff, M.; Grandi, P.; et al. Potent and selective chemical probe of hypoxic signalling downstream of HIF-α hydroxylation via VHL inhibition. Nat. Commun. 2016, 7, 13312. [Google Scholar] [CrossRef]
- Soares, P.; Gadd, M.S.; Frost, J.; Galdeano, C.; Ellis, L.; Epemolu, O.; Rocha, S.; Read, K.D.; Ciulli, A. Group-Based Optimization of Potent and Cell-Active Inhibitors of the von Hippel-Lindau (VHL) E3 Ubiquitin Ligase: Structure-Activity Relationships Leading to the Chemical Probe (2S,4R)-1-((S)-2-(1-Cyanocyclopropanecarboxamido)-3,3-dimethylbutanoyl)-4-hydroxy -N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (VH298). J. Med. Chem. 2018, 61, 599–618. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, S.; Fan, J.; Li, X.; Wen, Q.; Luo, N. New strategy for renal fibrosis: Targeting Smad3 proteins for ubiquitination and degradation. Biochem. Pharmacol. 2016, 116, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Su, H.; Li, Y.; Luo, C.; Xu, H.; Wang, Y.; Sun, H.; Wan, G.; Zhou, B.; Bu, X. Degradation of intracellular TGF-β1 by PROTACs efficiently reverses M2 macrophage induced malignant pathological events. Chem. Commun. 2020, 56, 2881–2884. [Google Scholar] [CrossRef] [PubMed]
- Girardini, M.; Maniaci, C.; Hughes, S.J.; Testa, A.; Ciulli, A. Cereblon versus VHL: Hijacking E3 ligases against each other using PROTACs. Bioorg. Med. Chem. 2019, 27, 2466–2479. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wan, L.; Liu, P.; Inuzuka, H.; Liu, J.; Wang, Z.; Wei, W. SCF(β-TRCP)-mediated degradation of NEDD4 inhibits tumorigenesis through modulating the PTEN/Akt signaling pathway. Oncotarget 2014, 5, 1026–1037. [Google Scholar] [CrossRef]
- Wang, X.; Jin, C.; Tang, Y.; Tang, L.Y.; Zhang, Y.E. Ubiquitination of tumor necrosis factor receptor-associated factor 4 (TRAF4) by Smad ubiquitination regulatory factor 1 (Smurf1) regulates motility of breast epithelial and cancer cells. J. Biol. Chem. 2013, 288, 21784–21792. [Google Scholar] [CrossRef]
- Maculins, T.; Carter, N.; Dorval, T.; Hudson, K.; Nissink, J.W.; Hay, R.T.; Alwan, H. A Generic Platform for Cellular Screening Against Ubiquitin Ligases. Sci. Rep. 2016, 6, 18940. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Broad Group of Ligase | Name | Type of Effect | Target | Effect | Associated Cancer | References |
|---|---|---|---|---|---|---|
| HECT E3 Ligase | SMURF1 | Negative | TβRI |
| pancreatic cancer, gastric cancer, prostate cancer, ovarian cancer colorectal cancer (CRC),renal cancer, head and neck squamous cell carcinoma (HNSCC), thyroid cancer | [120,121] |
| SMAD7 |
| [120,121,123] | ||||
| ||||||
| SMAD1/5/8 |
| [118,119] | ||||
| SMAD4 |
| [122] | ||||
| SMURF2 | Negative | TβRI/TβRII |
| breast cancer, hepatocellular carcinoma (HCC), CRC, esophageal squamous cell carcinoma, pancreatic cancer, prostate cancer | [31] | |
| SMAD7 |
| [31,120,121,123] | ||||
| SMAD2/3 |
| [124,125] | ||||
| SMAD1 |
| [124] | ||||
| SMAD4 |
| [122] | ||||
| SNoN |
| [162] | ||||
| NEDD4-2 | Negative | TβRI |
| lung cancer | [28] | |
| SMAD2 |
| [28] | ||||
| SMAD4 |
| [122] | ||||
| WWP1 | Negative | TβRI |
| breast cancer, prostate cancer, HCC, gastric cancer | [129] | |
| SMAD7 |
| [129] | ||||
| SMAD4 |
| [122] | ||||
| SMAD2 |
| [130] | ||||
| ITCH | Positive | SMAD2 |
| [131] | ||
| RING E3 Ligase | CBL-b | Positive | SMAD2 |
| [137] | |
| SMAD7 |
| [152] | ||||
| TRIM33 | Negative | SMAD4 |
| pancreatic cancer, HCC, breast cancer | [147,148] | |
| SMAD2/3 |
| [149] | ||||
| TRIM62 | Negative | SMAD3 |
| [138] | ||
| TRIM59 | Positive | SMAD2/3 |
| gastric, lung, breast cancer, bladder cancer | [163,164] | |
| TRAF4 | Positive | TβRI/TβRII |
| breast cancer, prostate cancer | [134] | |
| SMURF2 |
| [165] | ||||
| TRAF6 | Positive | TβRI |
| breast cancer, lung, prostate cancer | [133] | |
| PRAJA | Negative | SMAD3 |
| gastro-intestinal cancer, HCC | [139] | |
| ELF |
| [139,140] | ||||
| SCF–SKP2 | Negative | SMAD4 |
| [150] | ||
| SCF–βTrCP1 | Negative | SMAD4 |
| pancreatic cancer | [123,151] | |
| ROC1 | Negative | SMAD3 |
| [141] | ||
| APC | Positive | SNoN |
| [142,143] | ||
| VHL | Negative | TβRI |
| renal cell carcinoma | [136] | |
| ARKADIA/RNF111 | Positive | SMAD7 |
| lung cancer, breast cancer | [153] | |
| SNoN |
| [144,145] | ||||
| RNF12 | Positive | SMAD7 |
| breast cancer | [154] | |
| Other E3 Ligases | CHIP | Negative | SMAD1 |
| [156] | |
| SMAD3 |
| [158] | ||||
| SMAD4 |
| [157] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, A.; Iyengar, P.V.; ten Dijke, P. E3 Ubiquitin Ligases: Key Regulators of TGFβ Signaling in Cancer Progression. Int. J. Mol. Sci. 2021, 22, 476. https://doi.org/10.3390/ijms22020476
Sinha A, Iyengar PV, ten Dijke P. E3 Ubiquitin Ligases: Key Regulators of TGFβ Signaling in Cancer Progression. International Journal of Molecular Sciences. 2021; 22(2):476. https://doi.org/10.3390/ijms22020476
Chicago/Turabian StyleSinha, Abhishek, Prasanna Vasudevan Iyengar, and Peter ten Dijke. 2021. "E3 Ubiquitin Ligases: Key Regulators of TGFβ Signaling in Cancer Progression" International Journal of Molecular Sciences 22, no. 2: 476. https://doi.org/10.3390/ijms22020476
APA StyleSinha, A., Iyengar, P. V., & ten Dijke, P. (2021). E3 Ubiquitin Ligases: Key Regulators of TGFβ Signaling in Cancer Progression. International Journal of Molecular Sciences, 22(2), 476. https://doi.org/10.3390/ijms22020476