An Overview of Several Inhibitors for Alzheimer’s Disease: Characterization and Failure



Abstract
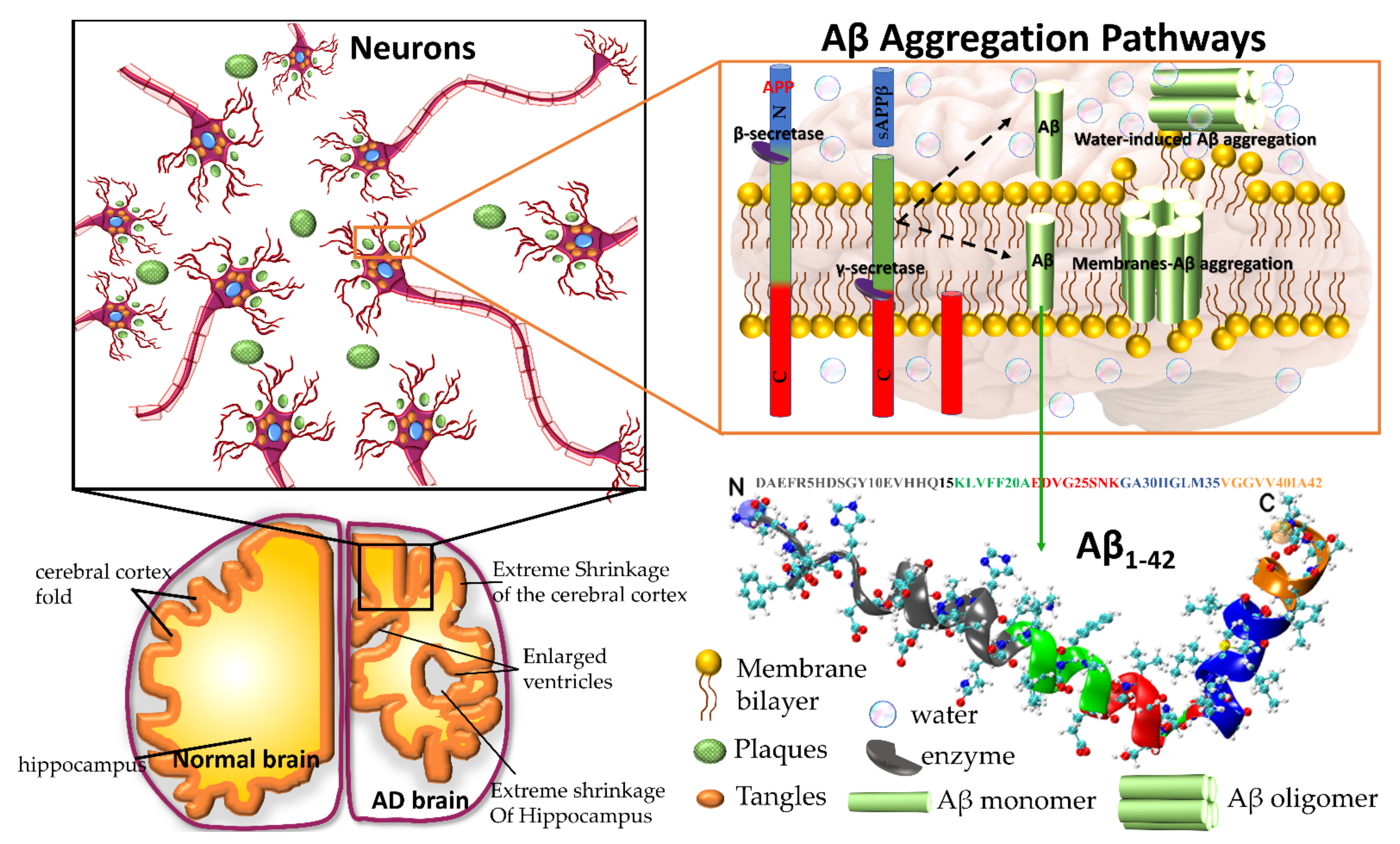
1. Introduction
2. Why Do Molecular Dynamics Simulations Cannot Accurately Quantify the Aβ Structural Ensemble?
3. Why Does Aβ Peptide Perturb Membrane Integrity?
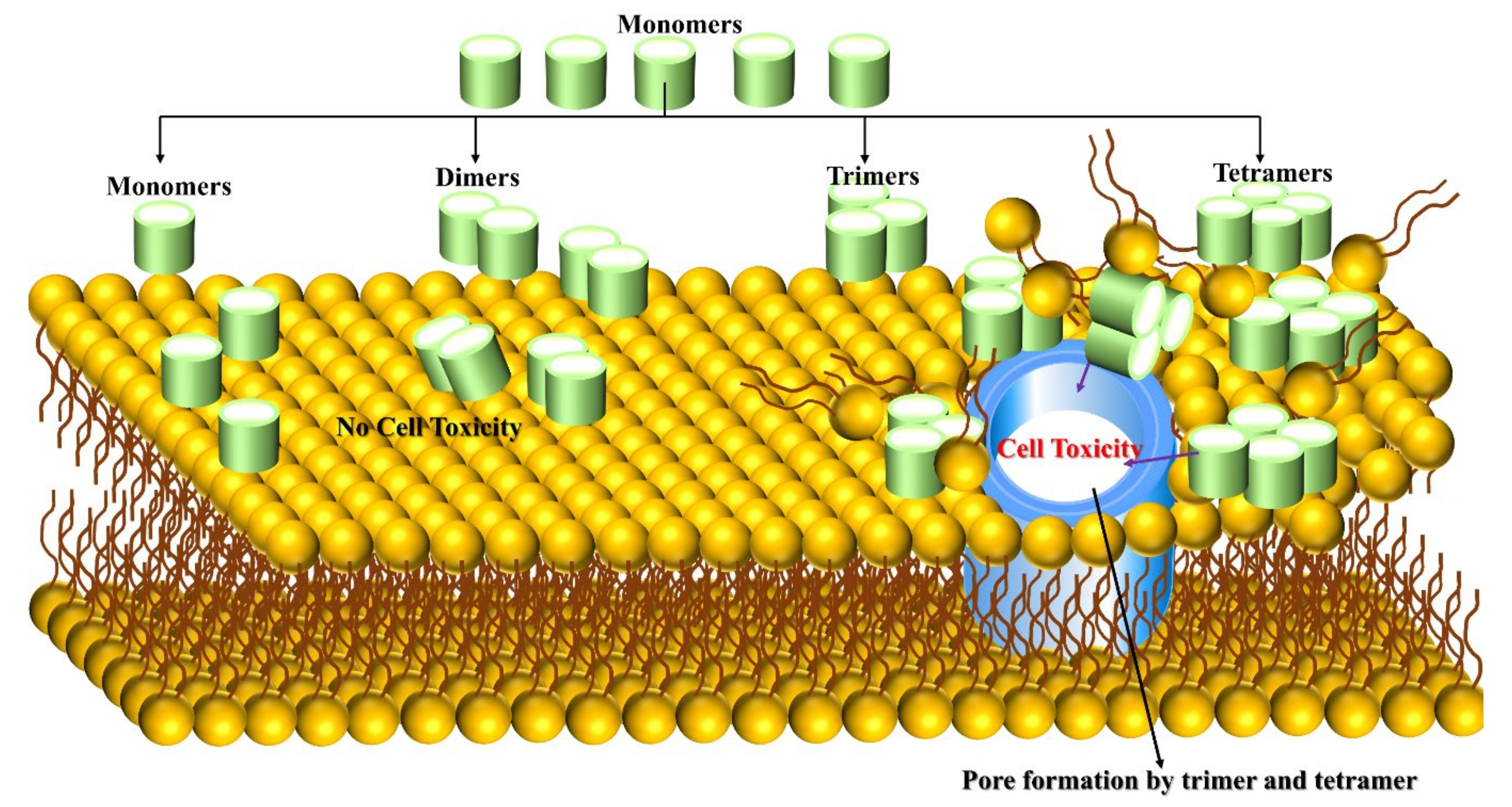
3.1. Aβ Monomer-Membrane
3.2. Aβ Dimer-Membrane
3.3. Aβ Trimer/Tetramer-Membrane
3.4. Aβ Oligomer-Membrane
4. How Do Metal Ions Govern Aβ Peptide Behavior?
4.1. Cu and Zn Ions Interactions with Aβ Peptides
4.2. Fe Interaction with Aβ Peptide
4.3. Al Interaction with Aβ Peptide
4.4. Ag Interaction with Aβ Peptide
4.5. Pb Interaction with Aβ Peptide
4.6. Hg Interaction with Aβ Peptide
4.7. Mn Interaction with Aβ Peptide
4.8. Li Interaction with Aβ Peptide
5. What Is the Role of Small Molecules in Inhibition Mechanism of Aβ Aggregation?
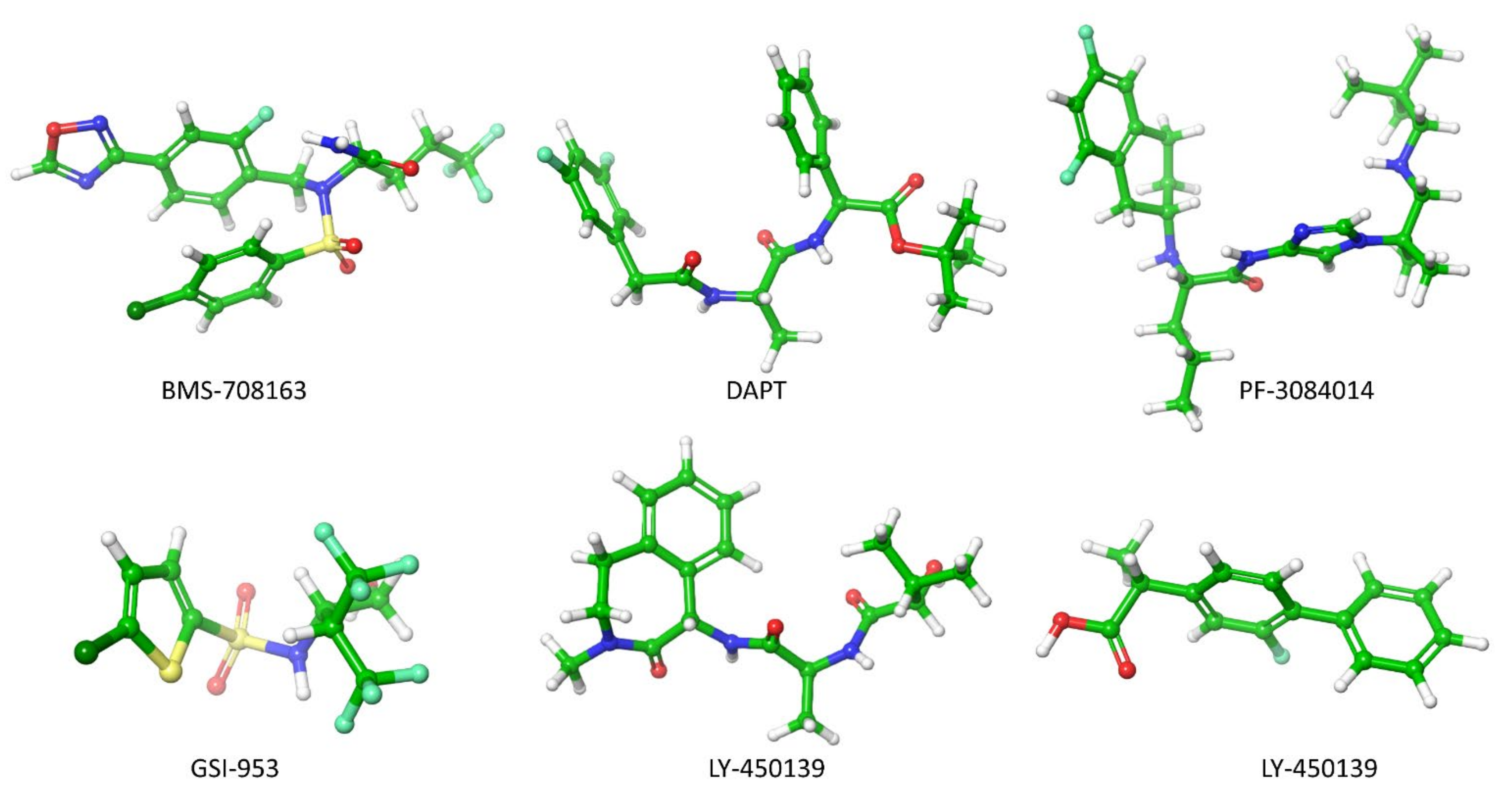
5.1. Secretase Inhibitors and Modulators
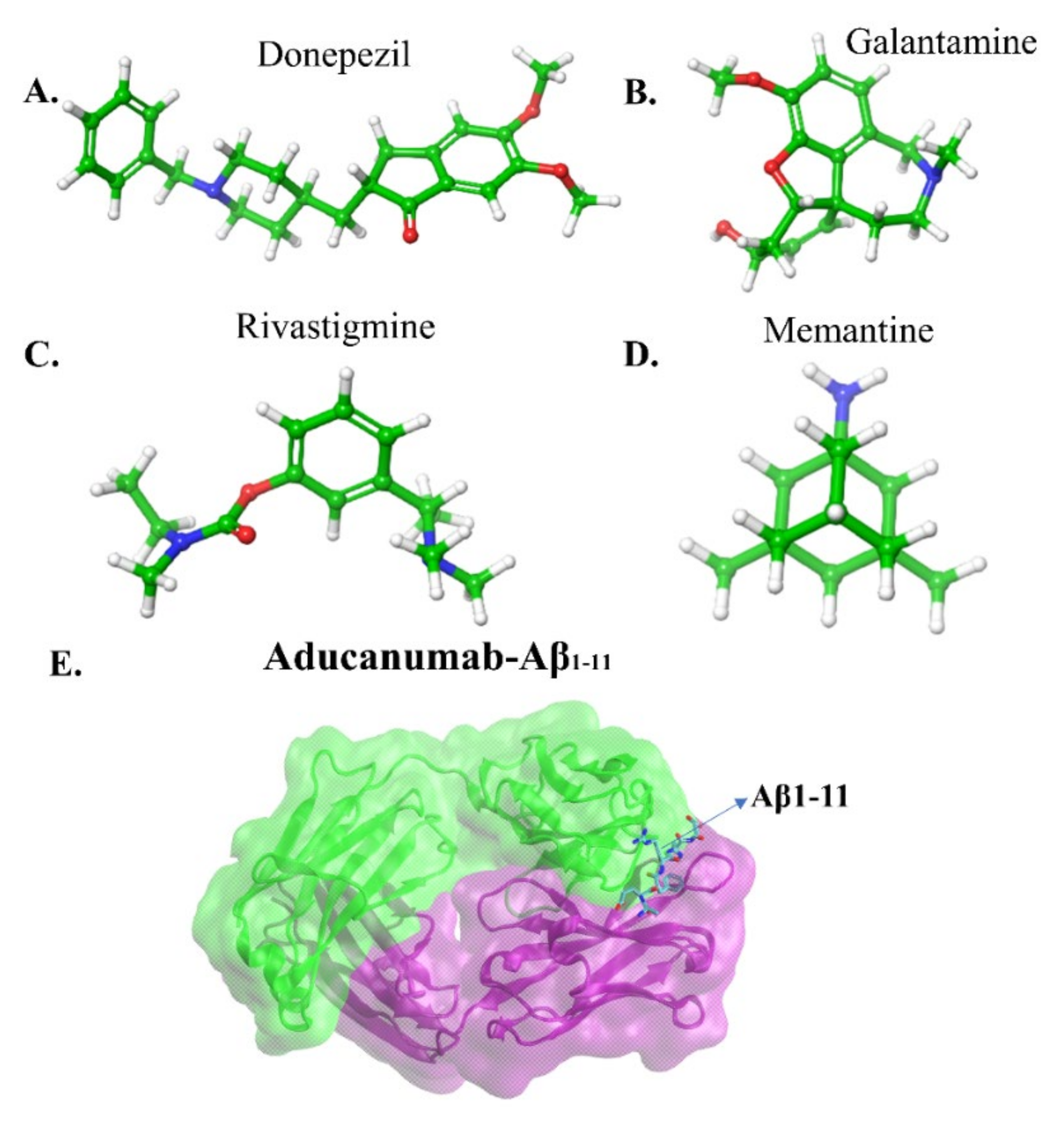
5.2. Immunotherapeutic Strategies
5.3. Peptide-Based Inhibitors
5.4. Small Molecular Inhibitors
6. Conclusion and Future Perspective
- Omega3 and Omega6 polyunsaturated fatty acids are buried in the neuronal membranes and are linked to a slowing down and increasing the risk of AD, respectively. Thus, it is essential to explore the dynamic mechanism of Aβ1-40/Aβ1-42 oligomers with and without interaction of metal ions in omega3 and omega6 phospholipids as these details are lacking in the AD research.
- Tyr10 residue in Aβ1-42 regulates the toxic β-sheet formation and leads to aggregation, but none of the studies in the literature regarding the role of Tyr10 in metal-bound Aβ1-42 peptide addressed this issue. In this perspective, investigating the interaction mechanism between metal-bound Aβ1-42 peptides and lipid membranes will be a crucial step to describe the toxicity of Aβ1-42 peptides.
- The inhibitory effect of the number of natural compounds in the aggregation pathways has been explained in the manuscript. However, their actions remain to be elucidated. The transition metal ions, Cu2+ and Zn2+, and their bindings induced a higher tendency of β-sheet formation in the Leu17-Met35 regions of Aβ1-42 peptide that can decrease solvent exposure (increase hydrophobicity) of the peptide which can lead to regulate toxicity [98]. To attenuate the hydrophobicity propensity of the peptide by the interaction of natural compounds, it is an essential investigation for anti-AD drug discovery.
- Large evidence [170,171] implicated the Aβ1-42 peptide aggregation controlled by lipid membranes, but in contrast, cholesterol in the lipid membrane significantly enhances the peptide aggregation by cholesterol, showing a high affinity for the peptide. However, the detailed interaction and its connection between the lipid bilayer and membrane remains elusive. The new idea is to study how free and metal ions bound to Aβ1-42 during aggregation changes in cholesterol-contained membranes.
- The Aβ25-35 cytotoxicity is well explained by the lipid bilayer mediated truncated Aβ25-35 peptide aggregation, which has made perforation in the membrane causing uncontrollable permeation of Ca2+ ions [82]. So far, exploring the full-length of Aβ1-42 peptide aggregation in the presence or absence of Cu2+, Zn2+ and Fe2+ metal ions in the membrane remains an open research problem. Here one can describe the cytotoxicity of the full-length Aβ1-42.
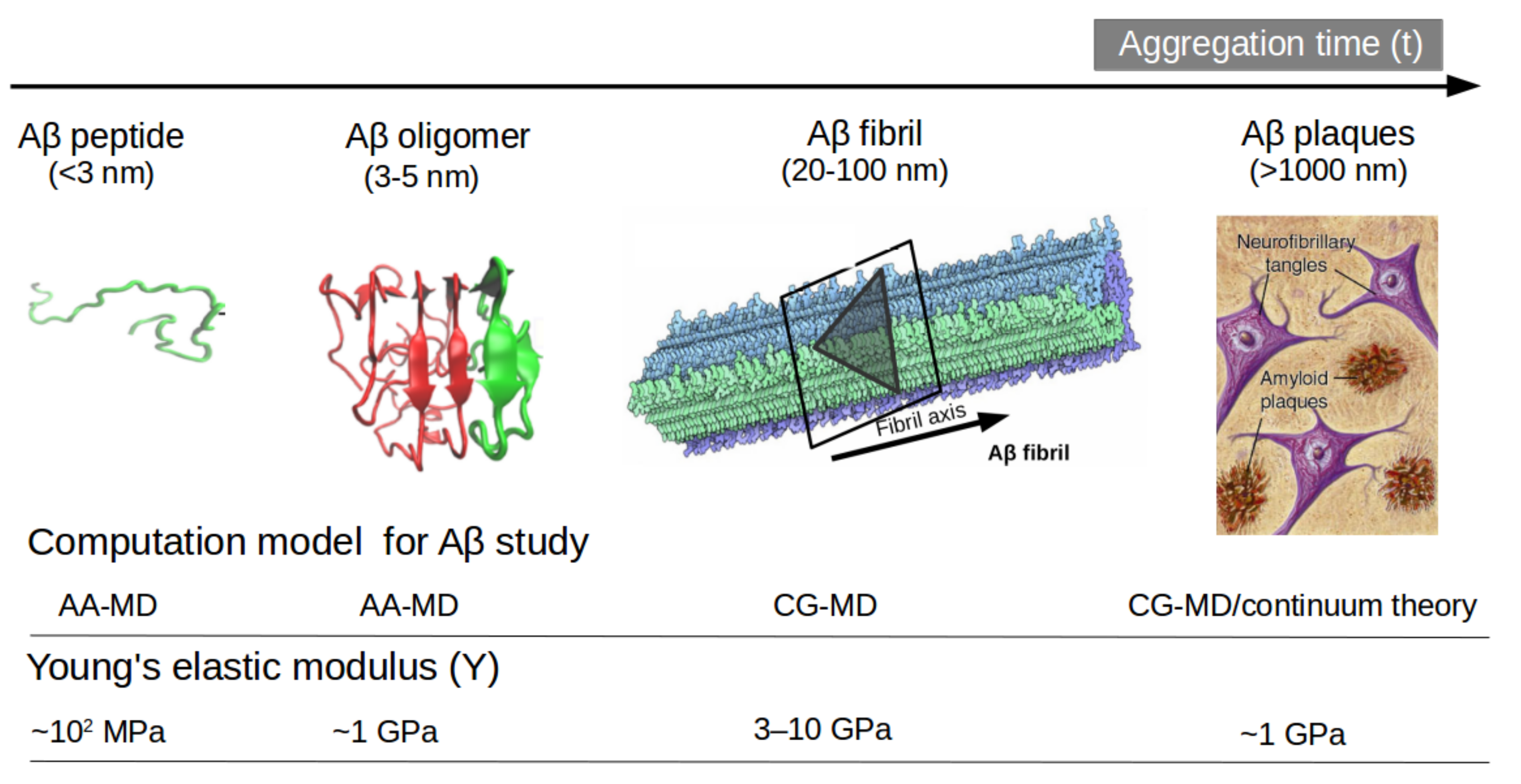
- Advanced MD simulation, aided by the improved version of coarse-grained force fields (e.g., MARTINI 3, UNRES, PRIMO, etc), can describe large-scale systems and overcome time-scale limitations, and improve our understanding of the connection between AD progression and initial stages of the disease by tracing the molecular pathways of full-length Aβ aggregation in complex with lipid bilayers. Similarly, it will help to unveil the fundamental role played by mechanical stability during amyloidogenesis and Aβ aggregate maturation.
- Our previous investigations have envisaged two molecules, M30 and Gabapentin, blocking multiple steps of the Aβ cascade: Aggregation, Synaptotoxicity, membrane pore formation, calcium dyshomeostasis and memory impairment. Thus, we strongly recommend using M30 and Gabapentin as part of the clinical trial as an alternative AD treatment.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | Amyloid beta |
| AD | Alzheimer’s disease |
| MD | Molecular dynamics |
| DFT | Density Functional Theory |
| IDP | Intrinsically disordered proteins |
| DMPC | Dimyristoylgylcerophosphocholine |
| DPPC | Dipalmitoylphosphatidylcholine |
| DOPS | Dioleoyl phosphatidylserine |
| POPC | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| POPE | Palmytoil-oleoyl-phosphatidylethanolamine |
References
- Alzheimer’s Association. 2021 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Alzheimer’s Disease. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Bai, X.C.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.W.; Shi, Y. An Atomic Structure of Human γ-Secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Colvin, M.T.; Silvers, R.; Ni, Q.Z.; Can, T.V.; Sergeyev, I.; Rosay, M.; Donovan, K.J.; Michael, B.; Wall, J.; Linse, S.; et al. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J. Am. Chem. Soc. 2016, 138, 9663–9674. [Google Scholar] [CrossRef] [PubMed]
- Wälti, M.A.; Ravotti, F.; Arai, H.; Glabe, C.G.; Wall, J.S.; Böckmann, A.; Güntert, P.; Meier, B.H.; Riek, R. Atomic-Resolution Structure of a Disease-Relevant Aβ(1-42) Amyloid Fibril. Proc. Natl. Acad. Sci. USA 2016, 113, E4976–E4984. [Google Scholar] [CrossRef]
- Gremer, L.; Schenk, C.; Reinartz, E.; Ravelli, R.B.G.; Tusche, M.; Lopez-iglesias, C.; Hoyer, W.; Heise, H.; Willbold, D. Fibril Structure of Amyloid-b (1-42) by Cryo–Electron Microscopy. Science 2017, 119, 116–119. [Google Scholar] [CrossRef]
- Roberts, B.R.; Lind, M.; Wagen, A.Z.; Rembach, A.; Frugier, T.; Li, Q.X.; Ryan, T.M.; McLean, C.A.; Doecke, J.D.; Rowe, C.C.; et al. Biochemically-Defined Pools of Amyloid-β in Sporadic Alzheimer’s Disease: Correlation with Amyloid PET. Brain 2017, 140, 1486–1498. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabel, C.G. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- O’Nuallain, B.; Wetzel, R. Conformational Abs Recognizing a Generic Amyloid Fibril Epitope. Proc. Natl. Acad. Sci. USA 2002, 99, 1485–1490. [Google Scholar] [CrossRef]
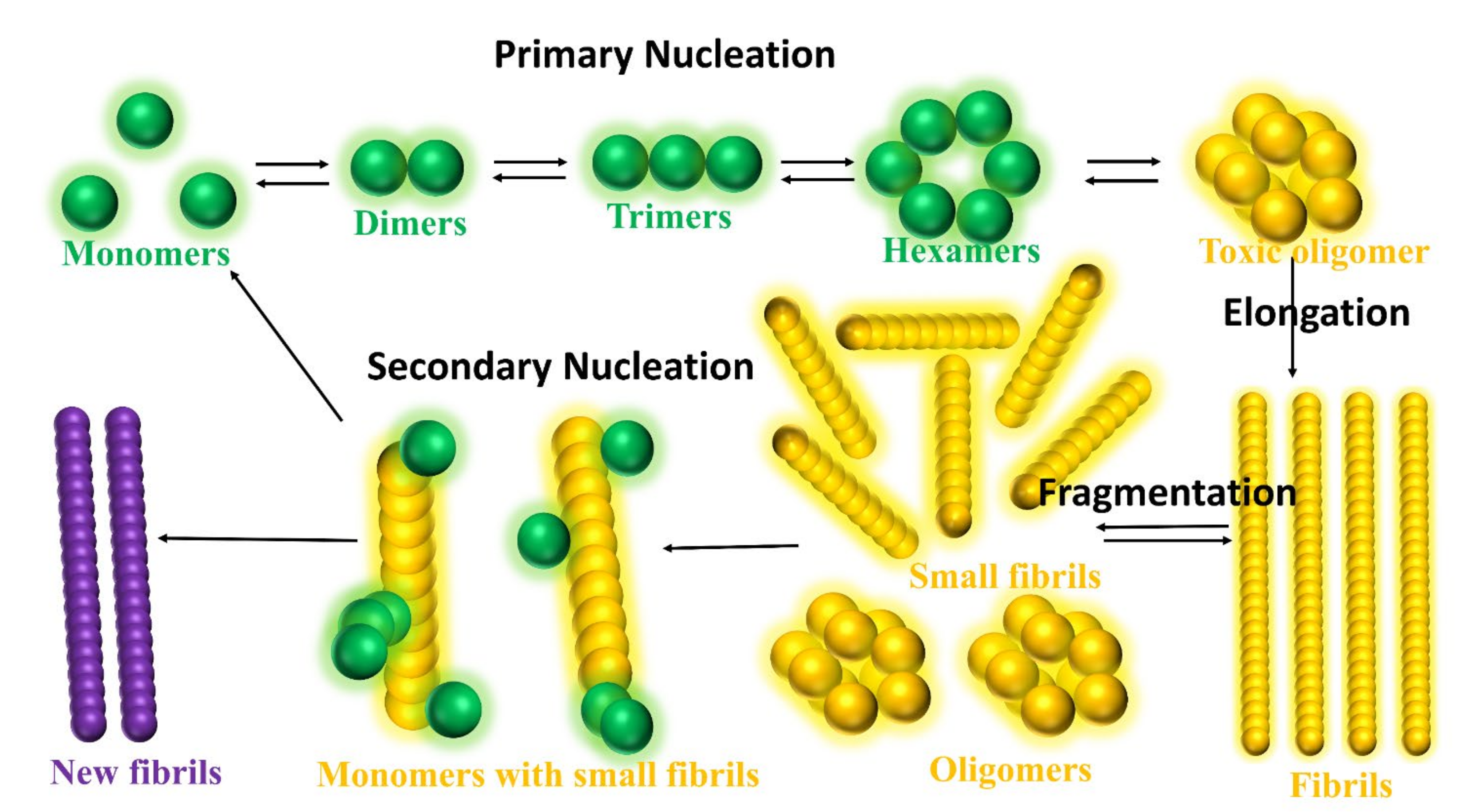
- Michaels, T.C.T.; Šarić, A.; Curk, S.; Bernfur, K.; Arosio, P.; Meisl, G.; Dear, A.J.; Cohen, S.I.A.; Dobson, C.M.; Vendruscolo, M.; et al. Dynamics of Oligomer Populations Formed during the Aggregation of Alzheimer’s Aβ42 Peptide. Nat. Chem. 2020, 12, 445–451. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of Amyloid and Tau with Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol. 2019, 76, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Ramamoorthy, A.; Sahoo, B.R.; Zheng, J.; Faller, P.; Straub, J.E.; Dominguez, L.; Shea, J.E.; Dokholyan, N.V.; de Simone, A.; et al. Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer’s Disease, Parkinson’s Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis. Chem. Rev. 2021, 121, 2545–2647. [Google Scholar] [CrossRef]
- Saravanan, K.M.; Zhang, H.; Zhang, H.; Xi, W.; Wei, Y. On the Conformational Dynamics of β-Amyloid Forming Peptides: A Computational Perspective. Front. Bioeng. Biotechnol. 2020, 8, 1–19. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a Molecular Dynamics Force Field for Both Folded and Disordered Protein States. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef]
- Krupa, P.; Pham, D.Q.H.; Li, M.S. Properties of Monomeric Aβ42 Probed by Different Sampling Methods and Force Fields: Role of Energy Components. J. Chem. Phys. 2019, 151, 055101. [Google Scholar] [CrossRef]
- Kirkitadze, M.D.; Condron, M.M.; Teplow, D.B. Identification and Characterization of Key Kinetic Intermediates in Amyloid β-Protein Fibrillogenesis. J. Mol. Biol. 2001, 312, 1103–1119. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.H.; Chatterjee, P.; Ham, S. Computer Simulations of Intrinsically Disordered Proteins. Annu. Rev. Phys. Chem. 2017, 68, 117–134. [Google Scholar] [CrossRef]
- Viet, M.H.; Nguyen, P.H.; Derreumaux, P.; Li, M.S. Effect of the English Familial Disease Mutation (H6R) on the Monomers and Dimers of Aβ40 and Aβ42. ACS Chem. Neurosci. 2014, 5, 646–657. [Google Scholar] [CrossRef]
- Huy, P.D.Q.; Vuong, Q.V.; La Penna, G.; Faller, P.; Li, M.S. Impact of Cu(II) Binding on Structures and Dynamics of Aβ42 Monomer and Dimer: Molecular Dynamics Study. ACS Chem. Neurosci. 2016, 7, 1348–1363. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Krupa, P.; Hai, N.M.; Linh, H.Q.; Li, M.S. Structure and Physicochemical Properties of the Aβ42 Tetramer: Multiscale Molecular Dynamics Simulations. J. Phys. Chem. B 2019, 123, 7253–7269. [Google Scholar] [CrossRef]
- Chong, S.H.; Ham, S. Assessing the Influence of Solvation Models on Structural Characteristics of Intrinsically Disordered Protein. Comput. Theor. Chem. 2013, 1017, 194–199. [Google Scholar] [CrossRef]
- Best, R.B.; Zheng, W.; Mittal, J. Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput. 2014, 10, 5113–5124. [Google Scholar] [CrossRef]
- Piana, S.; Donchev, A.G.; Robustelli, P.; Shaw, D.E. Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119, 5113–5123. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.U.; Rehman, A.U.; Liu, H.; Chen, H.F. Comparison and Evaluation of Force Fields for Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2020, 60, 4912–4923. [Google Scholar] [CrossRef]
- Ye, W.; Ji, D.; Wang, W.; Luo, R.; Chen, H.F. Test and Evaluation of Ff99IDPs Force Field for Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2015, 55, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Wang, W.; Ye, W.; Ji, D.; Luo, R.; Chen, H.F. Ff14IDPs Force Field Improving the Conformation Sampling of Intrinsically Disordered Proteins. Chem. Biol. Drug Des. 2017, 89, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Luo, R.; Chen, H.F. The IDP-Specific Force Field Ff14IDPSFF Improves the Conformer Sampling of Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2017, 57, 1166–1178. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, H.; Yang, S.; Luo, R.; Chen, H.F. Well-Balanced Force Field Ff03 CMAP for Folded and Disordered Proteins. J. Chem. Theory Comput. 2019, 15, 6769–6780. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Best, R.B.; Mittal, J. Protein Simulations with an Optimized Water Model: Cooperative Helix Formation and Temperature-Induced Unfolded State Collapse. J. Phys. Chem. B 2010, 114, 14916–14923. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2016, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Song, D.; Zhang, Y.; Yang, S.; Luo, R.; Chen, H.F. Extensive Tests and Evaluation of the CHARMM36IDPSFF Force Field for Intrinsically Disordered Proteins and Folded Proteins. Phys. Chem. Chem. Phys. 2019, 21, 21918–21931. [Google Scholar] [CrossRef]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D.E. How Robust Are Protein Folding Simulations with Respect to Force Field Parameterization? Biophys. J. 2011, 100, L47–L49. [Google Scholar] [CrossRef]
- Samantray, S.; Yin, F.; Kav, B.; Strodel, B. Different Force Fields Give Rise to Different Amyloid Aggregation Pathways in Molecular Dynamics Simulations. J. Chem. Inf. Model. 2020, 60, 6462–6475. [Google Scholar] [CrossRef]
- Mu, J.; Liu, H.; Zhang, J.; Luo, R.; Chen, H.F. Recent Force Field Strategies for Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2021, 61, 1037–1047. [Google Scholar] [CrossRef]
- Kmiecik, S.; Gront, D.; Kolinski, M.; Wieteska, L.; Dawid, A.E.; Kolinski, A. Coarse-Grained Protein Models and Their Applications. Chem. Rev. 2016, 116, 7898–7936. [Google Scholar] [CrossRef]
- Seo, M.; Rauscher, S.; Pomès, R.; Tieleman, D.P. Improving Internal Peptide Dynamics in the Coarse-Grained MARTINI Model: Toward Large-Scale Simulations of Amyloid- and Elastin-like Peptides. J. Chem. Theory Comput. 2012, 8, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Maisuradze, N.; Kachlishvili, K.; Scheraga, H.A.; Maisuradze, G.G. Elucidating Important Sites and the Mechanism for Amyloid Fibril Formation by Coarse-Grained Molecular Dynamics. ACS Chem. Neurosci. 2017, 8, 201–209. [Google Scholar] [CrossRef]
- Latshaw, D.C.; Cheon, M.; Hall, C.K. Effects of Macromolecular Crowding on Amyloid Beta (16-22) Aggregation Using Coarse-Grained Simulations. J. Phys. Chem. B 2014, 118, 13513–13526. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, A.; Xu, H.; Matysiak, S. Pathways of Amyloid-Beta Absorption and Aggregation in a Membranous Environment. Phys. Chem. Chem. Phys. 2019, 21, 8559–8568. [Google Scholar] [CrossRef]
- Souza, P.C.T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.H.; Wassenaar, T.A.; et al. Martini 3: A General Purpose Force Field for Coarse-Grained Molecular Dynamics. Nat. Methods 2021, 18, 382–388. [Google Scholar] [CrossRef]
- Mahmood, M.I.; Poma, A.B.; Okazak, K.I. Optimizing Gō-MARTINI Coarse-Grained Model for F-BAR Protein on Lipid Membrane. Front. Mol. Biosci. 2021, 8, 1–10. [Google Scholar] [CrossRef]
- Poma, A.B.; Cieplak, M.; Theodorakis, P.E. Combining the MARTINI and Structure-Based Coarse-Grained Approaches for the Molecular Dynamics Studies of Conformational Transitions in Proteins. J. Chem. Theory Comput. 2017, 13, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.B.; Li, M.S.; Theodorakis, P.E. Generalization of the Elastic Network Model for the Study of Large Conformational Changes in Biomolecules. Phys. Chem. Chem. Phys. 2018, 20, 17020–17028. [Google Scholar] [CrossRef] [PubMed]
- Ingólfsson, H.I.; Melo, M.N.; Van Eerden, F.J.; Arnarez, C.; Lopez, C.A.; Wassenaar, T.A.; Periole, X.; De Vries, A.H.; Tieleman, D.P.; Marrink, S.J. Lipid Organization of the Plasma Membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. [Google Scholar] [CrossRef]
- Poma, A.B.; Guzman, H.V.; Li, M.S.; Theodorakis, P.E. Mechanical and Thermodynamic Properties of Aβ42, Aβ40, and α-Synuclein Fibrils: A Coarse-Grained Method to Complement Experimental Studies. Beilstein J. Nanotechnol. 2019, 10, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Chwastyk, M.; Bernaola, A.P.; Cieplak, M. Statistical Radii Associated with Amino Acids to Determine the Contact Map: Fixing the Structure of a Type i Cohesin Domain in the Clostridium Thermocellum Cellulosome. Phys. Biol. 2015, 12, 46002. [Google Scholar] [CrossRef]
- Poma, A.B.; Chwastyk, M.; Cieplak, M. Elastic Moduli of Biological Fibers in a Coarse-Grained Model: Crystalline Cellulose and β-Amyloids. Phys. Chem. Chem. Phys. 2017, 19, 28195–28206. [Google Scholar] [CrossRef]
- Poma, A.B.; Thu, T.T.M.; Tri, L.T.M.; Nguyen, H.L.; Li, M.S. Nanomechanical Stability of Aβ Tetramers and Fibril-like Structures: Molecular Dynamics Simulations. J. Phys. Chem. B 2021, 125, 7628–7637. [Google Scholar] [CrossRef]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-beta oligomers in Toxicity, Propogation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.M.; Moeendarbary, E.; Sheridan, G.K. Mechanobiology of the Brain in Ageing and Alzheimer’s Disease. Eur. J. Neurosci. 2021, 53, 3851–3878. [Google Scholar] [CrossRef] [PubMed]
- Carballo-Pacheco, M.; Ismail, A.E.; Strodel, B. On the Applicability of Force Fields to Study the Aggregation of Amyloidogenic Peptides Using Molecular Dynamics Simulations. J. Chem. Theory Comput. 2018, 14, 6063–6075. [Google Scholar] [CrossRef] [PubMed]
- Senguen, F.T.; Doran, T.M.; Anderson, E.A.; Nilsson, B.L. Clarifying the Influence of Core Amino Acid Hydrophobicity, Secondary Structure Propensity, and Molecular Volume on Amyloid-β 16-22 Self-Assembly. Mol. Biosyst. 2011, 7, 497–510. [Google Scholar] [CrossRef]
- Lockhart, C.; Smith, A.K.; Klimov, D.K. Three Popular Force Fields Predict Consensus Mechanism of Amyloid β Peptide Binding to the Dimyristoylgylcerophosphocholine Bilayer. J. Chem. Inf. Model. 2020, 60, 2282–2293. [Google Scholar] [CrossRef] [PubMed]
- Akbayraka, I.Y.; Caglayanb, S.I.; Ozcanb, Z.; Uverskyc, V.N.; Coskuner-Weberb, O. Current Challenges and Limitations in the Studies of Intrinsically Disordered Proteins in Neurodegenerative Diseases by Computer Simulations. Curr. Alzheimer Res. 2020, 17, 805–818. [Google Scholar] [CrossRef]
- Press-Sandler, O.; Miller, Y. Molecular Mechanisms of Membrane-Associated Amyloid Aggregation: Computational Perspective and Challenges. Biochim. Biophys. Acta -Biomembr. 2018, 1860, 1889–1905. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.H.; Berkowitz, M.L. Interaction between Amyloid-β (1-42) Peptide and Phospholipid Bilayers: A Molecular Dynamics Study. Biophys. J. 2009, 96, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.H.; Berkowitz, M.L. Structure of the Amyloid-β (1-42) Monomer Absorbed to Model Phospholipid Bilayers: A Molecular Dynamics Study. J. Phys. Chem. B 2009, 113, 14480–14486. [Google Scholar] [CrossRef]
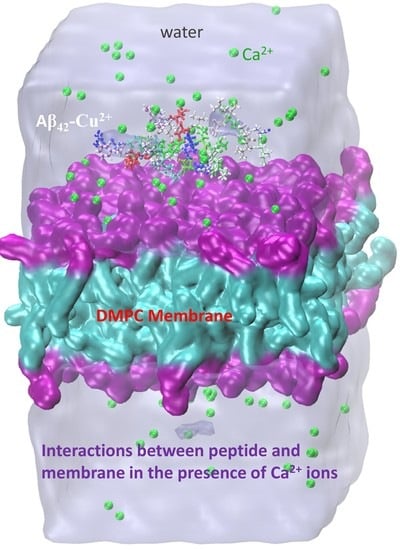
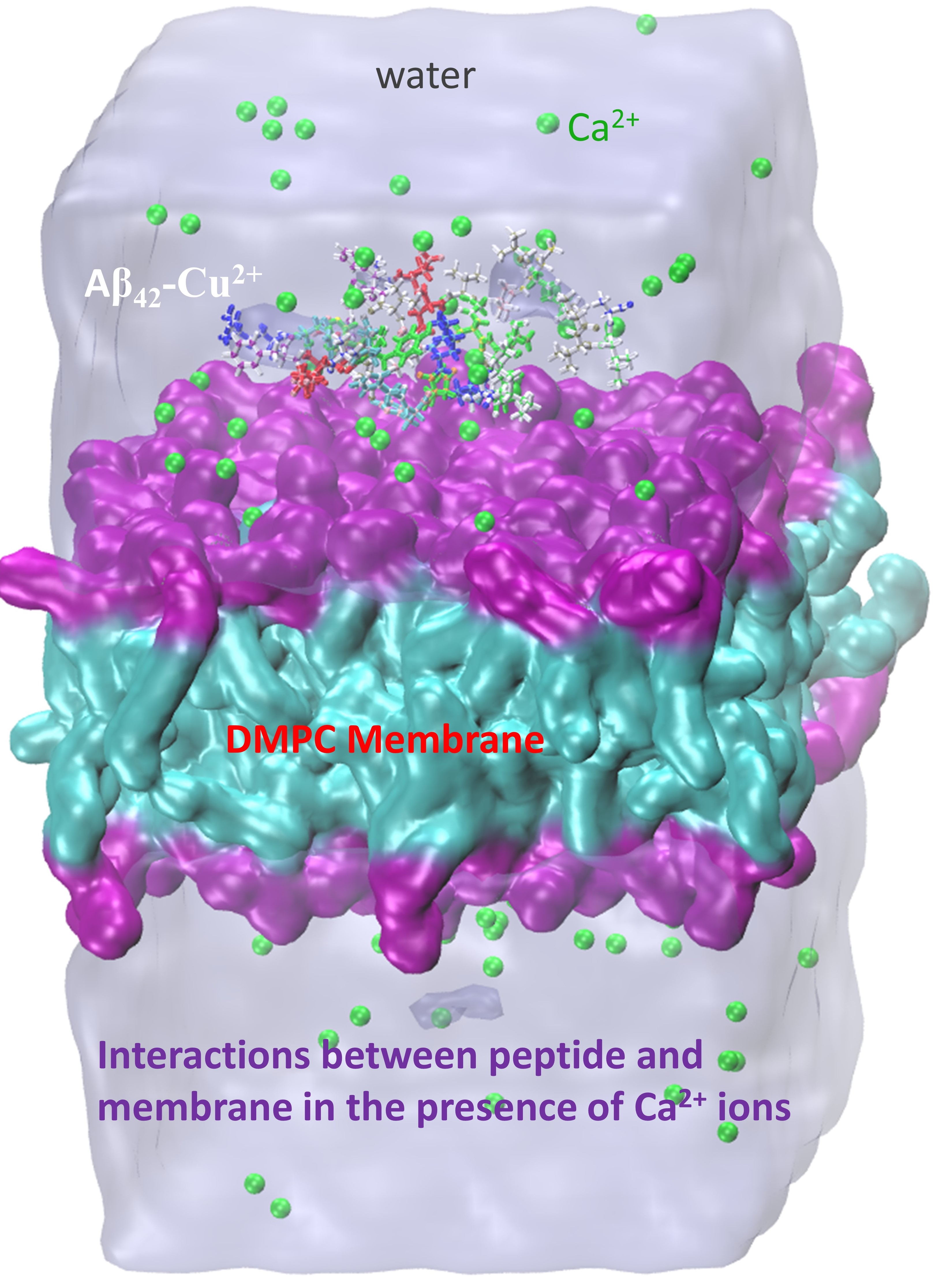
- Lockhart, C.; Klimov, D.K. Calcium Enhances Binding of Aβ Monomer to DMPC Lipid Bilayer. Biophys. J. 2015, 108, 1807–1818. [Google Scholar] [CrossRef][Green Version]
- Pike, C.J.; Burdick, D.; Walencewicz, A.J.; Glabe, C.G.; Cotman, C.W. Neurodegeneration Induced by β-Amyloid Peptides in Vitro: The Role of Peptide Assembly State. J. Neurosci. 1993, 13, 1676–1687. [Google Scholar] [CrossRef]
- Millucci, L.; Ghezzi, L.; Bernardini, G.; Santucci, A. Conformations and Biological Activities of Amyloid Beta Peptide 25–35. Curr. Protein Pept. Sci. 2009, 999, 1–6. [Google Scholar] [CrossRef]
- Smith, A.K.; Klimov, D.K. Binding of Cytotoxic Aβ25-35 Peptide to the Dimyristoylphosphatidylcholine Lipid Bilayer. J. Chem. Inf. Model. 2018, 58, 1053–1065. [Google Scholar] [CrossRef]
- Jang, H.; Connelly, L.; Teran Arce, F.; Ramachandran, S.; Kagan, B.L.; Lal, R.; Nussinov, R. Mechanisms for the Insertion of Toxic, Fibril-like β-Amyloid Oligomers into the Membrane. J. Chem. Theory Comput. 2013, 9, 822–833. [Google Scholar] [CrossRef]
- Davis, C.H.; Berkowitz, M.L. A Molecular Dynamics Study of the Early Stages of Amyloid-β(1-42) Oligomerization: The Role of Lipid Membranes. Proteins Struct. Funct. Bioinform. 2010, 78, 2533–2545. [Google Scholar] [CrossRef] [PubMed]
- Snowden, S.G.; Ebshiana, A.A.; Hye, A.; An, Y.; Pletnikova, O.; O’Brien, R.; Troncoso, J.; Legido-Quigley, C.; Thambisetty, M. Association between Fatty Acid Metabolism in the Brain and Alzheimer Disease Neuropathology and Cognitive Performance: A Nontargeted Metabolomic Study. PLoS Med. 2017, 14, e1002266. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Shi, X.F.; Nguyen, P.H.; Sterpone, F.; Salsbury, F.R.; Derreumaux, P. Amyloid-β(29-42) Dimeric Conformations in Membranes Rich in Omega-3 and Omega-6 Polyunsaturated Fatty Acids. J. Phys. Chem. B 2019, 123, 2687–2696. [Google Scholar] [CrossRef]
- Jana, M.K.; Cappai, R.; Pham, C.L.L.; Ciccotosto, G.D. Membrane-Bound Tetramer and Trimer Aβ Oligomeric Species Correlate with Toxicity towards Cultured Neurons. J. Neurochem. 2016, 136, 594–608. [Google Scholar] [CrossRef]
- Doig, A.J.; Castillo-frias, M.P.; Berthoumieu, O.; Tarus, B.; Nasica-labouze, J.; Sterpone, F.; Nguyen, P.H.; Hooper, N.M.; Faller, P.; Derreumaux, P. Why Is Research on Amyloid-β Failing to Give New Drugs for Alzheimer’s Disease? ACS Chem. Neurosci. 2017, 8, 1435–1437. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Hung, H.M.; Tran, K.N.; Nguyen, M.T. Replica Exchange Molecular Dynamics Study of the Amyloid Beta (11-40) Trimer Penetrating a Membrane. RSC Adv. 2017, 7, 7346–7357. [Google Scholar] [CrossRef]
- Ngo, S.T.; Nguyen, M.T.; Nguyen, N.T.; Vu, V.V. The Effects of A21G Mutation on Transmembrane Amyloid Beta (11-40) Trimer: An in Silico Study. J. Phys. Chem. B 2017, 121, 8467–8474. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Pan, F.; Tran, L.; Roland, C.; Sagui, C. The F19W Mutation Reduces the Binding Affinity of the Transmembrane Aβ11-40trimer to the Membrane Bilayer. RSC Adv. 2021, 11, 2664–2676. [Google Scholar] [CrossRef]
- Poojari, C.; Strodel, B. Stability of Transmembrane Amyloid β-Peptide and Membrane Integrity Tested by Molecular Modeling of Site-Specific Aβ42 Mutations. PLoS ONE 2013, 8, e78399. [Google Scholar] [CrossRef]
- Murakami, K.; Irie, K.; Morimoto, A.; Ohigashi, H.; Shindo, M.; Nagao, M.; Shimizu, T.; Shirasawa, T. Neurotoxicity and Physicochemical Properties of Aβ Mutant Peptides from Cerebral Amyloid Angiopathy: Implication for the Pathogenesis of Cerebral Amyloid Angiopathy and Alzheimer’s Disease. J. Biol. Chem. 2003, 278, 46179–46187. [Google Scholar] [CrossRef]
- Luheshi, L.M.; Tartaglia, G.G.; Brorsson, A.C.; Pawar, A.P.; Watson, I.E.; Chiti, F.; Vendruscolo, M.; Lomas, D.A.; Dobson, C.M.; Crowther, D.C. Systematic in Vivo Analysis of the Intrinsic Determinants of Amyloid β Pathogenicity. PLoS Biol. 2007, 5, e290. [Google Scholar] [CrossRef]
- Petkova, A.T.; Yau, W.M.; Tycko, R. Experimental Constraints on Quaternary Structure in Alzheimer’s β-Amyloid Fibrils. Biochemistry 2006, 45, 498–512. [Google Scholar] [CrossRef]
- Yung, S.K.; Liu, L.; Axelsen, P.H.; Hochstrasser, R.M. 2D IR Provides Evidence for Mobile Water Molecules in β-Amyloid Fibrils. Proc. Natl. Acad. Sci. USA 2009, 106, 17751–17756. [Google Scholar] [CrossRef]
- Wang, T.; Jo, H.; DeGrado, W.F.; Hong, M. Water Distribution, Dynamics, and Interactions with Alzheimer’s β-Amyloid Fibrils Investigated by Solid-State NMR. J. Am. Chem. Soc. 2017, 139, 6242–6252. [Google Scholar] [CrossRef] [PubMed]
- Xi, W.; Hansmann, U.H.E. Ring-like N-Fold Models of Aβ42 Fibrils. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Serra-Batiste, M.; Ninot-Pedrosa, M.; Bayoumi, M.; Gairí, M.; Maglia, G.; Carulla, N. Aβ42 Assembles into Specific β-Barrel Pore-Forming Oligomers in Membrane-Mimicking Environments. Proc. Natl. Acad. Sci. USA 2016, 113, 10866–10871. [Google Scholar] [CrossRef]
- Ciudad, S.; Puig, E.; Botzanowski, T.; Meigooni, M.; Arango, A.S.; Do, J.; Mayzel, M.; Bayoumi, M.; Chaignepain, S.; Maglia, G.; et al. Aβ(1-42) Tetramer and Octamer Structures Reveal Edge Conductivity Pores as a Mechanism for Membrane Damage. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.K.; Klimov, D.K. De Novo Aggregation of Alzheimer’s Aβ25-35 Peptides in a Lipid Bilayer. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tofoleanu, F.; Brooks, B.R.; Buchete, N.V. Modulation of Alzheimers Aβ Protofilament-Membrane Interactions by Lipid Headgroups. ACS Chem. Neurosci. 2015, 6, 446–455. [Google Scholar] [CrossRef]
- Qiang, W.; Doherty, K.E.; Klees, L.M.; Tobin-Miyaji, Y. Time-Dependent Lipid Dynamics, Organization and Peptide-Lipid Interaction in Phospholipid Bilayers with Incorporated β-Amyloid Oligomers. J. Phys. Chem. Lett. 2020, 11, 8329–8336. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Pérez, E.J.; Sepúlveda, F.J.; Peters, C.; Bascuñán, D.; Riffo-Lepe, N.O.; González-Sanmiguel, J.; Sánchez, S.A.; Peoples, R.W.; Vicente, B.; Aguayo, L.G. Effect of Cholesterol on Membrane Fluidity and Association of Aβ Oligomers and Subsequent Neuronal Damage: A Double-Edged Sword. Front. Aging Neurosci. 2018, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, G.; Vitiello, G.; Ortona, O.; Tedeschi, A.; Ramunno, A.; D’Ursi, A.M. Interaction between Alzheimer’s Aβ(25-35) Peptide and Phospholipid Bilayers: The Role of Cholesterol. Biochim. Biophys. Acta -Biomembr. 2008, 1778, 2710–2716. [Google Scholar] [CrossRef]
- Di Scala, C.; Troadec, J.D.; Lelièvre, C.; Garmy, N.; Fantini, J.; Chahinian, H. Mechanism of Cholesterol-Assisted Oligomeric Channel Formation by a Short Alzheimer β-Amyloid Peptide. J. Neurochem. 2014, 128, 186–195. [Google Scholar] [CrossRef]
- Di Scala, C.; Chahinian, H.; Yahi, N.; Garmy, N.; Fantini, J. Interaction of Alzheimer’s β-Amyloid Peptides with Cholesterol: Mechanistic Insights into Amyloid Pore Formation. Biochemistry 2014, 53, 4489–4502. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, C.; Yahi, N.; Garmy, N.; Chahinian, H.; Fantini, J. Biochemical Identi Fi Cation of a Linear Cholesterol-Binding Domain within Alzheimer’ s β Amyloid Peptide. ACS Chem. Neurosci. 2013, 4, 509–517. [Google Scholar] [CrossRef]
- Avdulov, N.A.; Chochina, S.V.; Igbavboa, U.; Warden, C.S.; Vassiliev, A.V.; Wood, W.G. Lipid Binding to Amyloid β-Peptide Aggregates: Preferential Binding of Cholesterol as Compared with Phosphatidylcholine and Fatty Acids. J. Neurochem. 1997, 69, 1746–1752. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, J. Cholesterol Promotes the Interaction of Alzheimer β-Amyloid Monomer with Lipid Bilayer. J. Mol. Biol. 2012, 421, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.N.; Chiu, S.W.; Benoit, J.; Chew, L.Y.; Mu, Y. Amyloid β Peptides Aggregation in a Mixed Membrane Bilayer: A Molecular Dynamics Study. J. Phys. Chem. B 2011, 115, 12247–12256. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Ionov, M.; Pavlov, E.; Duchen, M.R. Membrane Cholesterol Content Plays a Key Role in the Neurotoxicity of β-Amyloid: Implications for Alzheimer’s Disease. Aging Cell 2011, 10, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Dias, C.L.; Jalali, S.; Yang, Y.; Cruz, L. Role of Cholesterol on Binding of Amyloid Fibrils to Lipid Bilayers. J. Phys. Chem. B 2020, 124, 3036–3042. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, Iron and Zinc in Alzheimer’s Disease Senile Plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Candy, J.M.; Klinowski, J.; Perry, R.H.; Perry, E.K.; Fairbairn, A.; Oakley, A.E.; Carpenter, T.A.; Atack, J.R.; Blessed, G.; Edwardson, J.A. Aluminosilicates and Senile Plaque Formation in Alzheimer’S Disease. Lancet 1986, 327, 354–356. [Google Scholar] [CrossRef]
- Boopathi, S.; Kolandaivel, P. Role of Zinc and Copper Metal Ions in Amyloid β-Peptides Aβ 1–40 and Aβ 1–42 Aggregation. RSC Adv. 2014, 4, 38951–38965. [Google Scholar] [CrossRef]
- Boopathi, S.; Huy, P.D.Q.; González, W.; Theodorakis, P.E.; Li, M.S. Zinc Binding Promotes Greater Hydrophobicity in Alzheimer’s Aβ1–42 Peptide than Copper Binding: Molecular Dynamics and Solvation Thermodynamics Studies. Proteins 2020, 88, 1285–1302. [Google Scholar] [CrossRef]
- Miller, Y.; Ma, B.; Nussinov, R. Zinc Ions Promote Alzheimer A Aggregation via Population Shift of Polymorphic States. Proc. Natl. Acad. Sci. USA 2010, 107, 9490–9495. [Google Scholar] [CrossRef]
- Lee, M.; Kim, J.I.; Na, S.; Eom, K. Metal Ions Affect the Formation and Stability of Amyloid β Aggregates at Multiple Length Scales. Phys. Chem. Chem. Phys. 2018, 20, 8951–8961. [Google Scholar] [CrossRef]
- Hensley, K.; Maidt, M.L.; Yu, Z.; Sang, H.; Markesbery, W.R.; Floyd, R.A. Electrochemical Analysis of Protein Nitrotyrosine and Dityrosine in the Alzheimer Brain Indicates Region-Specific Accumulation. J. Neurosci. 1998, 18, 8126–8132. [Google Scholar] [CrossRef] [PubMed]
- Gu, M.; Bode, D.C.; Viles, J.H. Copper Redox Cycling Inhibits Aβ Fibre Formation and Promotes Fibre Fragmentation, While Generating a Dityrosine Aβ Dimer. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- De Santis, E.; Minicozzi, V.; Proux, O.; Rossi, G.; Silva, K.I.; Lawless, M.J.; Stellato, F.; Saxena, S.; Morante, S. Cu(II)−Zn(II) Cross-Modulation in Amyloid−Beta Peptide Binding: An X-ray Absorption Spectroscopy Study. J. Phys. Chem B 2015, 119, 15813–15820. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Pittman, J.M.; Zerweck, J.; Venkata, B.S.; Moore, P.C.; Sachleben, J.R.; Meredith, S.C. β-Amyloid Aggregation and Heterogeneous Nucleation. Protein Sci. 2019, 28, 1567–1581. [Google Scholar] [CrossRef]
- Vázquez De La Torre, A.; Gay, M.; Vilaprinyó-Pascual, S.; Mazzucato, R.; Serra-Batiste, M.; Vilaseca, M.; Carulla, N. Direct Evidence of the Presence of Cross-Linked Aβ Dimers in the Brains of Alzheimer’s Disease Patients. Anal. Chem. 2018, 90, 4552–4560. [Google Scholar] [CrossRef]
- Coskuner, O.; Uversky, V.N. Tyrosine Regulates β-Sheet Structure Formation in Amyloid-Β42: A New Clustering Algorithm for Disordered Proteins. J. Chem. Inf. Model. 2017, 57, 1342–1358. [Google Scholar] [CrossRef]
- Orr, M.E.; Garbarino, V.R.; Salinas, A.; Buffenstein, R. Sustained High Levels of Neuroprotective, High Molecular Weight, Phosphorylated Tau in the Longest-Lived Rodent. Neurobiol. Aging 2015, 36, 1496–1504. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; Paradis, M.D.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid Induction of Alzheimer Aβ Amyloid Formation by Zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef]
- Kozin, S.A.; Mezentsev, Y.V.; Kulikova, A.A.; Indeykina, M.I.; Golovin, A.V.; Ivanov, A.S.; Tsvetkov, P.O.; Makarov, A.A. Zinc-Induced Dimerization of the Amyloid-β Metal-Binding Domain 1–16 Is Mediated by Residues 11–14. Mol. Biosyst. 2011, 7, 1053–1055. [Google Scholar] [CrossRef]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.S.; et al. Treatment with a Copper-Zinc Chelator Markedly and Rapidly Inhibits β-Amyloid Accumulation in Alzheimer’s Disease Transgenic Mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef]
- Guilloreau, L.; Combalbert, S.; Sournia-Saquet, M.; Mazarguil, H.; Faller, P. Redox Chemistry of Copper-Amyloid-β: The Generation of Hydroxyl Radical in the Presence of Ascorbate Is Linked to Redox-Potentials and Aggregation State. ChemBioChem 2007, 8, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, S.; Kolandaivel, P. Fe2+ Binding on Amyloid β-Peptide Promotes Aggregation. Proteins Struct. Funct. Bioinform. 2016, 84, 1257–1274. [Google Scholar] [CrossRef]
- Tycko, R. Molecular Structure of Amyloid Fibrils: Insights from Solid-State NMR. Q. Rev. Biophys. 2006, 39, 1–55. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.; Mutter, S.T.; Kennedy-Britten, O.D.; Platts, J.A. Molecular Dynamics Simulation of Aluminium Binding to Amyloid-β and Its Effect on Peptide Structure. PLoS ONE 2019, 14, e0217992. [Google Scholar] [CrossRef] [PubMed]
- Platts, J.A. Quantum Chemical Molecular Dynamics and Metadynamics Simulation of Aluminium Binding to Amyloid-β and Related Peptides. R. Soc. Open Sci. 2020, 7, 191562. [Google Scholar] [CrossRef]
- Roldán-Martín, L.; Peccati, F.; Sciortino, G.; Sodupe, M.; Maréchal, J.-D. Impact of Cu(II) and Al(III) on the Conformational Landscape of Amyloidb1-42. Phys. Chem. Chem. Phys. 2021, 23, 13023–13032. [Google Scholar] [CrossRef]
- Wallin, C.; Jarvet, J.; Biverstål, H.; Wärmländer, S.; Danielsson, J.; Gräslund, A.; Abelein, A. Metal Ion Coordination Delays Amyloid-β Peptide Self-Assembly by Forming an Aggregation-Inert Complex. J. Biol. Chem. 2020, 295, 7224–7234. [Google Scholar] [CrossRef]
- Basha, M.R.; Wei, W.; Bakheet, S.A.; Benitez, N.; Siddiqi, H.K.; Ge, Y.W.; Lahiri, D.K.; Zawia, N.H. The Fetal Basis of Amyloidogenesis: Exposure to Lead and Latent Overexpression of Amyloid Precursor Protein and β-Amyloid in the Aging Brain. J. Neurosci. 2005, 25, 823–829. [Google Scholar] [CrossRef]
- Wu, J.; Basha, M.R.; Brock, B.; Cox, D.P.; Cardozo-Pelaez, F.; McPherson, C.A.; Harry, J.; Rice, D.C.; Maloney, B.; Chen, D.; et al. Alzheimer’s Disease (AD)-like Pathology in Aged Monkeys after Infantile Exposure to Environmental Metal Lead (Pb): Evidence for a Developmental Origin and Environmental Link for AD. J. Neurosci. 2008, 28, 3–9. [Google Scholar] [CrossRef]
- Wallin, C.; Sholts, S.B.; Österlund, N.; Luo, J.; Jarvet, J.; Roos, P.M.; Ilag, L.; Gräslund, A.; Wärmländer, S.K.T.S. Alzheimer’s Disease and Cigarette Smoke Components: Effects of Nicotine, PAHs, and Cd(II), Cr(III), Pb(II), Pb(IV) Ions on Amyloid-β Peptide Aggregation. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Charlet, L.; Chapron, Y.; Faller, P.; Kirsch, R.; Stone, A.T.; Baveye, P.C. Neurodegenerative Diseases and Exposure to the Environmental Metals Mn, Pb, and Hg. Coord. Chem. Rev. 2012, 256, 2147–2163. [Google Scholar] [CrossRef]
- Meleleo, D.; Notarachille, G.; Mangini, V.; Arnesano, F. Concentration-Dependent Effects of Mercury and Lead on Aβ42: Possible Implications for Alzheimer’s Disease. Eur. Biophys. J. 2019, 48, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Wallin, C.; Friedemann, M.; Sholts, S.B.; Noormägi, A.; Svantesson, T.; Jarvet, J.; Roos, P.M.; Palumaa, P.; Gräslund, A.; Wärmländer, S.K.T.S. Mercury and Alzheimer’s Disease: Hg(II) Ions Display Specific Binding to the Amyloid-β Peptide and Hinder Its Fibrillization. Biomolecules 2020, 10, 44. [Google Scholar] [CrossRef] [PubMed]
- Banta, R.G.; Markesbery, W.R. Elevated Manganese Levels Associated with Dementia and Extrapyramidal Signs. Neurology 1977, 27, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Wallin, C.; Kulkarni, Y.S.; Abelein, A.; Jarvet, J.; Liao, Q.; Strodel, B.; Olsson, L.; Luo, J.; Abrahams, J.P.; Sholts, S.B.; et al. Characterization of Mn(II) Ion Binding to the Amyloid-β Peptide in Alzheimer’s Disease. J. Trace Elem. Med. Biol. 2016, 38, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Zirah, S.; Kozin, S.A.; Mazur, A.K.; Blond, A.; Cheminant, M.; Ségalas-Milazzo, I.; Debey, P.; Rebuffat, S. Structural Changes of Region 1-16 of the Alzheimer Disease Amyloid β-Peptide upon Zinc Binding and in Vitro Aging. J. Biol. Chem. 2006, 281, 2151–2161. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Li, X.; Cheng, X.; Zhao, N.; Zheng, W. Manganese Exposure Aggravates β-Amyloid Pathology by Microglial Activation. Front. Aging Neurosci. 2020, 12, 1–15. [Google Scholar] [CrossRef]
- Hampel, H.; Lista, S.; Mango, D.; Nisticò, R.; Perry, G.; Avila, J.; Hernandez, F.; Geerts, H.; Vergallo, A. Lithium as a Treatment for Alzheimer’s Disease: The Systems Pharmacology Perspective. J. Alzheimer’s Dis. 2019, 69, 615–629. [Google Scholar] [CrossRef]
- Velosa, J.; Delgado, A.; Finger, E.; Berk, M.; Kapczinski, F.; de Azevedo Cardoso, T. Risk of Dementia in Bipolar Disorder and the Interplay of Lithium: A Systematic Review and Meta-Analyses. Acta Psychiatr. Scand. 2020, 141, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Short, J.L.; Newman, S.A.; Choy, K.H.C.; Tiwari, D.; Yap, C.; Senyschyn, D.; Banks, W.A.; Nicolazzo, J.A. Cognitive Benefits of Lithium Chloride in APP/PS1 Mice Are Associated with Enhanced Brain Clearance of β-Amyloid. Brain Behav. Immun. 2018, 70, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Habib, A.; Shytle, R.D.; Sawmiller, D.; Koilraj, S.; Munna, S.A.; Rongo, D.; Hou, H.; Borlongan, C.V.; Currier, G.; Tan, J. Comparing the Effect of the Novel Ionic Cocrystal of Lithium Salicylate Proline (LISPRO) with Lithium Carbonate and Lithium Salicylate on Memory and Behavior in Female APPswe/PS1dE9 Alzheimer’s Mice. J. Neurosci. Res. 2019, 97, 1066–1080. [Google Scholar] [CrossRef]
- Xiang, J.; Cao, K.; Dong, Y.T.; Xu, Y.; Li, Y.; Song, H.; Zeng, X.X.; Ran, L.Y.; Hong, W.; Guan, Z.Z. Lithium Chloride Reduced the Level of Oxidative Stress in Brains and Serums of APP/PS1 Double Transgenic Mice via the Regulation of GSK3β/Nrf2/HO-1 Pathway. Int. J. Neurosci. 2020, 130, 564–573. [Google Scholar] [CrossRef]
- Liu, M.; Qian, T.; Zhou, W.; Tao, X.; Sang, S.; Zhao, L. Beneficial Effects of Low-Dose Lithium on Cognitive Ability and Pathological Alteration of Alzheimer’s Disease Transgenic Mice Model. Neuroreport 2020, 31, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Berntsson, E.; Paul, S.; Vosough, F.; Sholts, S.B.; Jarvet, J.; Roos, P.M.; Barth, A.; Gräslund, A.; Wärmländer, S.K.T.S. Lithium Ions Display Weak Interaction with Amyloid-Beta (Aβ) Peptides and Have Minor Effects on Their Aggregation. Acta Biochim. Pol. 2021, 68, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Eketjäll, S.; Janson, J.; Kaspersson, K.; Bogstedt, A.; Jeppsson, F.; Fälting, J.; Haeberlein, S.B.; Kugler, A.R.; Alexander, R.C.; Cebers, G. AZD3293: A Novel, Orally Active BACE1 Inhibitor with High Potency and Permeability and Markedly Slow Off-Rate Kinetics. J. Alzheimer’s Dis. 2016, 50, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Athar, T.; Al Balushi, K.; Khan, S.A. Recent Advances on Drug Development and Emerging Therapeutic Agents for Alzheimer’s Disease. Mol. Biol. Rep. 2021, 48, 5629–5645. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Abbas, Q.; Seo, S.Y.; Shahzadi, S.; Al Ashwal, H.; Zaki, N.; Iqbal, Z.; Moustafa, A.A. Computational Modeling and Biomarker Studies of Pharmacological Treatment of Alzheimer’s Disease (Review). Mol. Med. Rep. 2018, 18, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Imbimbo, B.P. Alzheimer’s Disease: γ-Secretase Inhibitors. Drug Discov. Today Ther. Strateg. 2008, 5, 169–175. [Google Scholar] [CrossRef]
- Hopkins, C.R. ACS Chemical Neuroscience Molecule Spotlight on BMS-708163. ACS Chem. Neurosci. 2012, 3, 149–150. [Google Scholar] [CrossRef][Green Version]
- Hopkins, C.R. ACS Chemical Neuroscience Molecule Spotlight on Semagacestat (Ly450139). ACS Chem. Neurosci. 2010, 1, 533–534. [Google Scholar] [CrossRef][Green Version]
- Hopkins, C.R. ACS Chemical Neuroscience Molecule Spotlight on Begacestat (GSI- 953). ACS Chem. Neurosci. 2012, 3, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Siemers, E.R.; Sundell, K.L.; Carlson, C.; Case, M.; Sethuraman, G.; Liu-seifert, H.; Dowsett, S.A.; Pontecorvo, M.J.; Dean, R.A.; Demattos, R. Phase 3 Solanezumab Trials: Secondary Outcomes in Mild Alzheimer ’ s Disease Patients. Alzheimer’s Dement. 2016, 12, 110–120. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A Phase III Randomized Trial of Gantenerumab in Prodromal Alzheimer’ s Disease. Alzheimer’s Res. Ther. 2017, 9, 1–15. [Google Scholar] [CrossRef]
- Tucker, S.; Möller, C.; Tegerstedt, K.; Lord, A.; Laudon, H.; Sjödahl, J.; Söderberg, L.; Spens, E.; Sahlin, C.; Waara, E.R.; et al. The Murine Version of BAN2401 (MAb158) Selectively Reduces Amyloid-β Protofibrils in Brain and Cerebrospinal Fluid of Tg-ArcSwe Mice. J. Alzheimer’s Dis. 2015, 43, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Mueller-Steiner, S.; Zhou, Y.; Arai, H.; Roberson, E.D.; Sun, B.; Chen, J.; Wang, X.; Yu, G.; Esposito, L.; Mucke, L.; et al. Antiamyloidogenic and Neuroprotective Functions of Cathepsin B: Implications for Alzheimer’s Disease. Neuron 2006, 51, 703–714. [Google Scholar] [CrossRef] [PubMed]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Dodart, J.C.; Paul, S.M.; Holtzman, D.M. Peripheral Anti-Aβ Antibody Alters CNS and Plasma Aβ Clearance and Decreases Brain Aβ Burden in a Mouse Model of Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8850–8855. [Google Scholar] [CrossRef] [PubMed]
- Lannfelt, L.; Relkin, N.R.; Siemers, E.R. Amyloid-ß-Directed Immunotherapy for Alzheimer’s Disease. J. Intern. Med. 2014, 275, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Crespi, G.A.N.; Hermans, S.J.; Parker, M.W.; Miles, L.A. Molecular Basis for Mid-Region Amyloid-β Capture by Leading Alzheimer’s Disease Immunotherapies. Sci. Rep. 2015, 5, 2–6. [Google Scholar] [CrossRef]
- Schneider, L. A Resurrection of Aducanumab for Alzheimer’s Disease. Lancet Neurol. 2020, 19, 111–112. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.; Lemere, C.; Atri, A.; Sabbagh, M.; Salloway, S. Aducanumab Produced a Clinically Meaningful Benefit in Association with Amyloid Lowering. Alzheimer’s Res. Ther. 2021, 13, 10–12. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval for Alzheimer’s Drug. Available online: https://doi.org/https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug (accessed on 7 June 2021).
- Arndt, J.W.; Qian, F.; Smith, B.A.; Quan, C.; Kilambi, K.P.; Bush, M.W.; Walz, T.; Pepinsky, R.B.; Bussière, T.; Hamann, S.; et al. Structural and Kinetic Basis for the Selectivity of Aducanumab for Aggregated Forms of Amyloid-β. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Frost, C.V.; Zacharias, M. From Monomer to Fibril: Abeta-Amyloid Binding to Aducanumab Antibody Studied by Molecular Dynamics Simulation. Proteins Struct. Funct. Bioinform. 2020, 88, 1592–1606. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Fernandez-Perez, E.J.; Burgos, C.F.; Espinoza, M.P.; Castillo, C.; Urrutia, J.C.; Streltsov, V.A.; Opazo, C.; Aguayo, L.G. Inhibition of Amyloid Beta-Induced Synaptotoxicity by a Pentapeptide Derived from the Glycine Zipper Region of the Neurotoxic Peptide. Neurobiol. Aging 2013, 34, 2805–2814. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wu, C.; Liu, D.; Li, H.; Bitan, G.; Shea, J.E.; Bowers, M.T. Mechanism of C-Terminal Fragments of Amyloid β-Protein as Aβ Inhibitors: Do C-Terminal Interactions Play a Key Role in Their Inhibitory Activity? J. Phys. Chem. B 2016, 120, 1615–1623. [Google Scholar] [CrossRef]
- Cramer, P.E.; Cirrito, J.R.; Wesson, D.W.; Lee, C.Y.D.; Karlo, J.C.; Zinn, A.E.; Casali, B.T.; Restivo, J.L.; Goebel, W.D.; James, M.J.; et al. ApoE-Directed Therapeutics Rapidly Clear β-Amyloid and Reverse Deficits in AD Mouse Models. Science 2012, 335, 1503–1506. [Google Scholar] [CrossRef]
- Huy, P.D.Q.; Thai, N.Q.; Bednarikova, Z.; Phuc, L.H.; Linh, H.Q.; Gazova, Z.; Li, M.S. Bexarotene Does Not Clear Amyloid Beta Plaques but Delays Fibril Growth: Molecular Mechanisms. ACS Chem. Neurosci. 2017, 8, 1960–1969. [Google Scholar] [CrossRef]
- Pham, H.D.Q.; Thai, N.Q.; Bednarikova, Z.; Linh, H.Q.; Gazova, Z.; Li, M.S. Bexarotene Cannot Reduce Amyloid Beta Plaques through Inhibition of Production of Amyloid Beta Peptides: In Silico and in Vitro Study. Phys. Chem. Chem. Phys. 2018, 20, 24329–24338. [Google Scholar] [CrossRef]
- Mei, J.; Yang, H.; Sun, B.; Liu, C.; Ai, H. Small-Molecule Targeted Aβ42Aggregate Degradation: Negatively Charged Small Molecules Are More Promising than the Neutral Ones. ACS Chem. Neurosci. 2021, 12, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of Amyloid-Β42 Aggregates Occurs through a Secondary Nucleation Mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef]
- Heller, G.T.; Aprile, F.A.; Michaels, T.C.T.; Limbocker, R.; Perni, M.; Ruggeri, F.S.; Mannini, B.; Löhr, T.; Bonomi, M.; Camilloni, C.; et al. Small-Molecule Sequestration of Amyloid-β as a Drug Discovery Strategy for Alzheimer’s Disease. Sci. Adv. 2020, 6, 1–16. [Google Scholar] [CrossRef]
- Pagano, K.; Tomaselli, S.; Molinari, H.; Ragona, L. Natural Compounds as Inhibitors of Aβ Peptide Aggregation: Chemical Requirements and Molecular Mechanisms. Front. Neurosci. 2020, 14, 1–18. [Google Scholar] [CrossRef]
- Mahmoudinobar, F.; Nilsson, B.L.; Dia, C.L. Effects of Ions and Small Compounds on the Structure of Aβ42 Monomers. J. Phys. Chem. B 2021, 125, 1085–1097. [Google Scholar] [CrossRef]
- Ritter, C.; Adrian, M.; Riek-loher, D.; Bohrmann, B.; Do, H.; Schubert, D.; Riek, R. 3D Structure of Alzheimer’s Amyloid-Beta (1-42) Fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar]
- Boopathi, S.; Kolandaivel, P. Targeted Studies on the Interaction of Nicotine and Morin Molecules with Amyloid β-Protein. J. Mol. Model. 2014, 20, 2109. [Google Scholar] [CrossRef]
- Boopathi, S.; Kolandaivel, P. Study on the Inter- and Intra-Peptide Salt-Bridge Mechanism of Aβ 23–28 Oligomer Interaction with Small Molecules: QM/MM Method. Mol. Biosyst. 2015, 11, 2031–2041. [Google Scholar] [CrossRef]
- Aguayo, L.G.; Gonzalez-Sanmiguel, J.; Burgos, C.F.; Bascunan, D.; Fernandez-Perez, E.J.; Riffo-Lepe, N.; Boopathi, S.; Fernandez-Perez, A.; Bobadilla-Azocar, C.; Gonzalez, W.; et al. Gabapentin Inhibits Multiple Steps in the Amyloid Beta Toxicity Cascade. ACS Chem. Neurosci. 2020, 11, 3064–3076. [Google Scholar] [CrossRef]
- Peters, C.; Bascuñán, D.; Burgos, C.F.; Bobadilla, C.; González-Sanmiguel, J.; Boopathi, S.; Riffo, N.; Fernández-Pérez, E.J.; Tarnok, M.E.; Aguilar, L.F.; et al. Characterization of a New Molecule Capable of Inhibiting Several Steps of the Amyloid Cascade in Alzheimer’s Disease. Neurobiol. Dis. 2020, 141, 104938. [Google Scholar] [CrossRef]
- Ciuperca, I.S.; Dumont, M.; Lakmeche, A.; Mazzocco, P.; Pujo-Menjouet, L.; Rezaei, H.; Tine, L.M. Alzheimer’s Disease and Prion: An in Vitro Mathematical Model. Discret. Contin. Dyn. Syst. -Ser. B 2019, 24, 5225–5260. [Google Scholar] [CrossRef]
- Habchi, J.; Chia, S.; Galvagnion, C.; Michaels, T.C.T.; Bellaiche, M.M.J.; Ruggeri, F.S.; Sanguanini, M.; Idini, I.; Kumita, J.R.; Sparr, E.; et al. Cholesterol Catalyses Aβ42 Aggregation through a Heterogeneous Nucleation Pathway in the Presence of Lipid Membranes. Nat. Chem. 2018, 10, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Hashemi, M.; Zagorski, K.; Lyubchenko, Y.L. Cholesterol in Membranes Facilitates Aggregation of Amyloid β Protein at Physiologically Relevant Concentrations. ACS Chem. Neurosci. 2021, 12, 506–516. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Force Field | Parameter Set | Developments | Water Model | Reference |
|---|---|---|---|---|
| AMBER | ff99IDPs | Updated from ff99SBildn by adding a set of backbone torsion parameters of eight disordered promoting amino acids. | TIP3P | Wei Y et al. [26] 2015 |
| ff14IDPs | Updated from ff14SB by embedding a set of backbone torsion parameters of eight disordered promoting amino acids. | TIP3P | Song et al. [28] 2017 | |
| ff14IDPSFF | Updated from ff14SB by introducing a set of backbone torsion parameters for 20 amino acids | TIP3P | Song et al. [29] 2017 | |
| ff03CMAP | Updated from ff03 by introducing a correction maps (CMAP)-optimized force field | TIP4PD (Modified the dispersion interaction of the TIP4P) | Zhang et al. [30] 2019 | |
| ff14SB | Updated from ff99SB by improving the Accuracy of Protein Side Chain and Backbone Parameters | TIP3P | Maier et al. [31] 2015 | |
| ff03w | Updated from ff03 by adding slight backbone modification | TIP4P/2005 | Best et al. [32] 2010 | |
| A99SB_disp | Update from a99SB-ILDN by an introducing small change in the protein and water vdW interaction terms | TIP4P-D | Robustelli et al. [15] 2018 | |
| CHARMM | CHARMM36m | Updated from CHARMM36 by a refined backbone correction map potential | CHARMM-modified TIP3P | Huang et al. [33] 2017 |
| CHARMM36IDPSFF | Updated from CHARMM36m by CMAP corrections made for all 20 naturally occurring amino acids | CHARMM-modified TIP3P | Liu H et al. [34] 2019 | |
| CHARMM22* | Updated from CHARMM by introducing modifications in backbone torsion potential | CHARMM-modified TIP3P | Stefano Piana et al. [35] 2011 | |
| CHARMM36mW | Van der Waals interaction between protein and water are included in CHARMM36m | CHARMM-modified TIP3P | Samantray et al. [27] 2020 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boopathi, S.; Poma, A.B.; Garduño-Juárez, R. An Overview of Several Inhibitors for Alzheimer’s Disease: Characterization and Failure. Int. J. Mol. Sci. 2021, 22, 10798. https://doi.org/10.3390/ijms221910798
Boopathi S, Poma AB, Garduño-Juárez R. An Overview of Several Inhibitors for Alzheimer’s Disease: Characterization and Failure. International Journal of Molecular Sciences. 2021; 22(19):10798. https://doi.org/10.3390/ijms221910798
Chicago/Turabian StyleBoopathi, Subramanian, Adolfo B. Poma, and Ramón Garduño-Juárez. 2021. "An Overview of Several Inhibitors for Alzheimer’s Disease: Characterization and Failure" International Journal of Molecular Sciences 22, no. 19: 10798. https://doi.org/10.3390/ijms221910798
APA StyleBoopathi, S., Poma, A. B., & Garduño-Juárez, R. (2021). An Overview of Several Inhibitors for Alzheimer’s Disease: Characterization and Failure. International Journal of Molecular Sciences, 22(19), 10798. https://doi.org/10.3390/ijms221910798