
Wee1 Kinase: A Potential Target to Overcome Tumor Resistance to Therapy



Abstract
1. Introduction
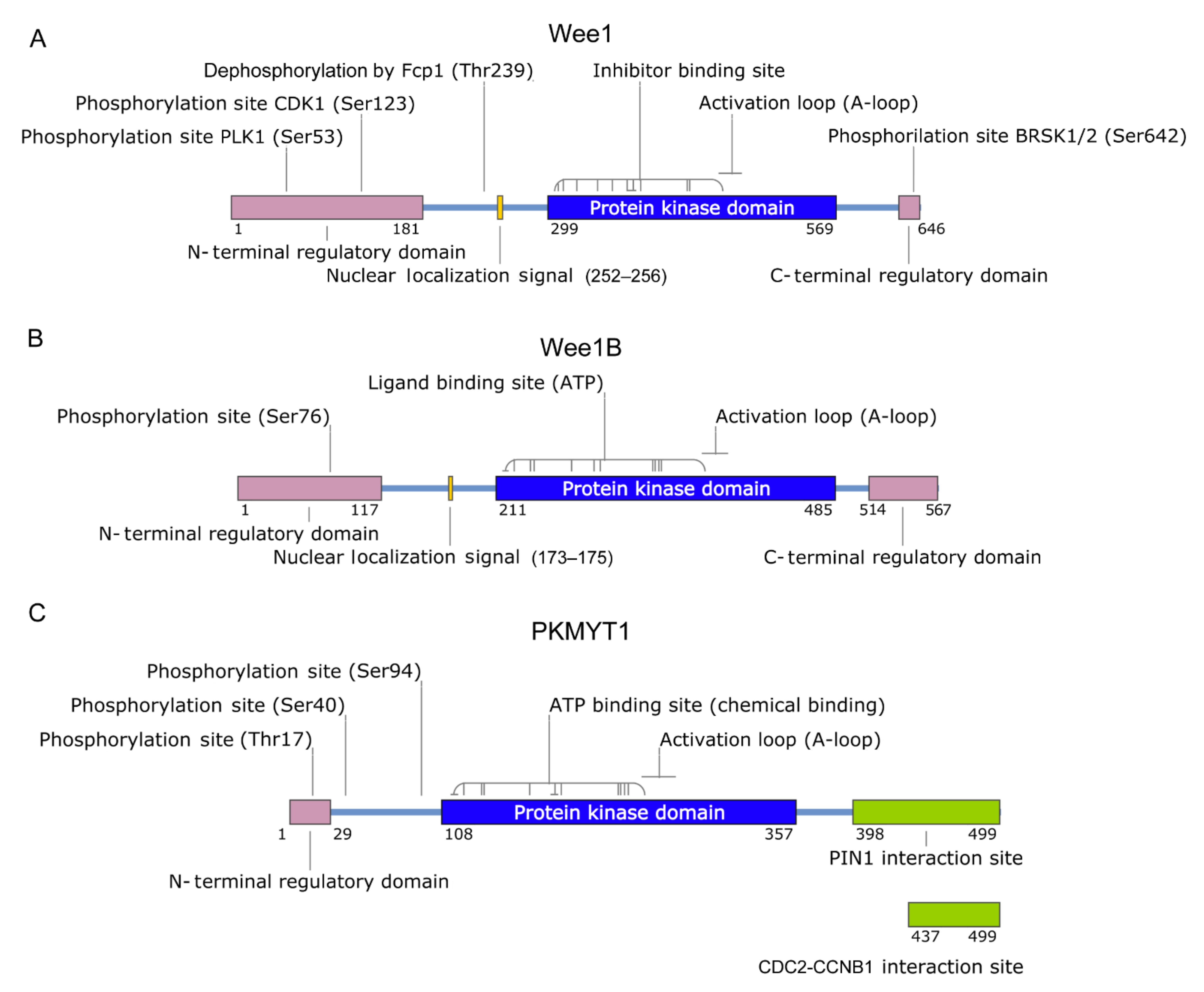
2. Wee1 Family
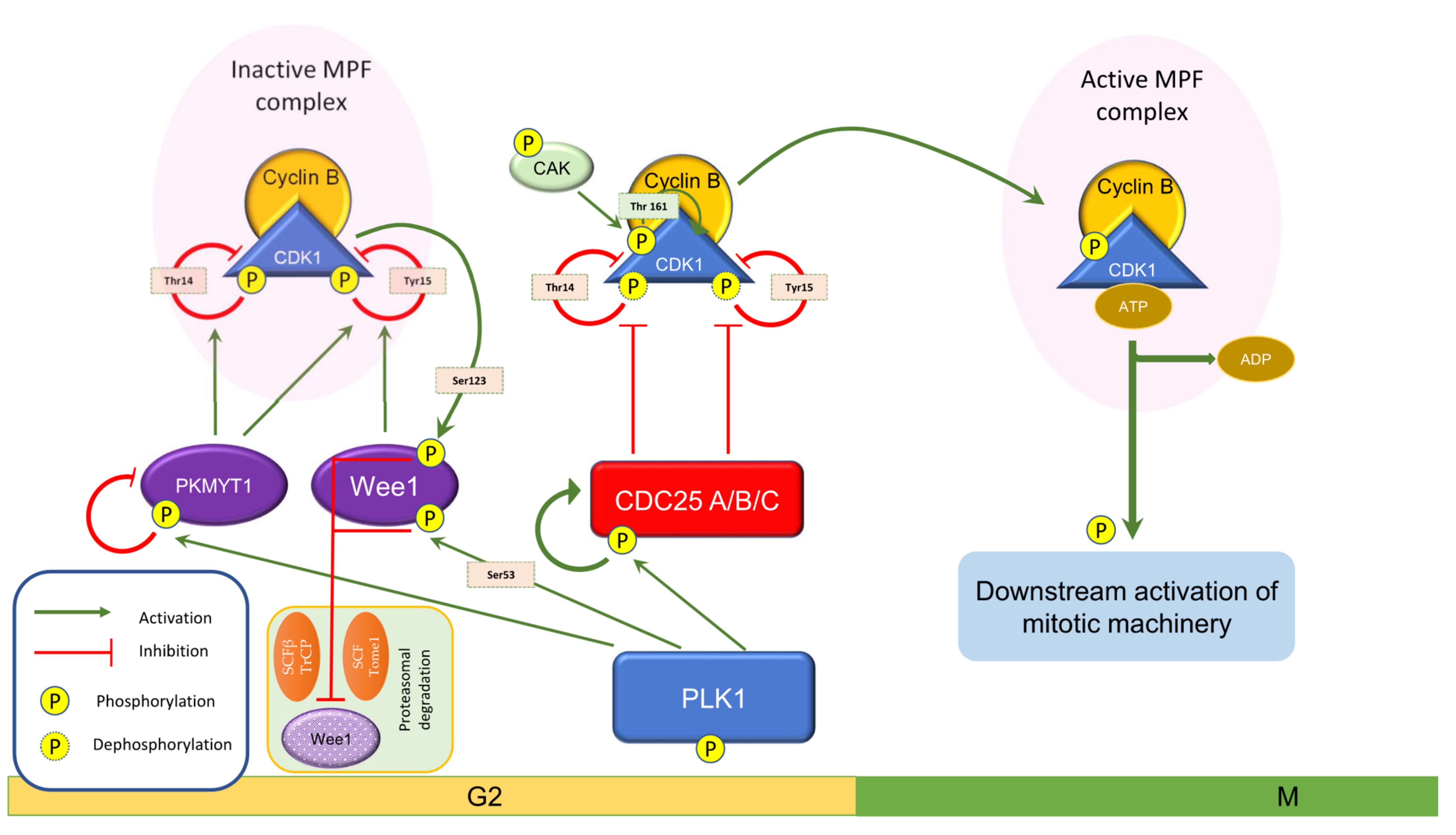
3. Wee1 in Cell Cycle Events
4. Role of Wee1 Kinase in Cancer Progression and Therapy
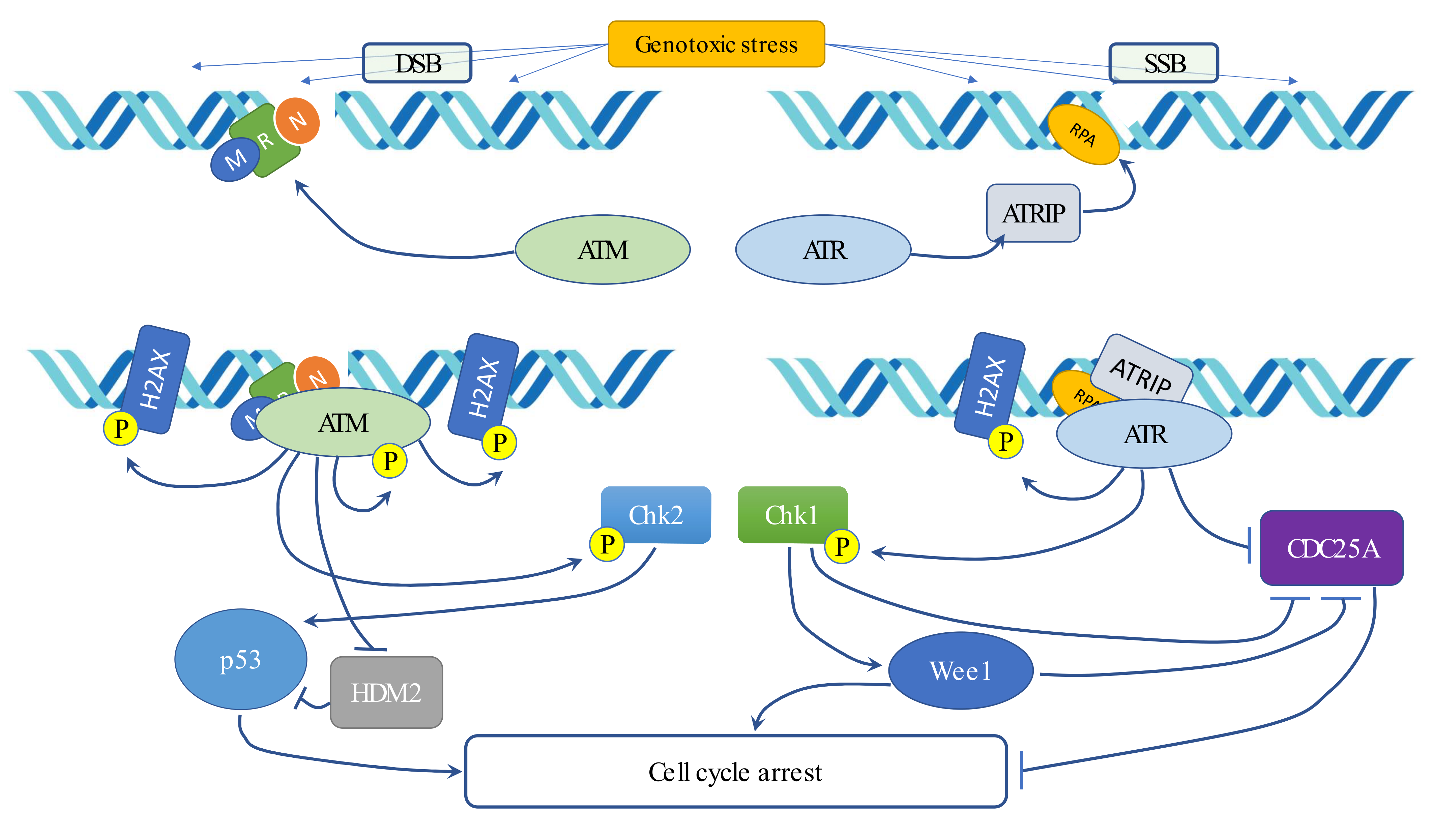
5. Radiotherapy and DNA Damage Response (DDR)
6. Cancer Stem Cells and WEE1 Involvement in Radiation Oncology
7. Wee1 Inhibitors
8. Wee1 Inhibition Sensitizes Response to Chemo/Radiotherapy
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef]
- Poon, R.Y.C. Cell Cycle Control: A System of Interlinking Oscillators. Methods Mol. Biol. 2016, 1342, 3–19. [Google Scholar] [CrossRef]
- Beetham, K.L.; Tolmach, L.J. The Action of Caffeine on X-Irradiated HeLa Cells. V. Identity of the Sector of Cells That Expresses Potentially Lethal Damage in G1 and G2. Radiat. Res. 1982, 91, 199–211. [Google Scholar] [CrossRef]
- Dixon, H.; Norbury, C.J. Therapeutic Exploitation of Checkpoint Defects in Cancer Cells Lacking p53 Function. Cell Cycle 2002, 1, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Benada, J.; Macurek, L. Targeting the Checkpoint to Kill Cancer Cells. Biomolecules 2015, 5, 1912–1937. [Google Scholar] [CrossRef] [PubMed]
- Santo, L.; Siu, K.T.; Raje, N. Targeting Cyclin-Dependent Kinases and Cell Cycle Progression in Human Cancers. Semin. Oncol. 2015, 42, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Rohe, A.; Platzer, C.; Najjar, A.; Erdmann, F.; Sippl, W. Regulation of G2/M Transition by Inhibition of WEE1 and PKMYT1 Kinases. Molecules 2017, 22, 2045. [Google Scholar] [CrossRef]
- Nakanishi, M.; Ando, H.; Watanabe, N.; Kitamura, K.; Ito, K.; Okayama, H.; Miyamoto, T.; Agui, T.; Sasaki, M. Identification and characterization of human Wee1B, a new member of the Wee1 family of Cdk-inhibitory kinases. Genes Cells 2000, 5, 839–847. [Google Scholar] [CrossRef]
- Russell, P.; Nurse, P. Negative regulation of mitosis by wee1+, a gene encoding a protein kinase homolog. Cell 1987, 49, 559–567. [Google Scholar] [CrossRef]
- Gould, K.; Nurse, P. Tyrosine phosphorylation of the fission yeast cdc2+ protein kinase regulates entry into mitosis. Nature 1989, 342, 39–45. [Google Scholar] [CrossRef]
- McGowan, C.; Russell, P. Cell cycle regulation of human WEE1. EMBO J. 1995, 14, 2166–2175. [Google Scholar] [CrossRef]
- Squire, C.; Dickson, J.M.; Ivanovic, I.; Baker, E. Structure and Inhibition of the Human Cell Cycle Checkpoint Kinase, Wee1A Kinase: An Atypical Tyrosine Kinase with a Key Role in CDK1 Regulation. Structure 2005, 13, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Mueller, P.R.; Coleman, T.R.; Kumagai, A.; Dunphy, W.G. Myt1: A Membrane-Associated Inhibitory Kinase That Phosphorylates Cdc2 on Both Threonine-14 and Tyrosine-15. Science 1995, 270, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Stanton, J.J.; Wu, Z.; Piwnica-Worms, H. The human Myt1 kinase preferentially phosphorylates Cdc2 on threonine 14 and localizes to the endoplasmic reticulum and Golgi complex. Mol. Cell. Biol. 1997, 17, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Wells, N.; Watanabe, N.; Tokusumi, T.; Jiang, W.; Verdecia, M.; Hunter, T. The C-terminal domain of the Cdc2 inhibitory kinase Myt1 interacts with Cdc2 complexes and is required for inhibition of G(2)/M progression. J. Cell Sci. 1999, 112, 3361–3371. [Google Scholar] [CrossRef]
- Booher, R.N.; Holman, P.S.; Fattaey, A. Human Myt1 Is a Cell Cycle-regulated Kinase That Inhibits Cdc2 but Not Cdk2 Activity. J. Biol. Chem. 1997, 272, 22300–22306. [Google Scholar] [CrossRef]
- Richard, C.W.; Boehnke, M.; Berg, D.J.; Lichy, J.H.; Meeker, T.C.; Hauser, E.; Myers, R.M.; Cox, D.R. A radiation hybrid map of the distal short arm of human chromosome 11, containing the Beckwith-Weidemann and associated embryonal tumor disease loci. Am. J. Hum. Genet. 1993, 52, 915–921. [Google Scholar]
- Thuriaux, P.; Nurse, P.; Carter, B. Mutants altered in the control co-ordinating cell division with cell growth in the fission yeast Schizosaccharomyces pombe. MGG Mol. Gen. Genet. 1978, 161, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Nurse, P.; Thuriaux, P. Regulatory genes controlling mitosis in the fission yeast Schizosaccharomyces pombe. Genetics 1980, 96, 627–637. [Google Scholar] [CrossRef]
- Igarashi, M.; Nagata, A.; Jinno, S.; Suto, K.; Okayama, H. Wee1+-like gene in human cells. Nature 1991, 353, 80–83. [Google Scholar] [CrossRef]
- McGowan, C.; Russell, P. Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 1993, 12, 75–85. [Google Scholar] [CrossRef]
- Nurse, P. Universal control mechanism regulating onset of M-phase. Nature 1990, 344, 503–508. [Google Scholar] [CrossRef]
- Mueller, P.R.; Coleman, T.R.; Dunphy, W.G. Cell cycle regulation of a Xenopus Wee1-like kinase. Mol. Biol. Cell 1995, 6, 119–134. [Google Scholar] [CrossRef]
- Booher, R.; Deshaies, R.; Kirschner, M. Properties of Saccharomyces cerevisiae wee1 and its differential regulation of p34CDC28 in response to G1 and G2 cyclins. EMBO J. 1993, 12, 3417–3426. [Google Scholar] [CrossRef]
- Campbell, S.D.; Sprenger, F.; Edgar, B.A.; O’Farrell, P.H. Drosophila Wee1 kinase rescues fission yeast from mitotic catastrophe and phosphorylates Drosophila Cdc2 in vitro. Mol. Biol. Cell 1995, 6, 1333–1347. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Ohba, Y.; Yasuda, H. Mouse p87wee1 kinase is regulated by M-phase specific phosphorylation. Chromosome Res. 1995, 3, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Boardman, P.E.; Sanz-Ezquerro, J.; Overton, I.M.; Burt, D.W.; Bosch, E.; Fong, W.T.; Tickle, C.; Brown, W.R.; Wilson, S.A.; Hubbard, S.J. A comprehensive collection of chicken cDNAs. Curr. Biol. 2002, 12, 1965–1969. [Google Scholar] [CrossRef]
- Hirayama, J.; Cardone, L.; Doi, M.; Sassone-Corsi, P. Common pathways in circadian and cell cycle clocks: Light-dependent activation of Fos/AP-1 in zebrafish controls CRY-1a and WEE-1. Proc. Natl. Acad. Sci. USA 2005, 102, 10194–10199. [Google Scholar] [CrossRef] [PubMed]
- Obsilova, V.; Obsil, T. The 14-3-3 Proteins as Important Allosteric Regulators of Protein Kinases. Int. J. Mol. Sci. 2020, 21, 8824. [Google Scholar] [CrossRef]
- Wang, Y.; Jacobs, C.; Hook, K.E.; Duan, H.; Booher, R.N.; Sun, Y. Binding of 14-3-3β to the carboxyl terminus of Wee1 increases Wee1 stability, kinase activity, and G2-M cell population. Cell Growth Differ. 2000, 11, 211–219. [Google Scholar] [PubMed]
- Rothblum-Oviatt, C.J.; E Ryan, C.; Piwnica-Worms, H. 14-3-3 binding regulates catalytic activity of human Wee1 kinase. Cell Growth Differ. 2001, 12, 581–589. [Google Scholar]
- Aligue, R.; Akhavan-Niak, H.; Russell, P. A role for Hsp90 in cell cycle control: Wee1 tyrosine kinase activity requires interaction with Hsp90. EMBO J. 1994, 13, 6099–6106. [Google Scholar] [CrossRef] [PubMed]
- Goes, F.S.; Martin, J. Hsp90 chaperone complexes are required for the activity and stability of yeast protein kinases Mik1, Wee1 and Swe1. Eur. J. Biochem. 2001, 268, 2281–2289. [Google Scholar] [CrossRef] [PubMed]
- Mollapour, M.; Tsutsumi, S.; Donnelly, A.C.; Beebe, K.; Tokita, M.J.; Lee, M.-J.; Lee, S.; Morra, G.; Bourboulia, D.; Scroggins, B.T.; et al. Swe1Wee1-Dependent Tyrosine Phosphorylation of Hsp90 Regulates Distinct Facets of Chaperone Function. Mol. Cell 2010, 37, 333–343. [Google Scholar] [CrossRef]
- Iwai, A.; Bourboulia, D.; Mollapour, M.; Jensen-Taubman, S.; Lee, S.; Donnelly, A.C.; Yoshida, S.; Miyajima, N.; Tsutsumi, S.; Smith, A.K.; et al. Combined inhibition of Wee1 and Hsp90 activates intrinsic apoptosis in cancer cells. Cell Cycle 2012, 11, 3649–3655. [Google Scholar] [CrossRef]
- Sasaki, M.; Terabayashi, T.; Weiss, S.M.; Ferby, I. The Tumor Suppressor MIG6 Controls Mitotic Progression and the G2/M DNA Damage Checkpoint by Stabilizing the WEE1 Kinase. Cell Rep. 2018, 24, 1278–1289. [Google Scholar] [CrossRef]
- Heald, R.; McLoughlin, M.; McKeon, F. Human wee1 maintains mitotic timing by protecting the nucleus from cytoplasmically activated cdc2 kinase. Cell 1993, 74, 463–474. [Google Scholar] [CrossRef]
- Watanabe, N.; Broome, M.; Hunter, T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995, 14, 1878–1891. [Google Scholar] [CrossRef]
- Dunphy, W.G.; Brizuela, L.; Beach, D.; Newport, J. The Xenopus cdc2 protein is a component of MPF, a cytoplasmic regulator of mitosis. Cell 1988, 54, 423–431. [Google Scholar] [CrossRef]
- Gautier, J.; Norbury, C.; Lohka, M.; Nurse, P.; Maller, J. Purified maturation-promoting factor contains the product of a Xenopus homolog of the fission yeast cell cycle control gene cdc2+. Cell 1988, 54, 433–439. [Google Scholar] [CrossRef]
- Murray, A.W. Turning on mitosis. Curr. Biol. 1993, 3, 291–293. [Google Scholar] [CrossRef]
- Parker, L.L.; Piwnica-Worms, H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992, 257, 1955–1957. [Google Scholar] [CrossRef] [PubMed]
- Solomon, M.J.; Lee, T.; Kirschner, M.W. Role of phosphorylation in p34cdc2 activation: Identification of an activating kinase. Mol. Biol. Cell 1992, 3, 13–27. [Google Scholar] [CrossRef]
- Larochelle, S.; Merrick, K.A.; Terret, M.-E.; Wohlbold, L.; Barboza, N.M.; Zhang, C.; Shokat, K.M.; Jallepalli, P.V.; Fisher, R.P. Requirements for Cdk7 in the Assembly of Cdk1/Cyclin B and Activation of Cdk2 Revealed by Chemical Genetics in Human Cells. Mol. Cell 2007, 25, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Ayad, N.G.; Rankin, S.; Murakami, M.; Jebanathirajah, J.; Gygi, S.; Kirschner, M.W. Tome-1, a Trigger of Mitotic Entry, Is Degraded during G1 via the APC. Cell 2003, 113, 101–113. [Google Scholar] [CrossRef]
- Watanabe, N.; Arai, H.; Nishihara, Y.; Taniguchi, M.; Watanabe, N.; Hunter, T.; Osada, H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFβ-TrCP. Proc. Natl. Acad. Sci. USA 2004, 101, 4419–4424. [Google Scholar] [CrossRef]
- Smith, A.; Simanski, S.; Fallahi, M.; Ayad, N.G. Redundant Ubiquitin Ligase Activities Regulate Wee1 Degradation and Mitotic Entry. Cell Cycle 2007, 6, 2795–2799. [Google Scholar] [CrossRef]
- Beck, H.; Nähse, V.; Larsen, M.S.Y.; Groth, P.; Clancy, T.; Lees, M.; Jørgensen, M.; Helleday, T.; Syljuåsen, R.G.; Sorensen, C. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J. Cell Biol. 2010, 188, 629–638. [Google Scholar] [CrossRef]
- Martín, Y.; Domínguez-Kelly, R.; Freire, R. Novel insights into maintaining genomic integrity: Wee1 regulating Mus81/Eme1. Cell Div. 2011, 6, 21. [Google Scholar] [CrossRef]
- Duda, H.; Arter, M.; Gloggnitzer, J.; Teloni, F.; Wild, P.; Blanco, M.G.; Altmeyer, M.; Matos, J. A Mechanism for Controlled Breakage of Under-replicated Chromosomes during Mitosis. Dev. Cell 2016, 39, 740–755. [Google Scholar] [CrossRef]
- Domínguez-Kelly, R.; Martín, Y.; Koundrioukoff, S.; Tanenbaum, M.E.; Smits, V.A.; Medema, R.; Debatisse, M.; Freire, R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Cell Biol. 2011, 194, 567–579. [Google Scholar] [CrossRef]
- Beck, H.; Nähse-Kumpf, V.; Larsen, M.S.Y.; O’Hanlon, K.A.; Patzke, S.; Holmberg, C.; Mejlvang, J.; Groth, A.; Nielsen, O.; Syljuåsen, R.G.; et al. Cyclin-Dependent Kinase Suppression by WEE1 Kinase Protects the Genome through Control of Replication Initiation and Nucleotide Consumption. Mol. Cell. Biol. 2012, 32, 4226–4236. [Google Scholar] [CrossRef]
- Técher, H.; Koundrioukoff, S.; Carignon, S.; Wilhelm, T.; Millot, G.A.; Lopez, B.; Brison, O.; Debatisse, M. Signaling from Mus81-Eme2-Dependent DNA Damage Elicited by Chk1 Deficiency Modulates Replication Fork Speed and Origin Usage. Cell Rep. 2016, 14, 1114–1127. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulos, A.; Tominaga, Y.; Kim, H.-S.; Lahusen, T.; Li, B.; Yu, H.; Gius, D.; Deng, C.-X. WEE1 murine deficiency induces hyper-activation of APC/C and results in genomic instability and carcinogenesis. Oncogene 2015, 34, 3023–3035. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, K.; Fang, B.; Koomen, J.M.; Mahajan, N.P. H2B Tyr37 phosphorylation suppresses expression of replication-dependent core histone genes. Nat. Struct. Mol. Biol. 2012, 19, 930–937. [Google Scholar] [CrossRef]
- Tominaga, Y.; Li, C.; Wang, R.-H.; Deng, C.-X. Murine Wee1 Plays a Critical Role in Cell Cycle Regulation and Pre-Implantation Stages of Embryonic Development. Int. J. Biol. Sci. 2006, 2, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Eom, M.; Han, A.; Lee, M.J.; Park, K.H. Expressional Difference of RHEB, HDAC1, and WEE1 Proteins in the Stromal Tumors of the Breast and Their Significance in Tumorigenesis. Korean J. Pathol. 2012, 46, 324–330. [Google Scholar] [CrossRef]
- Backert, S.; Gelos, M.; Kobalz, U.; Hanski, M.-L.; Mann, B.; Gratchev, A.; Mansmann, U.; Moyer, M.P.; Riecken, E.-O.; Hanski, C. Differential gene expression in colon carcinoma cells and tissues detected with a cDNA array. Int. J. Cancer 1999, 82, 868–874. [Google Scholar] [CrossRef]
- Yoshida, T.; Tanaka, S.; Mogi, A.; Shitara, Y.; Kuwano, H. The clinical significance of Cyclin B1 and Wee1 expression innon-small-cell lung cancer. Ann. Oncol. 2004, 15, 252–256. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, N. Downregulation of lncRNA NEAT1_2 radiosensitizes hepatocellular carcinoma cells through regulation of miR-101-3p/WEE1 axis. Cell Biol. Int. 2019, 43, 44–55. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Schmitz, U.; Wolkenhauer, O.; Schönherr, M.; Raatz, Y.; Kunz, M. Regulation of cell cycle checkpoint kinase WEE1 by miR-195 in malignant melanoma. Oncogene 2013, 32, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- Leary, A.; Auguste, A.; Mesnage, S. DNA damage response as a therapeutic target in gynecological cancers. Curr. Opin. Oncol. 2016, 28, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, Y.; Dang, Y.-Z.; Gao, H.-X.; Jiang, J.-L.; Chen, Z.-N. HAb18G/CD147 Promotes Radioresistance in Hepatocellular Carcinoma Cells: A Potential Role for Integrin β1 Signaling. Mol. Cancer Ther. 2015, 14, 553–563. [Google Scholar] [CrossRef]
- Yu, X.; Li, Z.; Zheng, H.; Chan, M.T.V.; Wu, W.K.K. NEAT1: A novel cancer-related long non-coding RNA. Cell Prolif. 2017, 50, e12329. [Google Scholar] [CrossRef]
- Tibes, R.; Bogenberger, J.M.; Chaudhuri, L.; Hagelstrom, R.T.; Chow, D.; Buechel, M.E.; Gonzales, I.M.; Demuth, T.; Slack, J.; Mesa, R.A.; et al. RNAi screening of the kinome with cytarabine in leukemias. Blood 2012, 119, 2863–2872. [Google Scholar] [CrossRef]
- Iorns, E.; Lord, C.; Grigoriadis, A.; McDonald, S.; Fenwick, K.; Mackay, A.; Mein, C.; Natrajan, R.; Savage, K.; Tamber, N.; et al. Integrated Functional, Gene Expression and Genomic Analysis for the Identification of Cancer Targets. PLoS ONE 2009, 4, e5120. [Google Scholar] [CrossRef]
- Mir, S.E.; Hamer, P.C.D.W.; Krawczyk, P.M.; Balaj, L.; Claes, A.; Niers, J.M.; Van Tilborg, A.A.; Zwinderman, A.H.; Geerts, D.; Kaspers, G.J.; et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell 2010, 18, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Masaki, T.; Shiratori, Y.; Rengifo, W.; Igarashi, K.; Yamagata, M.; Kurokohchi, K.; Uchida, N.; Miyauchi, Y.; Yoshiji, H.; Watanabe, S.; et al. Cyclins and cyclin-dependent kinases: Comparative study of hepatocellular carcinoma versus cirrhosis. Hepatology 2003, 37, 534–543. [Google Scholar] [CrossRef]
- Harris, P.S.; Venkataraman, S.; Alimova, I.; Birks, D.K.; Balakrishnan, I.; Cristiano, B.; Donson, A.M.; Dubuc, A.M.; Taylor, M.D.; Foreman, N.K.; et al. Integrated genomic analysis identifies the mitotic checkpoint kinase WEE1 as a novel therapeutic target in medulloblastoma. Mol. Cancer 2014, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Hashizume, R.; Yang, X.; Kolkowitz, I.; Olow, A.K.; Phillips, J.; Smirnov, I.; Tom, M.W.; Prados, M.D.; James, C.D.; et al. Targeting Wee1 for the treatment of pediatric high-grade gliomas. Neuro Oncol. 2014, 16, 352–360. [Google Scholar] [CrossRef]
- Music, D.; Dahlrot, R.; Hermansen, S.K.; Hjelmborg, J.; De Stricker, K.; Hansen, S.; Kristensen, B.W. Expression and prognostic value of the WEE1 kinase in gliomas. J. Neuro Oncol. 2016, 127, 381–389. [Google Scholar] [CrossRef]
- Slipicevic, A.; Holth, A.; Hellesylt, E.; Tropé, C.G.; Davidson, B.; Flørenes, V.A. Wee1 is a novel independent prognostic marker of poor survival in post-chemotherapy ovarian carcinoma effusions. Gynecol. Oncol. 2014, 135, 118–124. [Google Scholar] [CrossRef]
- Magnussen, G.I.; Hellesylt, E.; Nesland, J.M.; Tropé, C.G.; Flørenes, V.A.; Holm, R. High expression of wee1 is associated with malignancy in vulvar squamous cell carcinoma patients. BMC Cancer 2013, 13, 288. [Google Scholar] [CrossRef]
- Magnussen, G.I.; Holm, R.; Emilsen, E.; Rosnes, A.K.; Slipicevic, A.; Flørenes, V.A. High expression of Wee1 is associated with poor disease-free survival in malignant melanoma: Potential for targeted therapy. PLoS ONE 2012, 7, e38254. [Google Scholar] [CrossRef] [PubMed]
- Mak, J.P.Y.; Man, W.Y.; Chow, J.P.; Ma, H.T.; Poon, R.Y. Pharmacological inactivation of CHK1 and WEE1 induces mitotic catastrophe in nasopharyngeal carcinoma cells. Oncotarget 2015, 6, 21074–21084. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Sharpe, R.; Garcia-Murillas, I.; Gevensleben, H.; Hurd, M.S.; Shumway, S.D.; Toniatti, C.; Ashworth, A.; Turner, N.C. Forced Mitotic Entry of S-Phase Cells as a Therapeutic Strategy Induced by Inhibition of WEE1. Cancer Discov. 2012, 2, 524–539. [Google Scholar] [CrossRef] [PubMed]
- Kogiso, T.; Nagahara, H.; Hashimoto, E.; Ariizumi, S.; Yamamoto, M.; Shiratori, K. Efficient Induction of Apoptosis by Wee1 Kinase Inhibition in Hepatocellular Carcinoma Cells. PLoS ONE 2014, 9, e100495. [Google Scholar] [CrossRef] [PubMed]
- Pappano, W.N.; Zhang, Q.; Tucker, L.A.; Tse, C.; Wang, J. Genetic inhibition of the atypical kinase Wee1 selectively drives apoptosis of p53 inactive tumor cells. BMC Cancer 2014, 14, 430. [Google Scholar] [CrossRef]
- Hamer, P.C.D.W.; Mir, S.E.; Noske, D.; Van Noorden, C.J.F.; Würdinger, T. WEE1 Kinase Targeting Combined with DNA-Damaging Cancer Therapy Catalyzes Mitotic Catastrophe. Clin. Cancer Res. 2011, 17, 4200–4207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Dominguez, D.; Chen, S.; Fan, J.; Qin, L.; Long, A.; Li, X.; Zhang, Y.; Shi, H.; Zhang, B. WEE1 inhibition by MK1775 as a single-agent therapy inhibits ovarian cancer viability. Oncol. Lett. 2017, 14, 3580–3586. [Google Scholar] [CrossRef] [PubMed]
- Kreahling, J.M.; Gemmer, J.Y.; Reed, D.; Letson, D.; Bui, M.; Altiok, S. MK1775, a Selective Wee1 Inhibitor, Shows Single-Agent Antitumor Activity against Sarcoma Cells. Mol. Cancer Ther. 2012, 11, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.-B.S.; Bartek, J. Targeting the checkpoint kinases: Chemosensitization versus chemoprotection. Nat. Rev. Cancer 2004, 4, 216–225. [Google Scholar] [CrossRef]
- Ku, B.M.; Bae, Y.-H.; Koh, J.; Sun, J.-M.; Lee, S.-H.; Ahn, J.S.; Park, K.; Ahn, M.-J. Mutational status of TP53 defines the efficacy of Wee1 inhibitor AZD1775 in KRAS-mutant non-small cell lung cancer. Oncotarget 2017, 8, 67526–67537. [Google Scholar] [CrossRef]
- Bauman, J.E.; Chung, C.H. CHK It Out! Blocking WEE Kinase Routs TP53 Mutant Cancer. Clin. Cancer Res. 2014, 20, 4173–4175. [Google Scholar] [CrossRef][Green Version]
- Hirai, H.; Arai, T.; Okada, M.; Nishibata, T.; Kobayashi, M.; Sakai, N.; Imagaki, K.; Ohtani, J.; Sakai, T.; Yoshizumi, T.; et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol. Ther. 2010, 9, 514–522. [Google Scholar] [CrossRef]
- PosthumaDeBoer, J.; Würdinger, T.; Graat, H.C.; van Beusechem, V.W.; Helder, M.N.; van Royen, B.J.; Kaspers, G.J. WEE1 inhibition sensitizes osteosarcoma to radiotherapy. BMC Cancer 2011, 11, 156. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Booher, R.N.; Kraker, A.; Lawrence, T.; Leopold, W.R.; Sun, Y. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001, 61, 8211–8217. [Google Scholar] [PubMed]
- Li, J.; Wang, Y.; Sun, Y.; Lawrence, T.S. Wild-type TP53 inhibits G(2)-phase checkpoint abrogation and radiosensitization induced by PD0166285, a WEE1 kinase inhibitor. Radiat. Res. 2002, 157, 322–330. [Google Scholar] [CrossRef]
- Barbosa, R.S.; Dantonio, P.M.; Guimarães, T.; de Oliveira, M.B.; Alves, V.L.F.; Sandes, A.F.; Fernando, R.C.; Colleoni, G.W. Sequential combination of bortezomib and WEE1 inhibitor, MK-1775, induced apoptosis in multiple myeloma cell lines. Biochem. Biophys. Res. Commun. 2019, 519, 597–604. [Google Scholar] [CrossRef]
- Porter, C.C.; Kim, J.; Fosmire, S.; Gearheart, C.M.; Van Linden, A.; Baturin, D.; Zaberezhnyy, V.; Patel, P.R.; Gao, D.; Tan, A.C.; et al. Integrated genomic analyses identify WEE1 as a critical mediator of cell fate and a novel therapeutic target in acute myeloid leukemia. Leukemia 2012, 26, 1266–1276. [Google Scholar] [CrossRef]
- Van Linden, A.A.; Baturin, D.; Ford, J.B.; Fosmire, S.P.; Gardner, L.; Korch, C.; Reigan, P.; Porter, C.C. Inhibition of Wee1 Sensitizes Cancer Cells to Antimetabolite Chemotherapeutics In Vitro and In Vivo, Independent of p53 Functionality. Mol. Cancer Ther. 2013, 12, 2675–2684. [Google Scholar] [CrossRef]
- Atun, R.; Jaffray, D.; Barton, M.; Bray, F.; Baumann, M.; Vikram, B.; Hanna, T.; Knaul, F.M.; Lievens, Y.; Lui, T.; et al. Expanding global access to radiotherapy. Lancet Oncol. 2015, 16, 1153–1186. [Google Scholar] [CrossRef]
- Baumann, M.; Krause, M.; Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 2008, 8, 545–554. [Google Scholar] [CrossRef]
- Krause, M.; Yaromina, A.; Eicheler, W.; Koch, U.; Baumann, M. Cancer Stem Cells: Targets and Potential Biomarkers for Radiotherapy. Clin. Cancer Res. 2011, 17, 7224–7229. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Bütof, R.; Dubrovska, A.; Baumann, M. Clinical perspectives of cancer stem cell research in radiation oncology. Radiother. Oncol. 2013, 108, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Lawrence, T.S. Molecular Pathways: Overcoming Radiation Resistance by Targeting DNA Damage Response Pathways. Clin. Cancer Res. 2015, 21, 2898–2904. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Krause, M.; Overgaard, J.; Debus, J.; Bentzen, S.M.; Daartz, J.; Richter, C.; Zips, D.; Bortfeld, T. Radiation oncology in the era of precision medicine. Nat. Rev. Cancer 2016, 16, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Lee, P.W. The DNA-dependent protein kinase (DNA-PK): More than just a case of making ends meet? Cell Cycle 2010, 9, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605–611. [Google Scholar] [CrossRef]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in Checkpoint Signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.M.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM Phosphorylates Histone H2AX in Response to DNA Double-strand Breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef]
- Ammazzalorso, F.; Pirzio, L.M.; Bignami, M.; Franchitto, A.; Pichierri, P. ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 2010, 29, 3156–3169. [Google Scholar] [CrossRef]
- Johnson, N.; Cai, D.; Kennedy, R.D.; Pathania, S.; Arora, M.; Li, Y.-C.; D’Andrea, A.D.; Parvin, J.D.; Shapiro, G.I. Cdk1 Participates in BRCA1-Dependent S Phase Checkpoint Control in Response to DNA Damage. Mol. Cell 2009, 35, 327–339. [Google Scholar] [CrossRef]
- Liu, Q.; Guntuku, S.; Cui, X.-S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar]
- Matsuoka, S.; Rotman, G.; Ogawa, A.; Shiloh, Y.; Tamai, K.; Elledge, S.J. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 10389–10394. [Google Scholar] [CrossRef]
- Khosravi, R.; Maya, R.; Gottlieb, T.; Oren, M.; Shiloh, Y.; Shkedy, D. Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc. Natl. Acad. Sci. USA 1999, 96, 14973–14977. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 Checkpoint Pathway in Mammals: Linkage of DNA Damage to Cdk Regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar] [CrossRef]
- Xiao, Z.; Chen, Z.; Gunasekera, A.H.; Sowin, T.J.; Rosenberg, S.H.; Fesik, S.; Zhang, H. Chk1 Mediates S and G2 Arrests through Cdc25A Degradation in Response to DNA-damaging Agents. J. Biol. Chem. 2003, 278, 21767–21773. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.; Syljuåsen, R.G.; Falck, J.; Schroeder, T.; Rönnstrand, L.; Khanna, K.K.; Zhou, B.-B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar] [CrossRef]
- Syljuåsen, R.G.; Sorensen, C.; Hansen, L.T.; Fugger, K.; Lundin, C.; Johansson, F.; Helleday, T.; Sehested, M.; Lukas, J.; Bartek, J. Inhibition of Human Chk1 Causes Increased Initiation of DNA Replication, Phosphorylation of ATR Targets, and DNA Breakage. Mol. Cell. Biol. 2005, 25, 3553–3562. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.; Hansen, L.T.; Dziegielewski, J.; Syljuåsen, R.G.; Lundin, C.; Bartek, J.; Helleday, T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Zachos, G.; Black, E.J.; Walker, M.; Scott, M.; Vagnarelli, P.; Earnshaw, W.; Gillespie, D.A. Chk1 Is Required for Spindle Checkpoint Function. Dev. Cell 2007, 12, 247–260. [Google Scholar] [CrossRef]
- Carrassa, L.; Sanchez, Y.; Erba, E.; Damia, G. U2OS cells lacking Chk1 undergo aberrant mitosis and fail to activate the spindle checkpoint. J. Cell. Mol. Med. 2009, 13, 1565–1576. [Google Scholar] [CrossRef]
- Chilà, R.; Celenza, C.; Lupi, M.; Damia, G.; Carrassa, L. Chk1-Mad2 interaction: A crosslink between the DNA damage checkpoint and the mitotic spindle checkpoint. Cell Cycle 2013, 12, 1083–1090. [Google Scholar] [CrossRef]
- Jette, N.; Lees-Miller, S.P. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 2015, 117, 194–205. [Google Scholar] [CrossRef]
- Hill, R.; Milas, L. The proportion of stem cells in murine tumors. Int. J. Radiat. Oncol. 1989, 16, 513–518. [Google Scholar] [CrossRef]
- Baumann, M.; Dubois, W.; Suit, H.D. Response of Human Squamous Cell Carcinoma Xenografts of Different Sizes to Irradiation: Relationship of Clonogenic Cells, Cellular Radiation Sensitivity in Vivo, and Tumor Rescuing Units. Radiat. Res. 1990, 123, 325–330. [Google Scholar] [CrossRef]
- Koch, U.; Krause, M.; Baumann, M. Cancer stem cells at the crossroads of current cancer therapy failures—Radiation oncology perspective. Semin. Cancer Biol. 2010, 20, 116–124. [Google Scholar] [CrossRef]
- Puglisi, C.; Giuffrida, R.; Borzì, G.; Di Mattia, P.; Costa, A.; Colarossi, C.; Deiana, E.; Picardo, M.C.; Colarossi, L.; Mare, M.; et al. Radiosensitivity of Cancer Stem Cells Has Potential Predictive Value for Individual Responses to Radiotherapy in Locally Advanced Rectal Cancer. Cancers 2020, 12, 3672. [Google Scholar] [CrossRef]
- Yaromina, A.; Krause, M.; Thames, H.; Rosner, A.; Krause, M.; Hessel, F.; Grenman, R.; Zips, D.; Baumann, M. Pre-treatment number of clonogenic cells and their radiosensitivity are major determinants of local tumour control after fractionated irradiation. Radiother. Oncol. 2007, 83, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Maugeri-Saccà, M.; Bartucci, M.; De Maria, R. DNA Damage Repair Pathways in Cancer Stem Cells. Mol. Cancer Ther. 2012, 11, 1627–1636. [Google Scholar] [CrossRef]
- Chang, C.-H.; Zhang, M.; Rajapakshe, K.; Coarfa, C.; Edwards, D.; Huang, S.; Rosen, J.M. Mammary Stem Cells and Tumor-Initiating Cells Are More Resistant to Apoptosis and Exhibit Increased DNA Repair Activity in Response to DNA Damage. Stem Cell Rep. 2015, 5, 378–391. [Google Scholar] [CrossRef]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential Roles of Hypoxia-Inducible Factor 1α (HIF-1α) and HIF-2α in Hypoxic Gene Regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef]
- Covello, K.L.; Kehler, J.; Yu, H.; Gordan, J.D.; Arsham, A.M.; Hu, C.-J.; Labosky, P.; Simon, M.C.; Keith, B. HIF-2 regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006, 20, 557–570. [Google Scholar] [CrossRef]
- Gordan, J.D.; Bertout, J.A.; Hu, C.-J.; Diehl, J.A.; Simon, M.C. HIF-2α Promotes Hypoxic Cell Proliferation by Enhancing c-Myc Transcriptional Activity. Cancer Cell 2007, 11, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Cordes, N.; Seidler, J.; Durzok, R.; Geinitz, H.; Brakebusch, C. β1-integrin-mediated signaling essentially contributes to cell survival after radiation-induced genotoxic injury. Oncogene 2006, 25, 1378–1390. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Sarcar, B.; Kahali, S.; Prabhu, A.H.; Shumway, S.D.; Xu, Y.; DeMuth, T.; Chinnaiyan, P. Targeting Radiation-Induced G2 Checkpoint Activation with the Wee-1 Inhibitor MK-1775 in Glioblastoma Cell Lines. Mol. Cancer Ther. 2011, 10, 2405–2414. [Google Scholar] [CrossRef]
- Ronco, C.; Martin, A.R.; Demange, L.; Benhida, R. ATM, ATR, CHK1, CHK2 and WEE1 inhibitors in cancer and cancer stem cells. MedChemComm 2017, 8, 295–319. [Google Scholar] [CrossRef] [PubMed]
- Matheson, C.J.; Venkataraman, S.; Amani, V.; Harris, P.S.; Backos, D.; Donson, A.M.; Wempe, M.F.; Foreman, N.; Vibhakar, R.; Reigan, P. A WEE1 Inhibitor Analog of AZD1775 Maintains Synergy with Cisplatin and Demonstrates Reduced Single-Agent Cytotoxicity in Medulloblastoma Cells. ACS Chem. Biol. 2016, 11, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Matheson, C.J.; Casalvieri, K.A.; Backos, D.S.; Reigan, P. Development of Potent Pyrazolopyrimidinone-Based WEE1 Inhibitors with Limited Single-Agent Cytotoxicity for Cancer Therapy. ChemMedChem 2018, 13, 1681–1694. [Google Scholar] [CrossRef]
- Du, X.; Li, J.; Luo, X.; Li, R.; Li, F.; Zhang, Y.; Shi, J.; He, J. Structure-activity relationships of Wee1 inhibitors: A review. Eur. J. Med. Chem. 2020, 203, 112524. [Google Scholar] [CrossRef]
- Hashimoto, O.; Shinkawa, M.; Torimura, T.; Nakamura, T.; Selvendiran, K.; Sakamoto, M.; Koga, H.; Ueno, T.; Sata, M. Cell cycle regulation by the Wee1 Inhibitor PD0166285, Pyrido [2,3-d] pyimidine, in the B16 mouse melanoma cell line. BMC Cancer 2006, 6, 292. [Google Scholar] [CrossRef]
- Hashimoto, O.; Ueno, T.; Kimura, R.; Ohtsubo, M.; Nakamura, T.; Koga, H.; Torimura, T.; Uchida, S.; Yamashita, K.; Sata, M. Inhibition of proteasome-dependent degradation of Wee1 in G2-arrested Hep3B cells by TGFβ1. Mol. Carcinog. 2003, 36, 171–182. [Google Scholar] [CrossRef]
- Panek, R.L.; Lu, G.H.; Klutchko, S.R.; Batley, B.L.; Dahring, T.K.; Hamby, J.M.; Hallak, H.; Doherty, A.M.; Keiser, J.A. In vitro pharmacological characterization of PD 166285, a new nanomolar potent and broadly active protein tyrosine kinase inhibitor. J. Pharmacol. Exp. Ther. 1997, 283, 1433–1444. [Google Scholar]
- Palmer, B.D.; Smaill, J.B.; Rewcastle, G.W.; Dobrusin, E.M.; Kraker, A.; Moore, C.W.; Steinkampf, R.W.; Denny, W.A. Structure-activity relationships for 2-anilino-6-phenylpyrido[2,3-d]pyrimidin-7(8H)-ones as inhibitors of the cellular checkpoint kinase Wee1. Bioorgan. Med. Chem. Lett. 2005, 15, 1931–1935. [Google Scholar] [CrossRef] [PubMed]
- Hamby, J.M.; Connolly, C.J.C.; Schroeder, M.C.; Winters, R.T.; Showalter, H.D.H.; Panek, R.L.; Major, T.C.; Olsewski, B.; Ryan, M.J.; Dahring, T.; et al. Structure-Activity Relationships for a Novel Series of Pyrido[2,3-d]pyrimidine Tyrosine Kinase Inhibitors. J. Med. Chem. 1997, 40, 2296–2303. [Google Scholar] [CrossRef]
- Thompson, A.M.; Connolly, C.J.C.; Hamby, J.M.; Boushelle, S.; Hartl, B.G.; Amar, A.M.; Kraker, A.J.; Driscoll, D.L.; Steinkampf, R.W.; Patmore, S.J.; et al. 3-(3,5-Dimethoxyphenyl)-1,6-naphthyridine-2,7-diamines and Related 2-Urea Derivatives Are Potent and Selective Inhibitors of the FGF Receptor-1 Tyrosine Kinase. J. Med. Chem. 2000, 43, 4200–4211. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.D.; Thompson, A.M.; Booth, R.J.; Dobrusin, E.M.; Kraker, A.J.; Lee, H.H.; Lunney, E.A.; Mitchell, L.H.; Ortwine, D.F.; Smaill, J.B.; et al. 4-Phenylpyrrolo[3,4-c]carbazole-1,3(2H,6H)-dione Inhibitors of the Checkpoint Kinase Wee1. Structure-Activity Relationships for Chromophore Modification and Phenyl Ring Substitution. J. Med. Chem. 2006, 49, 4896–4911. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Bisanz, K.M.; Peralta, L.A.; Basu, G.D.; Choudhary, A.; Tibes, R.; Azorsa, D.O. RNAi screening of the kinome identifies modulators of cisplatin response in ovarian cancer cells. Gynecol. Oncol. 2010, 118, 220–227. [Google Scholar] [CrossRef]
- Smaill, J.B.; Baker, E.N.; Booth, R.J.; Bridges, A.J.; Dickson, J.M.; Dobrusin, E.M.; Ivanovic, I.; Kraker, A.J.; Lee, H.H.; Lunney, E.A.; et al. Synthesis and structure-activity relationships of N-6 substituted analogues of 9-hydroxy-4-phenylpyrrolo[3,4-c]carbazole-1,3(2H,6H)-diones as inhibitors of Wee1 and Chk1 checkpoint kinases. Eur. J. Med. Chem. 2008, 43, 1276–1296. [Google Scholar] [CrossRef]
- Smaill, J.B.; Lee, H.H.; Palmer, B.D.; Thompson, A.M.; Squire, C.J.; Baker, E.N.; Booth, R.J.; Kraker, A.; Hook, K.; Denny, W.A. Synthesis and structure-activity relationships of soluble 8-substituted 4-(2-chlorophenyl)-9-hydroxypyrrolo[3,4-c]carbazole-1,3(2H,6H)-diones as inhibitors of the Wee1 and Chk1 checkpoint kinases. Bioorgan. Med. Chem. Lett. 2008, 18, 929–933. [Google Scholar] [CrossRef]
- Tong, Y.; Torrent, M.; Florjancic, A.S.; Bromberg, K.D.; Buchanan, F.G.; Ferguson, D.C.; Johnson, E.F.; Lasko, L.M.; Maag, D.; Merta, P.J.; et al. Pyrimidine-Based Tricyclic Molecules as Potent and Orally Efficacious Inhibitors of Wee1 Kinase. ACS Med. Chem. Lett. 2014, 6, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Afaq, F.; Saleem, M.; Ahmad, N.; Mukhtar, H. Targeting Multiple Signaling Pathways by Green Tea Polyphenol (−)-Epigallocatechin-3-Gallate. Cancer Res. 2006, 66, 2500–2505. [Google Scholar] [CrossRef]
- Banerjee, S.; Li, Y.; Wang, Z.; Sarkar, F.H. Multi-targeted therapy of cancer by genistein. Cancer Lett. 2008, 269, 226–242. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, J.; Prasad, S.; Aggarwal, B.B. Curcumin and Cancer Cells: How Many Ways Can Curry Kill Tumor Cells Selectively? AAPS J. 2009, 11, 495–510. [Google Scholar] [CrossRef]
- Cuccioloni, M.; Mozzicafreddo, M.; Bonfili, L.; Cecarini, V.; Eleuteri, A.M.; Angeletti, M. Natural Occurring Polyphenols as Template for Drug Design. Focus on Serine Proteases. Chem. Biol. Drug Des. 2009, 74, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Korkina, L.; De Luca, C.; Kostyuk, V.; Pastore, S. Plant Polyphenols and Tumors: From Mechanisms to Therapies, Prevention, and Protection Against Toxicity of Anti-Cancer Treatments. Curr. Med. Chem. 2009, 16, 3943–3965. [Google Scholar] [CrossRef] [PubMed]
- Lamoral-Theys, D.; Pottier, L.; Dufrasne, F.; Neve, J.; Dubois, J.; Kornienko, A.; Kiss, R.; Ingrassia, L. Natural Polyphenols that Display Anticancer Properties through Inhibition of Kinase Activity. Curr. Med. Chem. 2010, 17, 812–825. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Eifes, S.; Dicato, M.; Aggarwal, B.B.; Diederich, M. Modulation of anti-apoptotic and survival pathways by curcumin as a strategy to induce apoptosis in cancer cells. Biochem. Pharmacol. 2008, 76, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Wang, X.; Lu, G.; Picinich, S.C. Cancer prevention by tea: Animal studies, molecular mechanisms and human relevance. Nat. Rev. Cancer 2009, 9, 429–439. [Google Scholar] [CrossRef]
- Lamoral-Theys, D.; Pottier, L.; Kerff, F.; Dufrasne, F.; Proutiere, F.; Wauthoz, N.; Neven, P.; Ingrassia, L.; Van Antwerpen, P.; Lefranc, F.; et al. Simple di- and trivanillates exhibit cytostatic properties toward cancer cells resistant to pro-apoptotic stimuli. Bioorgan. Med. Chem. 2010, 18, 3823–3833. [Google Scholar] [CrossRef]
- Bridges, K.A.; Hirai, H.; Buser, C.A.; Brooks, C.; Liu, H.; Buchholz, T.; Molkentine, J.; Mason, K.A.; Meyn, R.E. MK-1775, a Novel Wee1 Kinase Inhibitor, Radiosensitizes p53-Defective Human Tumor Cells. Clin. Cancer Res. 2011, 17, 5638–5648. [Google Scholar] [CrossRef]
- Do, K.; Doroshow, J.H.; Kummar, S. Wee1 kinase as a target for cancer therapy. Cell Cycle 2013, 12, 3348–3353. [Google Scholar] [CrossRef]
- Osman, A.A.; Monroe, M.M.; Alves, M.V.O.; Patel, A.A.; Katsonis, P.; Fitzgerald, A.L.; Neskey, D.M.; Frederick, M.J.; Woo, S.H.; Caulin, C.; et al. Wee-1 Kinase Inhibition Overcomes Cisplatin Resistance Associated with High-Risk TP53 Mutations in Head and Neck Cancer through Mitotic Arrest Followed by Senescence. Mol. Cancer Ther. 2015, 14, 608–619. [Google Scholar] [CrossRef]
- Caretti, V.; Hiddingh, L.; Lagerweij, T.; Schellen, P.; Koken, P.W.; Hulleman, E.; van Vuurden, D.G.; Vandertop, W.P.; Kaspers, G.J.; Noske, D.P.; et al. WEE1 Kinase Inhibition Enhances the Radiation Response of Diffuse Intrinsic Pontine Gliomas. Mol. Cancer Ther. 2013, 12, 141–150. [Google Scholar] [CrossRef][Green Version]
- Karnak, D.; Engelke, C.G.; Parsels, L.A.; Kausar, T.; Wei, D.; Robertson, J.R.; Marsh, K.B.; Davis, M.A.; Zhao, L.; Maybaum, J.; et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clin. Cancer Res. 2014, 20, 5085–5096. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| WEE1 Expression | Other Molecular Modulation | Cancer Type | Clinical Significance | Methods | Ref. |
|---|---|---|---|---|---|
| ↓ Wee1 | Stromal breast cancer (phyllodes tumor (PT)) human tissue samples | Wee1 reduction suggests a potential role of Wee1 as a tumor suppressor | Immunohistochemistry | [57] | |
| ↓ Wee1 | Colon carcinoma cell lines and human tissue samples | Wee1 suppression suggests a potential role of Wee1 in tumorigenesis | cDNA array, Northern blotting and semi-quantitative reverse transcription-PCR (RT-PCR) | [58] | |
| ↓ Wee1 | ↑ Cyclin B1/cdc2 complex | NSCLC human tissue samples | Wee1 reduction is associated with a poorer prognosis and a higher recurrence rate | Immunohistochemistry | [59] |
| ↓ Wee1 | ↑ miR-195 | Metastatic melanoma cell lines and human tissue samples | Wee1 expression in malignant melanoma is directly regulated by miR-195 | Immunoblotting, quantitative real-time PCR and immunohistochemistry | [61] |
| ↑ Wee1 | ↓ miR-101-3p | Hepatocellular carcinoma (HCC) cell lines and human tissue samples | Downregulation of Wee1 enhances the radiosensitivity of HCC cells | Quantitative real-time PCR, Western blotting and flowcytometry | [60] |
| ↑ Wee1 | Leukemia cell lines | Wee1 kinase inhibition by siRNA silencing or by specific inhibitors potently sensitizes myeloid and lymphoid leukemia cells to Ara-C | High-throughput siRNA screen and Western blotting | [65] | |
| ↑ Wee1 | Luminal breast cancer cell lines | Wee1 kinase inhibition by specific inhibitors has therapeutic potentials | High-throughput siRNA screen, Western blotting and immunohistochemistry | [66] | |
| ↑ Wee1 | Nasopharyngeal carcinoma (NPC) cell lines | Wee1 kinase inhibition by specific inhibitors has therapeutic potentials | Western blotting | [75] | |
| ↑ Wee1 | Glioblastoma cell lines and human tissue samples | Wee1 kinase inhibition by siRNA or specific inhibitors causes cell death and sensitizes glioblastoma to ionizing radiation in vivo, suggesting it has a potential therapeutic target | In silico analysis of microarray data, immunofluorescence staining, immunohistochemistry and Western blotting | [67] | |
| ↑ Wee1 kinase activity | ↑ Cyclin D1 | Hepatocellular carcinoma (HCC) human tissue samples | Activation of cyclin D1, Cdk4, cyclin E, cyclin A and Wee1 may play important roles in the process of malignant transformation of cirrhosis to HCC | Evaluation of WEE1 kinase activity using autoradiography | [68] |
| ↑ Wee1 | Medulloblastoma cell lines and human tissue samples | Wee1 kinase inhibition by siRNA or specific inhibitors (MK-1775) potently inhibits tumor growth in vivo and sensitizes medulloblastoma cells to cisplatin in vitro, suggesting Wee1 as a potential therapeutic target | Gene expression analysis, high-throughput siRNA screen and Western blotting | [69] | |
| ↑ Wee1 | Glioma human tissue samples | Wee1 expression is directly correlated with the malignancy grade in all types of gliomas, but it is inversely associated with prognosis in GBM | Immunohistochemistry | [71] | |
| ↑ Wee1 | Pediatric high-grade glioma | Wee1 expression positively correlates with the glioma grade; the Wee1 inhibitor MK-1775 increases the radiation cytotoxic effect and prolongs survival for mice with engrafted, orthotopic glioma | Gene expression analysis and immunohistochemistry | [70] | |
| ↑ Wee1 | Melanoma cell lines and human tissue samples | High expression of WEE1 is associated with poor prognosis and Wee1 silencing increases tumor cell death; Wee1 represents a potential therapeutic target in melanoma | Immunohistochemistry and Western blotting | [74] | |
| ↑ Wee1 | Vulvar squamous cell carcinomas human tissue samples | High Wee1 expression is associated with poor histological differentiation and lymph node metastases | Immunohistochemistry and Wee1 silencing using siRNA | [73] | |
| ↑ Wee1 | Acute myeloid leukemia (AML) cell lines and AML primary cells | Wee1 inhibition sensitizes AML cells to cytarabine in vitro; Wee1 represents a potential therapeutic target in AML | Integrated genomic analyses | [90] | |
| ↑ Wee1 | Osteosarcoma cell lines, human osteoblasts and tumor samples | Wee1 inhibition sensitizing OS cells to irradiation-induced cell death | Gene-expression data analysis, immunohistochemistry and Western blotting | [86] | |
| ↑ Wee1 | Multiple myeloma (MM) tissue samples and cell lines | Wee1 inhibition in combination with Bortezomib induces cell death in all cell lines more efficiently compared to the single agents | Quantitative real-time PCR (qPCR) | [89] | |
| ↑ Wee1 | Ovarian carcinoma (OC) peritoneal effusion samples and cell lines | Wee1 is overexpressed in post-chemotherapy disease, suggesting a role in mediating disease progression and as a prognostic marker of poor survival; Wee1 is a potential therapeutic target in OC | Immunohistochemistry and Western blotting | [72] |
| Core Chemical Structure | Lead Compounds | CHK1 IC50 (nM) | WEE1 IC50 (nM) | References | |
|---|---|---|---|---|---|
| Pyridopyrimidine derivatives |  | PD0166285 | 72 | 24 | [86] |
| Pyrazolopyrimidinone derivatives |  | CJM-061 | - | 2.8 | [133] |
AZD1775 | - | 1.9 | [133] | ||
| Pyrrolocarbazole derivatives |  | PD0407824 | 47 | 97 | [143] |
| Pyrimidine-based tricyclic molecules |  | 31 | - | H1299 EC = 230 | [147] |
| Vanillates |  | 32 | - | 20,000 | [156] |
33 | - | 7120 | [156] |
| Title | Conditions | Interventions | ClinicalTrial.govIdentifier Number | |
|---|---|---|---|---|
| 1 | WEE1 Inhibitor with Cisplatin and Radiotherapy: ATrial in Head and Neck Cancer |
|
| NCT03028766 |
| 2 | WEE1 Inhibitor AZD1775 with or without Cytarabine in Treating Patients with Advanced Acute Myeloid Leukemia or Myelodysplastic Syndrome |
|
| NCT02666950 |
| 3 | Phase Ib Study AZD1775 in Combination with Carboplatin and Paclitaxel in Adult Asian Patients with Solid Tumours | Advanced solid tumors |
| NCT02341456 |
| 4 | Cisplatin with or without WEE1 Inhibitor MK-1775 in Treating Patients with Recurrent or Metastatic Head and Neck Cancer |
|
| NCT02196168 |
| 5 | Ph II Trial of Carboplatin and Pemetrexed with or without AZD1775 for Untreated Lung Cancer | Previously untreated stage IV non-squamous non-small cell lung cancer |
| NCT02087241 |
| 6 | Dose Escalation Trial of AZD1775 and Gemcitabine (+Radiation) for Unresectable Adenocarcinoma of the Pancreas | Adenocarcinoma of the pancreas |
| NCT02037230 |
| 7 | A Study of MK-1775 in Combination with Topotecan/Cisplatin in Participants with Cervical Cancer (MK-1775-008) | Cervical cancer |
| NCT01076400 |
| 8 | A Dose Escalation Study of MK-1775 in Combination with Either Gemcitabine, Cisplatin, or Carboplatin in Adults with Advanced Solid Tumors (MK-1775-001) | Solid tumors |
| NCT00648648 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, F.; Giuffrida, R.; Raciti, G.; Puglisi, C.; Forte, S. Wee1 Kinase: A Potential Target to Overcome Tumor Resistance to Therapy. Int. J. Mol. Sci. 2021, 22, 10689. https://doi.org/10.3390/ijms221910689
Esposito F, Giuffrida R, Raciti G, Puglisi C, Forte S. Wee1 Kinase: A Potential Target to Overcome Tumor Resistance to Therapy. International Journal of Molecular Sciences. 2021; 22(19):10689. https://doi.org/10.3390/ijms221910689
Chicago/Turabian StyleEsposito, Francesca, Raffaella Giuffrida, Gabriele Raciti, Caterina Puglisi, and Stefano Forte. 2021. "Wee1 Kinase: A Potential Target to Overcome Tumor Resistance to Therapy" International Journal of Molecular Sciences 22, no. 19: 10689. https://doi.org/10.3390/ijms221910689
APA StyleEsposito, F., Giuffrida, R., Raciti, G., Puglisi, C., & Forte, S. (2021). Wee1 Kinase: A Potential Target to Overcome Tumor Resistance to Therapy. International Journal of Molecular Sciences, 22(19), 10689. https://doi.org/10.3390/ijms221910689