
Alterations in Leptin Signaling in Amyotrophic Lateral Sclerosis (ALS)

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Colony Maintenance and Mice Monitoring
2.2. Tissue Preparation
2.3. RNA Isolation and RT-qPCR
2.4. Protein Extraction and Western Blot Analysis
2.5. Measurement of Metabolic Markers in Plasma
2.6. Statistical Analysis
3. Results
3.1. Leptin Levels Are Altered in WAT of TDP-43A315T Mice
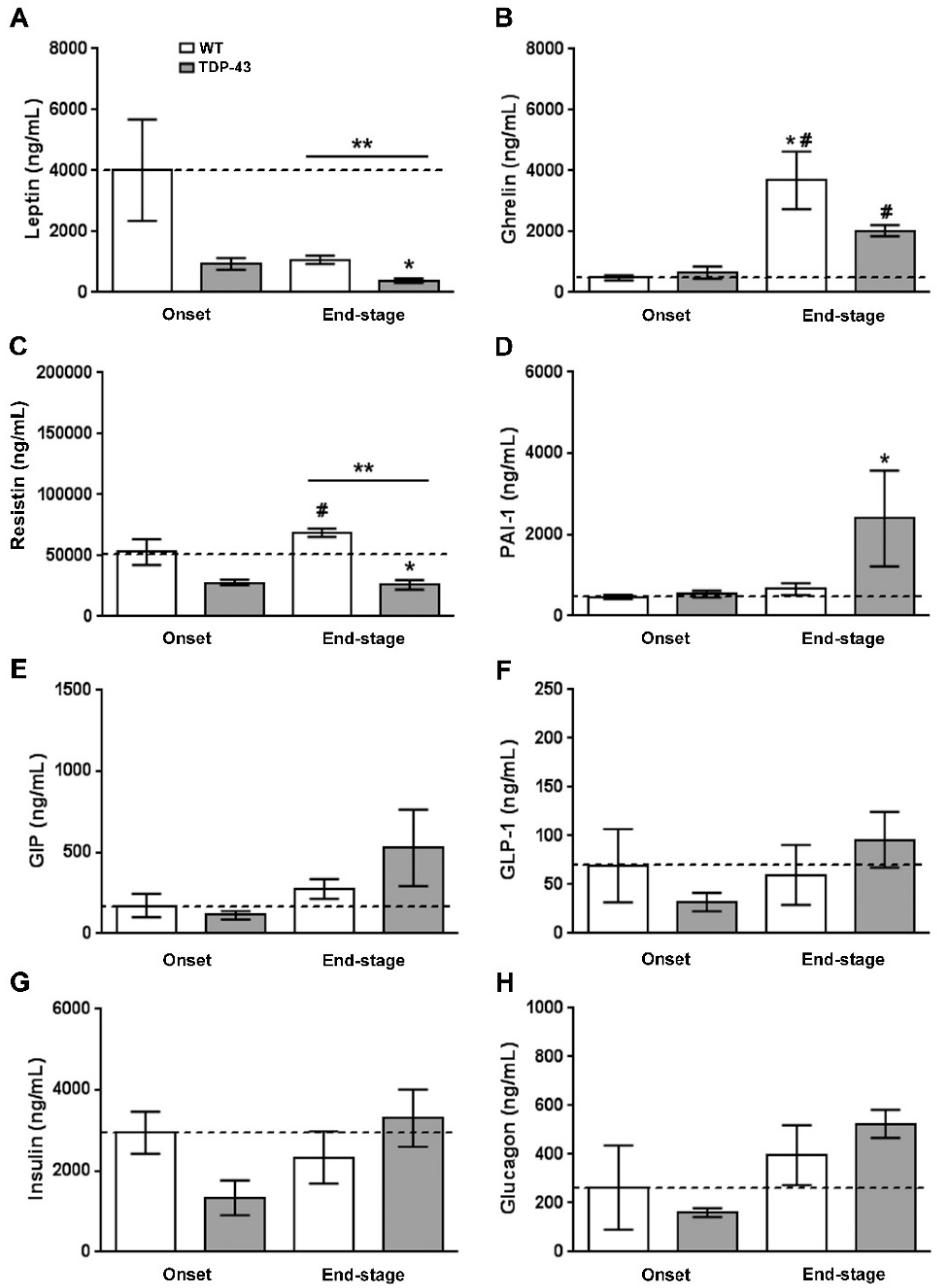
3.2. Peripheral Levels of Leptin, Ghrelin, and Resistin Are Altered in Plasma of TDP-43A315T Mice
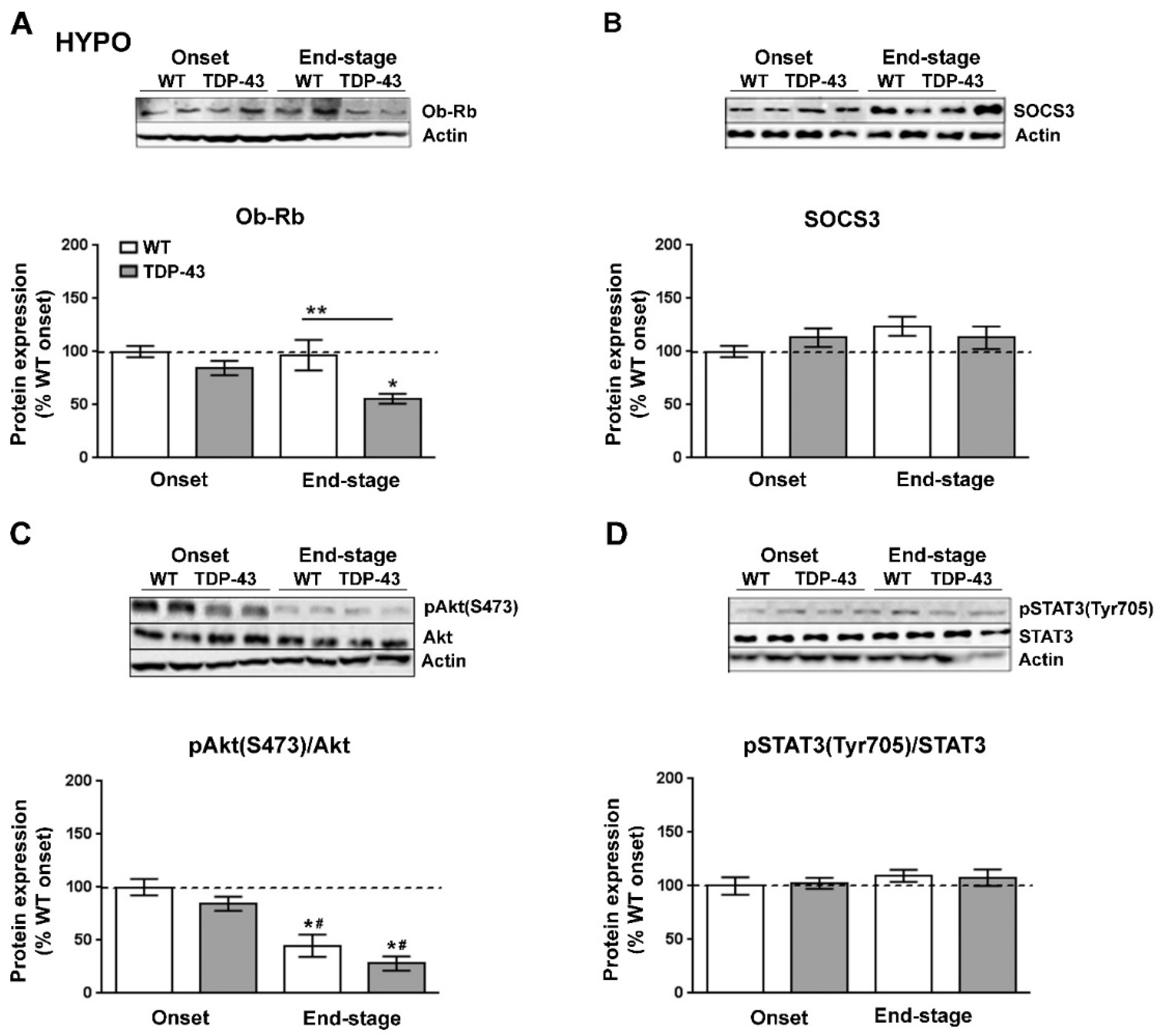
3.3. Hypothalamic Leptin Signaling in TDP-43A315T Mice
3.4. Leptin Signaling in the Spinal Cord of TDP-43A315T
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tapia, R. Cellular and molecular mechanisms of motor neuron death in amyotrophic lateral sclerosis: A perspective. Front. Cell Neurosci. 2014, 8, 241. [Google Scholar] [CrossRef]
- Stephens, T.W.; Basinski, M.B.; Bristow, P.K.; Bue-Valleskey, J.M.; Burgett, S.G.; Craft, L.S.; Hale, E.J.; Hoffmann, J.; Hsiung, H.M.; Kriauciunas, A.; et al. The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature 1995, 377, 530–532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Martos, C.M.; Gonzalez, P.; Rodriguez, F.J. Acute leptin treatment enhances functional recovery after spinal cord injury. PLoS ONE 2012, 7, e35594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wang, S.; Signore, A.P.; Chen, J. Neuroprotective effects of leptin against ischemic injury induced by oxygen-glucose deprivation and transient cerebral ischemia. Stroke 2007, 38, 2329–2336. [Google Scholar] [CrossRef]
- Fewlass, D.C.; Noboa, K.; Pi-Sunyer, F.X.; Johnston, J.M.; Yan, S.D.; Tezapsidis, N. Obesity-related leptin regulates Alzheimer’s Abeta. FASEB J. 2004, 18, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.J.; Sarkar, S.; Casadesus, G.; Zhu, X.; Smith, M.A.; Ashford, J.W.; Johnston, J.M.; Tezapsidis, N. Leptin inhibits glycogen synthase kinase-3beta to prevent tau phosphorylation in neuronal cells. Neurosci. Lett. 2009, 455, 191–194. [Google Scholar] [CrossRef]
- Greco, S.J.; Sarkar, S.; Johnston, J.M.; Tezapsidis, N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem. Biophys. Res. Commun. 2009, 380, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.A.; Bence, K.K.; Sandesara, I.; Andreux, P.; Auwerx, J.; Ishibashi, J.; Seale, P.; Kalb, R.G. Genetically altering organismal metabolism by leptin-deficiency benefits a mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2014, 23, 4995–5008. [Google Scholar] [CrossRef]
- Ngo, S.T.; Steyn, F.J.; Huang, L.; Mantovani, S.; Pfluger, C.M.; Woodruff, T.M.; O’Sullivan, J.D.; Henderson, R.D.; McCombe, P.A. Altered expression of metabolic proteins and adipokines in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2015, 357, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Nagel, G.; Peter, R.S.; Rosenbohm, A.; Koenig, W.; Dupuis, L.; Rothenbacher, D.; Ludolph, A.C. Adipokines, C-reactive protein and Amyotrophic Lateral Sclerosis—Results from a population- based ALS registry in Germany. Sci. Rep. 2017, 7, 4374. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.M.; Phan, K.; Highton-Williamson, E.; Strikwerda-Brown, C.; Caga, J.; Ramsey, E.; Zoing, M.; Devenney, E.; Kim, W.S.; Hodges, J.R.; et al. Eating peptides: Biomarkers of neurodegeneration in amyotrophic lateral sclerosis and frontotemporal dementia. Ann. Clin. Transl. Neurol. 2019, 6, 486–495. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Lee, V.M.; Trojanowski, J.Q. TAR DNA-binding protein 43 in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Morozova, N.; Weisskopf, M.G.; McCullough, M.L.; Munger, K.L.; Calle, E.E.; Thun, M.J.; Ascherio, A. Diet and amyotrophic lateral sclerosis. Epidemiology 2008, 19, 324–337. [Google Scholar] [CrossRef]
- Okamoto, K.; Kihira, T.; Kondo, T.; Kobashi, G.; Washio, M.; Sasaki, S.; Yokoyama, T.; Miyake, Y.; Sakamoto, N.; Inaba, Y.; et al. Nutritional status and risk of amyotrophic lateral sclerosis in Japan. Amyotroph. Lateral Scler. 2007, 8, 300–304. [Google Scholar] [CrossRef]
- Veldink, J.H.; Kalmijn, S.; Groeneveld, G.J.; Wunderink, W.; Koster, A.; de Vries, J.H.; van der Luyt, J.; Wokke, J.H.; Van den Berg, L.H. Intake of polyunsaturated fatty acids and vitamin E reduces the risk of developing amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2007, 78, 367–371. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cutler, R.G.; Camandola, S. Energy intake and amyotrophic lateral sclerosis. Neuromolecular Med. 2007, 9, 17–20. [Google Scholar] [CrossRef]
- Dupuis, L.; Oudart, H.; Rene, F.; Gonzalez de Aguilar, J.L.; Loeffler, J.P. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: Benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef]
- Zhao, W.; Varghese, M.; Vempati, P.; Dzhun, A.; Cheng, A.; Wang, J.; Lange, D.; Bilski, A.; Faravelli, I.; Pasinetti, G.M. Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PLoS ONE 2012, 7, e49191. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar] [CrossRef]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 18809–18814. [Google Scholar] [CrossRef]
- Hatzipetros, T.; Bogdanik, L.P.; Tassinari, V.R.; Kidd, J.D.; Moreno, A.J.; Davis, C.; Osborne, M.; Austin, A.; Vieira, F.G.; Lutz, C.; et al. C57BL/6J congenic Prp-TDP43A315T mice develop progressive neurodegeneration in the myenteric plexus of the colon without exhibiting key features of ALS. Brain Res. 2014, 1584, 59–72. [Google Scholar] [CrossRef]
- Rodriguez, A.; Ferrer-Donato, A.; Cabrera-Pinto, M.; Seseña, S.; Fernández, P.; Aranda, A.; Fernandez-Martos, C.M. Effect of ozone exposure on Amyotrophic Lateral Sclerosis (ALS) pathology using a mice model of TDP-43 proteinopathy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Fernandez, C.M.; Molto, E.; Gallardo, N.; del Arco, A.; Martinez, C.; Andres, A.; Ros, M.; Carrascosa, J.M.; Arribas, C. The expression of rat resistin isoforms is differentially regulated in visceral adipose tissues: Effects of aging and food restriction. Metabolism 2009, 58, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ortega Moreno, L.; Sanz-Garcia, A.; Fernandez de la Fuente, M.J.; Arroyo Solera, R.; Fernandez-Tome, S.; Marin, A.C.; Mora-Gutierrez, I.; Fernandez, P.; Baldan-Martin, M.; Chaparro, M.; et al. Serum adipokines as non-invasive biomarkers in Crohn’s disease. Sci. Rep. 2020, 10, 18027. [Google Scholar] [CrossRef]
- Paganoni, S.; Deng, J.; Jaffa, M.; Cudkowicz, M.E.; Wills, A.M. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve 2011, 44, 20–24. [Google Scholar] [CrossRef]
- Esmaeili, M.A.; Panahi, M.; Yadav, S.; Hennings, L.; Kiaei, M. Premature death of TDP-43 (A315T) transgenic mice due to gastrointestinal complications prior to development of full neurological symptoms of amyotrophic lateral sclerosis. Int. J. Exp. Pathol. 2013, 94, 56–64. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, Q.; Zhang, K.; An, T.; Shi, P.; Li, Z.; Duan, W.; Li, C. HO-1 induction in motor cortex and intestinal dysfunction in TDP-43 A315T transgenic mice. Brain Res. 2012, 1460, 88–95. [Google Scholar] [CrossRef]
- Medina, D.X.; Orr, M.E.; Oddo, S. Accumulation of C-terminal fragments of transactive response DNA-binding protein 43 leads to synaptic loss and cognitive deficits in human TDP-43 transgenic mice. Neurobiol. Aging 2014, 35, 79–87. [Google Scholar] [CrossRef]
- Munzberg, H.; Singh, P.; Heymsfield, S.B.; Yu, S.; Morrison, C.D. Recent advances in understanding the role of leptin in energy homeostasis. F1000Research 2020, 9. [Google Scholar] [CrossRef]
- Zhou, Y.; Rui, L. Leptin signaling and leptin resistance. Front. Med. 2013, 7, 207–222. [Google Scholar] [CrossRef]
- Tefera, T.W.; Borges, K. Metabolic Dysfunctions in Amyotrophic Lateral Sclerosis Pathogenesis and Potential Metabolic Treatments. Front. Neurosci. 2016, 10, 611. [Google Scholar] [CrossRef]
- Tefera, T.W.; Steyn, F.J.; Ngo, S.T.; Borges, K. CNS glucose metabolism in Amyotrophic Lateral Sclerosis: A therapeutic target? Cell Biosci. 2021, 11, 14. [Google Scholar] [CrossRef]
- Vandoorne, T.; De Bock, K.; Van Den Bosch, L. Energy metabolism in ALS: An underappreciated opportunity? Acta. Neuropathol. 2018, 135, 489–509. [Google Scholar] [CrossRef]
- Blasco, H.; Lanznaster, D.; Veyrat-Durebex, C.; Hergesheimer, R.; Vourch, P.; Maillot, F.; Andres, C.R.; Pradat, P.F.; Corcia, P. Understanding and managing metabolic dysfunction in Amyotrophic Lateral Sclerosis. Expert Rev. Neurother. 2020, 20, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.; Chiang, P.M.; Price, D.L.; Wong, P.C. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc. Natl. Acad. Sci. USA 2010, 107, 16325–16330. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [PubMed]
- Bejanin, A.; Tammewar, G.; Marx, G.; Cobigo, Y.; Iaccarino, L.; Kornak, J.; Staffaroni, A.M.; Dickerson, B.C.; Boeve, B.F.; Knopman, D.S.; et al. Longitudinal structural and metabolic changes in frontotemporal dementia. Neurology 2020, 95, e140–e154. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chua, S., Jr. Leptin Function and Regulation. Compr. Physiol. 2017, 8, 351–369. [Google Scholar]
- Cohen, S.; Danzaki, K.; MacIver, N.J. Nutritional effects on T-cell immunometabolism. Eur. J. Immunol. 2017, 47, 225–235. [Google Scholar] [CrossRef]
- Lopez-Gomez, J.J.; Ballesteros-Pomar, M.D.; Torres-Torres, B.; De la Maza, B.P.; Penacho-Lazaro, M.A.; Palacio-Mures, J.M.; Abreu-Padin, C.; Lopez-Guzman, A.; De Luis-Roman, D.A. Malnutrition at diagnosis in amyotrophic lateral sclerosis (als) and its influence on survival: Using glim criteria. Clin. Nutr. 2021, 40, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Ludolph, A.C.; Dorst, J.; Dreyhaupt, J.; Weishaupt, J.H.; Kassubek, J.; Weiland, U.; Meyer, T.; Petri, S.; Hermann, A.; Emmer, A.; et al. Effect of High-Caloric Nutrition on Survival in Amyotrophic Lateral Sclerosis. Ann. Neurol. 2020, 87, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Holm, T.; Maier, A.; Wicks, P.; Lang, D.; Linke, P.; Munch, C.; Steinfurth, L.; Meyer, R.; Meyer, T. Severe loss of appetite in amyotrophic lateral sclerosis patients: Online self-assessment study. Interact. J. Med. Res. 2013, 2, e8. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.; Coccurello, R. What is “Hyper” in the ALS Hypermetabolism? Mediat. Inflamm. 2017, 2017, 7821672. [Google Scholar] [CrossRef]
- Pundir, C.S.; Narwal, V. Biosensing methods for determination of triglycerides: A review. Biosens. Bioelectron. 2018, 100, 214–227. [Google Scholar] [CrossRef]
- Lee, G.H.; Proenca, R.; Montez, J.M.; Carroll, K.M.; Darvishzadeh, J.G.; Lee, J.I.; Friedman, J.M. Abnormal splicing of the leptin receptor in diabetic mice. Nature 1996, 379, 632–635. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Dembski, M.; Weng, X.; Deng, N.; Culpepper, J.; Devos, R.; Richards, G.J.; Campfield, L.A.; Clark, F.T.; Deeds, J.; et al. Identification and expression cloning of a leptin receptor, OB-R. Cell 1995, 83, 1263–1271. [Google Scholar] [CrossRef]
- Varela, L.; Horvath, T.L. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep. 2012, 13, 1079–1086. [Google Scholar] [CrossRef]
- Chiang, P.M.; Ling, J.; Jeong, Y.H.; Price, D.L.; Aja, S.M.; Wong, P.C. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 16320–16324. [Google Scholar] [CrossRef] [PubMed]
- Ohgomori, T.; Yamasaki, R.; Takeuchi, H.; Kadomatsu, K.; Kira, J.I.; Jinno, S. Differential activation of neuronal and glial STAT3 in the spinal cord of the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 2017, 46, 2001–2014. [Google Scholar] [CrossRef] [PubMed]
- Ohgomori, T.; Yamasaki, R.; Kira, J.I.; Jinno, S. Upregulation of Vesicular Glutamate Transporter 2 and STAT3 Activation in the Spinal Cord of Mice Receiving 3,3’-Iminodipropionitrile. Neurotox. Res. 2018, 33, 768–780. [Google Scholar] [CrossRef]
- Caron, A.; Dungan Lemko, H.M.; Castorena, C.M.; Fujikawa, T.; Lee, S.; Lord, C.C.; Ahmed, N.; Lee, C.E.; Holland, W.L.; Liu, C.; et al. POMC neurons expressing leptin receptors coordinate metabolic responses to fasting via suppression of leptin levels. Elife 2018, 7, e33710. [Google Scholar] [CrossRef] [PubMed]
- Pedroso, J.A.; Silveira, M.A.; Lima, L.B.; Furigo, I.C.; Zampieri, T.T.; Ramos-Lobo, A.M.; Buonfiglio, D.C.; Teixeira, P.D.; Frazao, R.; Donato, J., Jr. Changes in Leptin Signaling by SOCS3 Modulate Fasting-Induced Hyperphagia and Weight Regain in Mice. Endocrinology 2016, 157, 3901–3914. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.M.; Clark, R.M.; Hoyle, J.A.; Dickson, T.C. Pathogenic or protective? Neuropeptide Y in amyotrophic lateral sclerosis. J. Neurochem. 2021, 156, 273–289. [Google Scholar] [CrossRef]
- Vercruysse, P.; Sinniger, J.; El Oussini, H.; Scekic-Zahirovic, J.; Dieterle, S.; Dengler, R.; Meyer, T.; Zierz, S.; Kassubek, J.; Fischer, W.; et al. Alterations in the hypothalamic melanocortin pathway in amyotrophic lateral sclerosis. Brain 2016, 139, 1106–1122. [Google Scholar] [CrossRef]
- Butti, Z.; Patten, S.A. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front. Genet. 2018, 9, 712. [Google Scholar] [CrossRef]
- Xue, Y.C.; Ng, C.S.; Xiang, P.; Liu, H.; Zhang, K.; Mohamud, Y.; Luo, H. Dysregulation of RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2020, 13, 78. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrer-Donato, A.; Contreras, A.; Frago, L.M.; Chowen, J.A.; Fernandez-Martos, C.M. Alterations in Leptin Signaling in Amyotrophic Lateral Sclerosis (ALS). Int. J. Mol. Sci. 2021, 22, 10305. https://doi.org/10.3390/ijms221910305
Ferrer-Donato A, Contreras A, Frago LM, Chowen JA, Fernandez-Martos CM. Alterations in Leptin Signaling in Amyotrophic Lateral Sclerosis (ALS). International Journal of Molecular Sciences. 2021; 22(19):10305. https://doi.org/10.3390/ijms221910305
Chicago/Turabian StyleFerrer-Donato, Agueda, Ana Contreras, Laura M. Frago, Julie A. Chowen, and Carmen M. Fernandez-Martos. 2021. "Alterations in Leptin Signaling in Amyotrophic Lateral Sclerosis (ALS)" International Journal of Molecular Sciences 22, no. 19: 10305. https://doi.org/10.3390/ijms221910305
APA StyleFerrer-Donato, A., Contreras, A., Frago, L. M., Chowen, J. A., & Fernandez-Martos, C. M. (2021). Alterations in Leptin Signaling in Amyotrophic Lateral Sclerosis (ALS). International Journal of Molecular Sciences, 22(19), 10305. https://doi.org/10.3390/ijms221910305