
Design and Synthesis of Water-Soluble and Potent MMP-13 Inhibitors with Activity in Human Osteosarcoma Cells

 ,
,  , , ,
, , ,  , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
2. Results and Discussion
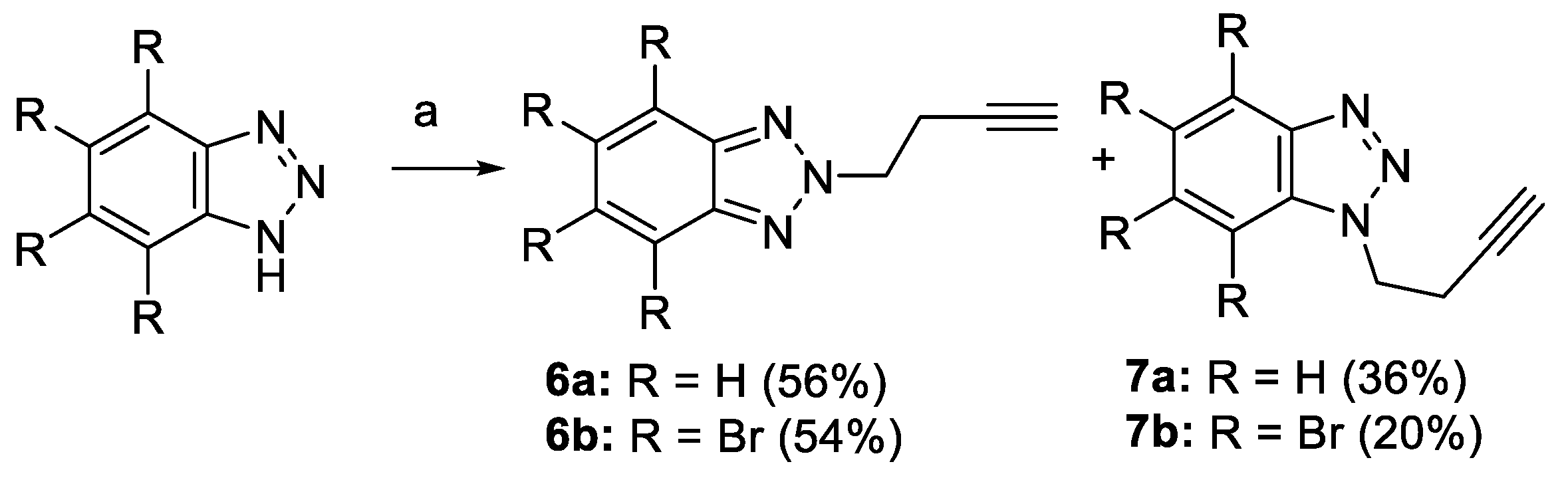
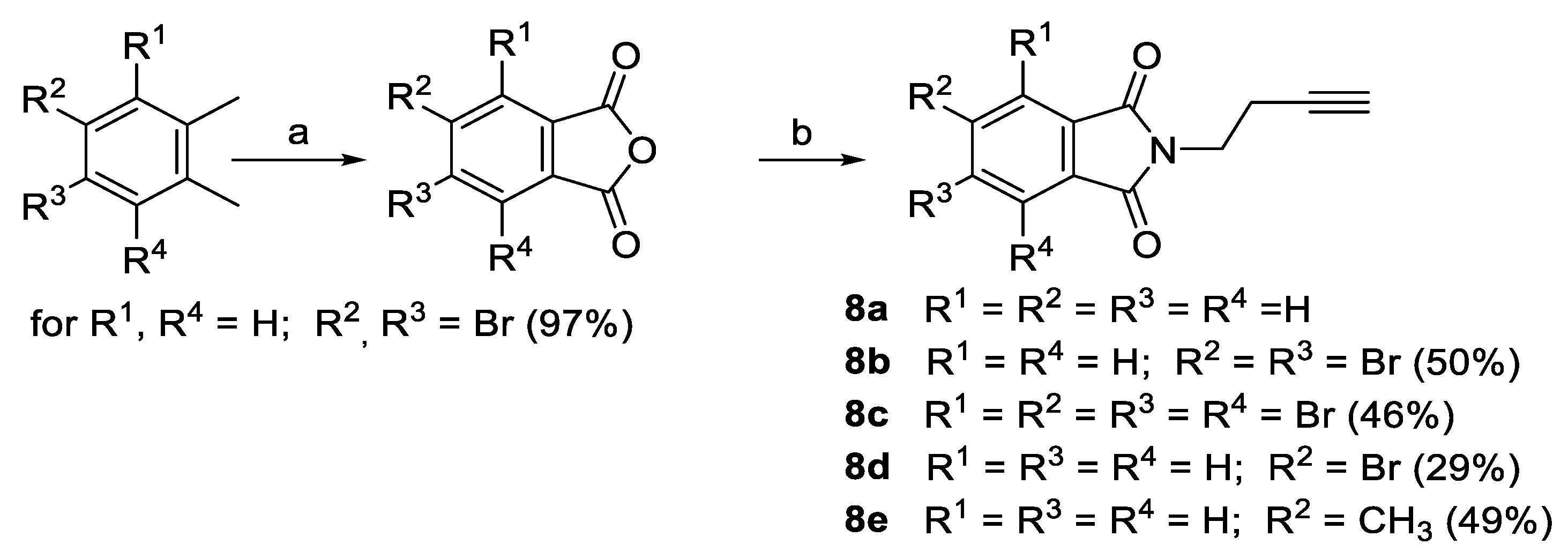
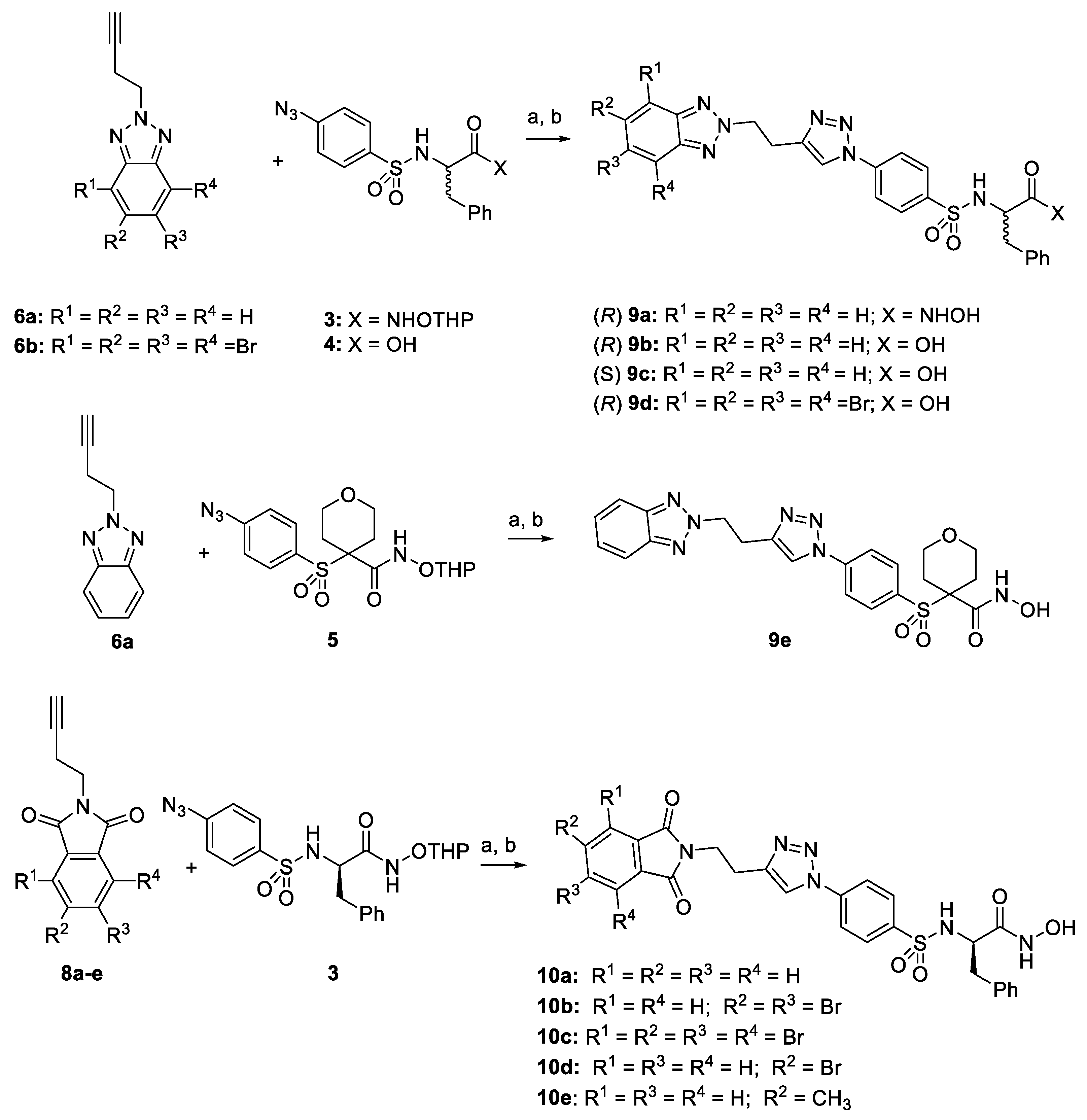
2.1. Synthesis
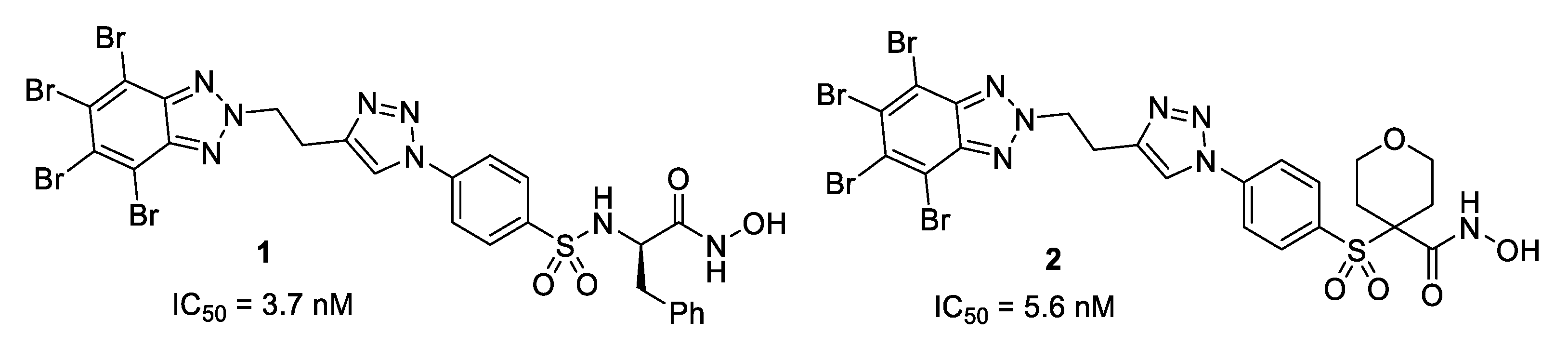
2.2. Activity Results
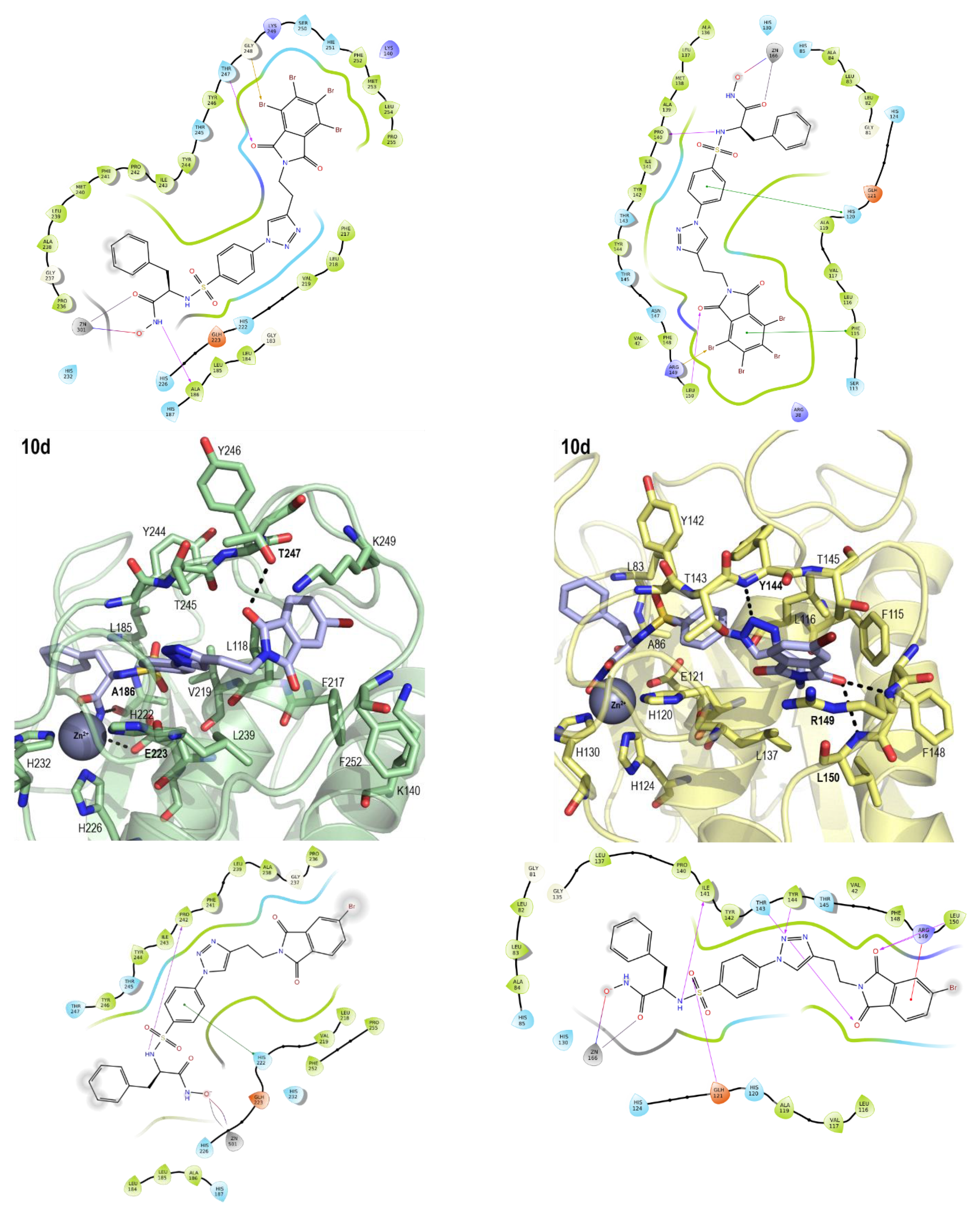
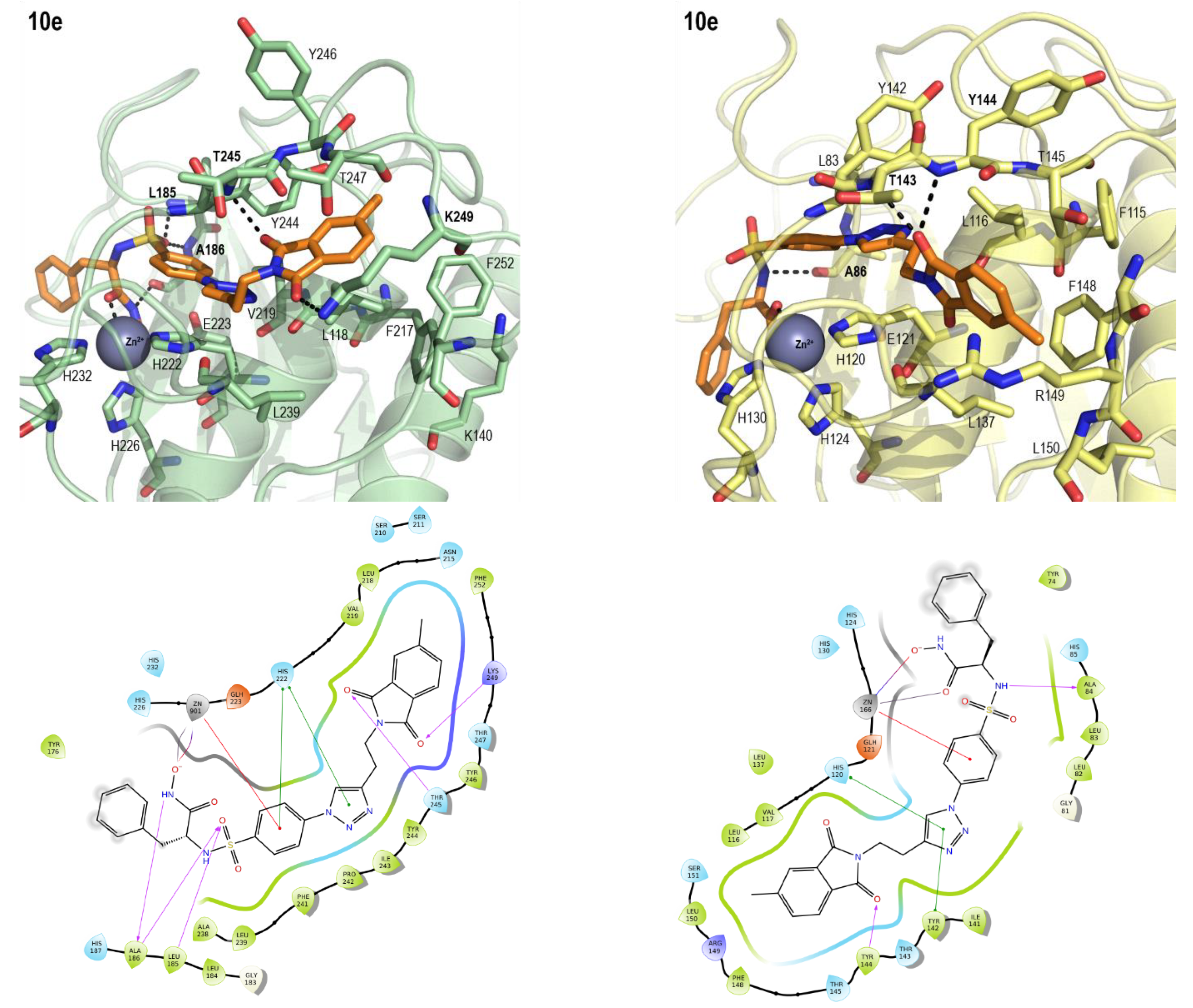
2.3. Molecular Modeling
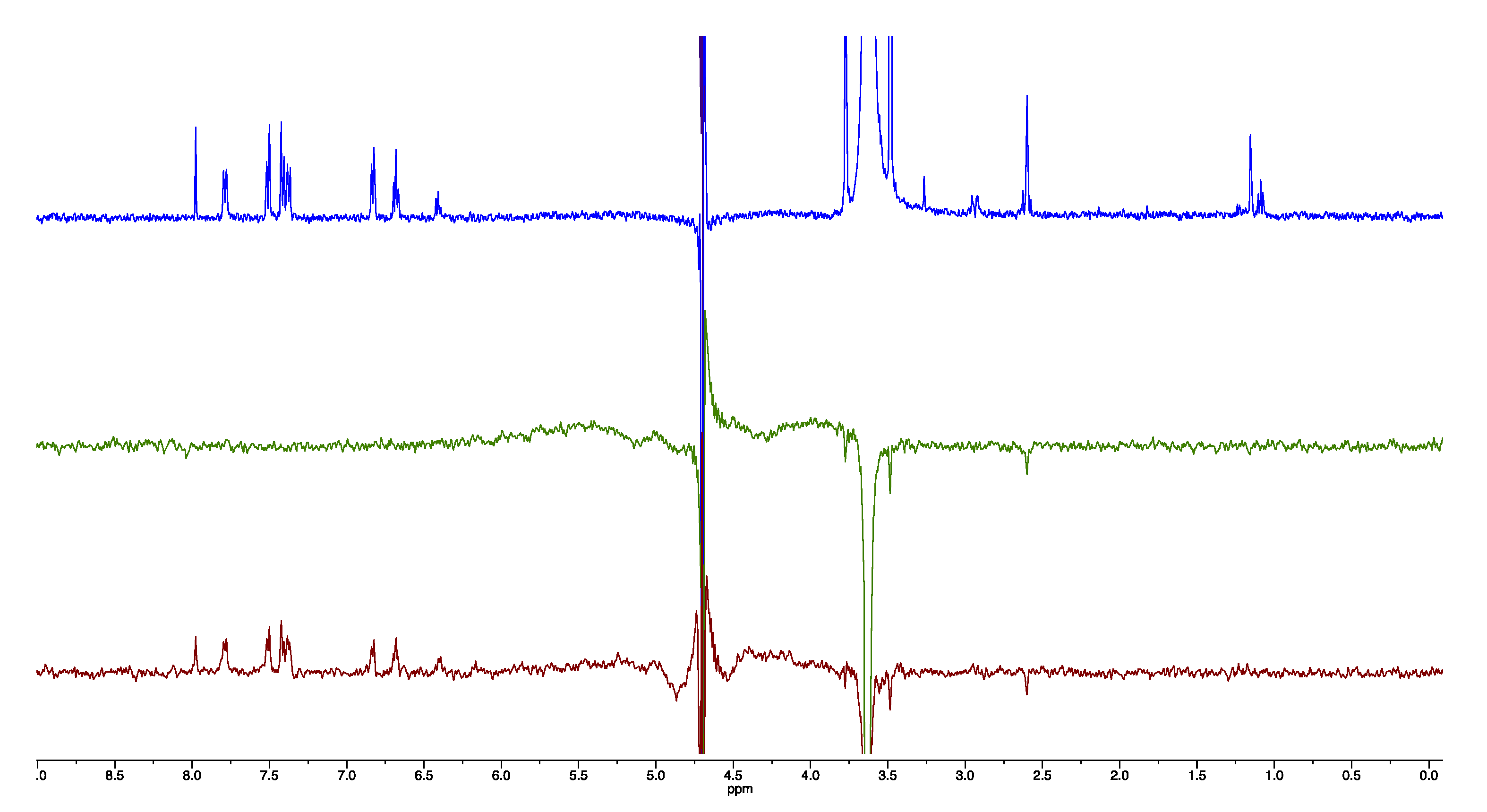
2.4. NMR Studies
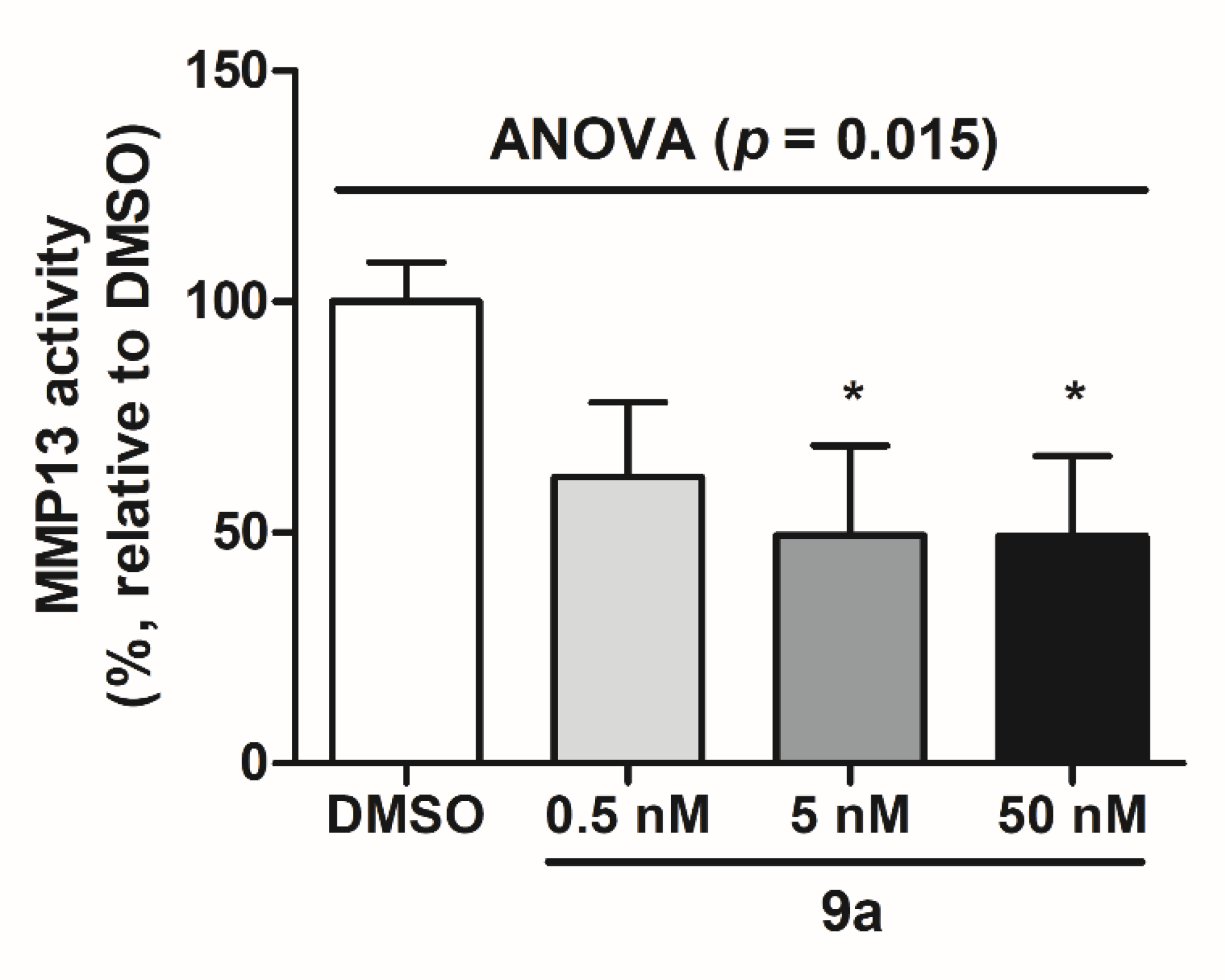
2.5. MMP-13 Inhibition in MG-63 Cells
3. Conclusions
4. Materials and Methods
4.1. Synthetic General Procedures
4.1.1. Materials
4.1.2. General Methods
4.1.3. General Synthetic Methods
- Preparation of triazoles by Copper-Catalyzed Azide-Alkyne Cycloadditions (CuAAC)
- 2.
- O-THP deprotection
4.2. MMP Inhibition Assays
4.3. Cell Culture
4.4. Thermodynamic Solubility (Shake Flask Method)
4.5. Recombinant Protein Production
4.6. NMR Experiments
4.7. Molecular Modeling
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACN | acrylonitrile |
| DCM | dichloromethane |
| DMF | dimethylformamide |
| DMSO | dimethylsulfoxide |
| MW | microwave |
| PDB | protein data bank |
| PK | pharmacokinetic |
| RT | room temperature |
| THF | tetrahydrofuran |
| THP | tetrahydropyrane |
| TBTA | (Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine) |
| TLC | thin layer chromatography |
References
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Senn, N.; Ott, M.; Lanz, J.; Riedl, R. Targeted Polypharmacology: Discovery of a Highly Potent Non-Hydroxamate Dual Matrix Metalloproteinase (MMP)-10/-13 Inhibitor. J. Med. Chem. 2017, 60, 9585–9598. [Google Scholar] [CrossRef]
- Ala-Aho, R.; Kahari, V.M. Collagenases in cancer. Biochimie 2005, 87, 273–286. [Google Scholar] [CrossRef]
- Li, H.; Wang, D.; Yuan, Y.; Min, J. New insights on the MMP-13 regulatory network in the pathogenesis of early osteoarthritis. Arthritis Res. Ther. 2017, 19, 248. [Google Scholar] [CrossRef]
- Li, N.G.; Shi, Z.H.; Tang, Y.P.; Wang, Z.J.; Song, S.L.; Qian, L.H.; Qian, D.W.; Duan, J.A. New Hope for the Treatment of Osteoarthritis Through Selective Inhibition of MMP-13. Curr. Med. Chem. 2011, 18, 977–1001. [Google Scholar] [CrossRef]
- Murphy, G.; Nagase, H. Reappraising metalloproteinases in rheumatoid arthritis and osteoarthritis: Destruction or repair? Nat. Clin. Pract. Rheumatol. 2008, 4, 128–135. [Google Scholar] [CrossRef]
- Chow, Y.Y.; Chin, K.-Y. The Role of Inflammation in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2020, 2020, 8293921. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.A.; Aznavoorian, S.; Engler, J.A.; Windsor, L.J. Induction of collagenase-3 (MMP-13) in rheumatoid arthritis synovial fibroblasts. Biochim. Biophys. Acta-Mol. Basis Dis. 2000, 1502, 307–318. [Google Scholar] [CrossRef]
- Shih, C.-L.M.; Ajuwon, K.M. Inhibition of MMP-13 prevents diet-induced obesity in mice and suppresses adipogenesis in 3T3-L1 preadipocytes. Mol. Biol. Rep. 2015, 42, 1225–1232. [Google Scholar] [CrossRef]
- Zamolo, G.; Grahovac, M.; Zauhar, G.; Vucinic, D.; Kovac, L.; Brajenic, N.; Grahovac, B. Matrix metalloproteinases MMP-1, MMP-2, and MMP-13 are overexpressed in primary nodular melanoma. J. Cutan. Pathol. 2020, 47, 139–145. [Google Scholar] [CrossRef]
- Airola, K.; Johansson, N.; Kariniemi, A.L.; Kahari, V.M.; SaarialhoKere, U.K. Human collagenase-3 is expressed in malignant squamous epithelium of the skin. J. Investig. Dermatol. 1997, 109, 225–231. [Google Scholar] [CrossRef]
- Johansson, N.; Airola, K.; Grenman, R.; Kariniemi, A.L.; SaarialhoKere, U.; Kahari, V.M. Expression of collagenase-3 (matrix metalloproteinase-13) in squamous cell carcinomas of the head and neck. Am. J. Pathol. 1997, 151, 499–508. [Google Scholar]
- Xie, X.-W.; Wan, R.-Z.; Liu, Z.-P. Recent Research Advances in Selective Matrix Metalloproteinase-13 Inhibitors as Anti-Osteoarthritis Agents. ChemMedChem 2017, 12, 1157–1168. [Google Scholar] [CrossRef]
- Lan, Q.; Lu, R.; Chen, H.; Pang, Y.; Xiong, F.; Shen, C.; Qin, Z.; Zheng, L.; Xu, G.; Zhao, J. MMP-13 enzyme and pH responsive theranostic nanoplatform for osteoarthritis. J. Nanobiotechnol. 2020, 18, 117. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, J.W.J.; Berenbaum, F.; Lafeber, F.P.J.G. Osteoarthritis: An update with relevance for clinical practice. Lancet 2011, 377, 2115–2126. [Google Scholar] [CrossRef]
- Brooks, P.M. Impact of osteoarthritis on individuals and society: How much disability? Social consequences and health economic implications. Curr. Opin. Rheumatol. 2002, 14, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Li, W.; Liao, Z.; Yan, M.; Chen, X.; Tang, Z. Selective MMP-13 Inhibitors: Promising Agents for the Therapy of Osteoarthritis. Curr. Med. Chem. 2020, 27, 3753–3769. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.-F.; Ali, A.; Verzijl, N.; Hanemaaijer, R.; TeKoppele, J.; Konttinen, Y.T.; Salo, J. Increased collagen degradation around loosened total hip replacement implants. Arthritis Rheum. 2006, 54, 2928–2933. [Google Scholar] [CrossRef] [PubMed]
- Arabelovic, S.; McAlindon, T.E. Considerations in the treatment of early osteoarthritis. Curr. Rheumatol. Rep. 2005, 7, 29–35. [Google Scholar] [CrossRef]
- Di Pizio, A.; Agamennone, M.; Tortorella, P. Non-Zinc-Binding Inhibitors of MMP-13: GRID-Based Approaches to Rationalize the Binding Process. Curr. Top. Med. Chem. 2016, 16, 449–459. [Google Scholar] [CrossRef]
- Amar, S.; Smith, L.; Fields, G.B. Matrix metalloproteinase collagenolysis in health and disease. Biochim. Biophys. Acta-Mol. Cell Res. 2017, 1864, 1940–1951. [Google Scholar] [CrossRef]
- Mehana, E.-S.E.; Khafaga, A.F.; El-Blehi, S.S. The role of matrix metalloproteinases in osteoarthritis pathogenesis: An updated review. Life Sci. 2019, 234, 116786. [Google Scholar] [CrossRef]
- Wang, M.; Sampson, E.R.; Jin, H.; Li, J.; Ke, Q.H.; Im, H.-J.; Chen, D. MMP13 is a critical target gene during the progression of osteoarthritis. Arthritis Res. Ther. 2013, 15, R5. [Google Scholar] [CrossRef]
- Hu, Q.; Ecker, M. Overview of MMP-13 as a Promising Target for the Treatment of Osteoarthritis. Int. J. Mol. Sci. 2021, 22, 1742. [Google Scholar] [CrossRef]
- Lane, N.E.; Corr, M. Anti-NGF treatments for pain—Two steps forward, one step back? Nat. Rev. Rheumatol. 2017, 13, 76–78. [Google Scholar] [CrossRef]
- Fobian, Y.; Freskos, J.; Barta, T.; Bedell, L.; Heintz, R.; Kassab, D.; Kiefer, J.; Mischke, B.; Molyneaux, J.; Mullins, P.; et al. MMP-13 selective alpha-sulfone hydroxamates: Identification of selective P1’ amides. Bioorganic Med. Chem. Lett. 2011, 21, 2823–2825. [Google Scholar] [CrossRef][Green Version]
- Nara, H.; Sato, K.; Naito, T.; Mototani, H.; Oki, H.; Yamamoto, Y.; Kuno, H.; Santou, T.; Kanzaki, N.; Terauchi, J.; et al. Thieno[2,3-d]pyrimidine-2-carboxamides bearing a carboxybenzene group at 5-position: Highly potent, selective, and orally available MMP-13 inhibitors interacting with the S1″ binding site. Bioorganic Med. Chem. 2014, 22, 5487–5505. [Google Scholar] [CrossRef] [PubMed]
- Bendele, A.M.; Neelagiri, M.; Neelagiri, V.; Sucholeiki, I. Development of a selective matrix metalloproteinase 13 (MMP-13) inhibitor for the treatment of Osteoarthritis. Eur. J. Med. Chem. 2021, 224, 113666. [Google Scholar] [CrossRef]
- El Ashry, E.S.H.; Awad, L.F.; Teleb, M.; Ibrahim, N.A.; Abu-Serie, M.M.; Abd Al Moaty, M.N. Structure-based design and optimization of pyrimidine- and 1,2,4-triazolo 4,3-a pyrimidine-based matrix metalloproteinase-10/13 inhibitors via Dimroth rearrangement towards targeted polypharmacology. Bioorganic Chem. 2020, 96, 103616. [Google Scholar] [CrossRef]
- Satish Kumar, K.; Velayutham, R.; Roy, K.K. A systematic computational analysis of human matrix metalloproteinase 13 (MMP-13) crystal structures and structure-based identification of prospective drug candidates as MMP-13 inhibitors repurposable for osteoarthritis. J. Biomol. Struct. Dyn. 2020, 38, 3074–3086. [Google Scholar] [CrossRef]
- Lee, S.; Park, K.; Lee, S.-Y.; Ryu, J.H.; Park, J.W.; Ahn, H.J.; Kwon, I.C.; Youn, I.-C.; Kim, K.; Choi, K. Dark Quenched Matrix Metalloproteinase Fluorogenic Probe for Imaging Osteoarthritis Development in Vivo. Bioconjugate Chem. 2008, 19, 1743–1747. [Google Scholar] [CrossRef]
- Hugenberg, V.; Wagner, S.; Kopka, K.; Schäfers, M.; Schuit, R.C.; Windhorst, A.D.; Hermann, S. Radiolabeled Selective Matrix Metalloproteinase 13 (MMP-13) Inhibitors: (Radio)Syntheses and in Vitro and First in Vivo Evaluation. J. Med. Chem. 2017, 60, 307–321. [Google Scholar] [CrossRef]
- Pastor, M.; Zapico, J.M.; Coderch, C.; Maslyk, M.; Panchuk, R.; de Pascual-Teresa, B.; Ramos, A. From a MMP2/CK2 multitarget approach to the identification of potent and selective MMP13 inhibitors. Org. Biomol. Chem. 2019, 17, 916–929. [Google Scholar] [CrossRef]
- Zapico, J.M.; Puckowska, A.; Filipiak, K.; Coderch, C.; de Pascual-Teresa, B.; Ramos, A. Design and synthesis of potent hydroxamate inhibitors with increased selectivity within the gelatinase family. Org. Biomol. Chem. 2015, 13, 142–156. [Google Scholar] [CrossRef]
- Zapico, J.M.; Serra, P.; Garcia-Sanmartin, J.; Filipiak, K.; Carbajo, R.J.; Schott, A.K.; Pineda-Lucena, A.; Martinez, A.; Martin-Santamaria, S.; de Pascual-Teresa, B.; et al. Potent “Clicked” MMP2 Inhibitors: Synthesis, Molecular Modeling and Biological Exploration. Org. Biomol. Chem. 2011, 9, 4587–4599. [Google Scholar] [CrossRef]
- Purwin, M.; Hernandez-Toribio, J.; Coderch, C.; Panchuk, R.; Skorokhyd, N.; Filipiak, K.; de Pascual-Teresa, B.; Ramos, A. Design and synthesis of novel dual-target agents for HDAC1 and CK2 inhibition. RSC Adv. 2016, 6, 66595–66608. [Google Scholar] [CrossRef]
- Fabre, B.; Filipiak, K.; Zapico, J.M.; Díaz, N.; Carbajo, R.J.; Schott, A.K.; Martínez-Alcázar, M.P.; Suárez, D.; Pineda-Lucena, A.; Ramos, A.; et al. Progress towards water-soluble triazole-based selective MMP-2 inhibitors. Org. Biomol. Chem. 2013, 11, 6623–6641. [Google Scholar] [CrossRef]
- Fabre, B.; Ramos, A.; de Pascual-Teresa, B. Targeting matrix metalloproteinases: Exploring the dynamics of the s1’ pocket in the design of selective, small molecule inhibitors. J. Med. Chem. 2014, 57, 10205–10219. [Google Scholar] [CrossRef]
- Tuccinardi, T.; Martinelli, A.; Nuti, E.; Carelli, P.; Balzano, F.; Uccello-Barretta, G.; Murphy, G.; Rossello, A. Amber force field implementation, molecular modelling study, synthesis and MMP-1/MMP-2 inhibition profile of (R)- and (S)-N-hydroxy-2-(N-isopropoxybiphenyl-4-ylsulfonamido)-3-methylbutanamides. Bioorganic Med. Chem. 2006, 14, 4260–4276. [Google Scholar] [CrossRef]
- Fabre, B.; Filipiak, K.; Díaz, N.; Zapico, J.M.; Suárez, D.; Ramos, A.; de Pascual-Teresa, B. An Integrated Computational and Experimental Approach to Gaining Selectivity for MMP-2 within the Gelatinase Subfamily. ChemBioChem 2014, 15, 399–412. [Google Scholar] [CrossRef]
- Fabre, B.; Filipiak, K.; Coderch, C.; Zapico, J.M.; Carbajo, R.J.; Schott, A.K.; Pineda-Lucena, A.; de Pascual-Teresa, B.; Ramos, A. New clicked thiirane derivatives as gelatinase inhibitors: The relevance of the P1′ segment. RSC Adv. 2014, 4, 17726–17735. [Google Scholar] [CrossRef]
- Prouillet, C.; Mazière, J.C.; Mazière, C.; Wattel, A.; Brazier, M.; Kamel, S. Stimulatory effect of naturally occurring flavonols quercetin and kaempferol on alkaline phosphatase activity in MG-63 human osteoblasts through ERK and estrogen receptor pathway. Biochem. Pharmacol. 2004, 67, 1307–1313. [Google Scholar] [CrossRef]
- Obłoza, M.; Łapok, Ł.; Solarski, J.; Pędziński, T.; Nowakowska, M. Facile Synthesis, Triplet-State Properties, and Electrochemistry of Hexaiodo-Subphthalocyanine. Chem. A Eur. J. 2018, 24, 17080–17090. [Google Scholar] [CrossRef]
- Schnute, M.E.; O’Brien, P.M.; Nahra, J.; Morris, M.; Howard Roark, W.; Hanau, C.E.; Ruminski, P.G.; Scholten, J.A.; Fletcher, T.R.; Hamper, B.C.; et al. Discovery of (pyridin-4-yl)-2H-tetrazole as a novel scaffold to identify highly selective matrix metalloproteinase-13 inhibitors for the treatment of osteoarthritis. Bioorganic Med. Chem. Lett. 2010, 20, 576–580. [Google Scholar] [CrossRef]
- Hiroshi, N.; Kenjiro, S.; Takako, N.; Hideyuki, M.; Hideyuki, O.; Yamamoto, Y.; Haruhiko, K.; Takashi, S.; Naoyuki, K.; Terauchi, J.; et al. Discovery of Novel, Highly Potent, and Selective Quinazoline-2-carboxamide-Based Matrix Metalloproteinase (MMP)-13 Inhibitors without a Zinc Binding Group Using a Structure-Based Design Approach. J. Med. Chem. 2014, 57, 8886–8902. [Google Scholar]
- Lovejoy, B.; Welch, A.R.; Carr, S.; Luong, C.; Broka, C.; Hendricks, R.T.; Campbell, J.A.; Walker, K.A.M.; Martin, R.; Van Wart, H.; et al. Crystal structures of MMP-1 and -13 reveal the structural basis for selectivity of collagenase inhibitors. Nat. Struct. Biol. 1999, 6, 217–221. [Google Scholar]
- Devel, L.; Beau, F.; Amoura, M.; Vera, L.; Cassar-Lajeunesse, E.; Garcia, S.; Czarny, B.; Stura, E.A.; Dive, V. Simple Pseudo-dipeptides with a P2′ Glutamate: A novel inhibitor family of matrix metalloproteinases and other metzincis. J. Biol. Chem. 2012, 287, 26647–26656. [Google Scholar] [CrossRef]
- Feng, Y.; Likos, J.J.; Zhu, L.; Woodward, H.; Munie, G.; McDonald, J.J.; Stevens, A.M.; Howard, C.P.; De Crescenzo, G.A.; Welsch, D.; et al. Solution structure and backbone dynamics of the catalytic domain of matrix metalloproteinase-2 complexed with a hydroxamic acid inhibitor. Biochim. Biophys. Acta 2002, 1598, 10–23. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Epik, S. Schrödinger Release 2018-2: Schrödinger Suite 2018-2 Protein Preparation Wizard; LLC: New York, NY, USA, 2016. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Glide, S. Schrödinger Release 2018-2; LLC: New York, NY, USA, 2018. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MMP-13 IC50 (nM) | MMP-2 IC50 (nM) | clogD 1 | Water Solubility LogS 1,2 | Experimental Water Solubility LogS 2,3 |
|---|---|---|---|---|---|
| 9a | 0.65 ± 0.30 | 3.42 ± 1.15 | 2.94 | −3.71 | −5.01 |
| 9b | >100 | >100 | 0.32 | 0.00 | n.d. |
| 9c | >100 | >100 | 0.32 | 0.00 | n.d. |
| 9d | >100 | >100 | 3.39 | −1.93 | n.d. |
| 9e | 3.3 ± 2.1 | 10.3 ± 2.8 | 1.37 | −2.72 | −4.47 |
| 10a | 6.9 ± 1.1 | 3.1 ± 0.9 | 2.37 | −5.17 | −5.57 |
| 10b | 46 ± 21 | 15 ± 5 | 3.90 | −6.36 | −5.44 |
| 10c | 61 ± 18 | 17 ± 4 | 5.44 | −7.16 | −6.85 |
| 10d | 15 ± 4 | 14 ± 7 | 3.14 | −5.81 | −5.64 |
| 10e | 9.2 ± 3.0 | 2.9 ± 1.1 | 2.88 | −5.61 | −5.08 |
| 1 | 3.70 ± 1.85 | 70 | 6.01 | −5.85 | −5.54 |
| 2 | 5.60 ± 2.66 | 100 | 4.44 | −5.03 | −5.62 |
| Enzyme | Compound 9a | Compound 1 |
|---|---|---|
| MMP-1 | >1000 | >1000 |
| MMP-2 | 3.42 ± 1.15 | 70 |
| MMP-3 | 31 ± 8 | 500 |
| MMP-7 | 47 ± 21 | >1000 |
| MMP-8 | 100 ± 19 | >500 |
| MMP-9 | 500 ± 74 | >500 |
| MMP-10 | 280 ± 22 | 434 |
| MMP-12 | 3.7 ± 2.0 | 17.8 |
| MMP-13 | 0.65 ± 0.30 | 3.7 |
| MMP-14 | >1000 | >1000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zapico, J.M.; Acosta, L.; Pastor, M.; Rangasamy, L.; Marquez-Cantudo, L.; Coderch, C.; Ortin, I.; Nicolau-Sanus, M.; Puchades-Carrasco, L.; Pineda-Lucena, A.; et al. Design and Synthesis of Water-Soluble and Potent MMP-13 Inhibitors with Activity in Human Osteosarcoma Cells. Int. J. Mol. Sci. 2021, 22, 9976. https://doi.org/10.3390/ijms22189976
Zapico JM, Acosta L, Pastor M, Rangasamy L, Marquez-Cantudo L, Coderch C, Ortin I, Nicolau-Sanus M, Puchades-Carrasco L, Pineda-Lucena A, et al. Design and Synthesis of Water-Soluble and Potent MMP-13 Inhibitors with Activity in Human Osteosarcoma Cells. International Journal of Molecular Sciences. 2021; 22(18):9976. https://doi.org/10.3390/ijms22189976
Chicago/Turabian StyleZapico, Jose Maria, Lourdes Acosta, Miryam Pastor, Loganathan Rangasamy, Laura Marquez-Cantudo, Claire Coderch, Irene Ortin, Maria Nicolau-Sanus, Leonor Puchades-Carrasco, Antonio Pineda-Lucena, and et al. 2021. "Design and Synthesis of Water-Soluble and Potent MMP-13 Inhibitors with Activity in Human Osteosarcoma Cells" International Journal of Molecular Sciences 22, no. 18: 9976. https://doi.org/10.3390/ijms22189976
APA StyleZapico, J. M., Acosta, L., Pastor, M., Rangasamy, L., Marquez-Cantudo, L., Coderch, C., Ortin, I., Nicolau-Sanus, M., Puchades-Carrasco, L., Pineda-Lucena, A., Majali-Martinez, A., Ramos, P., de Pascual-Teresa, B., & Ramos, A. (2021). Design and Synthesis of Water-Soluble and Potent MMP-13 Inhibitors with Activity in Human Osteosarcoma Cells. International Journal of Molecular Sciences, 22(18), 9976. https://doi.org/10.3390/ijms22189976