
Potential Role of Soluble Toll-like Receptors 2 and 4 as Therapeutic Agents in Stroke and Brain Hemorrhage





{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
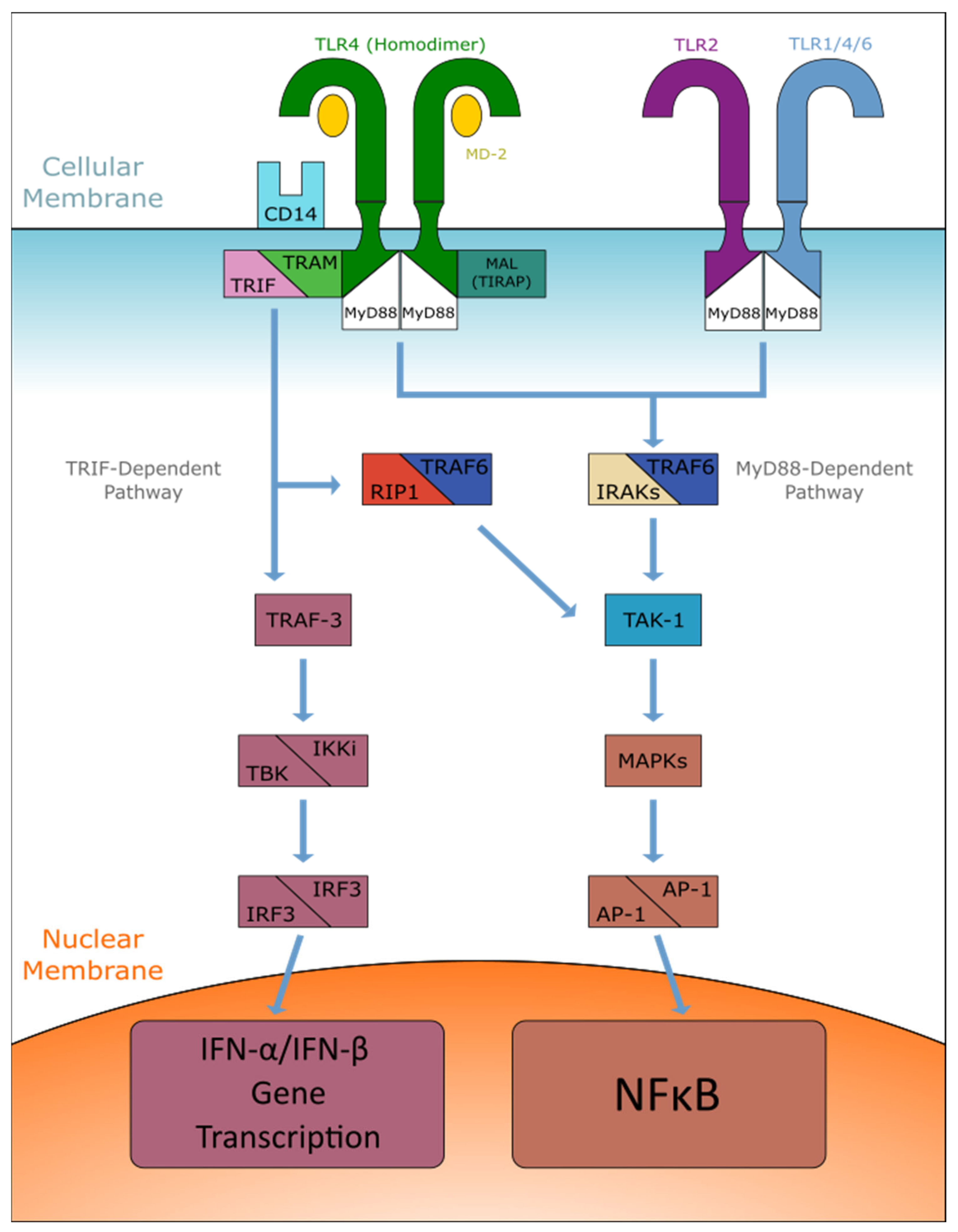
2. Characterization of TLR Pathways
2.1. MyD88-Dependent Pathway
2.2. TRIF-Dependent Pathway
2.3. Complications of the Signaling Pathway
3. Hemoglobin and Its Derivatives Interact with TLRs to Produce an Inflammatory Response in Multiple Cell Types
3.1. Microglia and Other Glial Cells
3.2. Neurons
3.3. Macrophages
3.4. Neutrophils and Other Immune Cells
3.5. Endothelial Cells
4. Alternative TLR Pathways
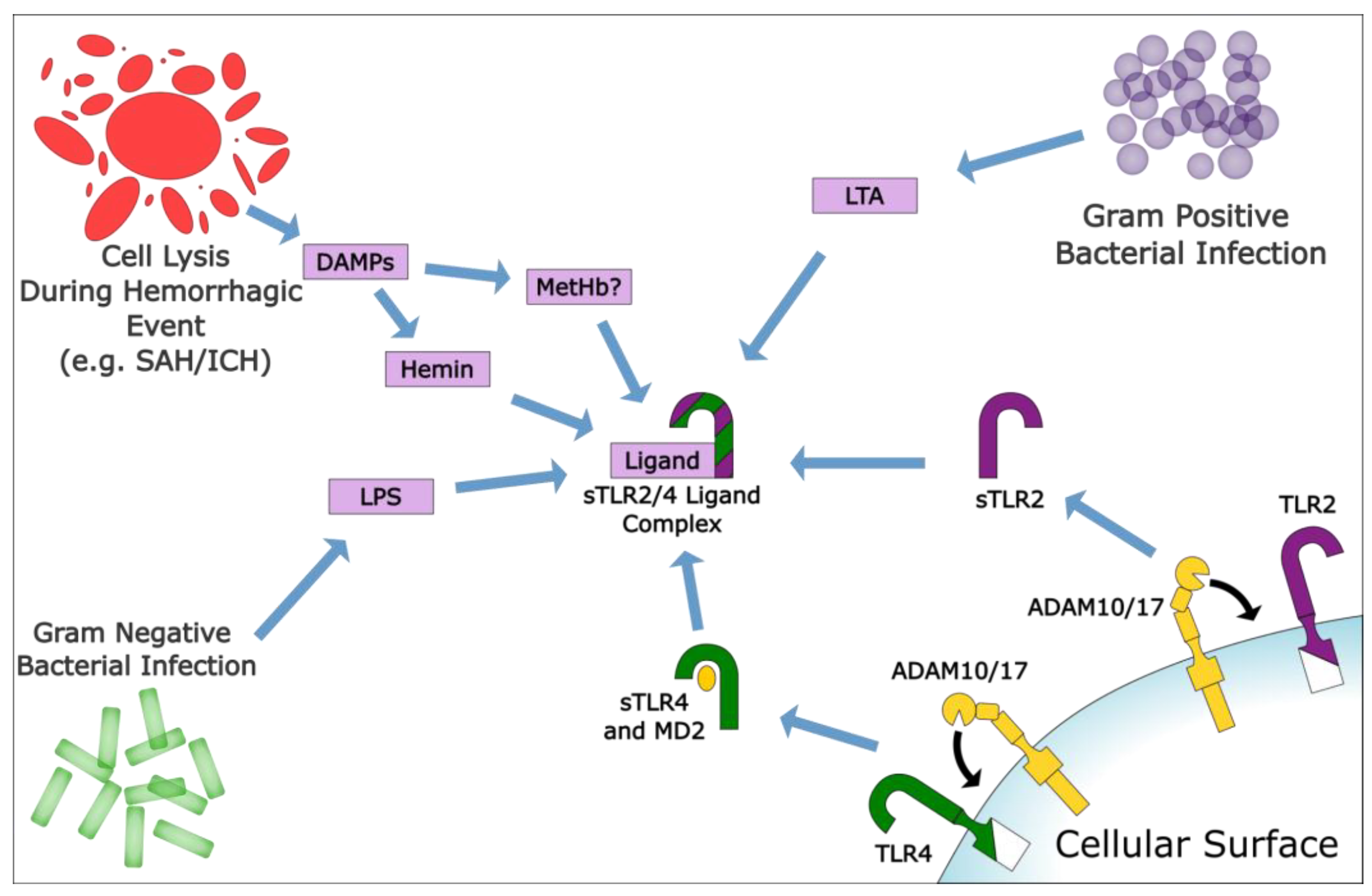
5. The Role of sTLRs in Reducing Hemorrhagic Inflammatory Responses
6. TLRs as a Therapeutic Target
7. Clinical Studies and Proteomics
8. Discussion
9. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, K.V.; Bokla, L.; Nüsslein-Volhard, C. Establishment of dorsal-ventral polarity in the Drosophila embryo: The induction of polarity by the Toll gene product. Cell 1985, 42, 791–798. [Google Scholar] [CrossRef]
- Buchanan, M.M.; Hutchinson, M.; Watkins, L.R.; Yin, H. Toll-like receptor 4 in CNS pathologies. J. Neurochem. 2010, 114, 13–27. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Janciauskiene, S.; Vijayan, V.; Immenschuh, S. TLR4 Signaling by Heme and the Role of Heme-Binding Blood Proteins. Front. Immunol. 2020, 11, 1964. [Google Scholar] [CrossRef] [PubMed]
- Blasius, A.L.; Beutler, B. Intracellular toll-like receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Rifkind, J.M.; Mohanty, J.G.; Nagababu, E. The pathophysiology of extracellular hemoglobin associated with enhanced oxidative reactions. Front. Physiol. 2014, 5, 500. [Google Scholar] [CrossRef] [Green Version]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef]
- Bashawri, L.A.M.; Mandil, A.A.; Bahnassy, A.A.; Ahmed, M.A. Malaria: Hematological aspects. Ann. Saudi Med. 2002, 22, 372–376. [Google Scholar] [CrossRef]
- Wagner, K.R.; Sharp, F.R.; Ardizzone, T.D.; Lu, A.; Clark, J.F. Heme and iron metabolism: Role in cerebral hemorrhage. J. Cereb. Blood Flow Metab. 2003, 23, 629–652. [Google Scholar] [CrossRef] [Green Version]
- Langjahr, P.; Díaz-Jiménez, D.; De la Fuente, M.; Rubio, E.; Golenbock, D.; Bronfman, F.C.; Quera, R.; González, M.-J.; Hermoso, M.A. Metalloproteinase-dependent TLR2 ectodomain shedding is involved in soluble toll-like receptor 2 (sTLR2) production. PLoS ONE 2014, 9, e104624. [Google Scholar]
- Iwami, K.I.; Matsuguchi, T.; Masuda, A.; Kikuchi, T.; Musikacharoen, T.; Yoshikai, Y. Cutting edge: Naturally occurring soluble form of mouse Toll-like receptor 4 inhibits lipopolysaccharide signaling. J. Immunol. 2000, 165, 6682–6686. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Kim, J.J.; Lee, M.J.; Lee, E.K.; Park, S.-K. ADAM17-Mediated Ectodomain Shedding of Toll-Like Receptor 4 as a Negative Feedback Regulation in Lipopolysaccharide-Activated Aortic Endothelial Cells. Cell Physiol. Biochem. 2018, 45, 1851–1862. [Google Scholar] [CrossRef]
- Hyakushima, N.; Mitsuzawa, H.; Nishitani, C.; Sano, H.; Kuronuma, K.; Konishi, M.; Himi, T.; Miyake, K.; Kuroki, Y. Interaction of soluble form of recombinant extracellular TLR4 domain with MD-2 enables lipopolysaccharide binding and attenuates TLR4-mediated signaling. J. Immunol. 2004, 173, 6949–6954. [Google Scholar] [CrossRef]
- LeBouder, E.; Rey-Nores, J.E.; Rushmere, N.K.; Grigorov, M.; Lawn, S.D.; Affolter, M.; Griffin, G.E.; Ferrara, P.; Schiffrin, E.J.; Morgan, B.P.; et al. Soluble forms of Toll-like receptor (TLR)2 capable of modulating TLR2 signaling are present in human plasma and breast milk. J. Immunol. 2003, 171, 6680–6689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokół, B.; Wąsik, N.; Jankowski, R.; Hołysz, M.; Więckowska, B.; Jagodziński, P. Soluble Toll-Like Receptors 2 and 4 in Cerebrospinal Fluid of Patients with Acute Hydrocephalus following Aneurysmal Subarachnoid Haemorrhage. PLoS ONE 2016, 11, e0156171. [Google Scholar]
- Ten Oever, J.; Kox, M.; van de Veerdonk, F.L.; Mothapo, K.M.; Slavcovici, A.; Jansen, T.L.; Tweehuysen, L.; Giamarellos-Bourboulis, E.J.; Schneeberger, P.M.; Wever, P.C.; et al. The discriminative capacity of soluble Toll-like receptor (sTLR)2 and sTLR4 in inflammatory diseases. BMC Immunol. 2014, 15, 55. [Google Scholar] [CrossRef] [Green Version]
- Zunt, S.L.; Burton, L.V.; Goldblatt, L.I.; Dobbins, E.E.; Srinivasan, M. Soluble forms of Toll-like receptor 4 are present in human saliva and modulate tumour necrosis factor-alpha secretion by macrophage-like cells. Clin. Exp. Immunol. 2009, 156, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Staller, S.; Lindsay, A.K.; Ramos, E.D.; Thomas, P.; Srinivasan, M. Changes in salivary microbial sensing proteins CD14 and TLR2 with aging. Clin. Oral Investig. 2020, 24, 2523–2528. [Google Scholar] [CrossRef]
- Al Qallaf, H.; Hamada, Y.; Blanchard, S.; Shin, D.; Gregory, R.; Srinivasan, M. Differential profiles of soluble and cellular toll like receptor (TLR)-2 and 4 in chronic periodontitis. PLoS ONE 2018, 13, e0200231. [Google Scholar]
- Oliveira-Nascimento, L.; Massari, P.; Wetzler, L.M. The role of TLR2 in infection and immunity. Front. Immunol. 2012, 3, 79. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.-G.; Lee, H.; Lee, J.-O. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kagan, J.C.; Medzhitov, R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell 2006, 125, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Rowe, D.C.; McGettrick, A.F.; Latz, E.; Monks, B.G.; Gay, N.J.; Yamamoto, M.; Akira, S.; O’Neill, L.A.; Fitzgerald, K.A.; Golenbock, D.T. The myristoylation of TRIF-related adaptor molecule is essential for Toll-like receptor 4 signal transduction. Proc. Natl. Acad. Sci. USA 2006, 103, 6299–6304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanimura, N.; Saitoh, S.; Matsumoto, F.; Akashi-Takamura, S.; Miyake, K. Roles for LPS-dependent interaction and relocation of TLR4 and TRAM in TRIF-signaling. Biochem. Biophys. Res. Commun. 2008, 368, 94–99. [Google Scholar] [CrossRef]
- Ma, C.; Zhou, W.; Yan, Z.; Qu, M.; Bu, X. Toll-like receptor 4 (TLR4) is correlated with delayed cerebral ischemia (DCI) and poor prognosis in aneurysmal subarachnoid hemorrhage. J. Neurol. Sci. 2015, 359, 67–71. [Google Scholar] [CrossRef]
- Huang, H.-F.; Zeng, Z.; Wang, K.-H.; Zhang, H.-Y.; Wang, S.; Zhou, W.-X.; Wang, Z.-B.; Xu, W.-G.; Duan, J. Heme oxygenase-1 protects rat liver against warm ischemia/reperfusion injury via TLR2/TLR4-triggered signaling pathways. World J. Gastroenterol. 2015, 21, 2937–2948. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graça-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Kim, H.P.; Geng, X.H.; Nakao, A.; Wang, X.; Murase, N.; Drain, P.F.; Wang, X.; Sasidhar, M.; Nabel, E.G.; et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J. Exp. Med. 2006, 203, 2377–2389. [Google Scholar] [CrossRef]
- Kim, H.P.; Ryter, S.W.; Choi, A.M.K. CO as a cellular signaling molecule. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 411–449. [Google Scholar] [CrossRef]
- Zhang, F.; Zhang, C. Rnf112 deletion protects brain against intracerebral hemorrhage (ICH) in mice by inhibiting TLR-4/NF-κB pathway. Biochem. Biophys. Res. Commun. 2018, 507, 43–50. [Google Scholar] [CrossRef]
- Fujimoto, M.; Shiba, M.; Kawakita, F.; Liu, L.; Shimojo, N.; Imanaka-Yoshida, K.; Yoshida, T.; Suzuki, H. Deficiency of tenascin-C and attenuation of blood-brain barrier disruption following experimental subarachnoid hemorrhage in mice. J. Neurosurg. 2016, 124, 1693–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Fujimoto, M.; Nakano, F.; Nishikawa, H.; Okada, T.; Kawakita, F.; Imanaka-Yoshida, K.; Yoshida, T.; Suzuki, H. Deficiency of Tenascin-C Alleviates Neuronal Apoptosis and Neuroinflammation After Experimental Subarachnoid Hemorrhage in Mice. Mol. Neurobiol. 2018, 55, 8346–8354. [Google Scholar] [CrossRef]
- Tsai, H.-L.; Chiu, W.-T.; Fang, C.-L.; Hwang, S.-M.; Renshaw, P.F.; Lai, W.-F.T. Different forms of tenascin-C with tenascin-R regulate neural differentiation in bone marrow-derived human mesenchymal stem cells. Tissue Eng. Part A 2014, 20, 1908–1921. [Google Scholar] [CrossRef] [Green Version]
- Vinchi, F.; Costa da Silva, M.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486. [Google Scholar] [CrossRef]
- García-Culebras, A.; Durán-Laforet, V.; Peña-Martínez, C.; Moraga, A.; Ballesteros, I.; Cuartero, M.I.; de la Parra, J.; Palma-Tortosa, S.; Hidalgo, A.; Corbí, A.L.; et al. Role of TLR4 (Toll-Like Receptor 4) in N1/N2 Neutrophil Programming After Stroke. Stroke 2019, 50, 2922–2932. [Google Scholar] [CrossRef] [Green Version]
- Philip, M.; Chiu, E.Y.; Hajjar, A.M.; Abkowitz, J.L. TLR stimulation dynamically regulates heme and iron export gene expression in macrophages. J. Immunol. Res. 2016, 2016, 4039038. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.; Kwak, Y.H.; Sammy, F.; He, P.; Thundivalappil, S.; Sun, G.; Chao, W.; Warren, H.S. Synergistic inflammation is induced by blood degradation products with microbial Toll-like receptor agonists and is blocked by hemopexin. J. Infect. Dis. 2010, 202, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Belcher, J.D.; Zhang, P.; Nguyen, J.; Kiser, Z.M.; Nath, K.A.; Hu, J.; Trent, J.O.; Vercellotti, G.M. Identification of a Heme Activation Site on the MD-2/TLR4 Complex. Front. Immunol. 2020, 11, 1370. [Google Scholar] [CrossRef]
- Fang, H.; Wang, P.-F.; Zhou, Y.; Wang, Y.-C.; Yang, Q.-W. Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. J. Neuroinflammation 2013, 10, 27. [Google Scholar] [CrossRef] [Green Version]
- Erridge, C. Endogenous ligands of TLR2 and TLR4: Agonists or assistants? J. Leukoc. Biol. 2010, 87, 989–999. [Google Scholar] [CrossRef]
- Silva, G.; Jeney, V.; Chora, A.; Larsen, R.; Balla, J.; Soares, M.P. Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J. Biol. Chem. 2009, 284, 29582–29595. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-K.; Goh, S.Y.; Wong, Y.Q.; Ding, J.L. Response of neutrophils to extracellular haemoglobin and LTA in human blood system. EBioMedicine 2015, 2, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-C.; Zhou, Y.; Fang, H.; Lin, S.; Wang, P.-F.; Xiong, R.-P.; Chen, J.; Xiong, X.-Y.; Lv, F.-L.; Liang, Q.-L.; et al. Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann. Neurol. 2014, 75, 876–889. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.S.; Woo, S.K.; Kurland, D.B.; Yoon, S.H.; Palmer, A.F.; Banerjee, U.; Iqbal, S.; Ivanova, S.; Gerzanich, V.; Simard, J.M. Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int. J. Mol. Sci. 2015, 16, 5028–5046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanafy, K.A. The role of microglia and the TLR4 pathway in neuronal apoptosis and vasospasm after subarachnoid hemorrhage. J. Neuroinflamm. 2013, 10, 83. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, R.L. Delayed neurological deterioration after subarachnoid haemorrhage. Nat. Rev. Neurol. 2014, 10, 44–58. [Google Scholar] [CrossRef]
- Gao, Y.; Zhuang, Z.; Lu, Y.; Tao, T.; Zhou, Y.; Liu, G.; Wang, H.; Zhang, D.; Wu, L.; Dai, H.; et al. Curcumin Mitigates Neuro-Inflammation by Modulating Microglia Polarization Through Inhibiting TLR4 Axis Signaling Pathway Following Experimental Subarachnoid Hemorrhage. Front. Neurosci. 2019, 13, 1223. [Google Scholar] [CrossRef]
- Yoshizaki, S.; Kijima, K.; Hara, M.; Saito, T.; Tamaru, T.; Tanaka, M.; Konno, D.-J.; Nakashima, Y.; Okada, S. Tranexamic acid reduces heme cytotoxicity via the TLR4/TNF axis and ameliorates functional recovery after spinal cord injury. J. Neuroinflamm. 2019, 16, 160. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Lu, Y.; Wu, Q.; Dai, H.; Li, W.; Lv, S.; Zhou, X.; Zhang, X.; Hang, C.; Wang, J. Astaxanthin mitigates subarachnoid hemorrhage injury primarily by increasing sirtuin 1 and inhibiting the Toll-like receptor 4 signaling pathway. FASEB J. 2019, 33, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhu, J.; Wang, Y.-H. RNF216 mediates neuronal injury following experimental subarachnoid hemorrhage through the Arc/Arg3.1-AMPAR pathway. FASEB J. 2020, 34, 15080–15092. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Zhou, K.; Liang, Q.-L.; Lin, S.; Wang, Y.-C.; Xiong, X.-Y.; Meng, Z.-Y.; Zhao, T.; Zhu, W.-Y.; Yang, Y.-R.; et al. Interleukin-23 Secreted by Activated Macrophages Drives γδT Cell Production of Interleukin-17 to Aggravate Secondary Injury After Intracerebral Hemorrhage. J. Am. Heart Assoc. 2016, 5, e004340. [Google Scholar] [CrossRef] [PubMed]
- Lisk, C.; Kominsky, D.; Ehrentraut, S.; Bonaventura, J.; Nuss, R.; Hassell, K.; Nozik-Grayck, E.; Irwin, D.C. Hemoglobin-induced endothelial cell permeability is controlled, in part, via a myeloid differentiation primary response gene-88-dependent signaling mechanism. Am. J. Respir. Cell Mol. Biol. 2013, 49, 619–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Guo, Z.; Tong, L.; Xue, F.; Krafft, P.R.; Budbazar, E.; Zhang, J.H.; Tang, J. TLR7 (Toll-Like Receptor 7) Facilitates Heme Scavenging Through the BTK (Bruton Tyrosine Kinase)-CRT (Calreticulin)-LRP1 (Low-Density Lipoprotein Receptor-Related Protein-1)-Hx (Hemopexin) Pathway in Murine Intracerebral Hemorrhage. Stroke 2018, 49, 3020–3029. [Google Scholar] [CrossRef]
- Liew, F.Y.; Xu, D.; Brint, E.K.; O’Neill, L.A.J. Negative regulation of toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 2005, 5, 446–458. [Google Scholar] [CrossRef]
- Chau, L.-Y. Heme oxygenase-1: Emerging target of cancer therapy. J. Biomed. Sci. 2015, 22, 22. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Saleem, S.; Zhen, G.; Cao, W.; Zhuang, H.; Lee, J.; Smith, A.; Altruda, F.; Tolosano, E.; Doré, S. Heme-hemopexin complex attenuates neuronal cell death and stroke damage. J. Cereb. Blood Flow Metab. 2009, 29, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Foresti, R.; Bains, S.K.; Pitchumony, T.S.; de Castro Brás, L.E.; Drago, F.; Dubois-Randé, J.-L.; Bucolo, C.; Motterlini, R. Small molecule activators of the Nrf2-HO-1 antioxidant axis modulate heme metabolism and inflammation in BV2 microglia cells. Pharmacol. Res. 2013, 76, 132–148. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Cui, Y.; Xu, J.; Gao, H. miR-140-5p Attenuates Neuroinflammation and Brain Injury in Rats Following Intracerebral Hemorrhage by Targeting TLR4. Inflammation 2019, 42, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-J.; Zhang, Q.-R.; Gao, X.; Wang, H.; Tao, T.; Gao, Y.-Y.; Zhou, Y.; Chen, X.-X.; Li, W.; Hang, C.-H. MiR-146a Ameliorates Hemoglobin-Induced Microglial Inflammatory Response via TLR4/IRAK1/TRAF6 Associated Pathways. Front. Neurosci. 2020, 14, 311. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, Y.; Anderson, E.M.; Birmingham, A.; Reynolds, A.; Karpilow, J.; Robinson, K.; Leake, D.; Marshall, W.S.; Khvorova, A. Off-target effects by siRNA can induce toxic phenotype. RNA 2006, 12, 1188–1196. [Google Scholar] [CrossRef] [Green Version]
- Andresen, L.; Theodorou, K.; Grünewald, S.; Czech-Zechmeister, B.; Könnecke, B.; Lühder, F.; Trendelenburg, G. Evaluation of the Therapeutic Potential of Anti-TLR4-Antibody MTS510 in Experimental Stroke and Significance of Different Routes of Application. PLoS ONE 2016, 11, e0148428. [Google Scholar] [CrossRef]
- Lin, S.; Yin, Q.; Zhong, Q.; Lv, F.-L.; Zhou, Y.; Li, J.-Q.; Wang, J.-Z.; Su, B.; Yang, Q.-W. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J. Neuroinflamm. 2012, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Sudan, K.; Vijayan, V.; Madyaningrana, K.; Gueler, F.; Igarashi, K.; Foresti, R.; Motterlini, R.; Immenschuh, S. TLR4 activation alters labile heme levels to regulate BACH1 and heme oxygenase-1 expression in macrophages. Free Radic. Biol. Med. 2019, 137, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Paule, R.; Leon, J.; Daugan, M.; Robe-Rybkine, T.; Poillerat, V.; Torset, C.; Frémeaux-Bacchi, V.; Dimitrov, J.D.; Roumenina, L.T. P-selectin drives complement attack on endothelium during intravascular hemolysis in TLR-4/heme-dependent manner. Proc. Natl. Acad. Sci. USA 2019, 116, 6280–6285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ii, M.; Matsunaga, N.; Hazeki, K.; Nakamura, K.; Takashima, K.; Seya, T.; Hazeki, O.; Kitazaki, T.; Iizawa, Y. A novel cyclohexene derivative, ethyl (6R)-6-[N-(2-Chloro-4-fluorophenyl)sulfamoyl]cyclohex-1-ene-1-carboxylate (TAK-242), selectively inhibits toll-like receptor 4-mediated cytokine production through suppression of intracellular signaling. Mol. Pharmacol. 2006, 69, 1288–1295. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharmacol. 2011, 79, 34–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Zhao, X.-D.; Zhuang, Z.; Xue, Y.-J.; Cheng, H.-L.; Yin, H.-X.; Shi, J.-X. Peroxisome proliferator-activated receptor gamma agonist rosiglitazone attenuates oxyhemoglobin-induced Toll-like receptor 4 expression in vascular smooth muscle cells. Brain Res. 2010, 1322, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Hua, F.; Tang, H.; Wang, J.; Prunty, M.C.; Hua, X.; Sayeed, I.; Stein, D.G. TAK-242, an antagonist for Toll-like receptor 4, protects against acute cerebral ischemia/reperfusion injury in mice. J. Cereb. Blood Flow Metab. 2015, 35, 536–542. [Google Scholar] [CrossRef]
- Galindo, D.C.; Banks, W.A.; Rhea, E.M. The impact of acute rosiglitazone on insulin pharmacokinetics at the blood-brain barrier. Endocrinol. Diab. Metab. 2020, 3, e00149. [Google Scholar] [CrossRef]
- Zhou, C.; Wang, C.; Xie, G.; Wu, L.; Wei, Y.; Wang, Q.; Zhang, H.; Hang, C.; Zhou, M.; Shi, J. Fisetin alleviates early brain injury following experimental subarachnoid hemorrhage in rats possibly by suppressing TLR 4/NF-κB signaling pathway. Brain Res. 2015, 1629, 250–259. [Google Scholar] [CrossRef]
- Yang, Y.; Tan, X.; Xu, J.; Wang, T.; Liang, T.; Xu, X.; Ma, C.; Xu, Z.; Wang, W.; Li, H.; et al. Luteolin alleviates neuroinflammation via downregulating the TLR4/TRAF6/NF-κB pathway after intracerebral hemorrhage. Biomed. Pharmacother. 2020, 126, 110044. [Google Scholar] [CrossRef]
- Li, T.; Jing, J.-J.; Yang, J.; Sun, L.-P.; Gong, Y.-H.; Xin, S.-J.; Yuan, Y. Serum levels of matrix metalloproteinase 9 and toll-like receptor 4 in acute aortic dissection: A case-control study. BMC Cardiovasc. Disord. 2018, 18, 219. [Google Scholar] [CrossRef]
- Hossain, M.J.; Morandi, E.; Tanasescu, R.; Frakich, N.; Caldano, M.; Onion, D.; Faraj, T.A.; Erridge, C.; Gran, B. The Soluble Form of Toll-Like Receptor 2 Is Elevated in Serum of Multiple Sclerosis Patients: A Novel Potential Disease Biomarker. Front. Immunol. 2018, 9, 457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Xia, Y.; Yang, J.; Guo, H.; Sun, G.; Sanyal, A.J.; Shah, V.H.; Lou, Y.; Zheng, X.; Chalasani, N.; et al. Immune Checkpoint Axes Are Dysregulated in Patients with Alcoholic Hepatitis. Hepatol. Commun. 2020, 4, 588–605. [Google Scholar] [CrossRef] [PubMed]
- Paarnio, K.; Tuomisto, A.; Väyrynen, S.A.; Väyrynen, J.P.; Klintrup, K.; Ohtonen, P.; Mäkinen, M.J.; Mäkelä, J.; Karttunen, T.J. Serum TLR2 and TLR4 levels in colorectal cancer and their association with systemic inflammatory markers, tumor characteristics, and disease outcome. APMIS 2019, 127, 561–569. [Google Scholar] [CrossRef]
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRx 2005, 2, 554–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beutler, B.A.; Peppel, K.; Crawford, D.F. DNA Encoding a Chimeric Polypeptide Comprising the Extracellular Domain of TNF Receptor Fused to IgG, Vectors, and Host Cells. U.S. Patent US5447851B1, 7 June 1999. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lua, J.; Ekanayake, K.; Fangman, M.; Doré, S. Potential Role of Soluble Toll-like Receptors 2 and 4 as Therapeutic Agents in Stroke and Brain Hemorrhage. Int. J. Mol. Sci. 2021, 22, 9977. https://doi.org/10.3390/ijms22189977
Lua J, Ekanayake K, Fangman M, Doré S. Potential Role of Soluble Toll-like Receptors 2 and 4 as Therapeutic Agents in Stroke and Brain Hemorrhage. International Journal of Molecular Sciences. 2021; 22(18):9977. https://doi.org/10.3390/ijms22189977
Chicago/Turabian StyleLua, Josh, Kanishka Ekanayake, Madison Fangman, and Sylvain Doré. 2021. "Potential Role of Soluble Toll-like Receptors 2 and 4 as Therapeutic Agents in Stroke and Brain Hemorrhage" International Journal of Molecular Sciences 22, no. 18: 9977. https://doi.org/10.3390/ijms22189977
APA StyleLua, J., Ekanayake, K., Fangman, M., & Doré, S. (2021). Potential Role of Soluble Toll-like Receptors 2 and 4 as Therapeutic Agents in Stroke and Brain Hemorrhage. International Journal of Molecular Sciences, 22(18), 9977. https://doi.org/10.3390/ijms22189977