
Angelman Syndrome and Angelman-like Syndromes Share the Same Calcium-Related Gene Signatures


Abstract
:1. Introduction
2. Results
2.1. AS Like Syndromes
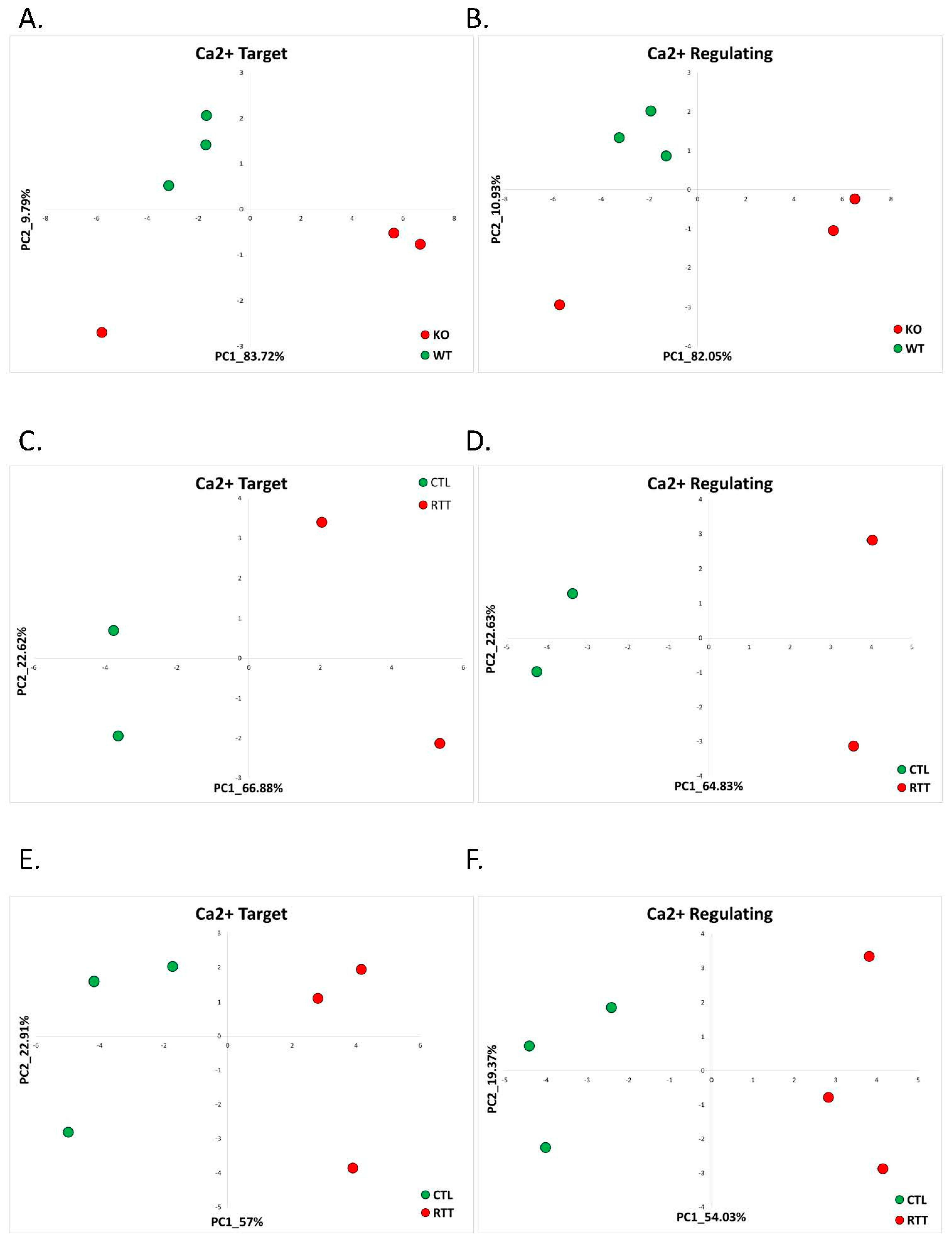
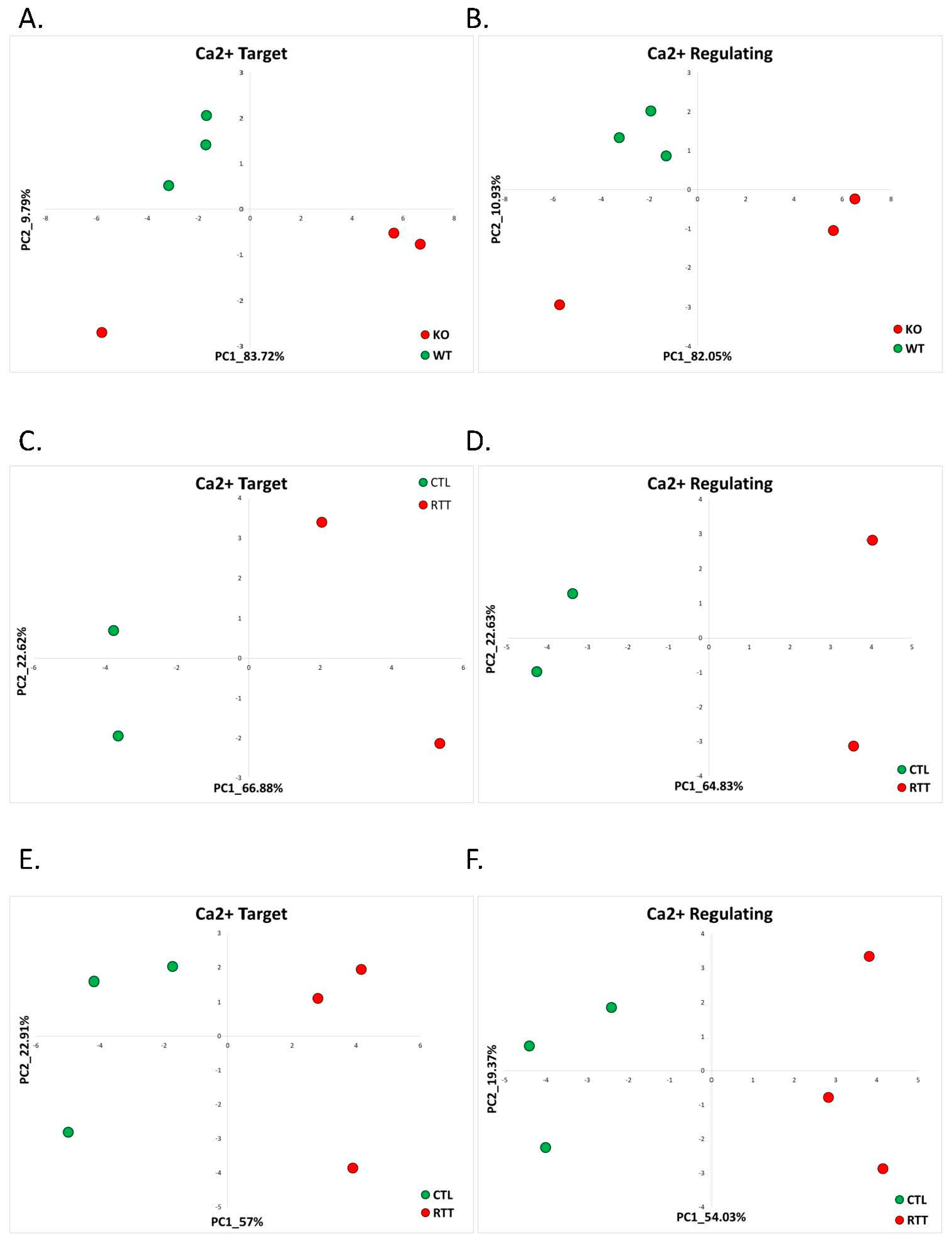
2.1.1. Rett Syndrome (GSE105045, GSE128380)
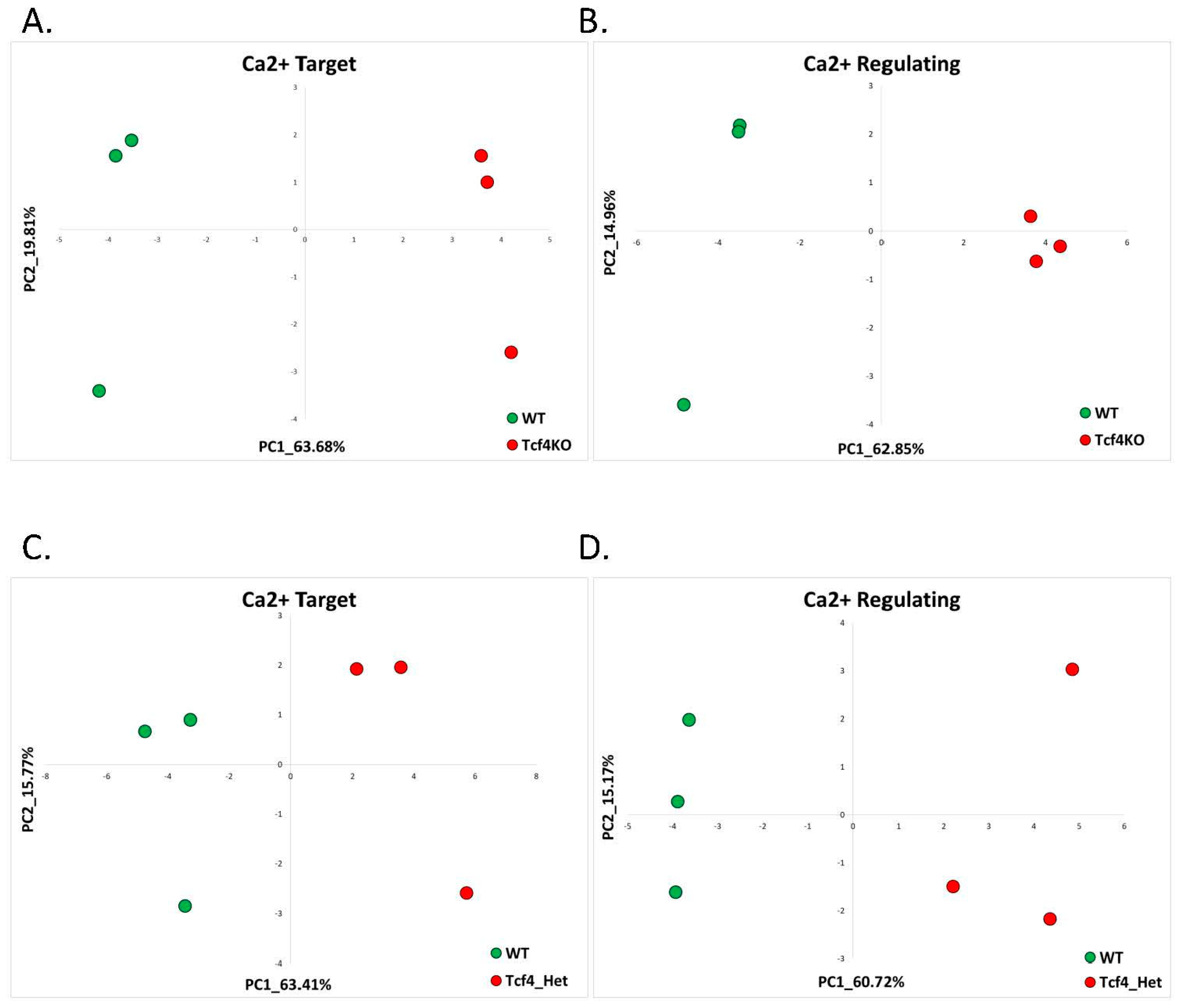
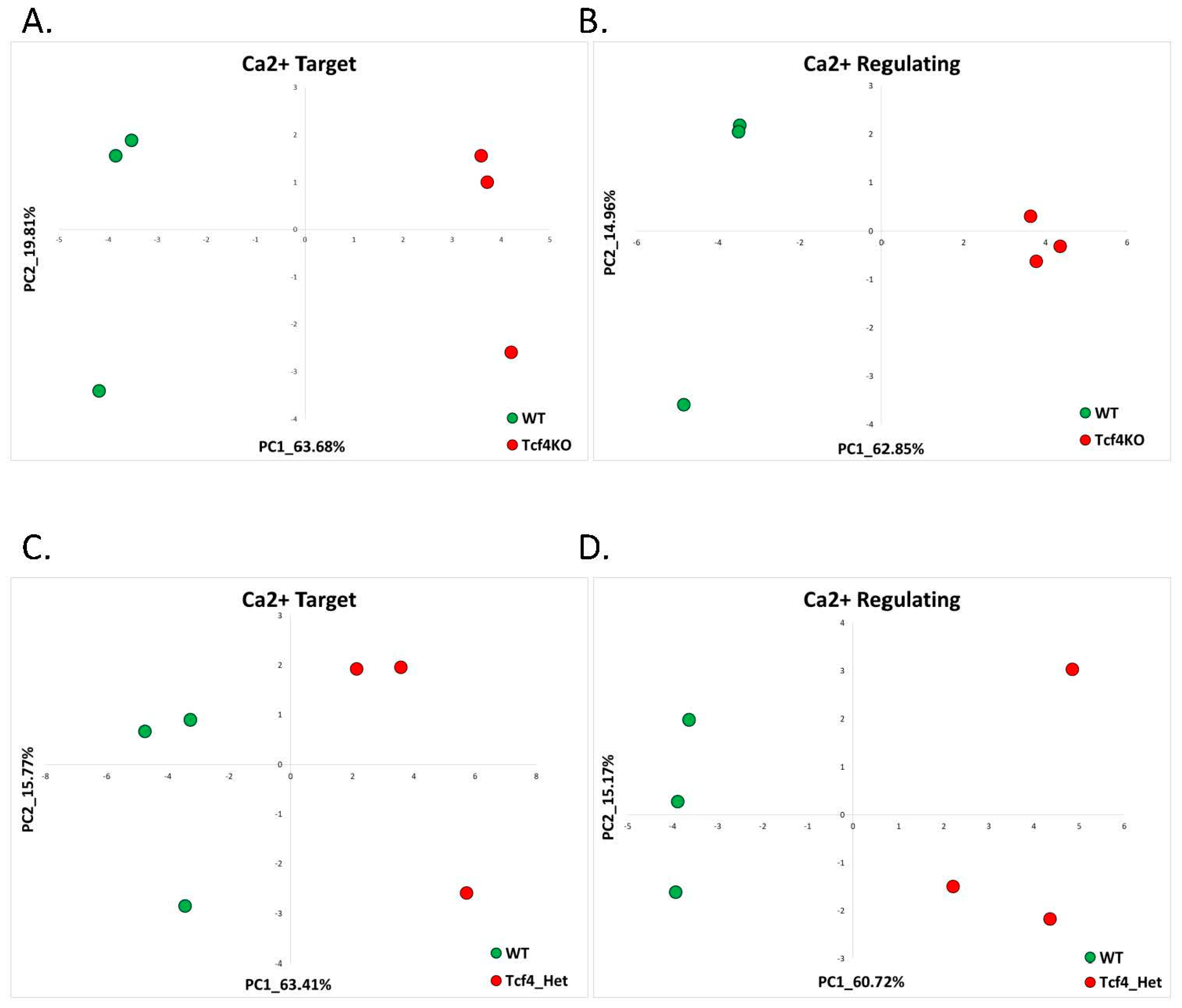
2.1.2. Pitt-Hopkins (GSE79663)
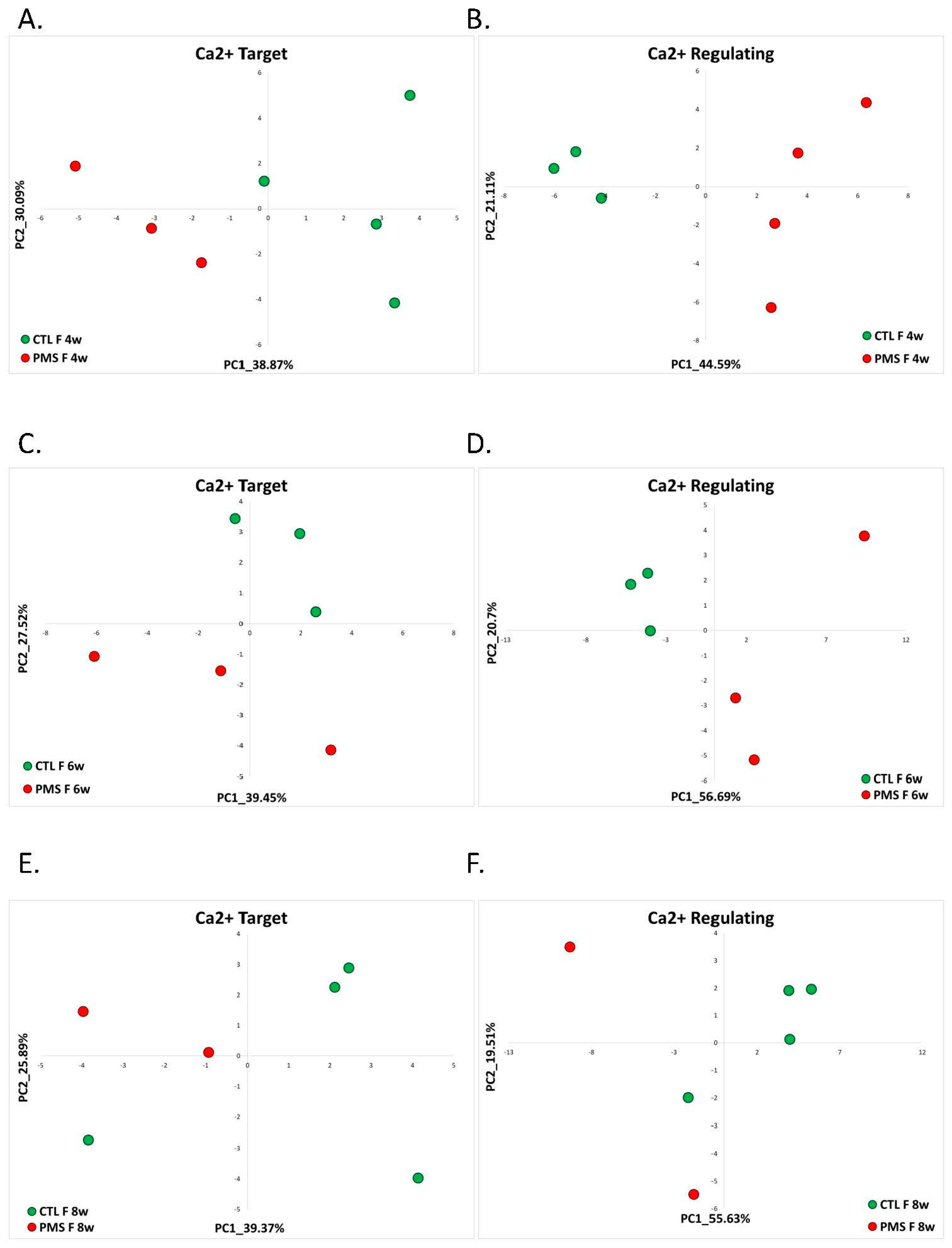
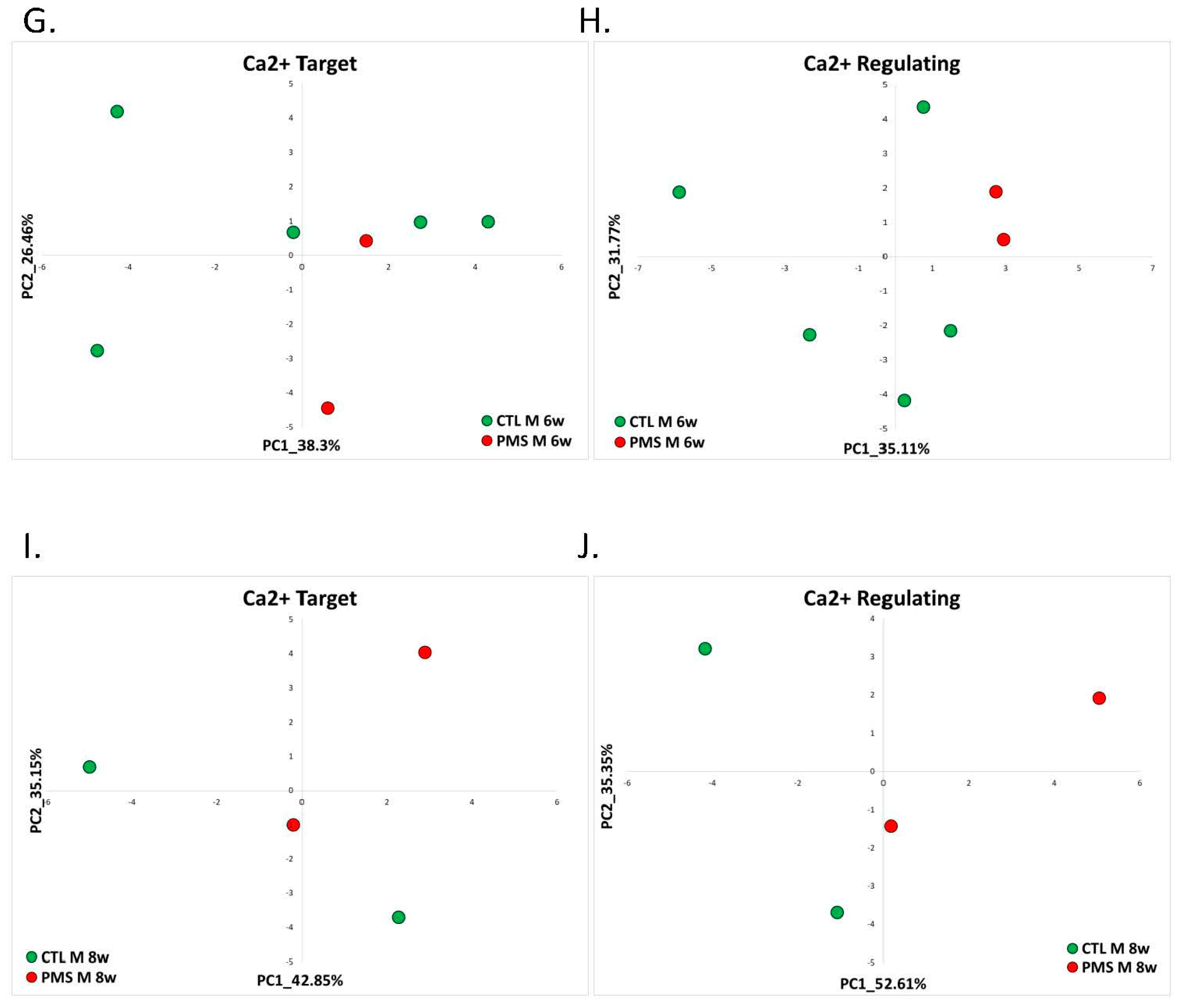
2.1.3. Phelan-McDermid Syndrome (GSE150429)
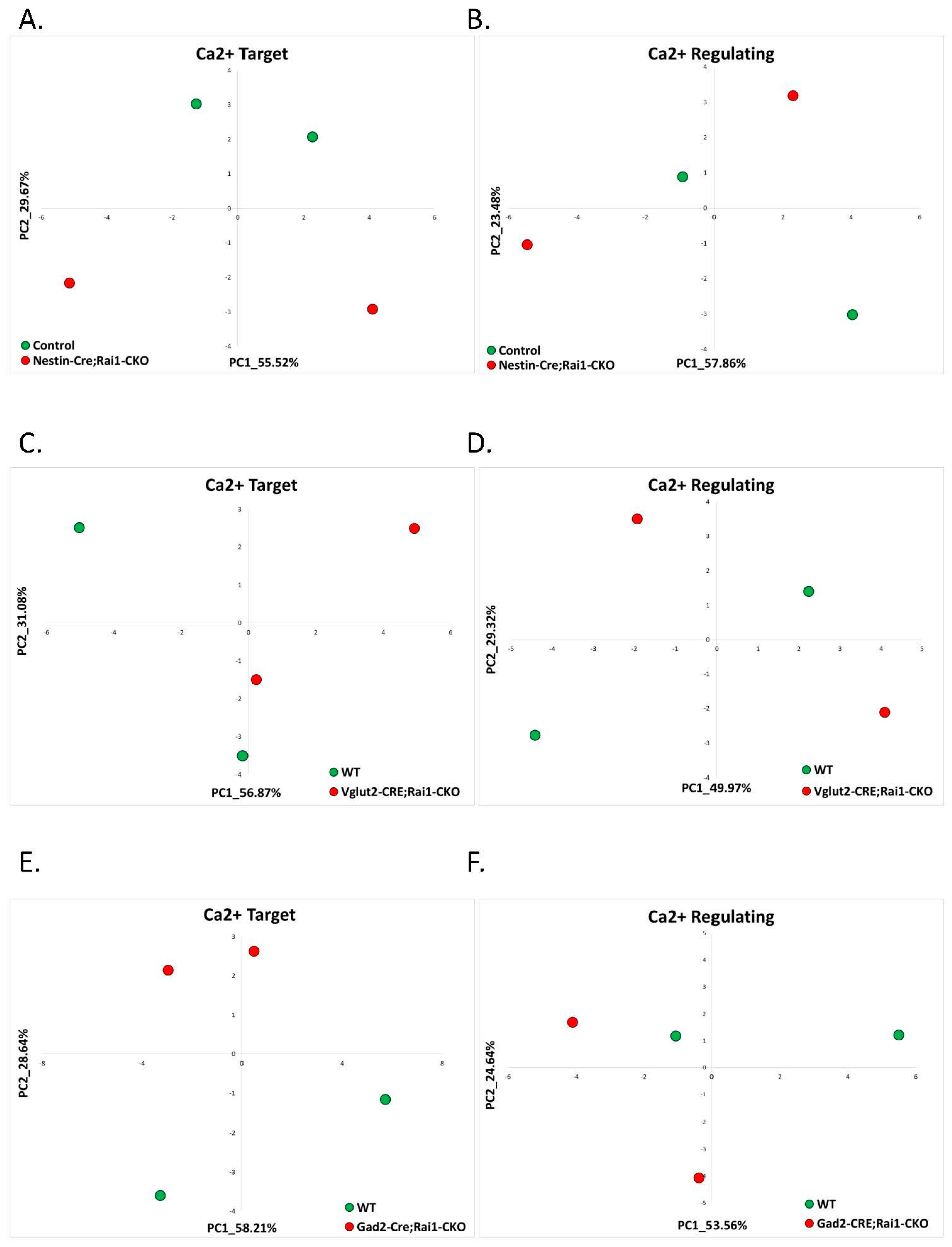
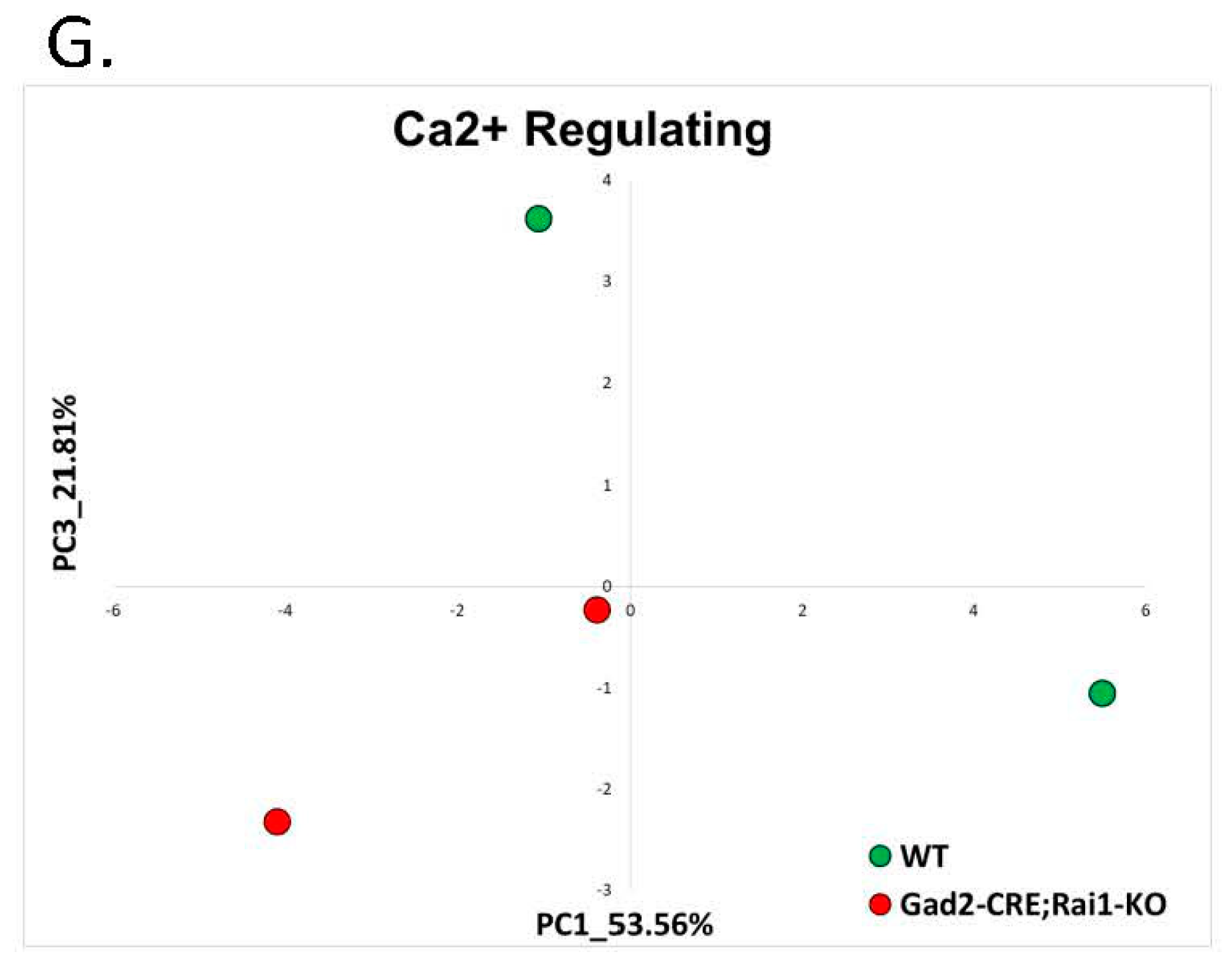
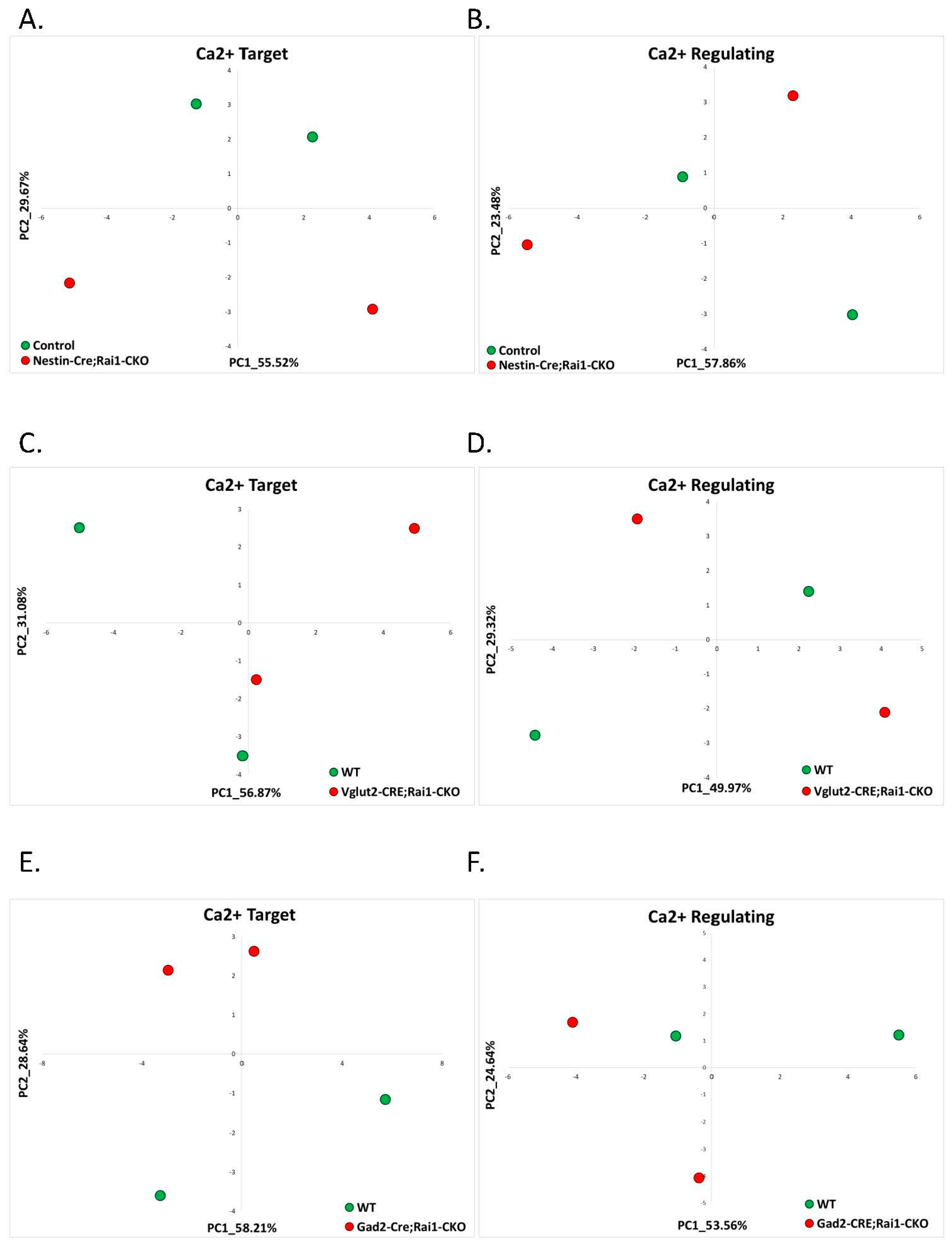
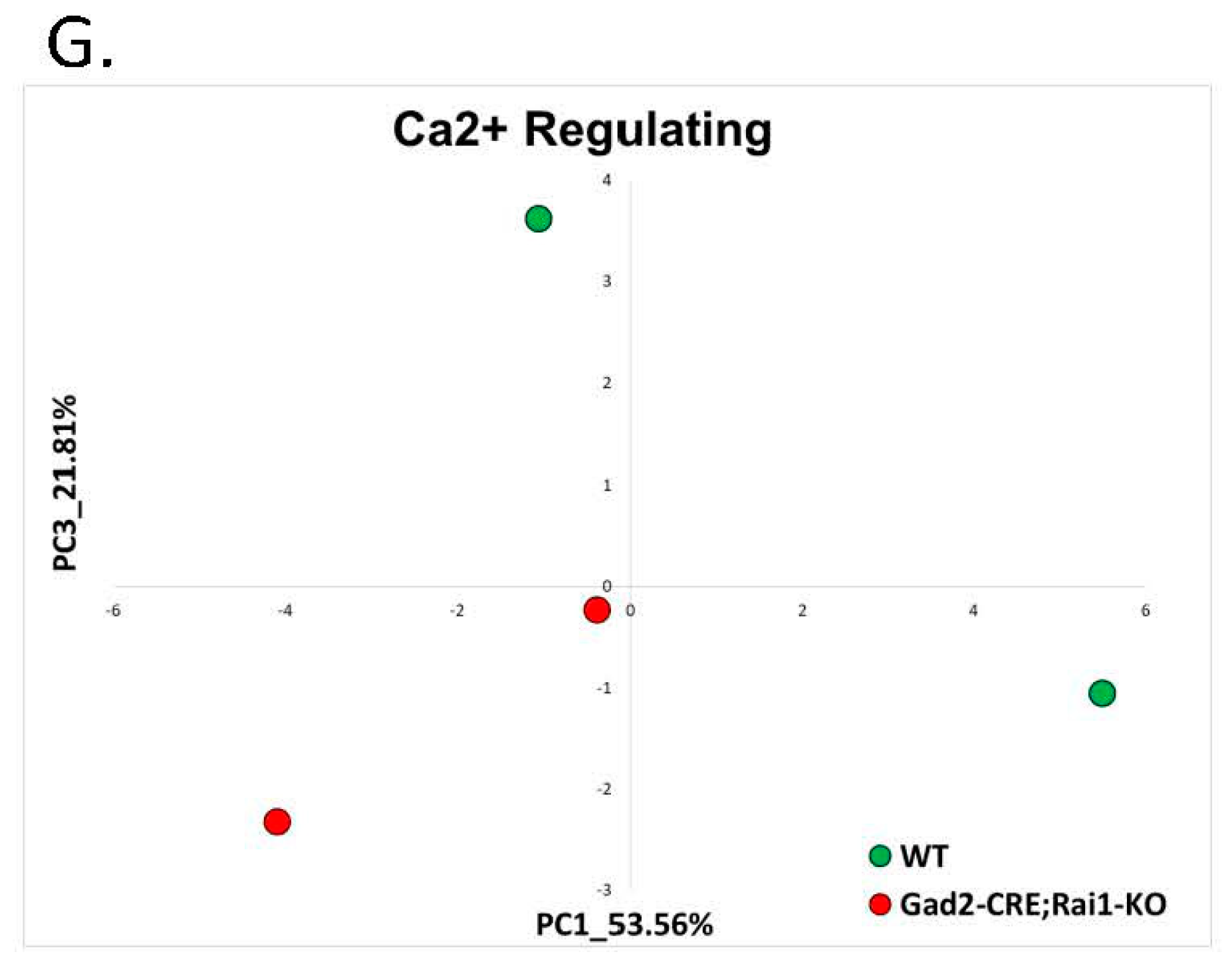
2.1.4. Smith-Magenis Syndrome (GSE81206)
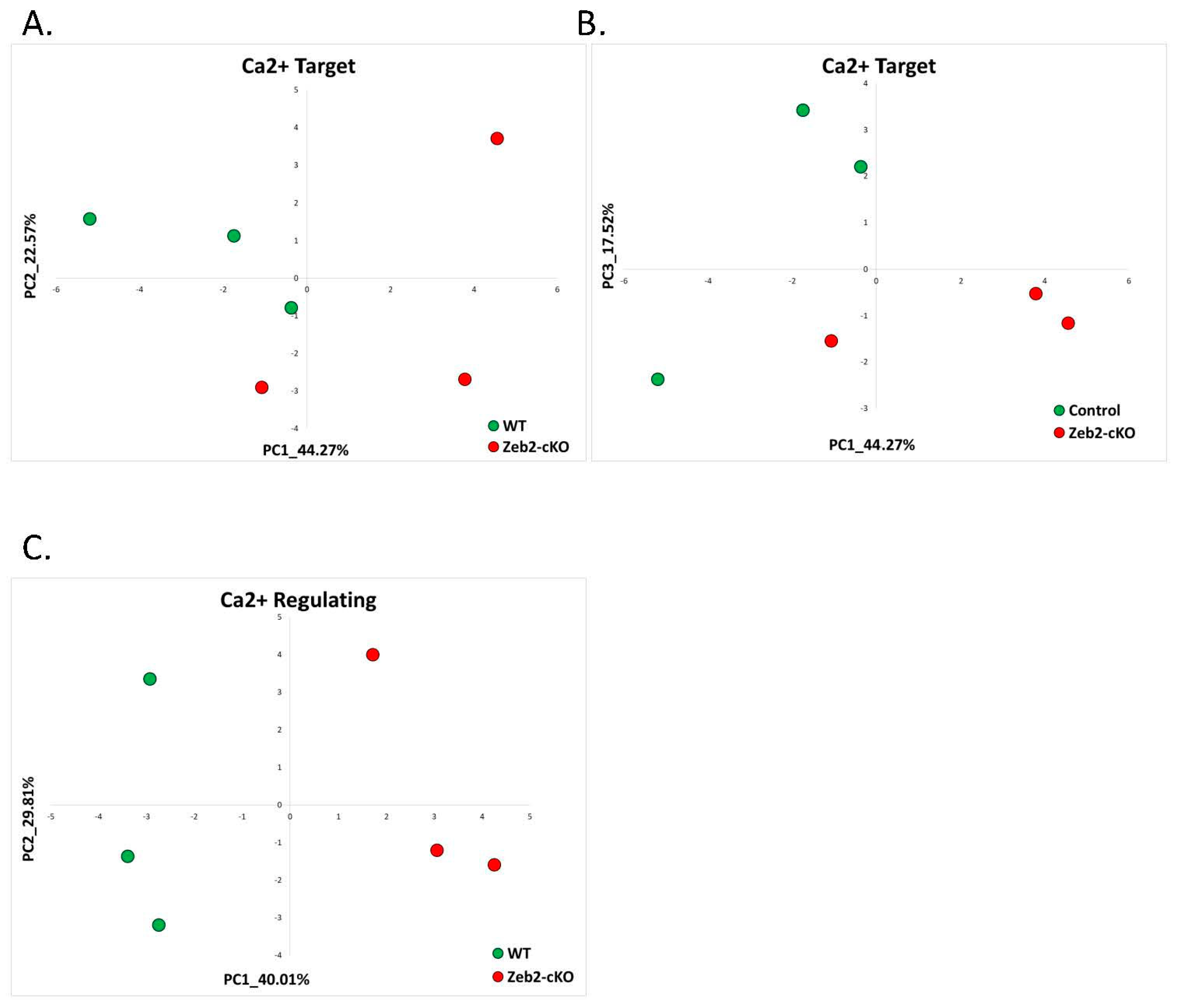
2.1.5. Mowat-Wilson Syndrome (GSE84098)
2.2. Non-AS Like Syndromes
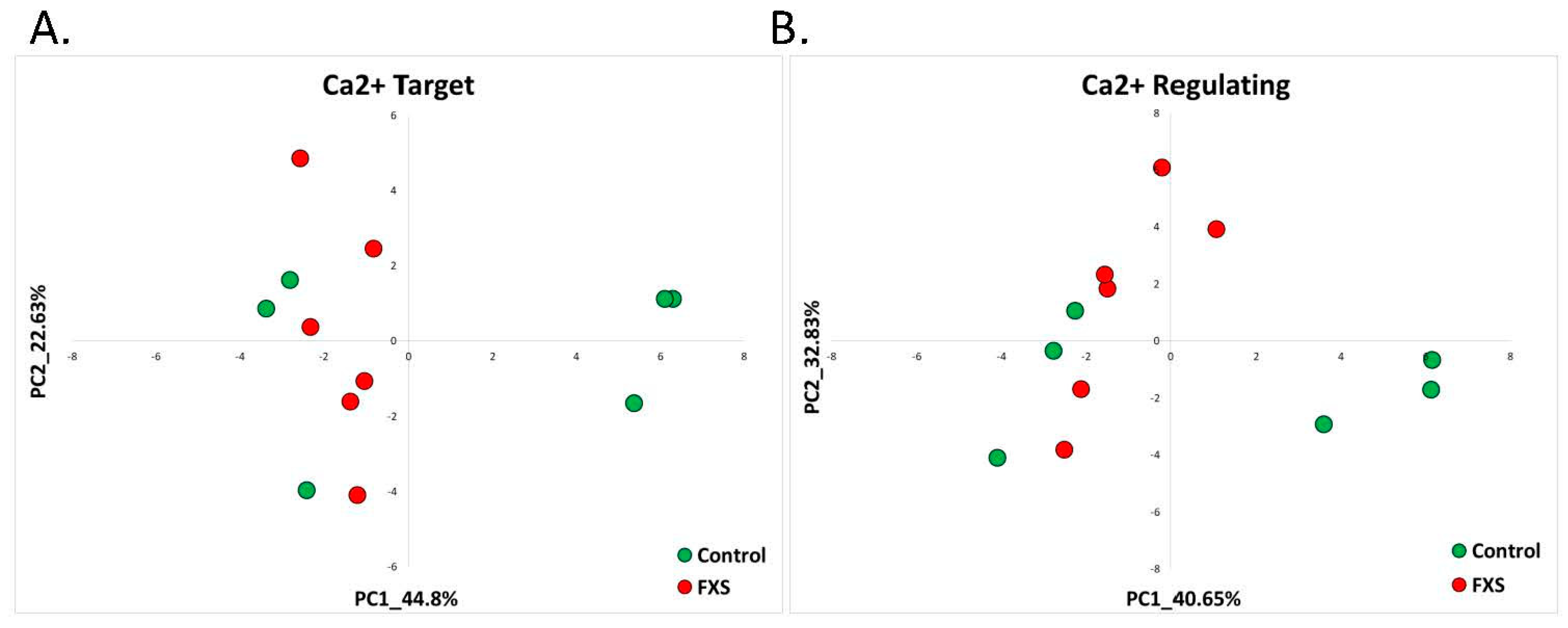
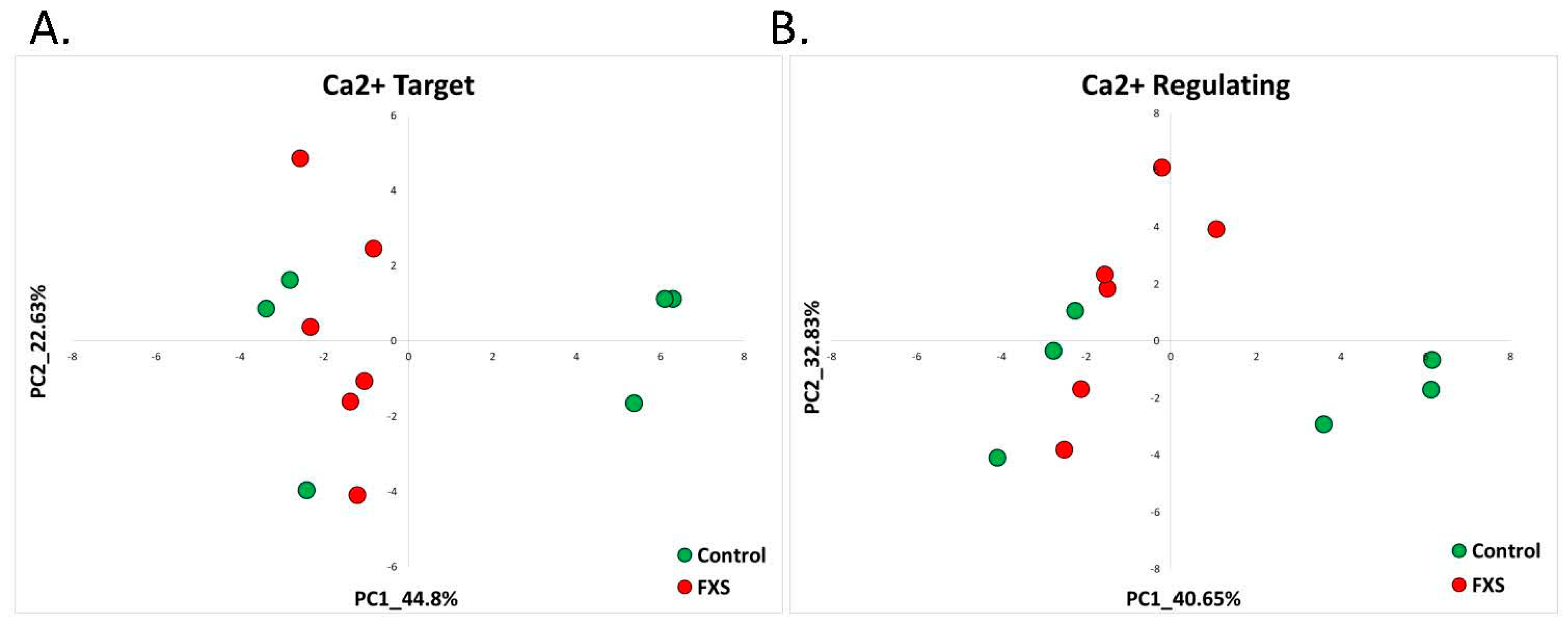
2.2.1. Fragile X Syndrome (GSE117248)
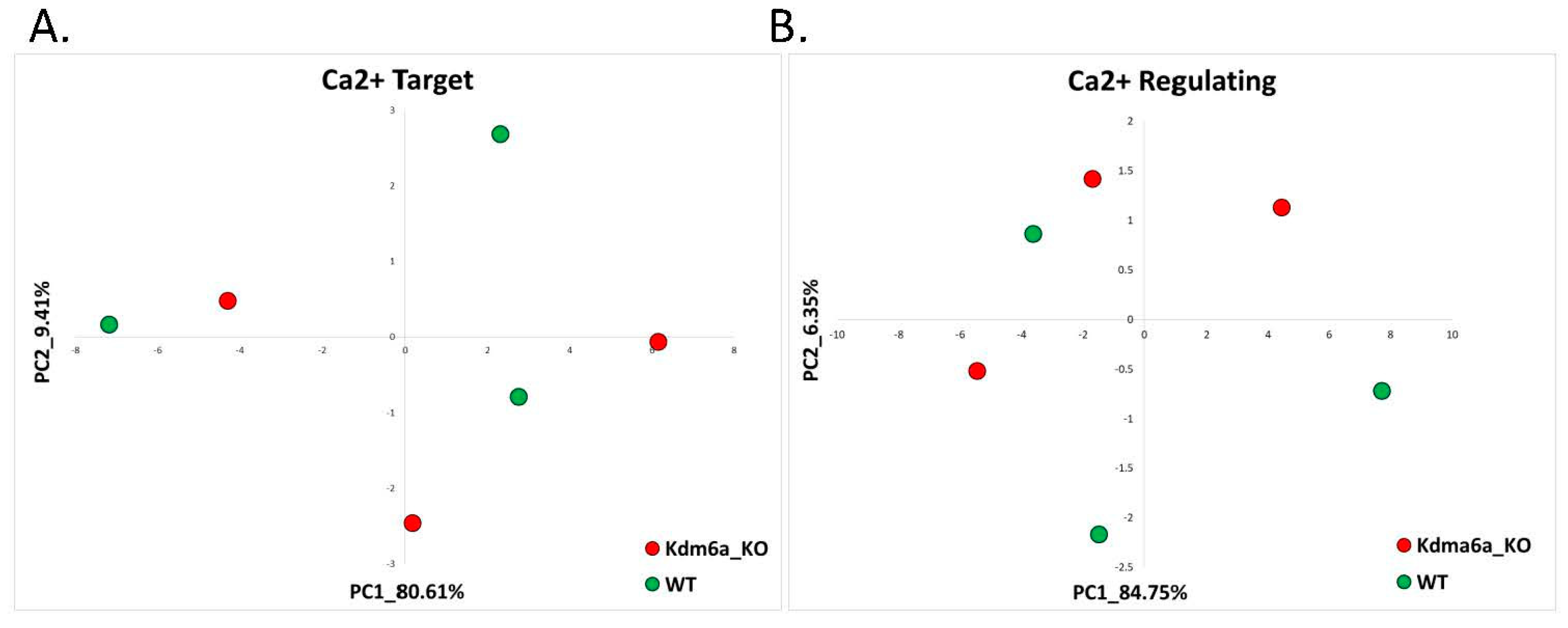
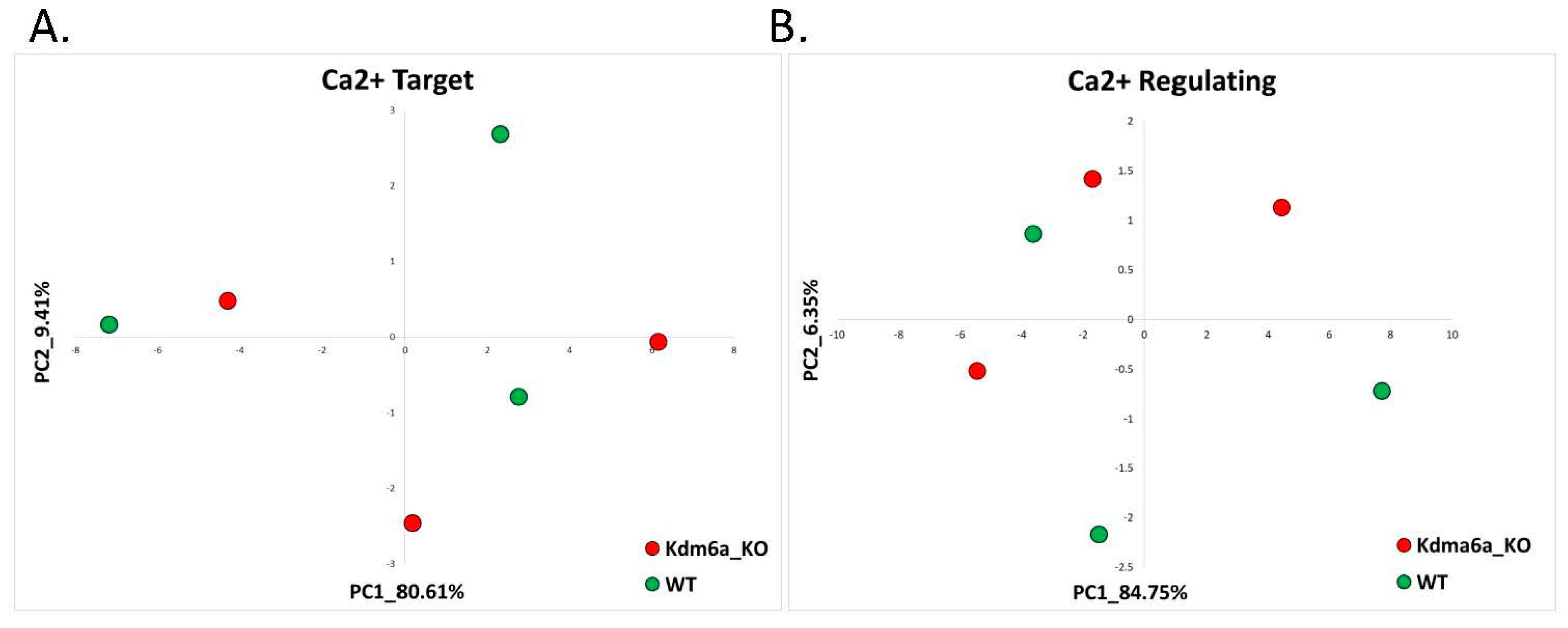
2.2.2. Kabuki Syndrome (GSE81251)
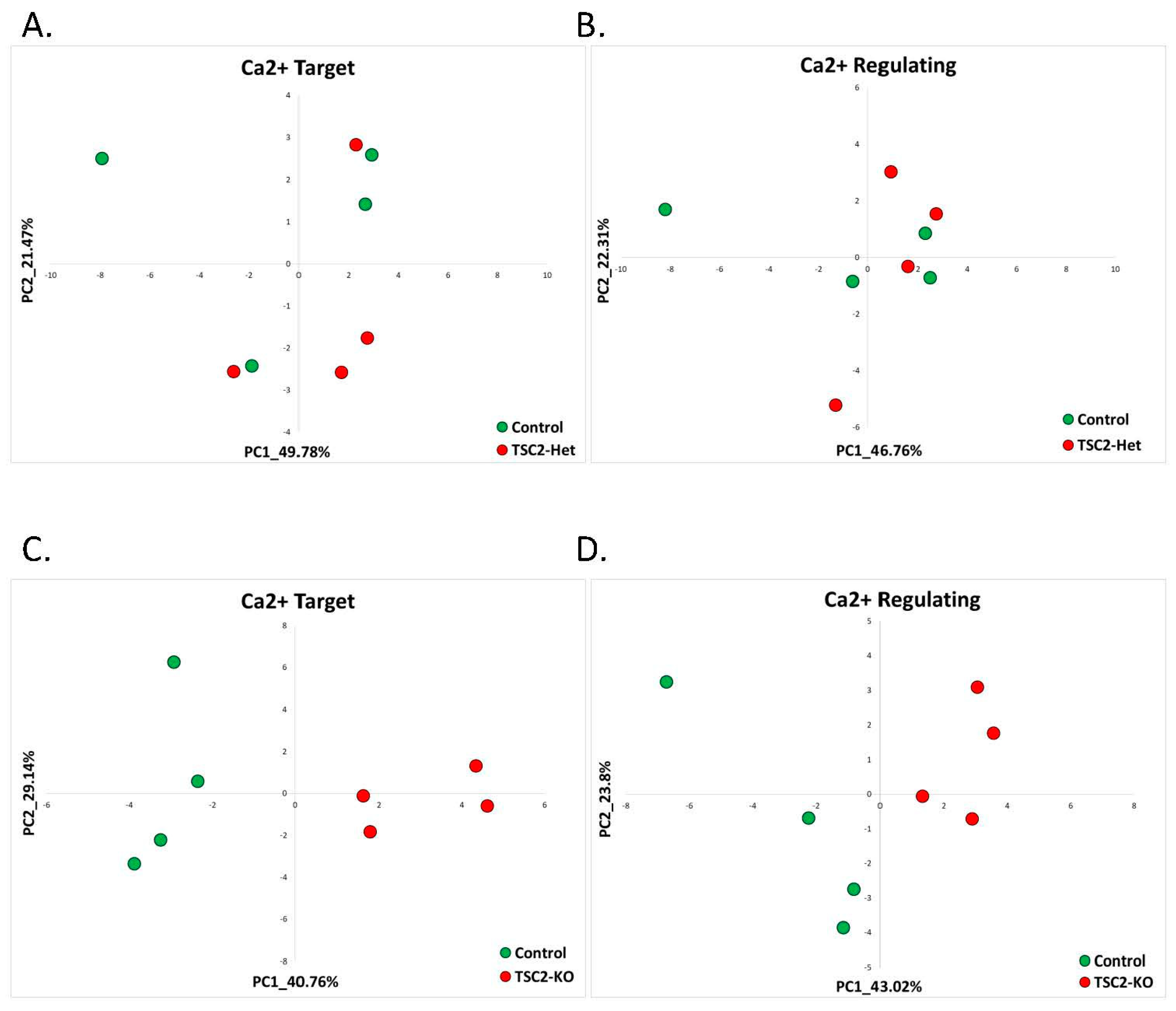
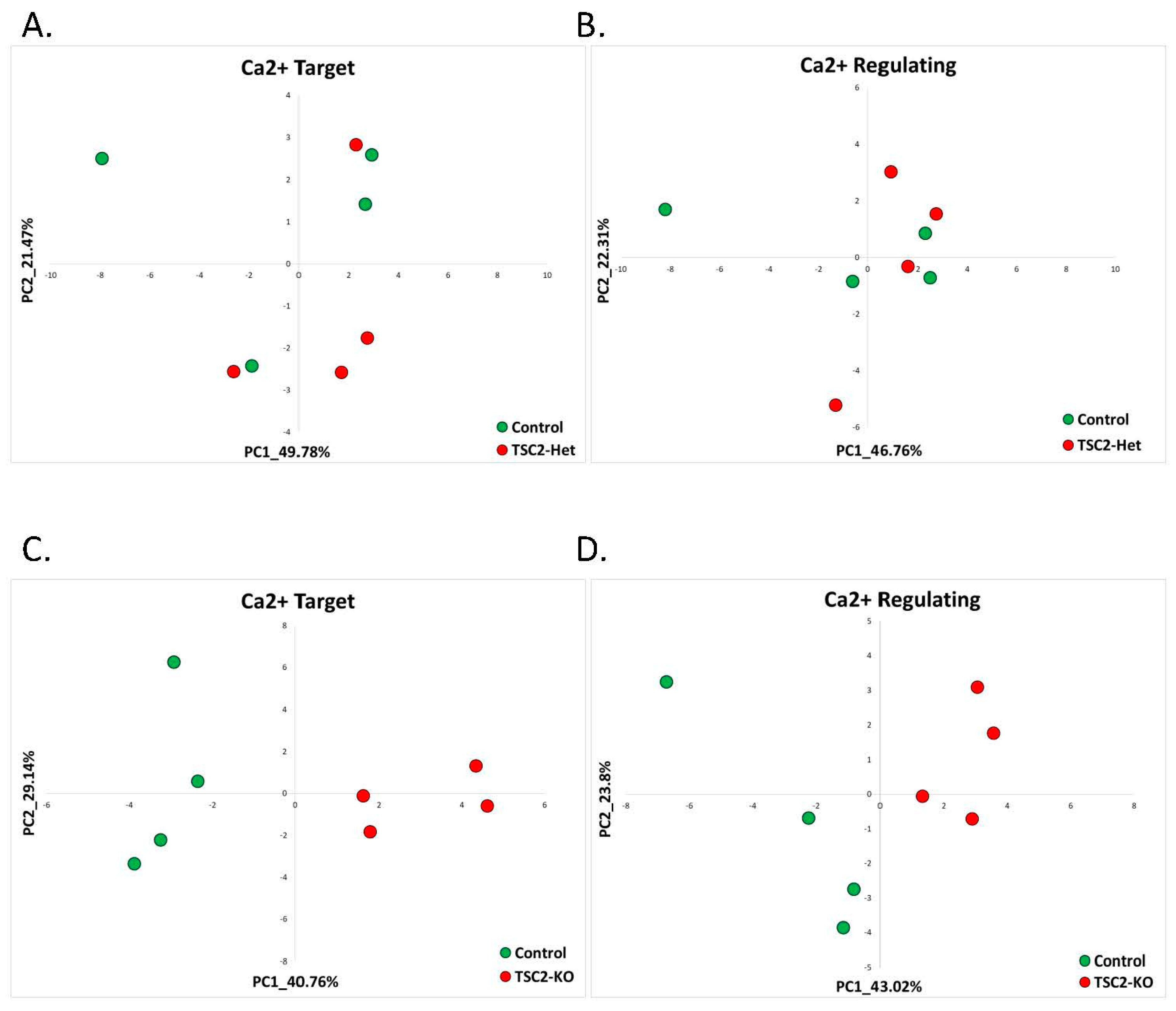
2.2.3. Tuberous Sclerosis (GSE78959)
3. Discussion
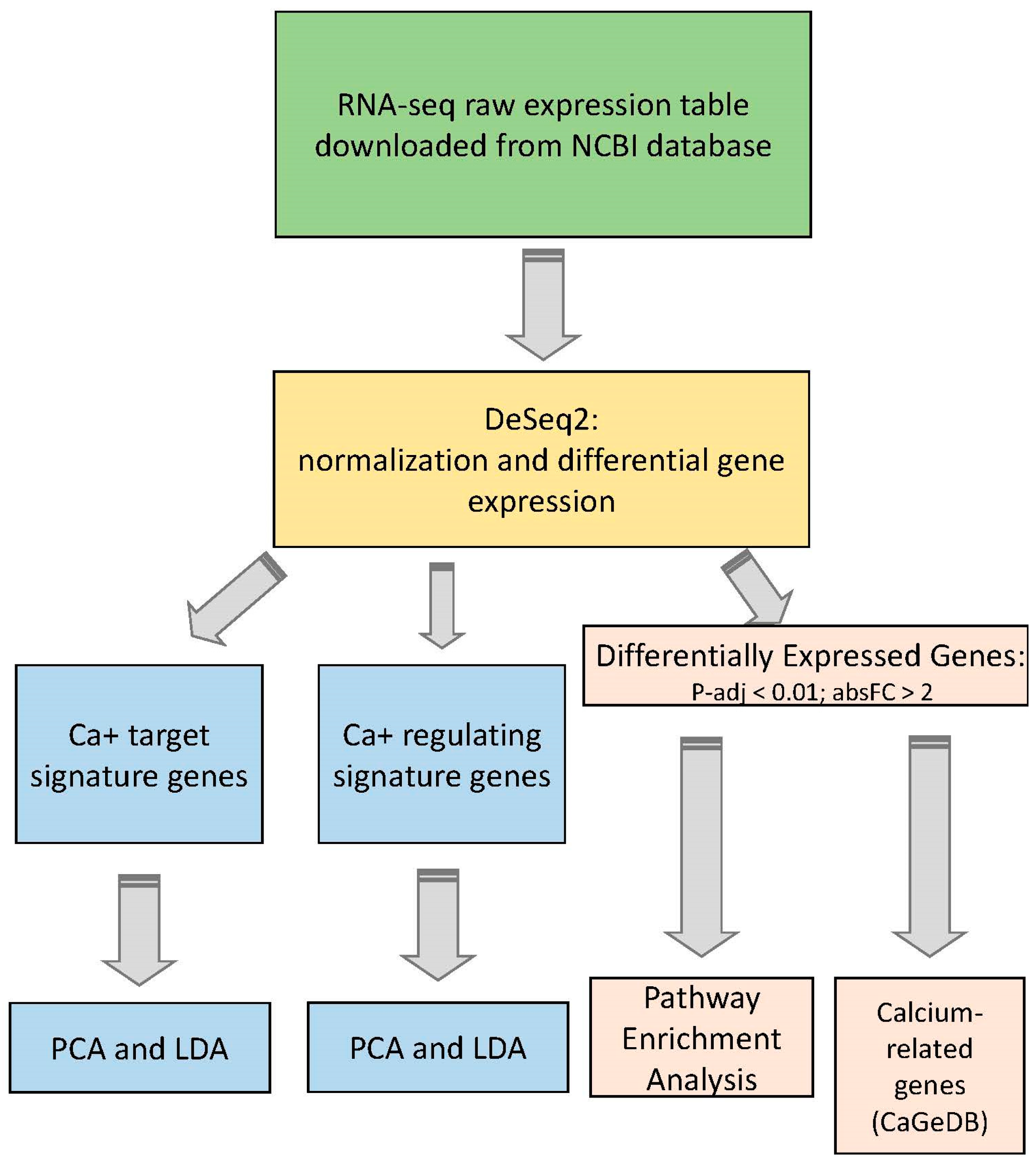
4. Materials and Methods
5. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Angelman, H. ‘Puppet’ children a report on three cases. Dev. Med. Child Neurol. 2008, 7, 681–688. [Google Scholar] [CrossRef]
- Williams, C.A.; Beaudet, A.L.; Clayton-Smith, J.; Knoll, J.H.; Kyllerman, M.; Laan, L.A.; Magenis, R.E.; Moncla, A.; Schinzel, A.A.; Summers, J.A.; et al. Angelman syndrome 2005: Updated consensus for diagnostic criteria. Am. J. Med. Genet. A 2006, 140, 413–418. [Google Scholar] [CrossRef]
- Peters, S.U.; Beaudet, A.L.; Madduri, N.; Bacino, C.A. Autism in Angelman syndrome: Implications for autism research. Clin. Genet. 2004, 66, 530–536. [Google Scholar] [CrossRef]
- Williams, C.A. Neurological aspects of the Angelman syndrome. Brain Dev. 2005, 27, 88–94. [Google Scholar] [CrossRef]
- Summers, J.A.; Allison, D.B.; Lynch, P.S.; Sandier, L. Behaviour problems in Angelman syndrome. J. Intellect. Disabil. Res. 1995, 39, 97–106. [Google Scholar] [CrossRef]
- Dan, B. Angelman syndrome: Current understanding and research prospects. Epilepsia 2009, 50, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Kishino, T.; Lalande, M.; Wagstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, T.; Sutcliffe, J.S.; Fang, P.; Galjaard, R.-J.; Jiang, Y.-H.; Benton, C.S.; Rommens, J.M.; Beaudet, A.L. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 1997, 15, 74–77. [Google Scholar] [CrossRef]
- Knoll, J.H.M.; Nicholls, R.D.; Magenis, R.E.; Graham, J.M.; Lalande, M.; Latt, S.A.; Opitz, J.M.; Reynolds, J.F. Angelman and Prader-Willi syndromes share a common chromosome 15 deletion but differ in parental origin of the deletion. Am. J. Med. Genet. 1989, 32, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Gustin, R.M.; Bichell, T.J.; Bubser, M.; Daily, J.; Filonova, I.; Mrelashvili, D.; Deutch, A.Y.; Colbran, R.J.; Weeber, E.J.; Haas, K.F. Tissue-specific variation of Ube3a protein expression in rodents and in a mouse model of Angelman syndrome. Neurobiol. Dis. 2010, 39, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, M.L.; Burette, A.C.; Weinberg, R.J.; Philpot, B.D. Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron 2012, 74, 793–800. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Stryker, M.P. Genomic imprinting of experience-dependent cortical plasticity by the ubiquitin ligase gene Ube3a. Proc. Natl. Acad. Sci. USA 2010, 107, 5611–5616. [Google Scholar] [CrossRef] [Green Version]
- Rayi, P.R.; Koyavski, L.; Chakraborty, D.; Bagrov, A.; Kaphzan, H. α1-Na/K-ATPase inhibition rescues aberrant dendritic calcium dynamics and memory deficits in the hippocampus of an Angelman syndrome mouse model. Prog. Neurobiol. 2019, 182, 101676. [Google Scholar] [CrossRef] [PubMed]
- Panov, J.; Kaphzan, H. Bioinformatics analyses show dysregulation of calcium-related genes in Angelman syndrome mouse model. Neurobiol. Dis. 2021, 148, 105180. [Google Scholar] [CrossRef] [PubMed]
- Hörtenhuber, M.; Toledo, E.; Smedler, E.; Arenas, E.; Malmersjö, S.; Louhivuori, L.; Uhlén, P. Mapping genes for calcium signaling and their associated human genetic disorders. Bioinformatics 2017, 33, 2547–2554. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.H.; Bird, L.M.; Thibert, R.L.; Williams, C.A. If not Angelman, what is it? A review of Angelman-like syndromes. Am. J. Med. Genet. Part A 2014, 164, 975–992. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jollife, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef] [PubMed]
- Jolliffe, I.T. Principal component analysis, second edition. Encycl. Stat. Behav. Sci. 2002, 30, 487. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; A Lempicki, R. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, R.E.; Van Den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl- CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Tillotson, R.; Bird, A. The molecular basis of MeCP2 function in the brain. J. Mol. Biol. 2020, 432, 1602–1623. [Google Scholar] [CrossRef] [PubMed]
- Raman, A.T.; Pohodich, A.E.; Wan, Y.-W.; Yalamanchili, H.K.; Lowry, W.E.; Zoghbi, H.Y.; Liu, Z. Apparent bias toward long gene misregulation in MeCP2 syndromes disappears after controlling for baseline variations. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Aldinger, K.A.; Timms, A.E.; Macdonald, J.W.; McNamara, H.K.; Herstein, J.S.; Bammler, T.K.; Evgrafov, O.V.; Knowles, J.A.; Levitt, P. Transcriptome data of temporal and cingulate cortex in the Rett syndrome brain. Sci. Data 2020, 7, 192. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [Green Version]
- Scaramuzza, L.; De Rocco, G.; Desiato, G.; Gigli, C.C.; Chiacchiaretta, M.; Mirabella, F.; Pozzi, D.; De Simone, M.; Conforti, P.; Pagani, M.; et al. The enhancement of activity rescues the establishment of Mecp2 null neuronal phenotypes. EMBO Mol. Med. 2021, 13, e12433. [Google Scholar] [CrossRef]
- Kennedy, A.J.; Rahn, E.J.; Paulukaitis, B.S.; Savell, K.; Kordasiewicz, H.B.; Wang, J.; Lewis, J.W.; Posey, J.; Strange, S.K.; Guzman-Karlsson, M.C.; et al. Tcf4 regulates synaptic plasticity, DNA methylation, and memory function. Cell Rep. 2016, 16, 2666–2685. [Google Scholar] [CrossRef] [Green Version]
- Zweier, C.; Peippo, M.M.; Hoyer, J.; Sousa, S.; Bottani, A.; Clayton-Smith, J.; Reardon, W.; Saraiva, J.M.; Cabral, A.; Göhring, I.; et al. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome). Am. J. Hum. Genet. 2007, 80, 994–1001. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhu, Y.; Morozov, Y.M.; Chen, X.; Page, S.C.; Rannals, M.D.; Maher, B.J.; Rakic, P. Disruption of TCF4 regulatory networks leads to abnormal cortical development and mental disabilities. Mol. Psychiatry 2019, 24, 1235–1246. [Google Scholar] [CrossRef]
- Phelan, K.; McDermid, H.E. The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol. Syndromol. 2012, 2, 186–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhar, S.; del Gaudio, D.; German, J.; Peters, S.; Ou, Z.; Bader, P.; Berg, J.; Blazo, M.; Brown, C.; Graham, B.; et al. 22q13.3 Deletion syndrome: Clinical and molecular analysis using array CGH. Am. J. Med. Genet. Part A 2010, 152, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarasua, S.M.; Dwivedi, A.; Boccuto, L.; Rollins, J.D.; Chen, C.-F.; Rogers, R.C.; Phelan, K.; Dupont, B.R.; Collins, J.S. Association between deletion size and important phenotypes expands the genomic region of interest in Phelan-McDermid syndrome (22q13 deletion syndrome). J. Med. Genet. 2011, 48, 761–766. [Google Scholar] [CrossRef]
- Naisbitt, S.; Kim, E.; Tu, J.C.; Xiao, B.; Sala, C.; Valtschanoff, J.; Weinberg, R.J.; Worley, P.F.; Sheng, M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 1999, 23, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, C.; Chen, Q.; Van Der Goes, M.-S.; Hawrot, J.; Yao, A.Y.; Gao, X.; Lu, C.; Zang, Y.; Zhang, Q.; et al. Striatopallidal dysfunction underlies repetitive behavior in Shank3-deficient model of autism. J. Clin. Investig. 2017, 127, 1978–1990. [Google Scholar] [CrossRef] [Green Version]
- Perfitt, T.L.; Wang, X.; Dickerson, M.T.; Stephenson, J.R.; Nakagawa, T.; Jacobson, D.A.; Colbran, R.J. Neuronal L-type calcium channel signaling to the nucleus requires a novel CaMKIIα-SHANK3 interaction. J. Neurosci. 2020, 40, 2000–2014. [Google Scholar] [CrossRef]
- Breen, M.S.; Browne, A.; Hoffman, G.E.; Stathopoulos, S.; Brennand, K.; Buxbaum, J.D.; Drapeau, E. Transcriptional signatures of participant-derived neural progenitor cells and neurons implicate altered Wnt signaling in Phelan-McDermid syndrome and autism. Mol. Autism 2020, 11, 53. [Google Scholar] [CrossRef]
- Koyavski, L.; Panov, J.; Simchi, L.; Rayi, P.R.; Sharvit, L.; Feuermann, Y.; Kaphzan, H. Sex-dependent sensory phenotypes and related transcriptomic expression profiles are differentially affected by Angelman syndrome. Mol. Neurobiol. 2019, 15, 5998–6016. [Google Scholar] [CrossRef]
- Elsea, S.H.; Girirajan, S.S. Smith-Magenis syndrome. Eur. J. Hum. Genet. 2008, 16, 412–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buiting, K. Prader-Willi syndrome and Angelman syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2010, 154, 365–376. [Google Scholar] [CrossRef]
- Smith Magenis Syndrome—NORD (National Organization for Rare Disorders) n.d. Available online: https://rarediseases.org/rare-diseases/smith-magenis-syndrome/ (accessed on 5 June 2021).
- Falco, M.; Amabile, S.; Acquaviva, F. The application of clinical genetics dovepress RAI1 gene mutations: Mechanisms of Smith-Magenis syndrome. Appl. Clin. Genet. 2017, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Garay, P.M.; Chen, A.; Tsukahara, T.; Díaz, J.C.R.; Kohen, R.; Althaus, J.C.; Wallner, M.A.; Giger, R.J.; Jones, K.S.; Sutton, M.A.; et al. RAI1 Regulates activity-dependent nascent transcription and synaptic scaling. Cell Rep. 2020, 32, 108002. [Google Scholar] [CrossRef]
- Bi, W.; Yan, J.; Shi, X.; Yuva-Paylor, L.A.; Antalffy, B.A.; Goldman, A.; Yoo, J.W.; Noebels, J.; Armstrong, D.L.; Paylor, R.; et al. Rai1 deficiency in mice causes learning impairment and motor dysfunction, whereas Rai1 heterozygous mice display minimal behavioral phenotypes. Hum. Mol. Genet. 2007, 16, 1802–1813. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-H.; Guenthner, C.J.; Xu, J.; Nguyen, T.; Schwarz, L.A.; Wilkinson, A.W.; Gozani, O.; Chang, H.Y.; Shamloo, M.; Luo, L. Molecular and neural functions of Rai1, the causal gene for Smith-Magenis syndrome. Neuron 2016, 92, 392–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanovski, I.; Djuric, O.; Caraffi, S.G.; Santodirocco, D.; Pollazzon, M.; Rosato, S.; Cordelli, D.M.; Abdalla, E.; Accorsi, P.; Adam, M.P.; et al. Phenotype and genotype of 87 patients with Mowat–Wilson syndrome and recommendations for care. Genet. Med. 2018, 20, 965–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkhoff, J.C.; Huylebroeck, D.; Conidi, A. ZEB2, the Mowat-Wilson syndrome transcription factor: Confirmations, novel functions, and continuing surprises. Genes 2021, 12, 1037. [Google Scholar] [CrossRef]
- Garavelli, L.; Ivanovski, I.; Caraffi, S.G.; Santodirocco, D.; Pollazzon, M.; Cordelli, D.M.; Abdalla, E.; Accorsi, P.; Adam, M.P.; Baldo, C.; et al. Neuroimaging findings in Mowat-Wilson syndrome: A study of 54 patients. Genet. Med. 2017, 19, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladka, M.; De Leeuw, A.; Kohela, A.; Molenaar, B.; Versteeg, D.; Kooijman, L.; Van Geldorp, M.; Van Rooij, E. ZEB2 regulates a transcriptional network of calcium-handling genes in the injured heart. Eur. Heart J. 2020, 41, ehaa946-3633. [Google Scholar] [CrossRef]
- He, L.; Yu, K.; Lu, F.; Wang, J.; Wu, L.N.; Zhao, C.; Li, Q.; Zhou, X.; Liu, H.; Mu, D.; et al. Transcriptional regulator ZEB2 is essential for bergmann glia development. J. Neurosci. 2018, 38, 1575–1587. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Hagerman, P.J. The fragile X premutation: Into the phenotypic fold. Curr. Opin. Genet. Dev. 2002, 12, 278–283. [Google Scholar] [CrossRef]
- Ciaccio, C.; Fontana, L.; Milani, D.; Tabano, S.; Miozzo, M.; Esposito, S. Fragile X syndrome: A review of clinical and molecular diagnoses. Ital. J. Pediatr. 2017, 43, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hagerman, P.J.; Stafstrom, C.E. Origins of epilepsy in fragile X syndrome. Epilepsy Curr. 2009, 9, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Salcedo-Arellano, M.J.; Dufour, B.; McLennan, Y.; Martinez-Cerdeno, V.; Hagerman, R. Fragile X syndrome and associated disorders: Clinical aspects and pathology. Neurobiol. Dis. 2020, 136, 104740. [Google Scholar] [CrossRef] [PubMed]
- Richter, J.D.; Zhao, X. The molecular biology of FMRP: New insights into fragile X syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Razak, K.A.; Dominick, K.C.; Erickson, C.A. Developmental studies in fragile X syndrome. J. Neurodev. Disord. 2020, 12. [Google Scholar] [CrossRef]
- Utami, K.H.; Skotte, N.H.; Colaço, A.R.; Yusof, N.A.B.M.; Sim, B.; Yeo, X.Y.; Bae, H.-G.; Garcia-Miralles, M.; Radulescu, C.I.; Chen, Q.; et al. Integrative analysis identifies key molecular signatures underlying neurodevelopmental deficits in fragile X syndrome. Biol. Psychiatry 2020, 88, 500–511. [Google Scholar] [CrossRef]
- Boniel, S.; Szymańska, K.; Śmigiel, R.; Szczałuba, K. Kabuki syndrome—Clinical review with molecular aspects. Genes 2021, 12, 468. [Google Scholar] [CrossRef]
- Zhang, L.; Pilarowski, G.; Pich, E.M.; Nakatani, A.; Dunlop, J.; Baba, R.; Matsuda, S.; Daini, M.; Hattori, Y.; Matsumoto, S.; et al. Inhibition of KDM1A activity restores adult neurogenesis and improves hippocampal memory in a mouse model of Kabuki syndrome. Mol. Ther. Methods Clin. Dev. 2021, 20, 779–791. [Google Scholar] [CrossRef]
- Van Laarhoven, P.M.; Neitzel, L.R.; Quintana, A.M.; Geiger, E.A.; Zackai, E.H.; Clouthier, D.E.; Artinger, K.B.; Ming, J.E.; Shaikh, T.H. Kabuki syndrome genes KMT2D and KDM6A: Functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum. Mol. Genet. 2015, 24, 4443–4453. [Google Scholar] [CrossRef] [Green Version]
- Feliciano, D.M. The neurodevelopmental pathogenesis of tuberous sclerosis complex (TSC). Front. Neuroanat. 2020, 14, 39. [Google Scholar] [CrossRef]
- Martin, K.; Zhou, W.; Bowman, M.; Shih, J.; Au, K.S.; Dittenhafer-Reed, K.; Sisson, K.A.; Koeman, J.; Weisenberger, D.J.; Cottingham, S.L.; et al. The genomic landscape of tuberous sclerosis complex. Nat. Commun. 2017, 8, 15816. [Google Scholar] [CrossRef]
- Switon, K.; Kotulska, K.; Janusz-Kaminska, A.; Zmorzynska, J.; Jaworski, J. Tuberous sclerosis complex: From molecular biology to novel therapeutic approaches. IUBMB Life 2016, 68, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Grabole, N.; Zhang, J.D.; Aigner, S.; Ruderisch, N.; Costa, V.; Weber, F.C.; Theron, M.; Berntenis, N.; Spleiss, O.; Ebeling, M.; et al. Genomic analysis of the molecular neuropathology of tuberous sclerosis using a human stem cell model. Genome Med. 2016, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Orlova, K.A.; Crino, P.B. The tuberous sclerosis complex. Ann. N. Y. Acad. Sci. 2010, 1184, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorder | Model | GEO | Number of Samples in Each Dataset |
|---|---|---|---|
| Rett Syndrome | RNA-seq of cerebellum of Mecp2 knockdown male mice and WT controls | GSE105045 | RTT = 3 Control = 3 |
| Rett Syndrome | RNA-seq of postmortem brain tissue samples of female patients clinically diagnosed with Rett syndrome and age-matched female donors | GSE128380 | Cingulate cortex: RTT = 2 and control = 2. Temporal cortex: RTT = 3 and control = 3 |
| Pitt-Hopkins | RNA sequencing dataset of dorsal telencephalons of Tcf4-knockout, Tcf4-heterozygous, and WT mice at P0 (immediately after birth) | GSE79663 | Tcf4-KO = 3; Tcf4-Het = 3; control = 3 |
| Phelan-McDermid Syndrome | RNA-seq data generated from human-induced pluripotent stem cell-based model (hiPSC-neurons) of PMS by reprogramming peripheral blood samples from individuals with PMS (n = 7) and their unaffected siblings (n = 6) | GSE150429 | Female 4w: PMS = 3 and control = 4. Female 6w: PMS = 3 and control = 3. Female 8w: PMS = 2 and control = 4. Male 6w: PMS = 2 and control = 5. Male 8w: PMS = 2 and control = 2. |
| Smith-Magenis Syndrome | RNA-seq of Rai1 conditional knockouts and wild-type mice | GSE81206 | Cortex: SMS = 2 and control = 2 Hypothalamus: SMS = 2 and control = 2 Striatum: SMS = 2 and control = 2 |
| Mowat-Wilson Syndrome | RNA-seq data generated from P0 cerebellum of Zeb2-cKO mice and of their WT controls | GSE84098 | Zeb-cKO = 3 Control = 3 |
| Fragile X Syndrome | RNA-seq dataset generated from human neural progenitor cells-derived hiPSCs with a midbrain-patterning differentiation protocol using CRISPR/Cas9 to introduce indels in exon 3 of FMR1 | GSE117248 | FXS = 6 Control = 6 |
| Kabuki Syndrome | RNA-seq data from brains of a neuron-specific Kdm6a-deficient mouse model | GSE81251 | Kdma6a-KO = 6 Control = 6 |
| Tuberous Sclerosis | RNA-seq generated from human neural stem cells derived from embryonic stem cells that have a deletion (either TSC2+/− (TSC2-Het) or TSC2−/− (TSC2-KO)) of the TSC2 gene | GSE78959 | TSC2-KO = 4 TSC2-Het = 4 Control = 4 |
| Disorder | Model | Calcium Target | Calcium Regulating | ||||
|---|---|---|---|---|---|---|---|
| Down Regulated (% from Downregulated) | Up Regulated (% from Upregulated) | Down Regulated (% from Downregulated) | Up Regulated (% from Upregulated) | All Calcium-Related DEG | |||
| 1 | 1.1 Rett Syndrome | Mecp2-KO model (GSE105045) | 3 (9.37%) | 0 (0%) | 1 (3.18%) | 0 (0%) | 12.5% |
| 2 | Postmortem brain cingulate cortex (GSE128380) | 28 (14.5%) | 28 (6.89%) | 13 (6.73%) | 17 (4.19%) | 14.35% | |
| 3 | Postmortem brain temporal cortex (GSE128380) | 46 (10.55%) | 63 (7.04%) | 33 (7.56%) | 35 (3.91%) | 13.31% | |
| 4 | 1.2 Pitt-Hopkins | Tcf4-KO (GSE79663) | 12 (15%) | 1 (2.86%) | 5 (6.25%) | 0 (0%) | 15.65% |
| 5 | Tcf4-Het (GSE79663) | 4 (6.89%) | 0 (0%) | 5 (8.62%) | 0 (0%) | 11.54% | |
| 6 | 1.3 Phelan-McDermid | PMS Female 4w hiPSC neurons (GSE150429) | 2 (2.89%) | 13 (4.58%) | 2 (2.89%) | 22 (8.39%) | 11.78% |
| 7 | PMS Female 6w hiPSC neurons(GSE150429) | 2 (1.92%) | 14 (7.57%) | 3 (2.88%) | 14 (7.57%) | 11.42% | |
| 8 | PMS Female 8w hiPSC neurons (GSE150429) | 1 (5%) | 0 (0%) | 0 (0%) | 3 (11.11%) | 8.5% | |
| 9 | PMS Male 6w hiPSC neurons (GSE150429) | 21 (8.36%) | 8 (14.55%) | 11 (4.38%) | 1 (1.82%) | 13.4% | |
| 10 | PMS Male 8w hiPSC neurons (GSE150429) | 14 (5.34%) | 12 (11.65%) | 12 (4.58%) | 2 (1.94%) | 10.96% | |
| 11 | 1.4 Smith-Magenis | Rai1-CKO Cortex (GSE81206) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0% |
| 12 | Rai1-CKO Hypothalamus (GSE81206) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0% | |
| 13 | Rai1-CKO Striatum (GSE81206) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0% | |
| 14 | 1.5 Mowat-Wilson | Zeb2-cKO cerebellum (P0) (GSE84098) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0% |
| 15 | 2.1 Fragile X | hiPSC indel exon3 FMR1 (GSE117248) | 10 (4.33%) | 15 (7.28%) | 7 (3.03%) | 12 (5.82%) | 10.07% |
| 16 | 2.2 Kabuki Syndrome | brains of neuron-specific Kdm6a deficient mice | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0% |
| 17 | 2.3 Tuberous Sclerosis | human neural stem cells derived from embryonic stem cells TSC2+/− (TSC2-Het) | 1 (25%) | 1 (16.67%) | 0 (0%) | 0 (0%) | 20% |
| 18 | human neural stem cells derived from embryonic stem cell sTSC2−/− (TSC2-KO) | 78 (8.76%) | 56 (6.59%) | 49 (5.50%) | 35 (4.12%) | 12.54% | |
| GeneSymbol | Ensemble_Mouse | Ensemble_Human |
|---|---|---|
| Syt11 | ENSMUSG00000068923 | ENSG00000132718 |
| Nedd4 | ENSMUSG00000032216 | ENSG00000069869 |
| Phkb | ENSMUSG00000036879 | ENSG00000102893 |
| Braf | ENSMUSG00000002413 | ENSG00000157764 |
| Macf1 | ENSMUSG00000028649 | ENSG00000127603 |
| Ahcyl1 | ENSMUSG00000027893 | ENSG00000168710 |
| Grm5 | ENSMUSG00000049583 | ENSG00000168959 |
| Atpif1 | ENSMUSG00000054428 | ENSG00000130770 |
| Mcfd2 | ENSMUSG00000024150 | ENSG00000180398 |
| Myo6 | ENSMUSG00000033577 | ENSG00000196586 |
| Cpne7 | ENSMUSG00000034796 | ENSG00000178773 |
| Adcy1 | ENSMUSG00000020431 | ENSG00000164742 |
| Agrn | ENSMUSG00000041936 | ENSG00000104490 |
| Ncald | ENSMUSG00000051359 | ENSG00000188157 |
| Dgkg | ENSMUSG00000022861 | ENSG00000058866 |
| Dst | ENSMUSG00000026131 | ENSG00000067715 |
| Syt1 | ENSMUSG00000035864 | ENSG00000151914 |
| Fus | ENSMUSG00000030795 | ENSG00000089280 |
| Chp1 | ENSMUSG00000014077 | ENSG00000187446 |
| Rbm22 | ENSMUSG00000024604 | ENSG00000086589 |
| Rab3gap1 | ENSMUSG00000036104 | ENSG00000115839 |
| Sptan1 | ENSMUSG00000057738 | ENSG00000197694 |
| Pdcd6ip | ENSMUSG00000032504 | ENSG00000170248 |
| Camk1d | ENSMUSG00000039145 | ENSG00000183049 |
| Usp32 | ENSMUSG00000000804 | ENSG00000143622 |
| Rit1 | ENSMUSG00000028057 | ENSG00000170832 |
| Sparc | ENSMUSG00000018593 | ENSG00000113140 |
| Hspa5 | ENSMUSG00000026864 | ENSG00000044574 |
| Spock1 | ENSMUSG00000056222 | ENSG00000152377 |
| Ppm1f | ENSMUSG00000026181 | ENSG00000100034 |
| GeneSymbol | Ensemble_Mouse | Ensemble_human |
|---|---|---|
| Cacna1g | ENSMUSG00000020866 | ENSG00000006283 |
| Ppp3r1 | ENSMUSG00000033953 | ENSG00000221823 |
| Fkbp1a | ENSMUSG00000032966 | ENSG00000088832 |
| Hsp90b1 | ENSMUSG00000020048 | ENSG00000166598 |
| Sri | ENSMUSG00000003161 | ENSG00000075142 |
| Calm1 | ENSMUSG00000001175 | ENSG00000198668 |
| Rgs4 | ENSMUSG00000038530 | ENSG00000117152 |
| Herpud1 | ENSMUSG00000031770 | ENSG00000051108 |
| Ywhae | ENSMUSG00000020849 | ENSG00000108953 |
| Gsto1 | ENSMUSG00000025068 | ENSG00000148834 |
| Opa1 | ENSMUSG00000038084 | ENSG00000198836 |
| Bnip3 | ENSMUSG00000078566 | ENSG00000176171 |
| Nrxn1 | ENSMUSG00000024109 | ENSG00000179915 |
| Arrb2 | ENSMUSG00000060216 | ENSG00000141480 |
| Adcy3 | ENSMUSG00000020654 | ENSG00000138031 |
| Fyn | ENSMUSG00000019843 | ENSG00000198947 |
| Dmd | ENSMUSG00000045103 | ENSG00000010810 |
| Calr | ENSMUSG00000003814 | ENSG00000179218 |
| Slc9a1 | ENSMUSG00000028854 | ENSG00000090020 |
| Stoml2 | ENSMUSG00000028455 | ENSG00000165283 |
| Ppp3cb | ENSMUSG00000021816 | ENSG00000107758 |
| Ddit3 | ENSMUSG00000025408 | ENSG00000175197 |
| Stim2 | ENSMUSG00000039156 | ENSG00000109689 |
| Micu1 | ENSMUSG00000020111 | ENSG00000107745 |
| Dlg4 | ENSMUSG00000020886 | ENSG00000132535 |
| Atp13a2 | ENSMUSG00000036622 | ENSG00000159363 |
| Nptn | ENSMUSG00000032336 | ENSG00000156642 |
| Gnb5 | ENSMUSG00000032192 | ENSG00000069966 |
| Sgk1 | ENSMUSG00000019970 | ENSG00000118515 |
| Tpt1 | ENSMUSG00000060126 | ENSG00000133112 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panov, J.; Kaphzan, H. Angelman Syndrome and Angelman-like Syndromes Share the Same Calcium-Related Gene Signatures. Int. J. Mol. Sci. 2021, 22, 9870. https://doi.org/10.3390/ijms22189870
Panov J, Kaphzan H. Angelman Syndrome and Angelman-like Syndromes Share the Same Calcium-Related Gene Signatures. International Journal of Molecular Sciences. 2021; 22(18):9870. https://doi.org/10.3390/ijms22189870
Chicago/Turabian StylePanov, Julia, and Hanoch Kaphzan. 2021. "Angelman Syndrome and Angelman-like Syndromes Share the Same Calcium-Related Gene Signatures" International Journal of Molecular Sciences 22, no. 18: 9870. https://doi.org/10.3390/ijms22189870
APA StylePanov, J., & Kaphzan, H. (2021). Angelman Syndrome and Angelman-like Syndromes Share the Same Calcium-Related Gene Signatures. International Journal of Molecular Sciences, 22(18), 9870. https://doi.org/10.3390/ijms22189870