
Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime?




Abstract
1. Introduction
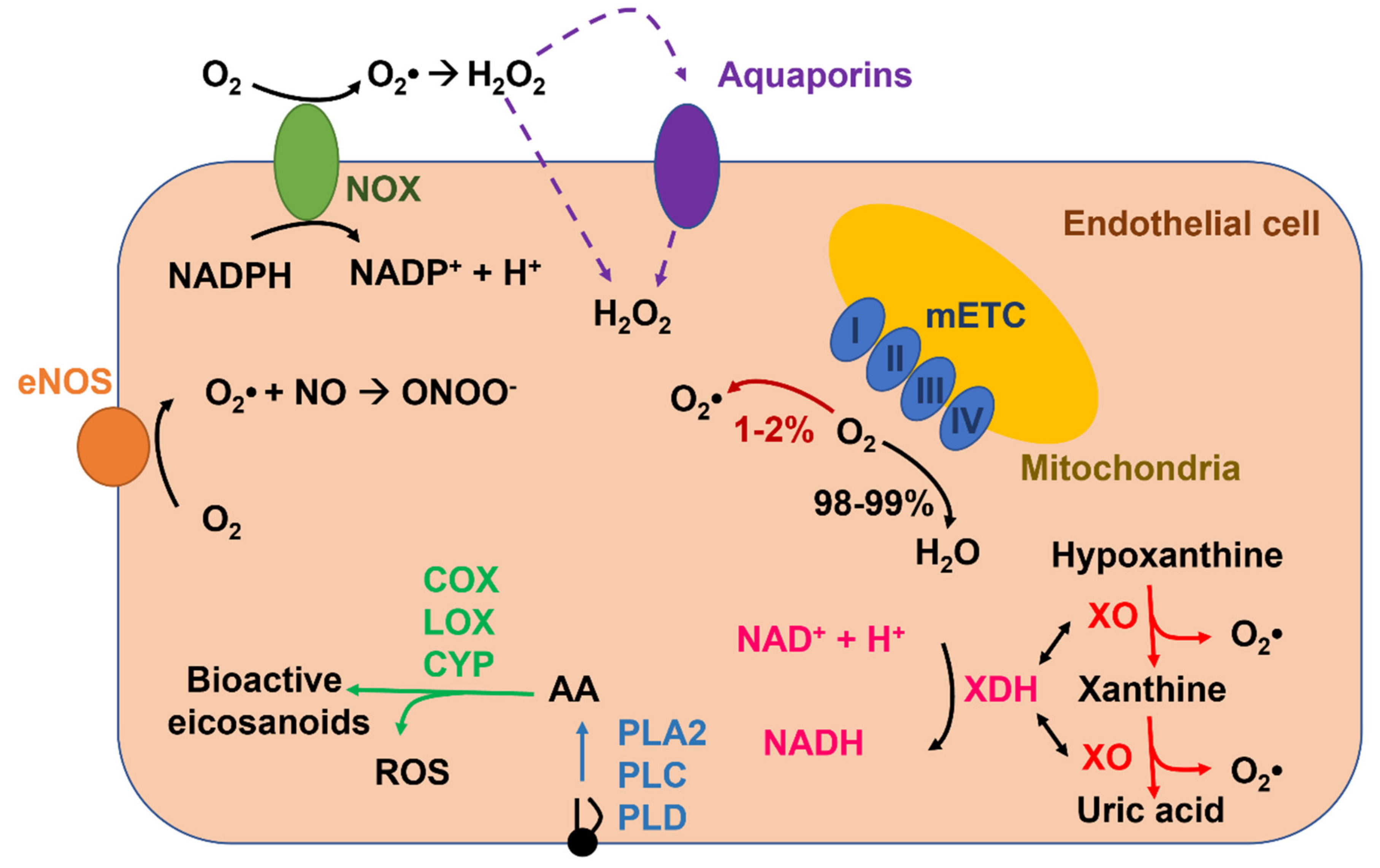
2. ROS Production and Elimination in Endothelial Cells
2.1. NADPH Oxidase-Mediated ROS Production in Endothelial Cells
2.2. Xanthine Oxidoreductase
2.3. Uncoupled eNOS
2.4. Mitochondria
2.5. Arachidonic-Acid-Metabolizing Enzymes
2.6. ROS Elimination
3. ROS Evoke or Modulate Intracellular Ca2+ Release in Endothelial Cells
3.1. Superoxide Anion, O2•−, and Hydroxyl Radical, OH•, Evoke Intracellular Ca2+ Release in Vascular Endothelial Cells
3.2. H2O2 Triggers InsP3-Induced ER Ca2+ Release in Vascular Endothelial Cells

{kind=link}
{kind=link}
{kind=link}
| ROS | Mechanism of ROS Stimulation | Dose of ROS or of ROS-Generating Enzymes | ROS Scavenger | Endothelial Cell Type | Effect on Intracellular Ca2+ Homeostasis | Reference |
|---|---|---|---|---|---|---|
| H2O2 | Acute exposure | 1-5-10 mM | Not used | CJVECs | ICR and ECI | [130] |
| H2O2 | Acute exposure | 100 µM | Not used | CPAECs | ICR and ECI | [131] |
| H2O2 | Acute exposure | 500 µM | Not used | SRLECs HUVECs | ICR | [129] |
| H2O2 | Acute exposure | 100 µM-10 mM | Not used | HAECs | ICR | [139] |
| H2O2 | Acute exposure | 10 µM | Not used | HUVECs | Not determined | [79] |
| H2O2 | Acute exposure | 1 mM | Not used | ICR | [140] | |
| H2O2 | Acute exposure | 5 mM | Cat, effect DMSO, no effect | MAECs MesAECs | ICI and ECI | [138] |
| H2O2 | Acute exposure | 100 µM | BAECs | ICI | [115] | |
| H2O2 | Acute exposure | 10-100 µM | Cat, effect NAC, effect | HUVECs | Increases agonists-induced Ca2+ signaling | [150] |
| H2O2 | HX/XO | 1 mM HX/2 mU/mL XO | Cat, effect SOD, no effect O-phen, no effect | SRLECs HUVECs | ICR | [129] |
| H2O2 | G/GO | 10 mM G/2 mU/mL GO | Cat, effect SOD, no effect O-phen, no effect | SRLECs HUVECs | ICR | [129] |
| H2O2 | HX/XO | 0.5 mM HX/50 mU/mL XO | Cat, effect SOD, no effect | CPAECs | ICR and ECI | [128] |
| H2O2 | G/GO | 10 nM G/[GO] →10 nM H2O2/mL/min | Not used | HUVECs | Not determined | [79] |
| H2O2, O2•− and •OH | HX/XO | 2 mM HX/[XO] → O2- nM/mL/min | Cat, effect SOD, effect O-phen, effect | HUVECs | ICI and ECI | [79,135] |
| H2O2 and O2•− | HX/XO | 200 µM HX/20 mU/mL XO | Cat, effect SOD, effect | MAECs MesAECs | ICI and ECI | [138] |
| O2•− | HX/XO | 1 mM HX/150 mU/mL XO | SOD, effect | PAECs | Increases agonist-induced ICI and SOCE | [136] |
| H2O2, O2•− and •OH | X/XO | 200 µM HX/2 mU/mL XO | Cat, effect SOD, effect O-phen and Def, effect | PAECs | ICI and ECI | [137] |
3.3. Evidence That ROS May Trigger Agonists-Induced Intracellular Ca2+ Release in Vascular Endothelial Cells
3.4. Evidence That ROS Can Modulate SERCA2B Activity during Agonists-Induced Ca2+ Signals in Vascular Endothelial Cells
4. ROS Modulate Store-Operated Ca2+ Entry in Vascular Endothelial Cells
4.1. H2O2 Modulates STIM and Orai Proteins: Direct and Indirect Mechanisms
4.2. Evidence That ROS May Modulate SOCE in Vascular Endothelial Cells
4.3. Prolonged Exposure to Oxidant Stress Impairs SOCE in Vascular Endothelial Cells
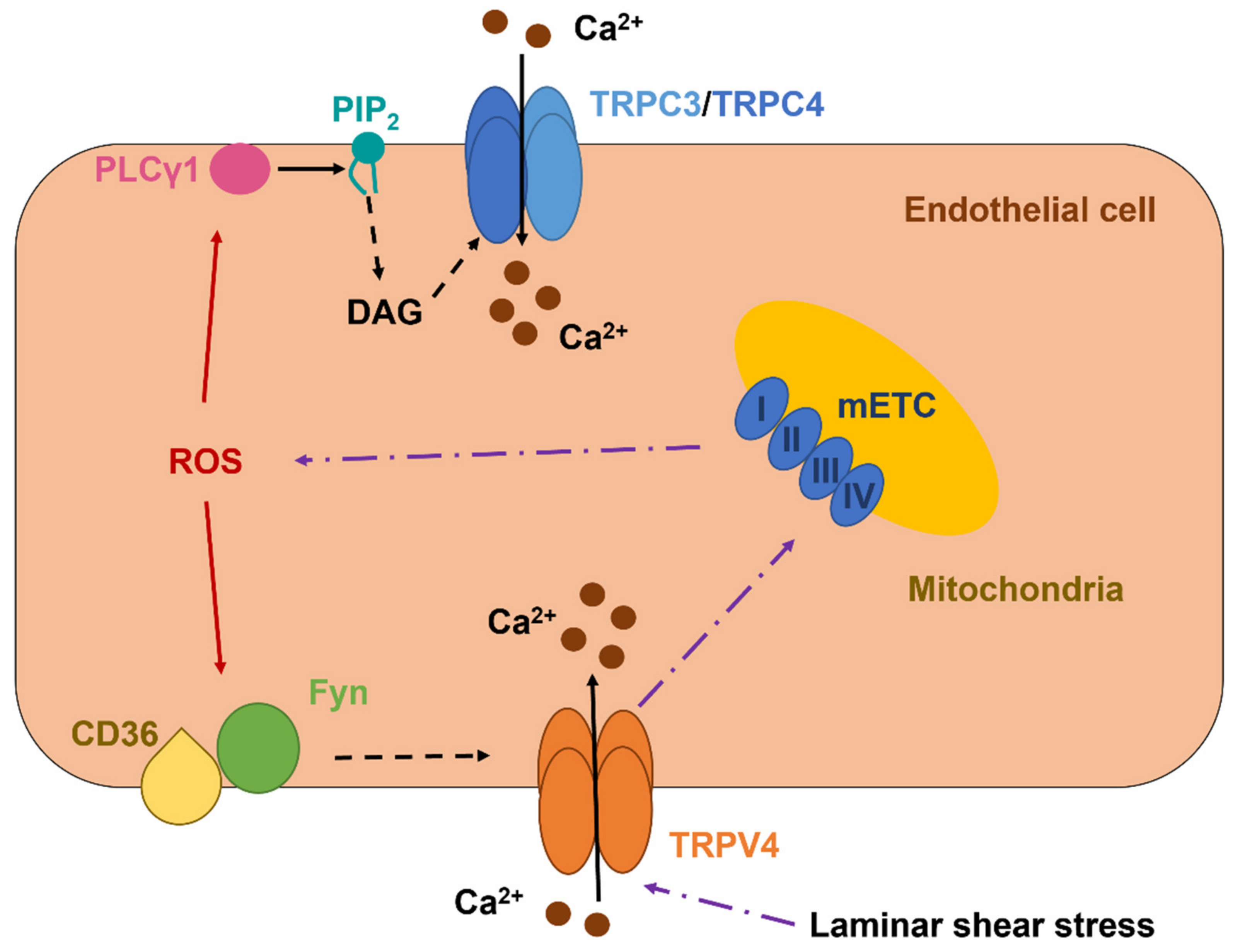
5. ROS Mediate Extracellular Ca2+ Influx through the Activation of Transient Receptor Potential (TRP) Channels
5.1. TRPC3 and TRPC4 Form a Redox-Sensitive Ca2+-Permeable Channel in Vascular Endothelial Cells
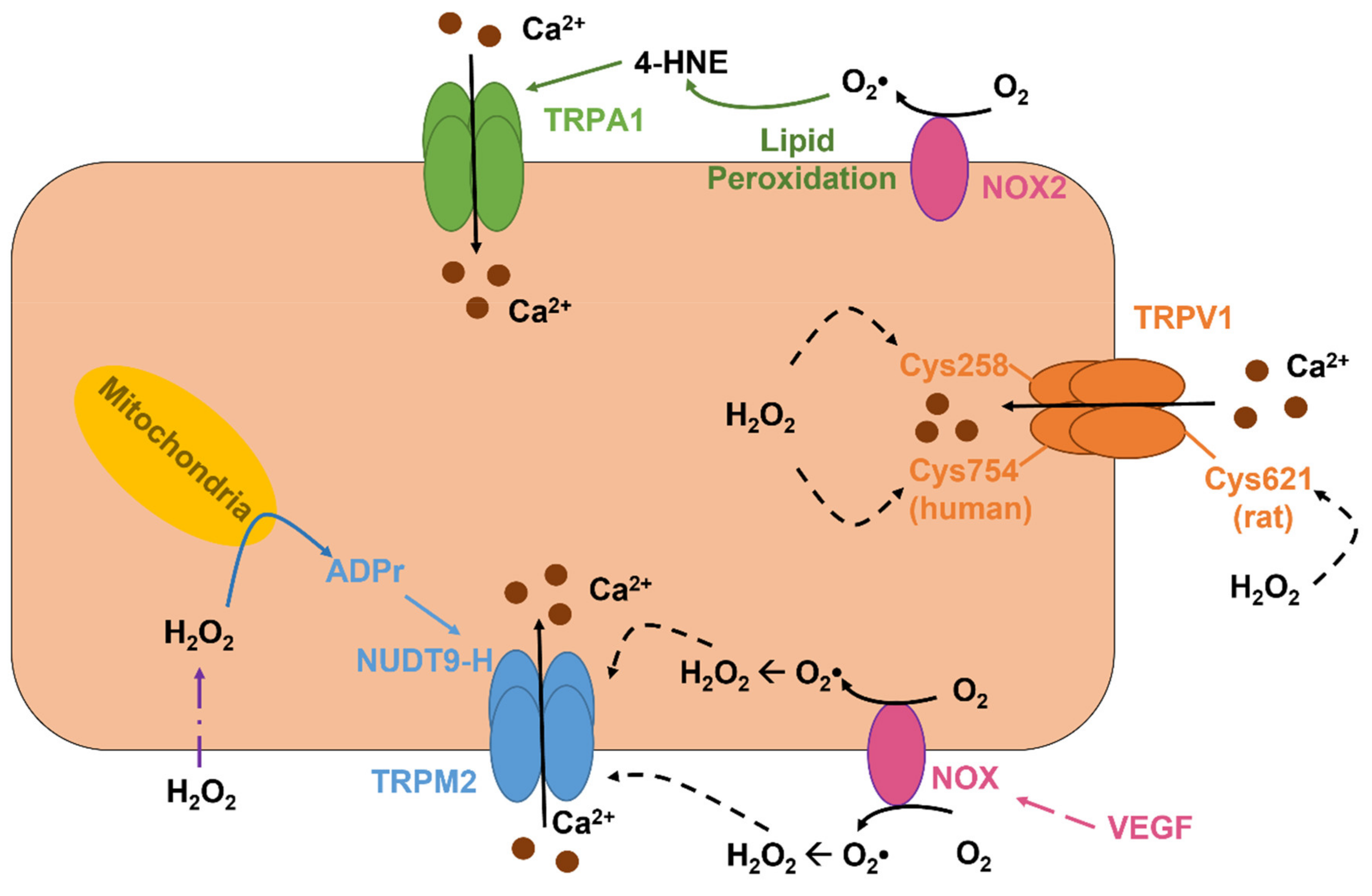
5.2. The Role of TRPV1 as a Novel Sensor in Redox Signaling in Vascular Endothelial Cells
5.3. The Role of TRPV4 in Vascular Endothelial Cells: A Sensor and an Inducer of Redox Signaling
5.4. The Role TRPM2 as an Indirect Sensor of Redox Signaling in Vascular Endothelial Cells
5.5. The Role of TRPM4 in ROS-Induced Angiogenesis
5.6. The Role of ROS-Sensitive Endothelial TRPA1 in Dilation of Cerebral Arteries and in Neurovascular Coupling
| ROS | Mechanism of ROS Stimulation | Dose of ROS or of ROS-Generating Enzymes | Endothelial Cell Type | TRP Targeted | Function | Ref. |
|---|---|---|---|---|---|---|
| t-BHQ | Acute exposure | 400 µM | PAECs | TRPC3 | Unknown | [207,210] |
| ChOx | Acute exposure | 0.5 u/mL | PAECs | TRPC3/TRPC4 | Unknown | [208] |
| H2O2 | Acute exposure | 250 µM | MCAECs and BAECs | TRPV1 | Vasodilation | [216] |
| H2O2 | Acute exposure | 250 µM | Human and mouse lung microvascular endothelial cells | TRPV4 | Barrier permeability | [223] |
| H2O2 | Acute exposure | 0–500 µM | HPAECs | TRPM2 | Decrease in barrier permeability, apoptosis | [58,236] |
| H2O2 | Acute exposure | 300 µM | Mouse lung microvascular endothelial cells | TRPM2 | Decrease in barrier permeability, neutrophil migration | [36] |
| H2O2 | Acute exposure | 0.5–1 mM | Mouse brain endothelial cells | TRPM2 | Aβ1-40 -induced endothelial dysfunction | [38] |
| H2O2 | Acute exposure | Not specified | MAECs | TRPM2 | Endothelial dysfunction | [54] |
| H2O2 | Acute exposure | 3 mM | H5V | TRPM2 | Apoptosis | [248] |
| H2O2 | Acute exposure | 1–10 µM | HUVECs | TRPM4 | Migration, spreading, and adhesion | [238] |
| 4-HNE | Acute exposure | 5–1000 nM | Mouse brain endothelial cells | TRPA1 | Vasorelaxation, neuroprotection, and NVC | [77,242,247] |
6. Therapeutic Applications and Pathological Implications of ROS-Induced Endothelial Ca2+ Signals
6.1. Exploiting ROS-Induced Endothelial Ca2+ Signals to Promote Therapeutic Angiogenesis and Rescue Blood Flow Perfusion
6.2. Exploiting ROS-Induced Endothelial Ca2+ Signals to Treat Cancer
6.3. Pathological Implications of ROS-Induced Endothelial Ca2+ Signaling
6.3.1. The Role of ROS-Induced Endothelial Ca2+ Signaling in the Inflammatory Response
6.3.2. The Role of ROS-Induced Endothelial Ca2+ Signals in Metabolic Disorders
6.3.3. The Role of TRPM Channels in ROS-Induced Endothelial Dysfunction
6.3.4. The Role of TRPV4 in Pulmonary Arterial Hypertension
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McCarron, J.G.; Wilson, C.; Heathcote, H.R.; Zhang, X.; Buckley, C.; Lee, M.D. Heterogeneity and emergent behaviour in the vascular endothelium. Curr. Opin. Pharmacol. 2019, 45, 23–32. [Google Scholar] [CrossRef]
- McCarron, J.G.; Lee, M.D.; Wilson, C. The endothelium solves problems that endothelial cells do not know exist. Trends Pharmacol. Sci. 2017, 38, 322–338. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Rosti, V.; Antognazza, M.R.; Lodola, F.; Moccia, F. Endothelial TRPV1 as an emerging molecular target to promote therapeutic angiogenesis. Cells 2020, 9, 1341. [Google Scholar] [CrossRef]
- Faris, P.; Negri, S.; Perna, A.; Rosti, V.; Guerra, G.; Moccia, F. Therapeutic potential of endothelial colony-forming cells in ischemic disease: Strategies to improve their regenerative efficacy. Int. J. Mol. Sci. 2020, 21, 7406. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Berra-Romani, R.; Guerra, G.; Moccia, F. Endothelial transient receptor potential channels and vascular remodeling: Extracellular Ca2+ entry for angiogenesis, arteriogenesis and vasculogenesis. Front. Physiol. 2019, 10, 1618. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.; Earley, S. Transient receptor potential channels and endothelial cell calcium signaling. Compr. Physiol. 2019, 9, 1249–1277. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, M.; Sonkusare, S.K. The calcium signaling mechanisms in arterial smooth muscle and endothelial cells. Compr. Physiol. 2021, 11, 1831–1869. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Berra-Romani, R. Targeting the endothelial Ca2+ tool kit to rescue endothelial dysfunction in obesity associated-hypertension. Curr. Med. Chem. 2019, 27, 240–257. [Google Scholar] [CrossRef]
- Moccia, F.; Bonetti, E.; Dragoni, S.; Fontana, J.; Lodola, F.; Romani, R.B.; Laforenza, U.; Rosti, V.; Tanzi, F. Hematopoietic progenitor and stem cells circulate by surfing on intracellular Ca2+ waves: A novel target for cell-based therapy and anti-cancer treatment? Curr. Signal Transd. T. 2012, 7, 161–176. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Pellavio, G.; Orgiu, M.; Negri, S.; Forcaia, G.; Var-Gaz-Guadarrama, V.; Garcia-Carrasco, M.; Botta, L.; Sancini, G.; et al. Histamine induces intracellular Ca2+ oscillations and nitric oxide release in endothelial cells from brain microvascular circulation. J. Cell. Physiol. 2020, 235, 1515–1530. [Google Scholar] [CrossRef]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca2+ oscillations and nitric oxide release by inducing NAADP- and InsP3 -dependent Ca2+ release in mouse brain endothelial cells. J. Cell. Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Baruffi, S.; Spaggiari, S.; Coltrini, D.; Berra-Romani, R.; Signorelli, S.; Castelli, L.; Taglietti, V.; Tanzi, F. P2y1 and P2y2 receptor-operated Ca2+ signals in primary cultures of cardiac microvascular endothelial cells. Microvasc. Res. 2001, 61, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; de Wit, C. Endothelium-derived hyperpolarizing factor and myoendothelial coupling: The in vivo perspective. Front. Physiol. 2020, 11, 602930. [Google Scholar] [CrossRef] [PubMed]
- Genova, T.; Gaglioti, D.; Munaron, L. Regulation of vessel permeability by TRP channels. Front. Physiol. 2020, 11, 421. [Google Scholar] [CrossRef]
- Smani, T.; Gomez, L.J.; Regodon, S.; Woodard, G.E.; Siegfried, G.; Khatib, A.M.; Rosado, J.A. TRP channels in angiogenesis and other endothelial functions. Front. Physiol. 2018, 9, 1731. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Raqeeb, A.; Torres-Jácome, J.; Guzman-Silva, A.; Guerra, G.; Tanzi, F.; Moccia, F. The mechanism of injury-induced intracellular calcium concentration oscillations in the endothelium of excised rat aorta. J. Vasc. Res. 2012, 49, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Raqeeb, A.; Avelino-Cruz, J.E.; Moccia, F.; Oldani, A.; Speroni, F.; Taglietti, V.; Tanzi, F. Ca2+ signaling in injured in situ endothelium of rat aorta. Cell Calcium 2008, 44, 298–309. [Google Scholar] [CrossRef]
- Noy, P.J.; Gavin, R.L.; Colombo, D.; Haining, E.J.; Reyat, J.S.; Payne, H.; Thielmann, I.; Lokman, A.B.; Neag, G.; Yang, J.; et al. Tspan18 is a novel regulator of the Ca2+ channel Orai1 and von Willebrand factor release in endothelial cells. Haematologica 2018, 104, 1892. [Google Scholar] [CrossRef] [PubMed]
- Esposito, B.; Gambara, G.; Lewis, A.M.; Palombi, F.; D’Alessio, A.; Taylor, L.X.; Genazzani, A.A.; Ziparo, E.; Galione, A.; Churchill, G.C.; et al. NAADP links histamine H1 receptors to secretion of von Willebrand factor in human endothelial cells. Blood 2011, 117, 4968–4977. [Google Scholar] [CrossRef] [PubMed]
- Rios, F.J.; Zou, Z.G.; Harvey, A.P.; Harvey, K.Y.; Nosalski, R.; Anyfanti, P.; Camargo, L.L.; Lacchini, S.; Ryazanov, A.G.; Ryazanova, L.; et al. Chanzyme TRPM7 protects against cardiovascular inflammation and fibrosis. Cardiovasc. Res. 2020, 116, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Weber, E.W.; Han, F.; Tauseef, M.; Birnbaumer, L.; Mehta, D.; Muller, W.A. TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J. Exp. Med. 2015, 212, 1883–1899. [Google Scholar] [CrossRef] [PubMed]
- Gandhirajan, R.K.; Meng, S.; Chandramoorthy, H.C.; Mallilankaraman, K.; Mancarella, S.; Gao, H.; Razmpour, R.; Yang, X.F.; Houser, S.R.; Chen, J.; et al. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J. Clin. Investig. 2013, 123, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, R.; Gegg, M.; Longbottom, R.; Adamson, P.; Turowski, P.; Greenwood, J. ICAM-1-mediated endothelial nitric oxide synthase activation via calcium and AMP-activated protein kinase is required for transendothelial lymphocyte migration. Mol. Biol. Cell 2009, 20, 995–1005. [Google Scholar] [CrossRef]
- Guerra, G.; Lucariello, A.; Perna, A.; Botta, L.; De Luca, A.; Moccia, F. The role of endothelial Ca2+ signaling in neurovascular coupling: A view from the Lumen. Int. J. Mol. Sci. 2018, 19, 938. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Soda, T.; Moccia, F. Endothelial signaling at the core of neurovascular coupling: The emerging role of endothelial inward-rectifier K+ (Kir2.1) channels and N-methyl-d-aspartate receptors in the regulation of cerebral blood flow. Int. J. Biochem. Cell Biol. 2021, 135, 105983. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Shekha, M.; Faris, P.; Guerra, G. Endothelial Ca2+ signaling, angiogenesis and vasculogenesis: Just what it takes to make a blood vessel. Int. J. Mol. Sci. 2019, 20, 3962. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Di Nezza, F.; Pellavio, G.; Faris, P.S.; Negri, S.; De Luca, A.; Laforenza, U.; Ambrosone, L.; Rosti, V.; et al. Nicotinic acid adenine dinucleotide phosphate activates two-pore channel TPC1 to mediate lysosomal Ca2+ release in endothelial colony-forming cells. J. Cell. Physiol. 2021, 236, 688–705. [Google Scholar] [CrossRef]
- Dong, Y.; Lee, Y.; Cui, K.; He, M.; Wang, B.; Bhattacharjee, S.; Zhu, B.; Yago, T.; Zhang, K.; Deng, L.; et al. Epsin-mediated degradation of IP3R1 fuels atherosclerosis. Nat. Commun. 2020, 11, 3984. [Google Scholar] [CrossRef]
- Wilson, C.; Zhang, X.; Buckley, C.; Heathcote, H.R.; Lee, M.D.; McCarron, J.G. Increased vascular contractility in hypertension results from impaired endothelial calcium signaling. Hypertension 2019, 74, 1200–1214. [Google Scholar] [CrossRef]
- Ottolini, M.; Hong, K.; Cope, E.L.; Daneva, Z.; DeLalio, L.J.; Sokolowski, J.D.; Marziano, C.; Nguyen, N.Y.; Altschmied, J.; Haendeler, J.; et al. Local peroxynitrite impairs endothelial transient receptor potential vanilloid 4 channels and elevates blood pressure in obesity. Circulation 2020, 141, 1318–1333. [Google Scholar] [CrossRef] [PubMed]
- Suresh, K.; Servinsky, L.; Jiang, H.; Bigham, Z.; Yun, X.; Kliment, C.; Huetsch, J.; Damarla, M.; Shimoda, L.A. Reactive oxygen species induced Ca2+ influx via TRPV4 and microvascular endothelial dysfunction in the SU5416/hypoxia model of pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L893–L907. [Google Scholar] [CrossRef]
- Wilson, C.; Zhang, X.; Lee, M.D.; MacDonald, M.; Heathcote, H.R.; Alorfi, N.M.N.; Buckley, C.; Dolan, S.; McCarron, J.G. Disrupted endothelial cell heterogeneity and network organization impair vascular function in prediabetic obesity. Metabolism 2020, 111, 154340. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Guzman-Silva, A.; Vargaz-Guadarrama, A.; Flores-Alonso, J.C.; Alonso-Romero, J.; Trevino, S.; Sanchez-Gomez, J.; Coyotl-Santiago, N.; Garcia-Carrasco, M.; Moccia, F. Type 2 diabetes alters intracellular Ca2+ handling in native endothelium of excised rat aorta. Int. J. Mol. Sci. 2019, 21, 250. [Google Scholar] [CrossRef] [PubMed]
- Propson, N.E.; Roy, E.R.; Litvinchuk, A.; Kohl, J.; Zheng, H. Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Komici, K.; Faris, P.; Negri, S.; Rosti, V.; Garcia-Carrasco, M.; Mendoza-Pinto, C.; Berra-Romani, R.; Cervera, R.; Guerra, G.; Moccia, F. Systemic lupus erythematosus, endothelial progenitor cells and intracellular Ca2+ signaling: A novel approach for an old disease. J. Autoimmun. 2020, 112, 102486. [Google Scholar] [CrossRef]
- Mittal, M.; Nepal, S.; Tsukasaki, Y.; Hecquet, C.M.; Soni, D.; Rehman, J.; Tiruppathi, C.; Malik, A.B. Neutrophil activation of endothelial cell-expressed TRPM2 mediates transendothelial neutrophil migration and vascular injury. Circ. Res. 2017, 121, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Tauseef, M.; Knezevic, N.; Chava, K.R.; Smith, M.; Sukriti, S.; Gianaris, N.; Obukhov, A.G.; Vogel, S.M.; Schraufnagel, D.E.; Dietrich, A.; et al. TLR4 activation of TRPC6-dependent calcium signaling mediates endotoxin-induced lung vascular permeability and inflammation. J. Exp. Med. 2012, 209, 1953–1968. [Google Scholar] [CrossRef]
- Park, L.; Wang, G.; Moore, J.; Girouard, H.; Zhou, P.; Anrather, J.; Iadecola, C. The key role of transient receptor potential melastatin-2 channels in amyloid-beta-induced neurovascular dysfunction. Nat. Commun. 2014, 5, 5318. [Google Scholar] [CrossRef]
- Shimizu, T.; Dietz, R.M.; Cruz-Torres, I.; Strnad, F.; Garske, A.K.; Moreno, M.; Venna, V.R.; Quillinan, N.; Herson, P.S. Extended therapeutic window of a novel peptide inhibitor of TRPM2 channels following focal cerebral ischemia. Exp. Neurol. 2016, 283, 151–156. [Google Scholar] [CrossRef]
- Ryu, H.J.; Kim, J.E.; Kim, Y.J.; Kim, J.Y.; Kim, W.I.; Choi, S.Y.; Kim, M.J.; Kang, T.C. Endothelial transient receptor potential conical channel (TRPC)-3 activation induces vasogenic edema formation in the rat piriform cortex following status epilepticus. Cell. Mol. Neurobiol. 2013, 33, 575–585. [Google Scholar] [CrossRef]
- Lionetti, V.; Bollini, S.; Coppini, R.; Gerbino, A.; Ghigo, A.; Iaccarino, G.; Madonna, R.; Mangiacapra, F.; Miragoli, M.; Moccia, F.; et al. Understanding the heart-brain axis response in COVID-19 patients: A suggestive perspective for therapeutic development. Pharmacol. Res. 2021, 168, 105581. [Google Scholar] [CrossRef]
- Moccia, F.; Tanzi, F.; Munaron, L. Endothelial remodelling and intracellular calcium machinery. Curr. Mol. Med. 2014, 14, 457–480. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F. Endothelial Ca2+ signaling and the resistance to anticancer treatments: Partners in crime. Int. J. Mol. Sci. 2018, 19, 217. [Google Scholar] [CrossRef]
- Costa, T.J.; Barros, P.R.; Arce, C.; Santos, J.D.; da Silva-Neto, J.; Egea, G.; Dantas, A.P.; Tostes, R.C.; Jimenez-Altayo, F. The homeostatic role of hydrogen peroxide, superoxide anion and nitric oxide in the vasculature. Free Radic. Biol. Med. 2021, 162, 615–635. [Google Scholar] [CrossRef]
- Santoro, M.M. Fashioning blood vessels by ROS signalling and metabolism. Semin. Cell Dev. Biol. 2018, 80, 35–42. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS signaling and redox biology in endothelial cells. Cell. Mol. Life Sci. 2015, 72, 3281–3303. [Google Scholar] [CrossRef]
- Shimokawa, H.; Godo, S. Nitric oxide and endothelium-dependent hyperpolarization mediated by hydrogen peroxide in health and disease. Basic Clin. Pharmacol. Toxicol. 2020, 127, 92–101. [Google Scholar] [CrossRef]
- Deliyanti, D.; Alrashdi, S.F.; Touyz, R.M.; Kennedy, C.R.; Jha, J.C.; Cooper, M.E.; Jandeleit-Dahm, K.A.; Wilkinson-Berka, J.L. Nox (NADPH Oxidase) 1, Nox4, and Nox5 promote vascular permeability and neovascularization in retinopathy. Hypertension 2020, 75, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Cook-Mills, J.M.; Marchese, M.E.; Abdala-Valencia, H. Vascular cell adhesion molecule-1 expression and signaling during disease: Regulation by reactive oxygen species and antioxidants. Antioxid. Redox Signal. 2011, 15, 1607–1638. [Google Scholar] [CrossRef] [PubMed]
- Avdonin, P.V.; Rybakova, E.Y.; Avdonin, P.P.; Trufanov, S.K.; Mironova, G.Y.; Tsitrina, A.A.; Goncharov, N.V. VAS2870 inhibits histamine-induced calcium signaling and vwf secretion in human umbilical vein endothelial cells. Cells 2019, 8, 196. [Google Scholar] [CrossRef]
- Frey, R.S.; Ushio-Fukai, M.; Malik, A.B. NADPH oxidase-dependent signaling in endothelial cells: Role in physiology and pathophysiology. Antioxid. Redox Signal. 2009, 11, 791–810. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Cross-talk between NADPH oxidase and mitochondria: Role in ROS signaling and angiogenesis. Cells 2020, 9, 1849. [Google Scholar] [CrossRef]
- O’Neill, K.M.; Campbell, D.C.; Edgar, K.S.; Gill, E.K.; Moez, A.; McLoughlin, K.J.; O’Neill, C.L.; Dellett, M.; Hargey, C.J.; Abudalo, R.A.; et al. NOX4 is a major regulator of cord blood-derived endothelial colony-forming cells which promotes post-ischaemic revascularization. Cardiovasc. Res. 2020, 116, 393–405. [Google Scholar] [CrossRef]
- Sun, L.; Liu, Y.L.; Ye, F.; Xie, J.W.; Zeng, J.W.; Qin, L.; Xue, J.; Wang, Y.T.; Guo, K.M.; Ma, M.M.; et al. Free fatty acid-induced H2O2 activates TRPM2 to aggravate endothelial insulin resistance via Ca2+-dependent PERK/ATF4/TRB3 cascade in obese mice. Free Radic. Biol. Med. 2019, 143, 288–299. [Google Scholar] [CrossRef]
- Suresh, K.; Shimoda, L.A. Endothelial cell reactive oxygen species and Ca2+ signaling in pulmonary hypertension. Adv. Exp. Med. Biol. 2017, 967, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Quintana, D.D.; Garcia, J.A.; Anantula, Y.; Rellick, S.L.; Engler-Chiurazzi, E.B.; Sarkar, S.N.; Brown, C.M.; Simpkins, J.W. Amyloid-beta causes mitochondrial dysfunction via a Ca2+-driven upregulation of oxidative phosphorylation and superoxide production in cerebrovascular endothelial cells. J. Alzheimers Dis. 2020, 75, 119–138. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, H.; Wu, W.; Shi, C.; Hu, S.; Yin, T.; Ma, Q.; Han, T.; Zhang, Y.; Tian, F.; et al. Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca2+-XO-ROS axis via activation of the GLP-1R/PI3K/Akt/survivin pathways. Free Radic. Biol. Med. 2016, 95, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Hecquet, C.M.; Zhang, M.; Mittal, M.; Vogel, S.M.; Di, A.; Gao, X.; Bonini, M.G.; Malik, A.B. Cooperative interaction of trp melastatin channel transient receptor potential (TRPM2) with its splice variant TRPM2 short variant is essential for endothelial cell apoptosis. Circ. Res. 2014, 114, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Weissmann, N.; Sydykov, A.; Kalwa, H.; Storch, U.; Fuchs, B.; Mederos y Schnitzler, M.; Brandes, R.P.; Grimminger, F.; Meissner, M.; Freichel, M.; et al. Activation of TRPC6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat. Commun. 2012, 3, 649. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.W.; Earley, S. Redox regulation of transient receptor potential channels in the endothelium. Microcirculation 2017, 24, e12329. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.L.; Norton, C.E.; Segal, S.S. Apoptosis in resistance arteries induced by hydrogen peroxide: Greater resilience of endothelium versus smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H1625–H1633. [Google Scholar] [CrossRef]
- Joseph, S.K. Role of thiols in the structure and function of inositol trisphosphate receptors. Curr. Top. Membr. 2010, 66, 299–322. [Google Scholar] [CrossRef]
- Tan, Y.; Mui, D.; Toan, S.; Zhu, P.; Li, R.; Zhou, H. SERCA overexpression improves mitochondrial quality control and attenuates cardiac microvascular ischemia-reperfusion injury. Mol. Ther. Nucleic Acids 2020, 22, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Sakurada, R.; Odagiri, K.; Hakamata, A.; Kamiya, C.; Wei, J.; Watanabe, H. Calcium release from endoplasmic reticulum involves calmodulin-mediated NADPH oxidase-derived reactive oxygen species production in endothelial cells. Int. J. Mol. Sci. 2019, 20, 1644. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Shah, A.M. ROS signalling between endothelial cells and cardiac cells. Cardiovasc. Res. 2014, 102, 249–257. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Veal, E.; Day, A. Hydrogen peroxide as a signaling molecule. Antioxid Redox Signal 2011, 15, 147–151. [Google Scholar] [CrossRef]
- Di, A.; Mehta, D.; Malik, A.B. ROS-activated calcium signaling mechanisms regulating endothelial barrier function. Cell Calcium 2016, 60, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Breton-Romero, R.; Lamas, S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. 2014, 2, 529–534. [Google Scholar] [CrossRef]
- Cai, H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc. Res. 2005, 68, 26–36. [Google Scholar] [CrossRef]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Cai, H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ. Res. 2005, 96, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef]
- Gough, D.R.; Cotter, T.G. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis. 2011, 2, e213. [Google Scholar] [CrossRef]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Zinkevich, N.S.; Gutterman, D.D. ROS-induced ROS release in vascular biology: Redox-redox signaling. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H647–H653. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.N.; Gonzales, A.L.; Pires, P.W.; Bruhl, A.; Leo, M.D.; Li, W.; Oulidi, A.; Boop, F.A.; Feng, Y.; Jaggar, J.H.; et al. Localized TRPA1 channel Ca2+ signals stimulated by reactive oxygen species promote cerebral artery dilation. Sci. Signal. 2015, 8, ra2. [Google Scholar] [CrossRef]
- DelloStritto, D.J.; Sinharoy, P.; Connell, P.J.; Fahmy, J.N.; Cappelli, H.C.; Thodeti, C.K.; Geldenhuys, W.J.; Damron, D.S.; Bratz, I.N. 4-Hydroxynonenal dependent alteration of TRPV1-mediated coronary microvascular signaling. Free Radic. Biol. Med. 2016, 101, 10–19. [Google Scholar] [CrossRef]
- Dreher, D.; Junod, A.F. Differential effects of superoxide, hydrogen peroxide, and hydroxyl radical on intracellular calcium in human endothelial cells. J. Cell. Physiol. 1995, 162, 147–153. [Google Scholar] [CrossRef]
- Kelley, E.E.; Khoo, N.K.; Hundley, N.J.; Malik, U.Z.; Freeman, B.A.; Tarpey, M.M. Hydrogen peroxide is the major oxidant product of xanthine oxidase. Free Radic. Biol. Med. 2010, 48, 493–498. [Google Scholar] [CrossRef]
- Furuhashi, M. New insights into purine metabolism in metabolic diseases: Role of xanthine oxidoreductase activity. Am. J. Physiol. Endocrinol. Metab. 2020, 319, E827–E834. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Khaddaj Mallat, R.; Mathew John, C.; Kendrick, D.J.; Braun, A.P. The vascular endothelium: A regulator of arterial tone and interface for the immune system. Crit. Rev. Clin. Lab. Sci. 2017, 54, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Mancardi, D.; Pla, A.F.; Moccia, F.; Tanzi, F.; Munaron, L. Old and new gasotransmitters in the cardiovascular system: Focus on the role of nitric oxide and hydrogen sulfide in endothelial cells and cardiomyocytes. Curr. Pharm. Biotechnol. 2011, 12, 1406–1415. [Google Scholar] [CrossRef]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Hare, J.M. Nitroso-redox balance in the cardiovascular system. N. Engl. J. Med. 2004, 351, 2112–2114. [Google Scholar] [CrossRef]
- Daneva, Z.; Marziano, C.; Ottolini, M.; Chen, Y.L.; Baker, T.M.; Kuppusamy, M.; Zhang, A.; Ta, H.Q.; Reagan, C.E.; Mihalek, A.D.; et al. Caveolar peroxynitrite formation impairs endothelial TRPV4 channels and elevates pulmonary arterial pressure in pulmonary hypertension. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Li, H.; Forstermann, U. Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharmacol. 2013, 13, 161–167. [Google Scholar] [CrossRef]
- Li, Q.; Youn, J.Y.; Cai, H. Mechanisms and consequences of endothelial nitric oxide synthase dysfunction in hypertension. J. Hypertens. 2015, 33, 1128–1136. [Google Scholar] [CrossRef]
- Elrod, J.W.; Duranski, M.R.; Langston, W.; Greer, J.J.; Tao, L.; Dugas, T.R.; Kevil, C.G.; Champion, H.C.; Lefer, D.J. eNOS gene therapy exacerbates hepatic ischemia-reperfusion injury in diabetes: A role for eNOS uncoupling. Circ. Res. 2006, 99, 78–85. [Google Scholar] [CrossRef]
- Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafa, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+ signaling in health, disease and therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef] [PubMed]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Finkel, T. Signal transduction by mitochondrial oxidants. J. Biol. Chem. 2012, 287, 4434–4440. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Methods Mol. Biol. 2009, 554, 165–181. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Negri, S.; Botta, L.; Genova, T.; Moccia, F. Arachidonic acid evokes an increase in intracellular Ca2+ concentration and nitric oxide production in endothelial cells from human brain microcirculation. Cells 2019, 8, 689. [Google Scholar] [CrossRef]
- Balducci, V.; Faris, P.; Balbi, C.; Costa, A.; Negri, S.; Rosti, V.; Bollini, S.; Moccia, F. The human amniotic fluid stem cell secretome triggers intracellular Ca2+ oscillations, NF-kappaB nuclear translocation and tube formation in human endothelial colony-forming cells. J. Cell. Mol. Med. 2021, 25, 8074–8086. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kim, J.Y.; Kim, J.H. Cytosolic phospholipase A (2), lipoxygenase metabolites, and reactive oxygen species. BMB Rep. 2008, 41, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, T.B.; Moon, K.A.; Kim, T.J.; Shin, D.; Cho, Y.S.; Moon, H.B.; Lee, K.Y. Regulation of pro-inflammatory responses by lipoxygenases via intracellular reactive oxygen species in vitro and in vivo. Exp. Mol. Med. 2008, 40, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Swindle, E.J.; Coleman, J.W.; DeLeo, F.R.; Metcalfe, D.D. FcepsilonRI- and Fcgamma receptor-mediated production of reactive oxygen species by mast cells is lipoxygenase- and cyclooxygenase-dependent and NADPH oxidase-independent. J. Immunol. 2007, 179, 7059–7071. [Google Scholar] [CrossRef]
- Hidalgo, C.; Donoso, P. Crosstalk between calcium and redox signaling: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 1275–1312. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Thomas, C.; Ristow, M. Interrelation between ROS and Ca2+ in aging and age-related diseases. Redox Biol. 2020, 36, 101678. [Google Scholar] [CrossRef]
- Wood, P.G.; Gillespie, J.I. Evidence for mitochondrial Ca2+-induced Ca2+ release in permeabilised endothelial cells. Biochem. Biophys. Res. Commun. 1998, 246, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.M.; Thompson, M.D.; Weisbrod, R.M.; Pimental, D.R.; Tong, X.; Bolotina, V.M.; Cohen, R.A. Redox regulation of SERCA2 is required for vascular endothelial growth factor-induced signaling and endothelial cell migration. Antioxid. Redox Signal. 2012, 17, 1099–1108. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Berra-Romani, R.; Tanzi, F. Update on vascular endothelial Ca2+ signalling: A tale of ion channels, pumps and transporters. World J. Biol. Chem. 2012, 3, 127–158. [Google Scholar] [CrossRef]
- Rozen, E.J.; Roewenstrunk, J.; Barallobre, M.J.; Di Vona, C.; Jung, C.; Figueiredo, A.F.; Luna, J.; Fillat, C.; Arbones, M.L.; Graupera, M.; et al. DYRK1A kinase positively regulates angiogenic responses in endothelial cells. Cell Rep. 2018, 23, 1867–1878. [Google Scholar] [CrossRef]
- Lin, Q.; Zhao, L.; Jing, R.; Trexler, C.; Wang, H.; Li, Y.; Tang, H.; Huang, F.; Zhang, F.; Fang, X.; et al. Inositol 1,4,5-trisphosphate receptors in endothelial cells play an essential role in vasodilation and blood pressure regulation. J. Am. Heart Assoc. 2019, 8, e011704. [Google Scholar] [CrossRef]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell. Physiol. 2019, 234, 4540–4562. [Google Scholar] [CrossRef]
- Zuccolo, E.; Lim, D.; Kheder, D.A.; Perna, A.; Catarsi, P.; Botta, L.; Rosti, V.; Riboni, L.; Sancini, G.; Tanzi, F.; et al. Acetylcholine induces intracellular Ca2+ oscillations and nitric oxide release in mouse brain endothelial cells. Cell Calcium 2017, 66, 33–47. [Google Scholar] [CrossRef]
- Dragoni, S.; Laforenza, U.; Bonetti, E.; Lodola, F.; Bottino, C.; Berra-Romani, R.; Carlo Bongio, G.; Cinelli, M.P.; Guerra, G.; Pedrazzoli, P.; et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells 2011, 29, 1898–1907. [Google Scholar] [CrossRef] [PubMed]
- Woll, K.A.; Van Petegem, F. Calcium release channels: Structure and function of IP3 receptors and ryanodine receptors. Physiol. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.K.; Young, M.P.; Alzayady, K.; Yule, D.I.; Ali, M.; Booth, D.M.; Hajnoczky, G. Redox regulation of type-I inositol trisphosphate receptors in intact mammalian cells. J. Biol. Chem. 2018, 293, 17464–17476. [Google Scholar] [CrossRef] [PubMed]
- Bansaghi, S.; Golenar, T.; Madesh, M.; Csordas, G.; RamachandraRao, S.; Sharma, K.; Yule, D.I.; Joseph, S.K.; Hajnoczky, G. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 2014, 289, 8170–8181. [Google Scholar] [CrossRef]
- Lock, J.T.; Sinkins, W.G.; Schilling, W.P. Protein S-glutathionylation enhances Ca2+-induced Ca2+ release via the IP3 receptor in cultured aortic endothelial cells. J. Physiol. 2012, 590, 3431–3447. [Google Scholar] [CrossRef]
- Lock, J.T.; Sinkins, W.G.; Schilling, W.P. Effect of protein S-glutathionylation on Ca2+ homeostasis in cultured aortic endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H493–H506. [Google Scholar] [CrossRef]
- Groschner, L.N.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Endothelial mitochondria—Less respiration, more integration. Pflugers Arch. 2012, 464, 63–76. [Google Scholar] [CrossRef]
- Zhang, X.; Lee, M.D.; Wilson, C.; McCarron, J.G. Hydrogen peroxide depolarizes mitochondria and inhibits IP3-evoked Ca2+ release in the endothelium of intact arteries. Cell Calcium 2019, 84, 102108. [Google Scholar] [CrossRef]
- Garcia-Carlos, C.A.; Camargo-Loaiza, J.A.; Garcia-Villa, D.; Lopez-Cervantes, J.G.; Dominguez-Avila, J.A.; Gonzalez-Aguilar, G.A.; Astiazaran-Garcia, H.; Montiel-Herrera, M. Angiotensin II, ATP and high extracellular potassium induced intracellular calcium responses in primary rat brain endothelial cell cultures. Cell Biochem. Funct. 2021, 39, 688–698. [Google Scholar] [CrossRef]
- Rusko, J.; Wang, X.; van Breemen, C. Regenerative caffeine-induced responses in native rabbit aortic endothelial cells. Br. J. Pharmacol. 1995, 115, 811–821. [Google Scholar] [CrossRef]
- Zhang, G.; Teggatz, E.G.; Zhang, A.Y.; Koeberl, M.J.; Yi, F.; Chen, L.; Li, P.L. Cyclic ADP ribose-mediated Ca2+ signaling in mediating endothelial nitric oxide production in bovine coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H1172–H1181. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Dragoni, S.; Poletto, V.; Catarsi, P.; Guido, D.; Rappa, A.; Reforgiato, M.; Lodola, F.; Lim, D.; Rosti, V.; et al. Arachidonic acid-evoked Ca2+ signals promote nitric oxide release and proliferation in human endothelial colony forming cells. Vascul. Pharmacol. 2016, 87, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Negri, S.; Faris, P.; Perna, A.; De Luca, A.; Soda, T.; Romani, R.B.; Guerra, G. Targeting endolysosomal two-pore channels to treat cardiovascular disorders in the novel CoronaVirus Disease 2019. Front. Physiol. 2021, 12, 629119. [Google Scholar] [CrossRef]
- Galione, A. A primer of NAADP-mediated Ca2+ signalling: From sea urchin eggs to mammalian cells. Cell Calcium 2015, 58, 27–47. [Google Scholar] [CrossRef]
- Faris, P.; Shekha, M.; Montagna, D.; Guerra, G.; Moccia, F. Endolysosomal Ca2+ signalling and cancer hallmarks: Two-pore channels on the move, TRPML1 lags behind! Cancers 2018, 11, 27. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Nusco, G.A.; Lim, D.; Kyozuka, K.; Santella, L. NAADP and InsP3 play distinct roles at fertilization in starfish oocytes. Dev. Biol. 2006, 294, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Lounsbury, K.M.; Hu, Q.; Ziegelstein, R.C. Calcium signaling and oxidant stress in the vasculature. Free Radic. Biol. Med. 2000, 28, 1362–1369. [Google Scholar] [CrossRef]
- Wesson, D.E.; Elliott, S.J. The H2O2-generating enzyme, xanthine oxidase, decreases luminal Ca2+ content of the IP3-sensitive Ca2+ store in vascular endothelial cells. Microcirculation 1995, 2, 195–203. [Google Scholar] [CrossRef]
- Volk, T.; Hensel, M.; Kox, W.J. Transient Ca2+ changes in endothelial cells induced by low doses of reactive oxygen species: Role of hydrogen peroxide. Mol. Cell. Biochem. 1997, 171, 11–21. [Google Scholar] [CrossRef]
- Doan, T.N.; Gentry, D.L.; Taylor, A.A.; Elliott, S.J. Hydrogen peroxide activates agonist-sensitive Ca2+-flux pathways in canine venous endothelial cells. Biochem. J. 1994, 297, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Siflinger-Birnboim, A.; Lum, H.; Del Vecchio, P.J.; Malik, A.B. Involvement of Ca2+ in the H2O2-induced increase in endothelial permeability. Am. J. Physiol. 1996, 270, L973–L978. [Google Scholar] [CrossRef] [PubMed]
- Gericke, M.; Droogmans, G.; Nilius, B. Thimerosal induced changes of intracellular calcium in human endothelial cells. Cell Calcium 1993, 14, 201–207. [Google Scholar] [CrossRef]
- Henschke, P.N.; Elliott, S.J. Oxidized glutathione decreases luminal Ca2+ content of the endothelial cell ins (1,4,5) P3-sensitive Ca2+ store. Biochem. J. 1995, 312, 485–489. [Google Scholar] [CrossRef][Green Version]
- Elliott, S.J.; Doan, T.N. Oxidant stress inhibits the store-dependent Ca2+-influx pathway of vascular endothelial cells. Biochem. J. 1993, 292, 385–393. [Google Scholar] [CrossRef]
- Dreher, D.; Jornot, L.; Junod, A.F. Effects of hypoxanthine-xanthine oxidase on Ca2+ stores and protein synthesis in human endothelial cells. Circ. Res. 1995, 76, 388–395. [Google Scholar] [CrossRef]
- Graier, W.F.; Hoebel, B.G.; Paltauf-Doburzynska, J.; Kostner, G.M. Effects of superoxide anions on endothelial Ca2+ signaling pathways. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Az-ma, T.; Saeki, N.; Yuge, O. Cytosolic Ca2+ movements of endothelial cells exposed to reactive oxygen intermediates: Role of hydroxyl radical-mediated redox alteration of cell-membrane Ca2+ channels. Br. J. Pharmacol. 1999, 126, 1462–1470. [Google Scholar] [CrossRef]
- Sun, L.; Yau, H.Y.; Lau, O.C.; Huang, Y.; Yao, X. Effect of hydrogen peroxide and superoxide anions on cytosolic Ca2+: Comparison of endothelial cells from large-sized and small-sized arteries. PLoS ONE 2011, 6, e25432. [Google Scholar] [CrossRef]
- Hu, Q.; Corda, S.; Zweier, J.L.; Capogrossi, M.C.; Ziegelstein, R.C. Hydrogen peroxide induces intracellular calcium oscillations in human aortic endothelial cells. Circulation 1998, 97, 268–275. [Google Scholar] [CrossRef]
- Zheng, Y.; Shen, X. H2O2 directly activates inositol 1,4,5-trisphosphate receptors in endothelial cells. Redox Rep. Commun. Free. Radic. Res. 2005, 10, 29–36. [Google Scholar] [CrossRef]
- Yuan, W.; Guo, J.; Li, X.; Zou, Z.; Chen, G.; Sun, J.; Wang, T.; Lu, D. Hydrogen peroxide induces the activation of the phospholipase C-gamma1 survival pathway in PC12 cells: Protective role in apoptosis. Acta Biochim. Biophys. Sin. 2009, 41, 625–630. [Google Scholar] [CrossRef][Green Version]
- Hong, J.H.; Moon, S.J.; Byun, H.M.; Kim, M.S.; Jo, H.; Bae, Y.S.; Lee, S.I.; Bootman, M.D.; Roderick, H.L.; Shin, D.M.; et al. Critical role of phospholipase Cgamma1 in the generation of H2O2-evoked [Ca2+] i oscillations in cultured rat cortical astrocytes. J. Biol. Chem. 2006, 281, 13057–13067. [Google Scholar] [CrossRef]
- Vais, H.; Siebert, A.P.; Ma, Z.; Fernandez-Mongil, M.; Foskett, J.K.; Mak, D.O. Redox-regulated heterogeneous thresholds for ligand recruitment among InsP3R Ca2+-release channels. Biophys. J. 2010, 99, 407–416. [Google Scholar] [CrossRef]
- Joseph, S.K.; Nakao, S.K.; Sukumvanich, S. Reactivity of free thiol groups in type-I inositol trisphosphate receptors. Biochem. J. 2006, 393, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Denniss, A.; Dulhunty, A.F.; Beard, N.A. Ryanodine receptor Ca2+ release channel post-translational modification: Central player in cardiac and skeletal muscle disease. Int. J. Biochem. Cell Biol. 2018, 101, 49–53. [Google Scholar] [CrossRef]
- Higo, T.; Hattori, M.; Nakamura, T.; Natsume, T.; Michikawa, T.; Mikoshiba, K. Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell 2005, 120, 85–98. [Google Scholar] [CrossRef]
- Enyedi, B.; Varnai, P.; Geiszt, M. Redox state of the endoplasmic reticulum is controlled by Ero1L-alpha and intraluminal calcium. Antioxid. Redox Signal. 2010, 13, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Jones, D.P. Redox compartmentalization in eukaryotic cells. Biochim. Biophys. Acta 2008, 1780, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. The endoplasmic reticulum: A multifunctional signaling organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Avdonin, P.V.; Nadeev, A.D.; Mironova, G.Y.; Zharkikh, I.L.; Avdonin, P.P.; Goncharov, N.V. Enhancement by hydrogen peroxide of calcium signals in endothelial cells induced by 5-HT1B and 5-HT2B receptor agonists. Oxid. Med. Cell. Longev. 2019, 2019, 1701478. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.Y.; Zhang, Y.H.; Xie, G.Q.; Liu, C.X.; Zhou, R.; Shi, W. Down-regulation of Homer1 attenuates t-BHP-induced oxidative stress through regulating calcium homeostasis and ER stress in brain endothelial cells. Biochem. Biophys. Res. Commun. 2016, 477, 970–976. [Google Scholar] [CrossRef]
- Hu, Q.; Zheng, G.; Zweier, J.L.; Deshpande, S.; Irani, K.; Ziegelstein, R.C. NADPH oxidase activation increases the sensitivity of intracellular Ca2+ stores to inositol 1,4,5-trisphosphate in human endothelial cells. J. Biol. Chem. 2000, 275, 15749–15757. [Google Scholar] [CrossRef]
- Hu, Q.; Yu, Z.X.; Ferrans, V.J.; Takeda, K.; Irani, K.; Ziegelstein, R.C. Critical role of NADPH oxidase-derived reactive oxygen species in generating Ca2+ oscillations in human aortic endothelial cells stimulated by histamine. J. Biol. Chem. 2002, 277, 32546–32551. [Google Scholar] [CrossRef]
- Dalal, P.J.; Muller, W.A.; Sullivan, D.P. Endothelial cell calcium signaling during barrier function and inflammation. Am. J. Pathol. 2020, 190, 535–542. [Google Scholar] [CrossRef]
- Avdonin, P.V.; Nadeev, A.D.; Tsitrin, E.B.; Tsitrina, A.A.; Avdonin, P.P.; Mironova, G.Y.; Zharkikh, I.L.; Goncharov, N.V. Involvement of two-pore channels in hydrogen peroxide-induced increase in the level of calcium ions in the cytoplasm of human umbilical vein endothelial cells. Doklady. Biochem. Biophys. 2017, 474, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Chidgey, J.; Fraser, P.A.; Aaronson, P.I. Reactive oxygen species facilitate the EDH response in arterioles by potentiating intracellular endothelial Ca2+ release. Free Radic. Biol. Med. 2016, 97, 274–284. [Google Scholar] [CrossRef]
- Munoz, M.; Lopez-Oliva, M.E.; Pinilla, E.; Martinez, M.P.; Sanchez, A.; Rodriguez, C.; Garcia-Sacristan, A.; Hernandez, M.; Rivera, L.; Prieto, D. CYP epoxygenase-derived H2O2 is involved in the endothelium-derived hyperpolarization (EDH) and relaxation of intrarenal arteries. Free Radic. Biol. Med. 2017, 106, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Burger, D.; Paravicini, T.M.; Chignalia, A.Z.; Yusuf, H.; Almasri, M.; He, Y.; Callera, G.E.; He, G.; Krause, K.H.; et al. Nicotinamide adenine dinucleotide phosphate reduced oxidase 5 (Nox5) regulation by angiotensin II and endothelin-1 is mediated via calcium/calmodulin-dependent, rac-1-independent pathways in human endothelial cells. Circ. Res. 2010, 106, 1363–1373. [Google Scholar] [CrossRef]
- Roscoe, J.M.; Sevier, C.S. Pathways for sensing and responding to hydrogen peroxide at the endoplasmic reticulum. Cells 2020, 9, 2314. [Google Scholar] [CrossRef]
- Lermant, A.; Murdoch, C.E. Cysteine glutathionylation acts as a redox switch in endothelial cells. Antioxidants 2019, 8, 315. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schoneich, C.; Cohen, R.A. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef]
- Tong, X.; Hou, X.; Jourd’heuil, D.; Weisbrod, R.M.; Cohen, R.A. Upregulation of Nox4 by TGF {beta}1 oxidizes SERCA and inhibits NO in arterial smooth muscle of the prediabetic Zucker rat. Circ Res. 2010, 107, 975–983. [Google Scholar] [CrossRef]
- Horakova, L.; Strosova, M.K.; Spickett, C.M.; Blaskovic, D. Impairment of calcium ATPases by high glucose and potential pharmacological protection. Free Radic. Res. 2013, 47, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.M.; Thompson, M.D.; Bolotina, V.M.; Tong, X.; Cohen, R.A. Nox4- and Nox2-dependent oxidant production is required for VEGF-induced SERCA cysteine-674 S-glutathiolation and endothelial cell migration. Free Radic. Biol. Med. 2012, 53, 2327–2334. [Google Scholar] [CrossRef]
- Mei, Y.; Thompson, M.D.; Shiraishi, Y.; Cohen, R.A.; Tong, X. Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase C674 promotes ischemia- and hypoxia-induced angiogenesis via coordinated endothelial cell and macrophage function. J. Mol. Cell. Cardiol. 2014, 76, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Blatter, L.A. Tissue specificity: SOCE: Implications for Ca2+ handling in endothelial cells. Adv. Exp. Med. Biol. 2017, 993, 343–361. [Google Scholar] [CrossRef]
- Groschner, K.; Shrestha, N.; Fameli, N. Cardiovascular and hemostatic disorders: SOCE in cardiovascular cells: Emerging targets for therapeutic intervention. Adv. Exp. Med. Biol. 2017, 993, 473–503. [Google Scholar] [CrossRef]
- Moccia, F.; Dragoni, S.; Lodola, F.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Store-dependent Ca2+ entry in endothelial progenitor cells as a perspective tool to enhance cell-based therapy and adverse tumour vascularization. Curr. Med. Chem. 2012, 19, 5802–5818. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, E.; Di Buduo, C.; Lodola, F.; Orecchioni, S.; Scarpellino, G.; Kheder, D.A.; Poletto, V.; Guerra, G.; Bertolini, F.; Balduini, A.; et al. Stromal cell-derived factor-1alpha promotes endothelial colony-forming cell migration through the Ca2+-dependent activation of the extracellular signal-regulated kinase 1/2 and phosphoinositide 3-kinase/AKT pathways. Stem Cells Dev. 2018, 27, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef]
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.L.; Taylor, H.; et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ. Res. 2011, 108, 1190–1198. [Google Scholar] [CrossRef]
- Zhou, M.H.; Zheng, H.; Si, H.; Jin, Y.; Peng, J.M.; He, L.; Zhou, Y.; Munoz-Garay, C.; Zawieja, D.C.; Kuo, L.; et al. Stromal interaction molecule 1 (STIM1) and Orai1 mediate histamine-evoked calcium entry and nuclear factor of activated T-cells (NFAT) signaling in human umbilical vein endothelial cells. J. Biol. Chem. 2014, 289, 29446–29456. [Google Scholar] [CrossRef]
- Daskoulidou, N.; Zeng, B.; Berglund, L.M.; Jiang, H.; Chen, G.L.; Kotova, O.; Bhandari, S.; Ayoola, J.; Griffin, S.; Atkin, S.L.; et al. High glucose enhances store-operated calcium entry by upregulating ORAI/STIM via calcineurin-NFAT signalling. J. Mol. Med. 2015, 93, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Di Giuro, C.M.L.; Shrestha, N.; Malli, R.; Groschner, K.; van Breemen, C.; Fameli, N. Na+/Ca2+ exchangers and Orai channels jointly refill endoplasmic reticulum (ER) Ca2+ via ER nanojunctions in vascular endothelial cells. Pflugers Arch. 2017, 469, 1287–1299. [Google Scholar] [CrossRef]
- Antigny, F.; Jousset, H.; Konig, S.; Frieden, M. Thapsigargin activates Ca2+ entry both by store-dependent, STIM1/Orai1-mediated, and store-independent, TRPC3/PLC/PKC-mediated pathways in human endothelial cells. Cell Calcium 2011, 49, 115–127. [Google Scholar] [CrossRef]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.L.; de Souza, L.B.; Zheng, C.; Cheng, K.T.; Liu, X.; Goldsmith, C.M.; Feske, S.; Ambudkar, I.S. STIM2 enhances receptor-stimulated Ca2+ signaling by promoting recruitment of STIM1 to the endoplasmic reticulum-plasma membrane junctions. Sci. Signal. 2015, 8, ra3. [Google Scholar] [CrossRef] [PubMed]
- Emrich, S.M.; Yoast, R.E.; Xin, P.; Arige, V.; Wagner, L.E.; Hempel, N.; Gill, D.L.; Sneyd, J.; Yule, D.I.; Trebak, M. Omnitemporal choreographies of all five STIM/Orai and IP3Rs underlie the complexity of mammalian Ca2+ signaling. Cell Rep. 2021, 34, 108760. [Google Scholar] [CrossRef] [PubMed]
- Vaeth, M.; Yang, J.; Yamashita, M.; Zee, I.; Eckstein, M.; Knosp, C.; Kaufmann, U.; Karoly Jani, P.; Lacruz, R.S.; Flockerzi, V.; et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun. 2017, 8, 14714. [Google Scholar] [CrossRef] [PubMed]
- Eckstein, M.; Vaeth, M.; Aulestia, F.J.; Costiniti, V.; Kassam, S.N.; Bromage, T.G.; Pedersen, P.; Issekutz, T.; Idaghdour, Y.; Moursi, A.M.; et al. Differential regulation of Ca2+ influx by ORAI channels mediates enamel mineralization. Sci. Signal. 2019, 12, 578. [Google Scholar] [CrossRef]
- Yoast, R.E.; Emrich, S.M.; Zhang, X.; Xin, P.; Johnson, M.T.; Fike, A.J.; Walter, V.; Hempel, N.; Yule, D.I.; Sneyd, J.; et al. The native ORAI channel trio underlies the diversity of Ca2+ signaling events. Nat. Commun. 2020, 11, 2444. [Google Scholar] [CrossRef] [PubMed]
- Kito, H.; Yamamura, H.; Suzuki, Y.; Yamamura, H.; Ohya, S.; Asai, K.; Imaizumi, Y. Regulation of store-operated Ca2+ entry activity by cell cycle dependent up-regulation of Orai2 in brain capillary endothelial cells. Biochem. Biophys. Res. Commun. 2015, 459, 457–462. [Google Scholar] [CrossRef]
- Gibhardt, C.S.; Cappello, S.; Bhardwaj, R.; Schober, R.; Kirsch, S.A.; Bonilla Del Rio, Z.; Gahbauer, S.; Bochicchio, A.; Sumanska, M.; Ickes, C.; et al. Oxidative stress-induced STIM2 cysteine modifications suppress store-operated calcium entry. Cell Rep. 2020, 33, 108292. [Google Scholar] [CrossRef]
- Bhardwaj, R.; Hediger, M.A.; Demaurex, N. Redox modulation of STIM-ORAI signaling. Cell Calcium 2016, 60, 142–152. [Google Scholar] [CrossRef]
- Bogeski, I.; Kummerow, C.; Al-Ansary, D.; Schwarz, E.C.; Koehler, R.; Kozai, D.; Takahashi, N.; Peinelt, C.; Griesemer, D.; Bozem, M.; et al. Differential redox regulation of ORAI ion channels: A mechanism to tune cellular calcium signaling. Sci. Signal. 2010, 3, ra24. [Google Scholar] [CrossRef]
- Niemeyer, B.A. The STIM-orai pathway: Regulation of STIM and orai by thiol modifications. Adv. Exp. Med. Biol. 2017, 993, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.J.; Irrinki, K.M.; Mallilankaraman, K.; Lien, Y.C.; Wang, Y.; Bhanumathy, C.D.; Subbiah, R.; Ritchie, M.F.; Soboloff, J.; Baba, Y.; et al. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J. Cell Biol. 2010, 190, 391–405. [Google Scholar] [CrossRef]
- Prins, D.; Groenendyk, J.; Touret, N.; Michalak, M. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 2011, 12, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S. Store-operated calcium channels: From function to structure and back again. Cold Spring Harb. Perspect. Biol. 2020, 12, a035055. [Google Scholar] [CrossRef] [PubMed]
- Alansary, D.; Schmidt, B.; Dorr, K.; Bogeski, I.; Rieger, H.; Kless, A.; Niemeyer, B.A. Thiol dependent intramolecular locking of Orai1 channels. Sci. Rep. 2016, 6, 33347. [Google Scholar] [CrossRef] [PubMed]
- Holzmann, C.; Kilch, T.; Kappel, S.; Dorr, K.; Jung, V.; Stockle, M.; Bogeski, I.; Peinelt, C. Differential Redox regulation of Ca2+ signaling and viability in normal and malignant prostate cells. Biophys. J. 2015, 109, 1410–1419. [Google Scholar] [CrossRef] [PubMed]
- Grupe, M.; Myers, G.; Penner, R.; Fleig, A. Activation of store-operated I(CRAC) by hydrogen peroxide. Cell Calcium 2010, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Santiago, E.; Climent, B.; Munoz, M.; Garcia-Sacristan, A.; Rivera, L.; Prieto, D. Hydrogen peroxide activates store-operated Ca2+ entry in coronary arteries. Br. J. Pharmacol. 2015, 172, 5318–5332. [Google Scholar] [CrossRef] [PubMed]
- Martinotti, S.; Laforenza, U.; Patrone, M.; Moccia, F.; Ranzato, E. Honey-mediated wound healing: H2O2 entry through aqp3 determines extracellular Ca2+ influx. Int. J. Mol. Sci. 2019, 20, 764. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium oscillations. Biochem. Soc. Symp. 2007, 74, 1–7. [Google Scholar] [CrossRef]
- Yoon, M.N.; Kim, D.K.; Kim, S.H.; Park, H.S. Hydrogen peroxide attenuates refilling of intracellular calcium store in mouse pancreatic acinar cells. Korean J. Physiol. Pharmacol. 2017, 21, 233–239. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martinotti, S.; Patrone, M.; Balbo, V.; Mazzucco, L.; Ranzato, E. Endothelial response boosted by platelet lysate: The involvement of calcium toolkit. Int. J. Mol. Sci. 2020, 21, 808. [Google Scholar] [CrossRef] [PubMed]
- Ranzato, E.; Bonsignore, G.; Patrone, M.; Martinotti, S. Endothelial and vascular health: A tale of honey, H2O2 and calcium. Cells 2021, 10, 1071. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting stim and orai proteins as an alternative approach in anticancer therapy. Curr. Med. Chem. 2016, 23, 3450–3480. [Google Scholar] [CrossRef]
- Florea, S.M.; Blatter, L.A. The effect of oxidative stress on Ca2+ release and capacitative Ca2+ entry in vascular endothelial cells. Cell Calcium 2008, 43, 405–415. [Google Scholar] [CrossRef]
- Yamamura, H.; Suzuki, Y.; Asai, K.; Imaizumi, Y.; Yamamura, H. Oxidative stress facilitates cell death by inhibiting Orai1-mediated Ca2+ entry in brain capillary endothelial cells. Biochem. Biophys. Res. Commun. 2020, 523, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Tamareille, S.; Mignen, O.; Capiod, T.; Rucker-Martin, C.; Feuvray, D. High glucose-induced apoptosis through store-operated calcium entry and calcineurin in human umbilical vein endothelial cells. Cell Calcium 2006, 39, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Galeano-Otero, I.; Del Toro, R.; Khatib, A.M.; Rosado, J.A.; Ordonez-Fernandez, A.; Smani, T. SARAF and Orai1 contribute to endothelial cell activation and angiogenesis. Front. Cell Dev. Biol. 2021, 9, 639952. [Google Scholar] [CrossRef]
- Earley, S.; Brayden, J.E. Transient receptor potential channels in the vasculature. Physiol. Rev. 2015, 95, 645–690. [Google Scholar] [CrossRef]
- Gees, M.; Colsoul, B.; Nilius, B. The role of transient receptor potential cation channels in Ca2+ signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a003962. [Google Scholar] [CrossRef]
- Moccia, F.; Lucariello, A.; Guerra, G. TRPC3-mediated Ca2+ signals as a promising strategy to boost therapeutic angiogenesis in failing hearts: The role of autologous endothelial colony forming cells. J. Cell. Physiol. 2018, 233, 3901–3917. [Google Scholar] [CrossRef]
- Balzer, M.; Lintschinger, B.; Groschner, K. Evidence for a role of Trp proteins in the oxidative stress-induced membrane conductances of porcine aortic endothelial cells. Cardiovasc. Res. 1999, 42, 543–549. [Google Scholar] [CrossRef][Green Version]
- Poteser, M.; Graziani, A.; Rosker, C.; Eder, P.; Derler, I.; Kahr, H.; Zhu, M.X.; Romanin, C.; Groschner, K. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J. Biol. Chem. 2006, 281, 13588–13595. [Google Scholar] [CrossRef]
- Antigny, F.; Girardin, N.; Frieden, M. Transient receptor potential canonical channels are required for in vitro endothelial tube formation. J. Biol. Chem. 2012, 287, 5917–5927. [Google Scholar] [CrossRef] [PubMed]
- Groschner, K.; Rosker, C.; Lukas, M. Role of TRP channels in oxidative stress. Novartis Found. Symp. 2004, 258, 222–230. [Google Scholar]
- Susankova, K.; Tousova, K.; Vyklicky, L.; Teisinger, J.; Vlachova, V. Reducing and oxidizing agents sensitize heat-activated vanilloid receptor (TRPV1) current. Mol. Pharmacol. 2006, 70, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.H.; Lin, S. Oxidative challenges sensitize the capsaicin receptor by covalent cysteine modification. Proc. Natl. Acad. Sci. USA 2009, 106, 20097–20102. [Google Scholar] [CrossRef]
- Pantke, S.; Fricke, T.C.; Eberhardt, M.J.; Herzog, C.; Leffler, A. Gating of the capsaicin receptor TRPV1 by UVA-light and oxidants are mediated by distinct mechanisms. Cell Calcium 2021, 96, 102391. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chuang, H.H. C-terminal dimerization activates the nociceptive transduction channel transient receptor potential vanilloid 1. J. Biol. Chem. 2011, 286, 40601–40607. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, N.; Kurokawa, T.; Fujiwara, K.; Polat, O.K.; Badr, H.; Takahashi, N.; Mori, Y. Functional and structural divergence in human TRPV1 channel subunits by oxidative cysteine modification. J. Biol. Chem. 2016, 291, 4197–4210. [Google Scholar] [CrossRef] [PubMed]
- DelloStritto, D.J.; Connell, P.J.; Dick, G.M.; Fancher, I.S.; Klarich, B.; Fahmy, J.N.; Kang, P.T.; Chen, Y.R.; Damron, D.S.; Thodeti, C.K.; et al. Differential regulation of TRPV1 channels by H2O2: Implications for diabetic microvascular dysfunction. Basic Res. Cardiol. 2016, 111, 21. [Google Scholar] [CrossRef]
- Chen, M.; Li, X. Role of TRPV4 channel in vasodilation and neovascularization. Microcirculation 2021, 28, e12703. [Google Scholar] [CrossRef]
- Liu, L.; Guo, M.; Lv, X.; Wang, Z.; Yang, J.; Li, Y.; Yu, F.; Wen, X.; Feng, L.; Zhou, T. Role of transient receptor potential vanilloid 4 in vascular function. Front. Mol. Biosci. 2021, 8, 677661. [Google Scholar] [CrossRef]
- Suresh, K.; Servinsky, L.; Reyes, J.; Baksh, S.; Undem, C.; Caterina, M.; Pearse, D.B.; Shimoda, L.A. Hydrogen peroxide-induced calcium influx in lung microvascular endothelial cells involves TRPV4. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 309, L1467–L1477. [Google Scholar] [CrossRef]
- Noble, M.; Mayer-Proschel, M.; Li, Z.; Dong, T.; Cui, W.; Proschel, C.; Ambeskovic, I.; Dietrich, J.; Han, R.; Yang, Y.M.; et al. Redox biology in normal cells and cancer: Restoring function of the redox/Fyn/c-Cbl pathway in cancer cells offers new approaches to cancer treatment. Free Radic. Biol. Med. 2015, 79, 300–323. [Google Scholar] [CrossRef]
- Suresh, K.; Servinsky, L.; Reyes, J.; Undem, C.; Zaldumbide, J.; Rentsendorj, O.; Modekurty, S.; Dodd, O.J.; Scott, A.; Pearse, D.B.; et al. CD36 mediates H2O2-induced calcium influx in lung microvascular endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L143–L153. [Google Scholar] [CrossRef]
- Bubolz, A.H.; Mendoza, S.A.; Zheng, X.; Zinkevich, N.S.; Li, R.; Gutterman, D.D.; Zhang, D.X. Activation of endothelial TRPV4 channels mediates flow-induced dilation in human coronary arterioles: Role of Ca2+ entry and mitochondrial ROS signaling. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H634–H642. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, S.A.; Fang, J.; Gutterman, D.D.; Wilcox, D.A.; Bubolz, A.H.; Li, R.; Suzuki, M.; Zhang, D.X. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H466–H476. [Google Scholar] [CrossRef]
- Ellinsworth, D.C.; Sandow, S.L.; Shukla, N.; Liu, Y.; Jeremy, J.Y.; Gutterman, D.D. Endothelium-derived hyperpolarization and coronary vasodilation: Diverse and integrated roles of epoxyeicosatrienoic acids, hydrogen peroxide, and gap junctions. Microcirculation 2016, 23, 15–32. [Google Scholar] [CrossRef]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Naziroglu, M. TRPM2 cation channels, oxidative stress and neurological diseases: Where are we now? Neurochem. Res. 2011, 36, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Sumoza-Toledo, A.; Penner, R. TRPM2: A multifunctional ion channel for calcium signalling. J. Physiol. 2011, 589, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Ding, R.; Yin, Y.L.; Jiang, L.H. Reactive oxygen species-induced TRPM2-mediated Ca2+ signalling in endothelial cells. Antioxidants 2021, 10, 718. [Google Scholar] [CrossRef] [PubMed]
- Prata, C.; Hrelia, S.; Fiorentini, D. Peroxiporins in cancer. Int. J. Mol. Sci. 2019, 20, 1371. [Google Scholar] [CrossRef]
- Perraud, A.L.; Takanishi, C.L.; Shen, B.; Kang, S.; Smith, M.K.; Schmitz, C.; Knowles, H.M.; Ferraris, D.; Li, W.; Zhang, J.; et al. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. J. Biol. Chem. 2005, 280, 6138–6148. [Google Scholar] [CrossRef]
- Dolle, C.; Rack, J.G.; Ziegler, M. NAD and ADP-ribose metabolism in mitochondria. FEBS J. 2013, 280, 3530–3541. [Google Scholar] [CrossRef]
- Naziroglu, M.; Luckhoff, A. A calcium influx pathway regulated separately by oxidative stress and ADP-Ribose in TRPM2 channels: Single channel events. Neurochem. Res. 2008, 33, 1256–1262. [Google Scholar] [CrossRef]
- Csanady, L.; Torocsik, B. Four Ca2+ ions activate TRPM2 channels by binding in deep crevices near the pore but intracellularly of the gate. J. Gen. Physiol. 2009, 133, 189–203. [Google Scholar] [CrossRef]
- Kolisek, M.; Beck, A.; Fleig, A.; Penner, R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol. Cell 2005, 18, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Fliegert, R.; Riekehr, W.M.; Guse, A.H. Does cyclic ADP-ribose (cADPR) activate the non-selective cation channel TRPM2? Front. Immunol. 2020, 11, 2018. [Google Scholar] [CrossRef]
- Hecquet, C.M.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ. Res. 2008, 102, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Urao, N.; Hecquet, C.M.; Zhang, M.; Sudhahar, V.; Gao, X.P.; Komarova, Y.; Ushio-Fukai, M.; Malik, A.B. Novel role of reactive oxygen species-activated Trp melastatin channel-2 in mediating angiogenesis and postischemic neovascularization. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, D.; Montorfano, I.; Cerda, O.; Caceres, M.; Becerra, A.; Cabello-Verrugio, C.; Elorza, A.A.; Riedel, C.; Tapia, P.; Velasquez, L.A.; et al. Increases in reactive oxygen species enhance vascular endothelial cell migration through a mechanism dependent on the transient receptor potential melastatin 4 ion channel. Microvasc. Res. 2015, 98, 187–196. [Google Scholar] [CrossRef]
- Foreman, M.A.; Smith, J.; Publicover, S.J. Characterisation of serum-induced intracellular Ca2+ oscillations in primary bone marrow stromal cells. J. Cell. Physiol. 2006, 206, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Pellavio, G.; Ferulli, F.; Di Nezza, F.; Shekha, M.; Lim, D.; Maestri, M.; Guerra, G.; Ambrosone, L.; Pedrazzoli, P.; et al. Nicotinic acid adenine dinucleotide phosphate (NAADP) induces intracellular Ca2+ release through the two-pore channel TPC1 in metastatic colorectal cancer cells. Cancers 2019, 11, E542. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, M.G.; Thakore, P.; Earley, S. Transient receptor potential channel ankyrin 1: A unique regulator of vascular function. Cells 2021, 10, 1167. [Google Scholar] [CrossRef]
- Thakore, P.; Alvarado, M.G.; Ali, S.; Mughal, A.; Pires, P.W.; Yamasaki, E.; Pritchard, H.A.; Isakson, B.E.; Tran, C.H.T.; Earley, S. Brain endothelial cell TRPA1 channels initiate neurovascular coupling. eLife 2021, 10, e63040. [Google Scholar] [CrossRef]
- Stoica, R.; Rusu, C.M.; Staicu, C.E.; Burlacu, A.E.; Radu, M.; Radu, B.M. Ca2+ homeostasis in brain microvascular endothelial cells. Int. Rev. Cell Mol. Biol. 2021, 362, 55–110. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Li, Y.; Attwell, D. Diverse mechanisms regulating brain energy supply at the capillary level. Curr. Opin. Neurobiol. 2021, 69, 41–50. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Duchen, M.R. The role of an astrocytic NADPH oxidase in the neurotoxicity of amyloid beta peptides. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005, 360, 2309–2314. [Google Scholar] [CrossRef] [PubMed]
- Tapella, L.; Soda, T.; Mapelli, L.; Bortolotto, V.; Bondi, H.; Ruffinatti, F.A.; Dematteis, G.; Stevano, A.; Dionisi, M.; Ummarino, S.; et al. Deletion of calcineurin from GFAP-expressing astrocytes impairs excitability of cerebellar and hippocampal neurons through astroglial Na+/K+ ATPase. Glia 2020, 68, 543–560. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.W.; Earley, S. Neuroprotective effects of TRPA1 channels in the cerebral endothelium following ischemic stroke. eLife 2018, 7, e35316. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yau, H.Y.; Wong, W.Y.; Li, R.A.; Huang, Y.; Yao, X. Role of TRPM2 in H (2)O(2)-induced cell apoptosis in endothelial cells. PLoS ONE 2012, 7, e43186. [Google Scholar] [CrossRef]
- Hixon, K.R.; Klein, R.C.; Eberlin, C.T.; Linder, H.R.; Ona, W.J.; Gonzalez, H.; Sell, S.A. A critical review and perspective of honey in tissue engineering and clinical wound healing. Adv. Wound Care 2019, 8, 403–415. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, J.; Qi, J.; Jin, Y.; Tong, L. Activation of NADPH/ROS pathway contributes to angiogenesis through JNK signaling in brain endothelial cells. Microvasc. Res. 2020, 131, 104012. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, D.; Huang, H.; Lei, Y.; Han, Y.; Han, W. Extracellular signal-regulated kinase 5 is required for low-concentration H2O2-induced angiogenesis of human umbilical vein endothelial cells. BioMed Res. Int. 2017, 2017, 6895730. [Google Scholar] [CrossRef]
- Mu, P.; Liu, Q.; Zheng, R. Biphasic regulation of H2O2 on angiogenesis implicated NADPH oxidase. Cell Biol. Int. 2010, 34, 1013–1020. [Google Scholar] [CrossRef]
- Anasooya Shaji, C.; Robinson, B.D.; Yeager, A.; Beeram, M.R.; Davis, M.L.; Isbell, C.L.; Huang, J.H.; Tharakan, B. The tri-phasic role of hydrogen peroxide in blood-brain barrier endothelial cells. Sci. Rep. 2019, 9, 133. [Google Scholar] [CrossRef]
- Park, K.M.; Park, K.D. In situ cross-linkable hydrogels as a dynamic matrix for tissue regenerative medicine. Tissue Eng. Regen. Med. 2018, 15, 547–557. [Google Scholar] [CrossRef]
- Lee, Y.; Son, J.Y.; Kang, J.I.; Park, K.M.; Park, K.D. Hydrogen peroxide-releasing hydrogels for enhanced endothelial cell activities and neovascularization. ACS Appl. Mater. Interfaces 2018, 10, 18372–18379. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Antognazza, M.R.; Lodola, F. Towards novel geneless approaches for therapeutic angiogenesis. Front. Physiol. 2020, 11, 616189. [Google Scholar] [CrossRef] [PubMed]
- Lodola, F.; Rosti, V.; Tullii, G.; Desii, A.; Tapella, L.; Catarsi, P.; Lim, D.; Moccia, F.; Antognazza, M.R. Conjugated polymers optically regulate the fate of endothelial colony-forming cells. Sci. Adv. 2019, 5, eaav4620. [Google Scholar] [CrossRef] [PubMed]
- Busselberg, D.; Florea, A.M. Targeting intracellular calcium signaling ([Ca(2+)]i) to overcome acquired multidrug resistance of cancer cells: A mini-overview. Cancers 2017, 9, 48. [Google Scholar] [CrossRef]
- Al-Taweel, N.; Varghese, E.; Florea, A.M.; Busselberg, D. Cisplatin (CDDP) triggers cell death of MCF-7 cells following disruption of intracellular calcium ([Ca2+]i) homeostasis. J. Toxicol. Sci. 2014, 39, 765–774. [Google Scholar] [CrossRef]
- Astesana, V.; Faris, P.; Ferrari, B.; Siciliani, S.; Lim, D.; Biggiogera, M.; De Pascali, S.A.; Fanizzi, F.P.; Roda, E.; Moccia, F.; et al. [Pt(O,O’-acac)(gamma-acac)(DMS)]: Alternative strategies to overcome cisplatin-induced side effects and resistance in T98G glioma cells. Cell. Mol. Neurobiol. 2020, 41, 563–587. [Google Scholar] [CrossRef]
- Noh, J.; Kwon, B.; Han, E.; Park, M.; Yang, W.; Cho, W.; Yoo, W.; Khang, G.; Lee, D. Amplification of oxidative stress by a dual stimuli-responsive hybrid drug enhances cancer cell death. Nat. Commun. 2015, 6, 6907. [Google Scholar] [CrossRef]
- Kwon, B.; Han, E.; Yang, W.; Cho, W.; Yoo, W.; Hwang, J.; Kwon, B.M.; Lee, D. Nano-fenton reactors as a new class of oxidative stress amplifying anticancer therapeutic agents. ACS Appl. Mater. Interfaces 2016, 8, 5887–5897. [Google Scholar] [CrossRef]
- Bernardini, M.; Brossa, A.; Chinigo, G.; Grolez, G.P.; Trimaglio, G.; Allart, L.; Hulot, A.; Marot, G.; Genova, T.; Joshi, A.; et al. Transient receptor potential channel expression signatures in tumor-derived endothelial cells: Functional roles in prostate cancer angiogenesis. Cancers 2019, 11, E956. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Li, C.; Yin, S.; Liu, J.; Gao, C.; Lin, Z.; Huang, R.; Huang, J.; Li, Z. Novel role of TRPV2 in promoting the cytotoxicity of H2O2-mediated oxidative stress in human hepatoma cells. Free Radic. Biol. Med. 2015, 89, 1003–1013. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, T.T.; Kirillov, A.M.; Liu, W.S.; Tang, Y. NIR light/H2O2-triggered nanocomposites for a highly efficient and selective synergistic photodynamic and photothermal therapy against hypoxic tumor cells. Chem. Commun. 2016, 52, 7939–7942. [Google Scholar] [CrossRef] [PubMed]
- Nimalasena, S.; Gothard, L.; Anbalagan, S.; Allen, S.; Sinnett, V.; Mohammed, K.; Kothari, G.; Musallam, A.; Lucy, C.; Yu, S.; et al. Intratumoral hydrogen peroxide with radiation therapy in locally advanced breast cancer: Results from a phase 1 clinical trial. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 1019–1029. [Google Scholar] [CrossRef]
- An, Q.; Sun, C.; Li, D.; Xu, K.; Guo, J.; Wang, C. Peroxidase-like activity of Fe3O4@carbon nanoparticles enhances ascorbic acid-induced oxidative stress and selective damage to PC-3 prostate cancer cells. ACS Appl. Mater. Interfaces 2013, 5, 13248–13257. [Google Scholar] [CrossRef] [PubMed]
- Verde, V.; Longo, A.; Cucci, L.M.; Sanfilippo, V.; Magri, A.; Satriano, C.; Anfuso, C.D.; Lupo, G.; La Mendola, D. Anti-angiogenic and anti-proliferative graphene oxide nanosheets for tumor cell therapy. Int. J. Mol. Sci. 2020, 21, 5571. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Xiao, W.; Song, X.; Wang, W.; Dong, X. Recent advances in tumor microenvironment hydrogen peroxide-responsive materials for cancer photodynamic therapy. Nano-Micro Lett. 2020, 12, 1–27. [Google Scholar] [CrossRef]
- Faris, P.; Ferulli, F.; Vismara, M.; Tanzi, M.; Negri, S.; Rumolo, A.; Lefkimmiatis, K.; Maestri, M.; Shekha, M.; Pedrazzoli, P.; et al. Hydrogen sulfide-evoked intracellular Ca2+ signals in primary cultures of metastatic colorectal cancer cells. Cancers 2020, 12, 3338. [Google Scholar] [CrossRef]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sinica. B 2017, 7, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Malko, P.; Jiang, L.H. TRPM2 channel-mediated cell death: An important mechanism linking oxidative stress-inducing pathological factors to associated pathological conditions. Redox Biol. 2020, 37, 101755. [Google Scholar] [CrossRef]
- Madesh, M.; Hawkins, B.J.; Milovanova, T.; Bhanumathy, C.D.; Joseph, S.K.; Ramachandrarao, S.P.; Sharma, K.; Kurosaki, T.; Fisher, A.B. Selective role for superoxide in InsP3 receptor-mediated mitochondrial dysfunction and endothelial apoptosis. J. Cell Biol. 2005, 170, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Jin, Q.; Li, Y.; Ma, Q.; Wang, J.; Li, D.; Zhou, H.; Chen, Y. Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca2+]c/VDAC-[Ca2+]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones 2017, 23, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Mazzucchelli, I.; Lisini, D.; Garofoli, F.; Dragoni, S.; Angelini, M.; Pozzi, M.; Bonetti, E.; Tzialla, C.; Kramer, B.W.; Spinillo, A.; et al. Expression and function of toll-like receptors in human circulating endothelial colony forming cells. Immunol. Lett. 2015, 168, 98–104. [Google Scholar] [CrossRef]
- Tocchetti, C.G.; Molinaro, M.; Angelone, T.; Lionetti, V.; Madonna, R.; Mangiacapra, F.; Moccia, F.; Penna, C.; Sartiani, L.; Quaini, F.; et al. Nitroso-redox balance and modulation of basal myocardial function: An update from the italian society of cardiovascular research (SIRC). Curr. Drug Targets 2015, 16, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Deruy, E.; Peugnet, V.; Turkieh, A.; Pinet, F. Oxidative stress in cardiovascular diseases. Antioxidants 2020, 9, 864. [Google Scholar] [CrossRef]
- Papaharalambus, C.A.; Griendling, K.K. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends Cardiovasc. Med. 2007, 17, 48–54. [Google Scholar] [CrossRef]
- Adams, J.A.; Uryash, A.; Lopez, J.R.; Sackner, M.A. The endothelium as a therapeutic target in diabetes: A narrative review and perspective. Front. Physiol. 2021, 12, 638491. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.Z.; Wang, D.; Braun, A.P. DAF-FM (4-amino-5-methylamino-2′,7′-difluorofluorescein) diacetate detects impairment of agonist-stimulated nitric oxide synthesis by elevated glucose in human vascular endothelial cells: Reversal by vitamin C and L-sepiapterin. J. Pharmacol. Exp. Ther. 2005, 315, 931–940. [Google Scholar] [CrossRef]
- Ding, H.; Triggle, C.R. Endothelial dysfunction in diabetes: Multiple targets for treatment. Pflugers Arch. 2010, 459, 977–994. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Feng, Q.; Arnold, M.; Peng, T. Calpain activation contributes to hyperglycaemia-induced apoptosis in cardiomyocytes. Cardiovasc. Res. 2009, 84, 100–110. [Google Scholar] [CrossRef]
- Martines, A.; Stifanese, R.; Faelli, E.L.; Perasso, L.; Melloni, I.; Ruggeri, P.; Averna, M. Calpain-1 resident in lipid raft/caveolin-1 membrane microdomains plays a protective role in endothelial cells. Biochimie 2017, 133, 20–27. [Google Scholar] [CrossRef]
- Stalker, T.J.; Gong, Y.; Scalia, R. The calcium-dependent protease calpain causes endothelial dysfunction in type 2 diabetes. Diabetes 2005, 54, 1132–1140. [Google Scholar] [CrossRef]
- Brechard, S.; Tschirhart, E.J. Regulation of superoxide production in neutrophils: Role of calcium influx. J. Leukoc. Biol. 2008, 84, 1223–1237. [Google Scholar] [CrossRef]
- Brechard, S.; Plancon, S.; Melchior, C.; Tschirhart, E.J. STIM1 but not STIM2 is an essential regulator of Ca2+ influx-mediated NADPH oxidase activity in neutrophil-like HL-60 cells. Biochem. Pharmacol. 2009, 78, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.; Gori, T.; Munzel, T. Oxidative stress and endothelial dysfunction in hypertension. Hypertens. Res. 2011, 34, 665–673. [Google Scholar] [CrossRef]
- Norton, C.E.; Jacobsen, N.L.; Sinkler, S.Y.; Manrique-Acevedo, C.; Segal, S.S. Female sex and Western-style diet protect mouse resistance arteries during acute oxidative stress. Am. J. Physiol. Cell Physiol. 2020, 318, C627–C639. [Google Scholar] [CrossRef] [PubMed]
- Socha, M.J.; Boerman, E.M.; Behringer, E.J.; Shaw, R.L.; Domeier, T.L.; Segal, S.S. Advanced age protects microvascular endothelium from aberrant Ca2+ influx and cell death induced by hydrogen peroxide. J. Physiol. 2015, 593, 2155–2169. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, L.; Moreno-Vinasco, L.; Lang, G.D.; Siegler, J.H.; Mathew, B.; Usatyuk, P.V.; Samet, J.M.; Geyh, A.S.; Breysse, P.N.; et al. Particulate matter air pollution disrupts endothelial cell barrier via calpain-mediated tight junction protein degradation. Part. Fibre Toxicol. 2012, 9, 35. [Google Scholar] [CrossRef]
- Deweirdt, J.; Quignard, J.F.; Crobeddu, B.; Baeza-Squiban, A.; Sciare, J.; Courtois, A.; Lacomme, S.; Gontier, E.; Muller, B.; Savineau, J.P.; et al. Involvement of oxidative stress and calcium signaling in airborne particulate matter—induced damages in human pulmonary artery endothelial cells. Toxicol. Vitr. 2017, 45, 340–350. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, S.B.; Park, J.K.; Yoo, Y.D. TNF-alpha-induced ROS production triggering apoptosis is directly linked to Romo1 and Bcl-X(L). Cell Death Differ. 2010, 17, 1420–1434. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Iadecola, C. The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
- Zhang, L.; Papadopoulos, P.; Hamel, E. Endothelial TRPV4 channels mediate dilation of cerebral arteries: Impairment and recovery in cerebrovascular pathologies related to Alzheimer’s disease. Br. J. Pharmacol. 2013, 170, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhang, R.; Wang, S.; Zhang, D.; Leung, C.K.; Yang, G.; Li, Y.; Liu, L.; Xu, Y.; Lin, S.; et al. Methamphetamine and HIV-tat protein synergistically induce oxidative stress and blood-brain barrier damage via transient receptor potential melastatin 2 channel. Front. Pharmacol. 2021, 12, 619436. [Google Scholar] [CrossRef]
- Raghunatha, P.; Vosoughi, A.; Kauppinen, T.M.; Jackson, M.F. Microglial NMDA receptors drive pro-inflammatory responses via PARP-1/TRMP2 signaling. Glia 2020, 68, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Negri, S.; Faris, P.; Maniezzi, C.; Pellavio, G.; Spaiardi, P.; Botta, L.; Laforenza, U.; Biella, G.; Moccia, D.F. NMDA receptors elicit flux-independent intracellular Ca2+ signals via metabotropic glutamate receptors and flux-dependent nitric oxide release in human brain microvascular endothelial cells. Cell Calcium 2021, 99, 102454. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liang, T.; Luo, Q.; Li, P.; Zhang, R.; Xu, M.; Su, J.; Xu, T.; Wu, Q. H9N2 swine influenza virus infection-induced damage is mediated by TRPM2 channels in mouse pulmonary microvascular endothelial cells. Microb. Pathog. 2020, 148, 104408. [Google Scholar] [CrossRef] [PubMed]
- Abuarab, N.; Munsey, T.S.; Jiang, L.H.; Li, J.; Sivaprasadarao, A. High glucose-induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn2+-mediated mitochondrial fission. Sci. Signal. 2017, 10, eaal4161. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, K.; Wang, G.; Park, L. Endothelial dysfunction and amyloid-beta-induced neurovascular alterations. Cell. Mol. Neurobiol. 2016, 36, 155–165. [Google Scholar] [CrossRef]
- Becerra, A.; Echeverria, C.; Varela, D.; Sarmiento, D.; Armisen, R.; Nunez-Villena, F.; Montecinos, M.; Simon, F. Transient receptor potential melastatin 4 inhibition prevents lipopolysaccharide-induced endothelial cell death. Cardiovasc. Res. 2011, 91, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.X.; Zhang, Y.Y.; Wu, X.Y.; Tang, H.X.; Liang, X.Q.; Xue, Z.M.; Xue, Y.D.; Li, J.; Zhu, H.; Huo, R.; et al. Transient receptor potential melastatin 4 contributes to early-stage endothelial injury induced by arsenic trioxide. Toxicol. Lett. 2019, 312, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Vineetha, V.P.; Raghu, K.G. An overview on arsenic trioxide-induced cardiotoxicity. Cardiovasc. Toxicol. 2019, 19, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Bertoni, G.; Pla, A.F.; Dragoni, S.; Pupo, E.; Merlino, A.; Mancardi, D.; Munaron, L.; Tanzi, F. Hydrogen sulfide regulates intracellular Ca2+ concentration in endothelial cells from excised rat aorta. Curr. Pharm. Biotechnol. 2011, 12, 1416–1426. [Google Scholar] [CrossRef]
- Kurakula, K.; Smolders, V.; Tura-Ceide, O.; Jukema, J.W.; Quax, P.H.A.; Goumans, M.J. Endothelial dysfunction in pulmonary hypertension: Cause or consequence? Biomedicines 2021, 9, 57. [Google Scholar] [CrossRef]
- Ranchoux, B.; Harvey, L.D.; Ayon, R.J.; Babicheva, A.; Bonnet, S.; Chan, S.Y.; Yuan, J.X.; Perez, V.J. Endothelial dysfunction in pulmonary arterial hypertension: An evolving landscape (2017 Grover Conference Series). Pulm. Circ. 2018, 8, 2045893217752912. [Google Scholar] [CrossRef]
- Duong, H.T.; Comhair, S.A.; Aldred, M.A.; Mavrakis, L.; Savasky, B.M.; Erzurum, S.C.; Asosingh, K. Pulmonary artery endothelium resident endothelial colony-forming cells in pulmonary arterial hypertension. Pulm. Circ. 2011, 1, 475–486. [Google Scholar] [CrossRef]
- Weise-Cross, L.; Resta, T.C.; Jernigan, N.L. Redox regulation of ion channels and receptors in pulmonary hypertension. Antioxid. Redox Signal. 2019, 31, 898–915. [Google Scholar] [CrossRef]
- Suresh, K.; Servinsky, L.; Jiang, H.; Bigham, Z.; Zaldumbide, J.; Huetsch, J.C.; Kliment, C.; Acoba, M.G.; Kirsch, B.J.; Claypool, S.M.; et al. Regulation of mitochondrial fragmentation in microvascular endothelial cells isolated from the SU5416/hypoxia model of pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L639–L652. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Negri, S.; Faris, P.; Moccia, F. Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime? Int. J. Mol. Sci. 2021, 22, 9821. https://doi.org/10.3390/ijms22189821
Negri S, Faris P, Moccia F. Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime? International Journal of Molecular Sciences. 2021; 22(18):9821. https://doi.org/10.3390/ijms22189821
Chicago/Turabian StyleNegri, Sharon, Pawan Faris, and Francesco Moccia. 2021. "Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime?" International Journal of Molecular Sciences 22, no. 18: 9821. https://doi.org/10.3390/ijms22189821
APA StyleNegri, S., Faris, P., & Moccia, F. (2021). Reactive Oxygen Species and Endothelial Ca2+ Signaling: Brothers in Arms or Partners in Crime? International Journal of Molecular Sciences, 22(18), 9821. https://doi.org/10.3390/ijms22189821