
MicroRNAs and Calcium Signaling in Heart Disease



Abstract
:1. Introduction
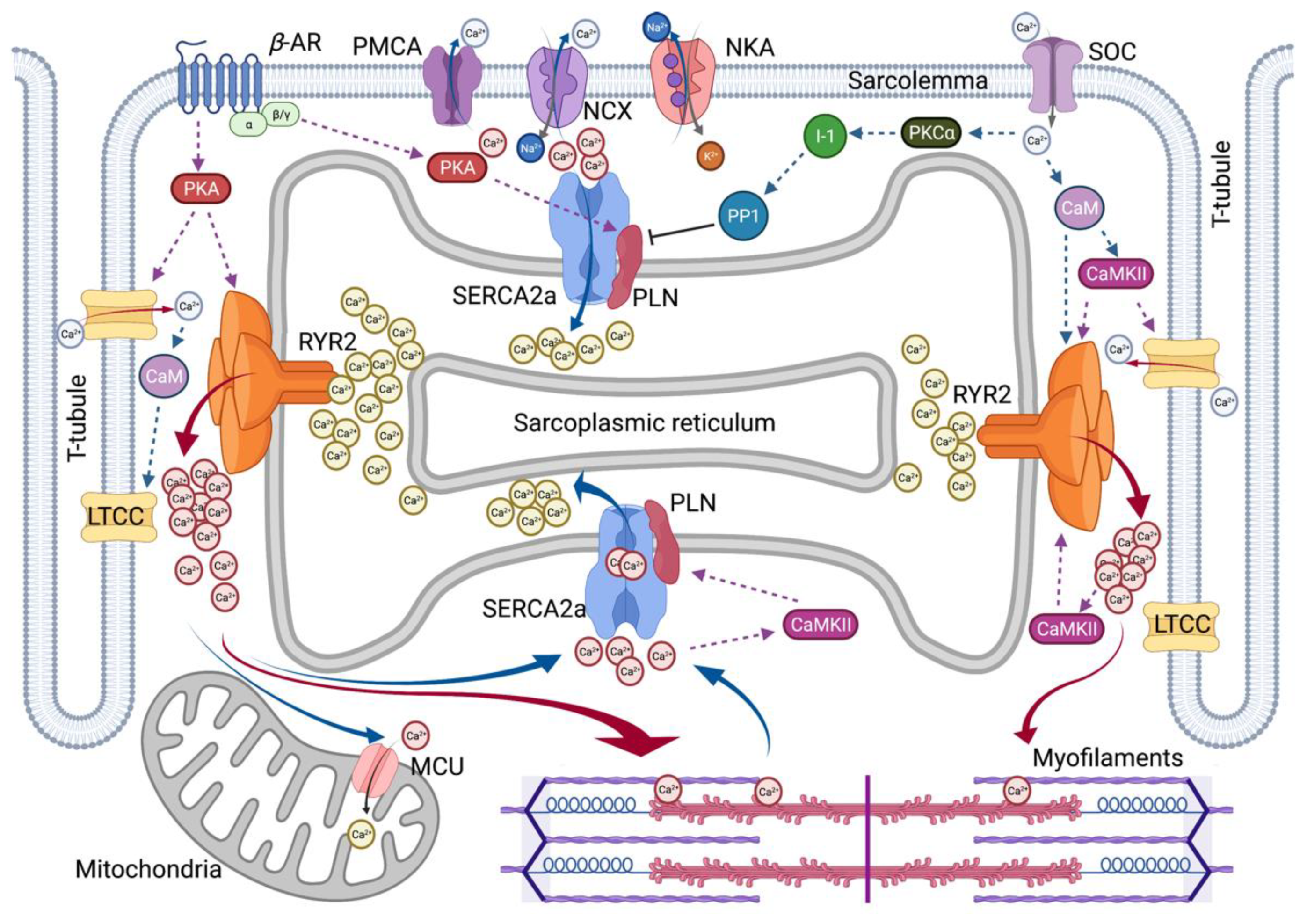
2. Roles of Calcium in Heart Contractility
2.1. Excitation–Contraction Coupling
2.2. Two Major Players in SR Calcium Flux in Heart Disease
2.2.1. SERCA2a Calcium Pump
2.2.2. RyR2 Calcium Release Channel
2.3. Calcium Signaling through Protein Kinases
2.3.1. β–Adrenergic Receptor-Mediated PAK Regulation
2.3.2. Calcium-Calmodulin Mediated CaMKII Regulation
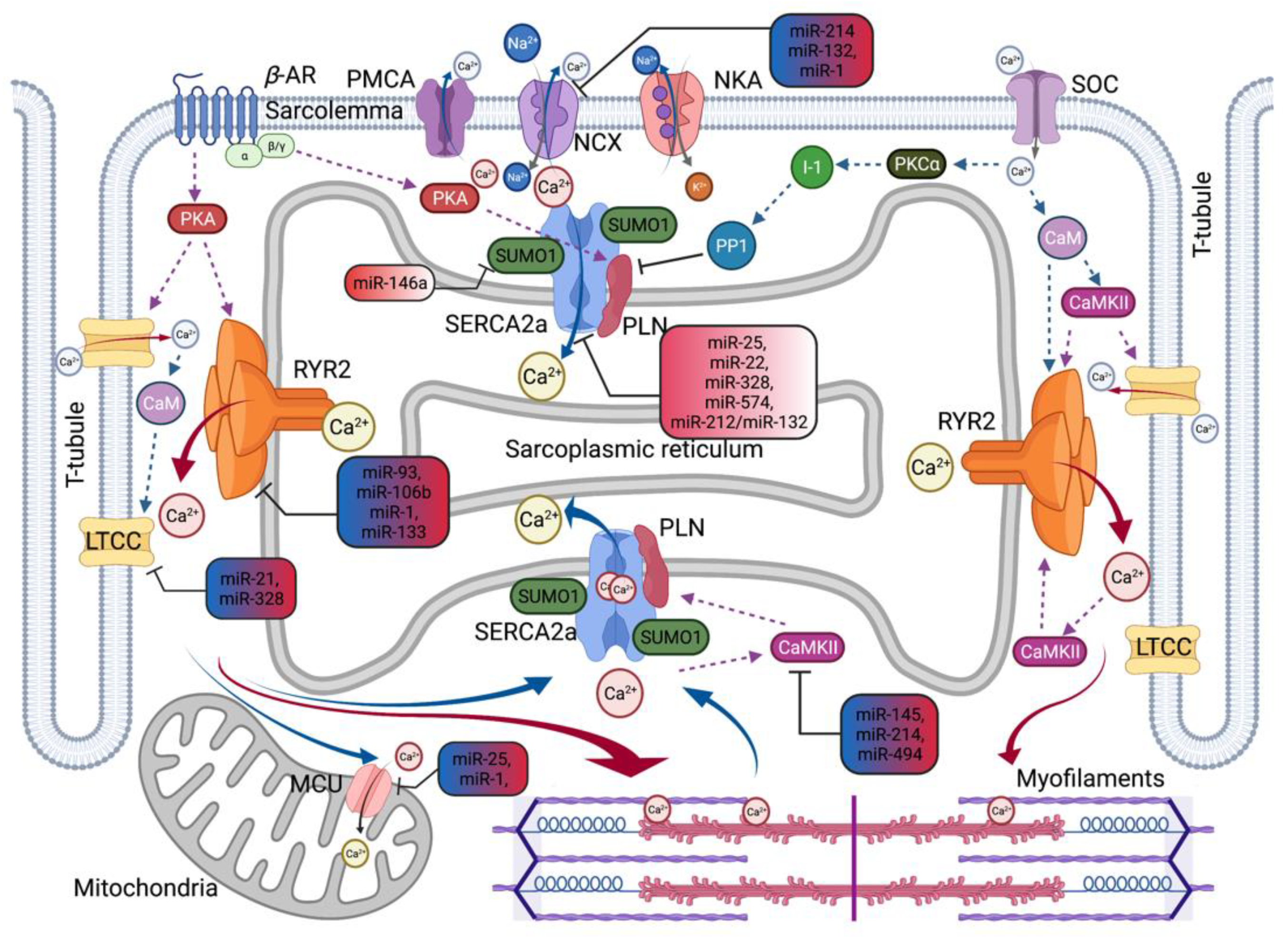
2.4. MicroRNAs as a New Modulator of Calcium Signaling Pathway
2.4.1. Calcium Regulating MicroRNAs Related to Hypertrophy and Heart Failure
2.4.2. Calcium Regulating MicroRNAs Related to Ischemic Heart Disease
2.4.3. Calcium Regulating MicroRNAs Related to Atrial Fibrillation
2.5. miRNAs as Potential Therapeutics for Heart Disease
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef]
- Mohammadzadeh, N.; Safdari, R.; Rahimi, A. Multi-agent system as a new approach to effective chronic heart failure management: Key considerations. Healthc. Inform. Res. 2013, 19, 162–166. [Google Scholar] [CrossRef] [Green Version]
- Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Heart disease and stroke statistics—2012 update: A report from the American Heart Association. Circulation 2012, 125, e2–e220. [Google Scholar]
- Clerkin, K.J.; Fried, J.A.; Raikhelkar, J.; Sayer, G.; Griffin, J.M.; Masoumi, A.; Jain, S.S.; Burkhoff, D.; Kumaraiah, D.; Rabbani, L.; et al. COVID-19 and Cardiovascular Disease. Circulation 2020, 141, 1648–1655. [Google Scholar] [CrossRef] [Green Version]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.B.; Anderson, R.N. The Leading Causes of Death in the US for 2020. JAMA 2021, 325, 1829–1830. [Google Scholar] [CrossRef]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef] [Green Version]
- Fearnley, C.J.; Roderick, H.L.; Bootman, M.D. Calcium Signaling in Cardiac Myocytes. Cold Spring Harb. Perspect. Biol. 2011, 3, a004242. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M. Cardiac excitation—Contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Dewenter, M.; von der Lieth, A.; Katus, H.A.; Backs, J. Calcium Signaling and Transcriptional Regulation in Cardiomyocytes. Circ. Res. 2017, 121, 1000–1020. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Napolitano, C.; Tiso, N.; Memmi, M.; Vignati, G.; Bloise, R.; Sorrentino, V.; Danieli, G.A. Danieli Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001, 103, 196–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiso, N.; Stephan, D.A.; Nava, A.; Bagattin, A.; Devaney, J.M.; Stanchi, F.; Larderet, G.; Brahmbhatt, B.; Brown, K.; Bauce, B.; et al. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Hum. Mol. Genet. 2001, 10, 189–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dridi, H.; Kushnir, A.; Zalk, R.; Yuan, Q.; Melville, Z.; Marks, A.R. Intracellular calcium leak in heart failure and atrial fibrillation: A unifying mechanism and therapeutic target. Nat. Rev. Cardiol. 2020, 17, 732–747. [Google Scholar] [CrossRef]
- Hasenfuss, G.; Reinecke, H.; Studer, R.; Meyer, M.; Pieske, B.; Holtz, J.; Holubarsch, C.; Posival, H.; Just, H.; Drexler, H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca(2+)-ATPase in failing and nonfailing human myocardium. Circ. Res. 1994, 75, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Schillinger, W.; Pieske, B.; Holubarsch, C.; Heilmann, C.; Posival, H.; Kuwajima, G.; Mikoshiba, K.; Just, H.; Hasenfuss, G. Alterations of sarcoplasmic reticulum proteins in failing human dilated cardiomyopathy. Circulation 1995, 92, 778–784. [Google Scholar] [CrossRef]
- Flesch, M.; Schwinger, R.H.; Schnabel, P.; Schiffer, F.; van Gelder, I.; Bavendiek, U.; Südkamp, M.; Kuhn-Regnier, F.; Böhm, M. Sarcoplasmic reticulum Ca2+ATPase and phospholamban mRNA and protein levels in end-stage heart failure due to ischemic or dilated cardiomyopathy. J. Mol. Med. 1996, 74, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Van der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 199–207. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Jordan, E. Dilated Cardiomyopathy Overview. GeneReviews [Internet]. 2007. Available online: https://pubmed.ncbi.nlm.nih.gov/20301486/ (accessed on 27 September 2021).
- Lahat, H.; Pras, E.; Olender, T.; Avidan, N.; Ben-Asher, E.; Man, O.; Levy-Nissenbaum, E.; Khoury, A.; Lorber, A.; Goldman, B.; et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am. J. Hum. Genet. 2001, 69, 1378–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postma, A.V.; Denjoy, I.; Hoorntje, T.M.; Lupoglazoff, J.M.; da Costa, A.; Sebillon, P.; Mannens, M.M.; Wilde, A.A.; Guicheney, P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2002, 91, e21–e26. [Google Scholar] [CrossRef]
- Chazin, W.J.; Johnson, C.N. Calmodulin Mutations Associated with Heart Arrhythmia: A Status Report. Int. J. Mol. Sci. 2020, 21, 1418. [Google Scholar] [CrossRef] [Green Version]
- Roux-Buisson, N.; Cacheux, M.; Fourest-Lieuvin, A.; Fauconnier, J.; Brocard, J.; Denjoy, I.; Durand, P.; Guicheney, P.; Kyndt, F.; Leenhardt, A.; et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum. Mol. Genet. 2012, 21, 2759–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooryck, C.; Kyndt, F.; Bozon, D.; Roux-Buisson, N.; Sacher, F.; Probst, V.; Thambo, J.B. New Family With Catecholaminergic Polymorphic Ventricular Tachycardia Linked to the Triadin Gene. J. Cardiovasc. Electrophysiol. 2015, 26, 1146–1150. [Google Scholar] [CrossRef]
- Gergs, U.; Berndt, T.; Buskase, J.; Jones, L.R.; Kirchhefer, U.; Müller, F.U.; Schlüter, K.D.; Schmitz, W.; Neumann, J. On the role of junctin in cardiac Ca2+ handling, contractility, and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H728–H734. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, D.A.; Sanoudou, D.; Kolokathis, F.; Vafiadaki, E.; Papalouka, V.; Kontrogianni-Konstantopoulos, A.; Theodorakis, G.N.; Paraskevaidis, I.A.; Adamopoulos, S.; Dorn, G.W., 2nd; et al. The Ser96Ala variant in histidine-rich calcium-binding protein is associated with life-threatening ventricular arrhythmias in idiopathic dilated cardiomyopathy. Eur. Heart J. 2008, 29, 2514–2525. [Google Scholar] [CrossRef] [Green Version]
- Amioka, M.; Nakano, Y.; Ochi, H.; Onohara, Y.; Sairaku, A.; Tokuyama, T.; Motoda, C.; Matsumura, H.; Tomomori, S.; Hironobe, N.; et al. Ser96Ala genetic variant of the human histidine-rich calcium-binding protein is a genetic predictor of recurrence after catheter ablation in patients with paroxysmal atrial fibrillation. PLoS ONE 2019, 14, e0213208. [Google Scholar] [CrossRef] [PubMed]
- Del Monte, F.; Harding, S.E.; Schmidt, U.; Matsui, T.; Kang, Z.B.; Dec, G.W.; Gwathmey, J.K.; Rosenzweig, A.; Hajjar, R.J. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 1999, 100, 2308–2311. [Google Scholar] [CrossRef] [Green Version]
- Lyon, A.R.; Bannister, M.L.; Collins, T.; Pearce, E.; Sepehripour, A.H.; Dubb, S.S.; Garcia, E.; O’Gara, P.; Liang, L.; Kohlbrenner, E.; et al. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ. Arrhythm. Electrophysiol. 2011, 4, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Kawase, Y.; Ly, H.Q.; Prunier, F.; Lebeche, D.; Shi, Y.; Jin, H.; Hadri, L.; Yoneyama, R.; Hoshino, K.; Takewa, Y.; et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J. Am. Coll. Cardiol. 2008, 51, 1112–1119. [Google Scholar] [CrossRef] [Green Version]
- Hajjar, R.J.; Zsebo, K.; Deckelbaum, L.; Thompson, C.; Rudy, J.; Yaroshinsky, A.; Ly, H.; Kawase, Y.; Wagner, K.; Borow, K.; et al. Design of a phase 1/2 trial of intracoronary administration of AAV1/SERCA2a in patients with heart failure. J. Card. Fail. 2008, 14, 355–367. [Google Scholar] [CrossRef]
- Jaski, B.E.; Jessup, M.L.; Mancini, D.M.; Cappola, T.P.; Pauly, D.F.; Greenberg, B.; Borow, K.; Dittrich, H.; Zsebo, K.M.; Hajjar, R.J. Calcium Up-Regulation by Percutaneous Administration of Gene Therapy In Cardiac Disease (CUPID) Trial Investigators. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J. Card. Fail. 2009, 15, 171–181. [Google Scholar]
- Hulot, J.S.; Salem, J.E.; Redheuil, A.; Collet, J.P.; Varnous, S.; Jourdain, P.; Logeart, D.; Gandjbakhch, E.; Bernard, C.; Hatem, S.N.; et al. AGENT-HF Investigators. Effect of intracoronary administration of AAV1/SERCA2a on ventricular remodelling in patients with advanced systolic heart failure: Results from the AGENT-HF randomized phase 2 trial. Eur. J. Heart Fail. 2017, 19, 1534–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, A.R.; Babalis, D.; Morley-Smith, A.C.; Hedger, M.; Suarez Barrientos, A.; Foldes, G.; Couch, L.S.; Chowdhury, R.A.; Tzortzis, K.N.; Peters, N.S.; et al. Investigation of the safety and feasibility of AAV1/SERCA2a gene transfer in patients with chronic heart failure supported with a left ventricular assist device—The SERCA-LVAD TRIAL. Gene Ther. 2020, 27, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, U.; Hajjar, R.J.; Kim, C.S.; Lebeche, D.; Doye, A.A.; Gwathmey, J.K. Human heart failure: cAMP stimulation of SR Ca(2+)-ATPase activity and phosphorylation level of phospholamban. Am. J. Physiol. 1999, 277, H474–H480. [Google Scholar] [CrossRef] [PubMed]
- Schwinger, R.H.; Munch, G.; Bolck, B.; Karczewski, P.; Krause, E.G.; Erdmann, E. Reduced Ca(2+)-sensitivity of SERCA 2a in failing human myocardium due to reduced serin-16 phospholamban phosphorylation. J. Mol. Cell. Cardiol. 1999, 31, 479–491. [Google Scholar] [CrossRef]
- Haghighi, K.; Bidwell, P.; Kranias, E.G. Phospholamban interactome in cardiac contractility and survival: A new vision of an old friend. J. Mol. Cell. Cardiol. 2014, 77, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Nelson, B.R.; Makarewich, C.A.; Anderson, D.M.; Winders, B.R.; Troupes, C.D.; Wu, F.; Reese, A.L.; McAnally, J.R.; Chen, X.; Kavalali, E.T.; et al. A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 2016, 351, 271–275. [Google Scholar] [CrossRef] [Green Version]
- Karakikes, I.; Stillitano, F.; Nonnenmacher, M.; Tzimas, C.; Sanoudou, D.; Termglinchan, V.; Kong, C.W.; Rushing, S.; Hansen, J.; Ceholski, D.; et al. Correction of human phospholamban R14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat. Commun. 2015, 6, 6955. [Google Scholar] [CrossRef]
- Doevendans, P.A.; Glijnis, P.C.; Kranias, E.G. Leducq Transatlantic Network of Excellence to Cure Phospholamban-Induced Cardiomyopathy (CURE-PLaN). Circ. Res. 2019, 125, 720–724. [Google Scholar] [CrossRef]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Chaanine, A.H.; Kizana, E.; Park, W.J.; Hajjar, R.J. SUMO1-dependent modulation of SERCA2a in heart failure. Nature 2011, 477, 601–605. [Google Scholar] [CrossRef]
- Lee, A.; Jeong, D.; Mitsuyama, S.; Oh, J.G.; Liang, L.; Ikeda, Y.; Sadoshima, J.; Hajjar, R.J.; Kho, C. The role of SUMO-1 in cardiac oxidative stress and hypertrophy. Antioxid. Redox Signal. 2014, 21, 1986–2001. [Google Scholar] [CrossRef] [Green Version]
- Tilemann, L.; Lee, A.; Ishikawa, K.; Aguero, J.; Rapti, K.; Santos-Gallego, C.; Kohlbrenner, E.; Fish, K.M.; Kho, C.; Hajjar, R.J. SUMO-1 gene transfer improves cardiac function in a large-animal model of heart failure. Sci. Transl. Med. 2013, 5, 211ra159. [Google Scholar] [CrossRef] [PubMed]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Gorski, P.A.; Fish, K.; Sanchez, R.; deVita, R.J.; Christensen, G.; Dahl, R.; et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 2015, 6, 7229. [Google Scholar] [CrossRef] [PubMed]
- Gorski, P.A.; Jang, S.P.; Jeong, D.; Lee, A.; Lee, P.; Oh, J.G.; Chepurko, V.; Yang, D.K.; Kwak, T.H.; Eom, S.H.; et al. Role of SIRT1 in Modulating Acetylation of the Sarco-Endoplasmic Reticulum Ca2+-ATPase in Heart Failure. Circ. Res. 2019, 124, e63–e80. [Google Scholar] [CrossRef] [PubMed]
- Zalk, R.; Lehnart, S.E.; Marks, A.R. Modulation of the ryanodine receptor and intracellular calcium. Annu. Rev. Biochem. 2007, 76, 367–385. [Google Scholar] [CrossRef] [PubMed]
- Federico, M.; Valverde, C.A.; Mattiazzi, A.; Palomeque, J. Unbalance Between Sarcoplasmic Reticulum Ca2+ Uptake and Release: A First Step Toward Ca2+ Triggered Arrhythmias and Cardiac Damage. Front. Physiol. 2020, 10, 1630. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, D.; Wehrens, X.H. Role of RyR2 Phosphorylation in Heart Failure and Arrhythmias. Circ. Res. 2014, 114, 1311–1319. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Kranias, E.G.; Mignery, G.A.; Bers, D.M. Protein kinase A phosphorylation of the ryanodine receptor does not affect calcium sparks in mouse ventricular myocytes. Circ. Res. 2002, 90, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Jayaraman, T.; Burkhoff, D.; Rosemblit, N.; Marks, A.R. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): Defective regulation in failing hearts. Cell 2000, 101, 365–376. [Google Scholar] [CrossRef] [Green Version]
- Belevych, A.E.; Radwanski, P.B.; Carnes, C.A.; Gyorke, S. ‘Ryanopathy’: Causes and manifestations of RyR2 dysfunction in heart failure. Cardiovasc. Res. 2013, 98, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Bongianino, R.; Denegri, M.; Mazzanti, A.; Lodola, F.; Vollero, A.; Boncompagni, S.; Fasciano, S.; Rizzo, G.; Mangione, D.; Barbaro, S.; et al. Allele-Specific Silencing of Mutant mRNA Rescues Ultrastructural and Arrhythmic Phenotype in Mice Carriers of the R4496C Mutation in the Ryanodine Receptor Gene (RYR2). Circ. Res. 2017, 121, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Patrick, C.; Tarah, A.W.; Xander, H.T.W. Targeting Pathological Leak of Ryanodine Receptors: Preclinical Progress and the Potential Impact on Treatments for Cardiac Arrhythmias and Heart Failure. Expert Opin. Ther. Targets 2020, 24, 25–36. [Google Scholar]
- Packer, M. The neurohormonal hypothesis: A theory to explain the mechanism of disease progression in heart failure. J. Am. Coll. Cardiol. 1992, 20, 248–254. [Google Scholar] [CrossRef] [Green Version]
- El-Armouche, A.; Boknik, P.; Eschenhagen, T.; Carrier, L.; Knaut, M.; Ravens, U.; Dobrev, D. Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation 2006, 114, 670–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakhary, D.R.; Moravec, C.S.; Stewart, R.W.; Bond, M. Protein kinase A (PKA)-dependent troponin-I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy. Circulation 1999, 99, 505–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacques, A.M.; Copeland, O.; Messer, A.E.; Gallon, C.E.; King, K.; McKenna, W.J.; Tsang, V.T.; Marston, S.B. Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle. J. Mol. Cell. Cardiol. 2008, 45, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Arneja, A.S.; Dhalla, N.S. Alterations in protein kinase A and protein kinase C levels in heart failure due to genetic cardiomyopathy. Can. J. Cardiol. 1999, 15, 683–690. [Google Scholar]
- Maier, L.S.; Zhang, T.; Chen, L.; DeSantiago, J.; Brown, J.H.; Bers, D.M. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: Reduced SR Ca2+ load and activated SR Ca2+ release. Circ. Res. 2003, 92, 904–911. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Hagenmueller, M.; Riffel, J.H.; Kreusser, M.M.; Bernhold, E.; Fan, J.; Katus, H.A.; Backs, J.; Hardt, S.E. Calcium/calmodulin-dependent protein kinase II couples Wnt signaling with histone deacetylase 4 and mediates dishevelled-induced cardiomyopathy. Hypertension 2015, 65, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Erickson, J.R. Mechanisms of CaMKII activation in the heart. Front. Pharmacol. 2014, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, P.D.; Purohit, A.; Hund, T.J.; Anderson, M.E. CaMKII: Linking heart failure and arrhythmias. Circ. Res. 2012, 110, 1661–1677. [Google Scholar] [CrossRef] [Green Version]
- Bell, J.R.; Vila-Petroff, M.; Delbridge, L.M. CaMKII-dependent responses to ischemia and reperfusion challenges in the heart. Front. Pharmacol. 2014, 5, 96. [Google Scholar] [CrossRef]
- Hegyi, B.; Bers, D.M.; Bossuyt, J. CaMKII signaling in heart diseases: Emerging role in diabetic cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 127, 246–259. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barwari, T.; Joshi, A.; Mayr, M. MicroRNAs in Cardiovascular Disease. J. Am. Coll. Cardiol. 2016, 68, 2577–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, M.; Luo, X.; Murohara, T.; Yang, B.; Dobrev, D.; Nattel, S. MicroRNA regulation and cardiac calcium signaling: Role in cardiac disease and therapeutic potential. Circ. Res. 2014, 114, 689–705. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhang, Y.; Wang, Y.; Zhao, Y.; Ding, H.; Li, P. Circular RNAs: Functions and Clinical Significance in Cardiovascular Disease. Front. Cell Dev. Biol. 2020, 8, 584051. [Google Scholar] [CrossRef] [PubMed]
- Kura, B.; Parikh, M.; Slezak, J.; Pierce, G.N. The Influence of Diet on MicroRNAs that Impact Cardiovascular Disease. Molecules 2019, 24, 1509. [Google Scholar] [CrossRef] [Green Version]
- Cannataro, R.; Perri, M.; Gallelli, L.; Caroleo, M.C.; de Sarro, G.; Cione, E. Ketogenic Diet Acts on Body Remodeling and MicroRNAs Expression Profile. Microrna 2019, 8, 116–126. [Google Scholar] [CrossRef]
- Watson, C.J.; Gupta, S.K.; O’Connell, E.; Thum, S.; Glezeva, N.; Fendrich, J.; Gallagher, J.; Ledwidge, M.; Grote-Levi, L.; McDonald, K.; et al. MicroRNA signatures differentiate preserved from reduced ejection fraction heart failure. Eur. J. Heart Fail. 2015, 17, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Quan, X.; Ji, Y.; Zhang, C.; Guo, X.; Zhang, Y.; Jia, S.; Ma, W.; Fan, Y.; Wang, C. Circulating MiR-146a May be a Potential Biomarker of Coronary Heart Disease in Patients with Subclinical Hypothyroidism. Cell. Physiol. Biochem. 2018, 45, 226–236. [Google Scholar] [CrossRef]
- Oh, J.G.; Watanabe, S.; Lee, A.; Gorski, P.A.; Lee, P.; Jeong, D.; Liang, L.; Liang, Y.; Baccarini, A.; Sahoo, S.; et al. miR-146a Suppresses SUMO1 Expression and Induces Cardiac Dysfunction in Maladaptive Hypertrophy. Circ. Res. 2018, 123, 673–685. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Houser, S.R.; Molkentin, J.D. Does contractile Ca2+ control calcineurin-NFAT signaling and pathological hypertrophy in cardiac myocytes? Sci. Signal. 2008, 1, pe31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Costa Martins, P.A.; Bourajjaj, M.; Gladka, M.; Kortland, M.; van Oort, R.J.; Pinto, Y.M.; Molkentin, J.D.; de Windt, L.J. Conditional Dicer Gene Deletion in the Postnatal Myocardium Provokes Spontaneous Cardiac Remodeling. Circulation 2008, 118, 1567–1576. [Google Scholar] [CrossRef] [Green Version]
- Van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kura, B.; Kalocayova, B.; Devaux, Y.; Bartekova, M. Potential Clinical Implications of miR-1 and miR-21 in Heart Disease and Cardioprotection. Int. J. Mol. Sci. 2020, 21, 700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, S.; He, A.; Kong, S.W.; Lu, J.; Bejar, R.; Bodyak, N.; Lee, K.H.; Ma, Q.; Kang, P.M.; Golub, T.R.; et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol. Cell. Biol. 2009, 29, 2193–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakikes, I.; Chaanine, A.H.; Kang, S.; Mukete, B.N.; Jeong, D.; Zhang, S.; Hajjar, R.J.; Lebeche, D. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J. Am. Heart Assoc. 2013, 2, e000078. [Google Scholar] [CrossRef] [Green Version]
- Icli, B.; Dorbala, P.; Feinberg, M.W. An emerging role for the miR-26 family in cardiovascular disease. Trends Cardiovasc. Med. 2014, 6, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Gurha, P.; Abreu-Goodger, C.; Wang, T.; Ramirez, M.O.; Drumond, A.L.; van Dongen, S.; Chen, Y.; Bartonicek, N.; Enright, A.J.; Lee, B.; et al. Targeted Deletion of MicroRNA-22 Promotes Stress-Induced Cardiac Dilation and Contractile Dysfunction. Circulation 2012, 125, 2751–2761. [Google Scholar] [CrossRef]
- Gurha, P.; Wang, T.; Larimore, A.H.; Sassi, Y.; Abreu-Goodger, C.; Ramirez, M.O.; Reddy, A.K.; Engelhardt, S.; Taffet, G.E.; Wehrens, X.H.; et al. microRNA-22 Promotes Heart Failure through Coordinate Suppression of PPAR/ERR-Nuclear Hormone Receptor Transcription. PLoS ONE 2013, 8, e75882. [Google Scholar] [CrossRef]
- Wahlquist, C.; Jeong, D.; Rojas-Muñoz, A.; Kho, C.; Lee, A.; Mitsuyama, S.; van Mil, A.; Park, W.J.; Sluijter, J.P.; Doevendans, P.A.; et al. Inhibition of miR-25 Improves Cardiac Contractility in the Failing Heart. Nature 2014, 508, 531–535. [Google Scholar] [CrossRef]
- Jeong, D.; Yoo, J.; Lee, P.; Kepreotis, S.V.; Lee, A.; Wahlquist, C.; Brown, B.D.; Kho, C.; Mercola, M.; Hajjar, R.J. miR-25 Tough Decoy Enhances Cardiac Function in Heart Failure. Mol. Ther. 2018, 26, 718–729. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Lu, Y.; Song, X.; Gong, X.; Li, Y. Inhibition of microRNA-146a attenuated heart failure in myocardial infarction rats. Biosci. Rep. 2019, 39, BSR20191732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira-Costa, L.; Barros, A.S.; Lourenço, A.P.; Leite-Moreira, A.F.; Nogueira-Ferreira, R.; Thongboonkerd, V.; Vitorino, R. Exosome-Derived Mediators as Potential Biomarkers for Cardiovascular Diseases: A Network Approach. Proteomes 2021, 9, 8. [Google Scholar] [CrossRef]
- Li, C.; Li, X.; Gao, X.; Zhang, R.; Zhang, Y.; Liang, H.; Xu, C.; Du, W.; Zhang, Y.; Liu, X.; et al. MicroRNA-328 as a regulator of cardiac hypertrophy. Int. J. Cardiol. 2014, 173, 268–276. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, Y.; Wang, N.; Pan, Z.; Gao, X.; Zhang, F.; Zhang, Y.; Shan, H.; Luo, X.; Bai, Y.; et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation 2010, 122, 2378–2387. [Google Scholar] [CrossRef] [PubMed]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat. Commun. 2012, 3, 1078. [Google Scholar] [CrossRef]
- Lei, Z.; Wahlquist, C.; El Azzouzi, H.; Deddens, J.C.; Kuster, D.; van Mil, A.; Rojas-Munoz, A.; Huibers, M.M.; Mercola, M.; de Weger, R.; et al. miR-132/212 Impairs Cardiomyocytes Contractility in the Failing Heart by Suppressing SERCA2a. Front. Cardiovasc. Med. 2021, 8, 592362. [Google Scholar] [CrossRef] [PubMed]
- Foinquinos, A.; Batkai, S.; Genschel, C.; Viereck, J.; Rump, S.; Gyöngyösi, M.; Traxler, D.; Riesenhuber, M.; Spannbauer, A.; Lukovic, D.; et al. Preclinical development of a miR-132 inhibitor for heart failure treatment. Nat. Commun. 2020, 11, 633. [Google Scholar] [CrossRef] [PubMed]
- Talukder, M.A.; Zweier, J.L.; Periasamy, M. Targeting calcium transport in ischaemic heart disease. Cardiovasc. Res. 2009, 84, 345–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.B.; Eisenhardt, S.U.; Stark, G.B.; Bode, C.; Moser, M.; Grundmann, S. MicroRNAs in ischemia-reperfusion injury. Am. J. Cardiovasc. Dis. 2012, 2, 237–247. [Google Scholar]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction). J. Mol. Cell. Cardiol. 2016, 94, 107–121. [Google Scholar] [CrossRef]
- Aurora, A.B.; Mahmoud, A.I.; Luo, X.; Johnson, B.A.; van Rooij, E.; Matsuzaki, S.; Humphries, K.M.; Hill, J.A.; Bassel-Duby, R.; Sadek, H.A.; et al. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J. Clin. Investig. 2012, 122, 1222–1232. [Google Scholar] [CrossRef]
- Cha, M.J.; Jang, J.K.; Ham, O.; Song, B.W.; Lee, S.Y.; Lee, C.Y.; Park, J.H.; Lee, J.; Seo, H.H.; Choi, E.; et al. MicroRNA-145 suppresses ROS-induced Ca2+ overload of cardiomyocytes by targeting CaMKIIδ. Biochem. Biophys. Res. Commun. 2013, 435, 720–726. [Google Scholar] [CrossRef]
- Nattel, S.; Heijman, J.; Zhou, L.; Dobrev, D. Molecular Basis of Atrial Fibrillation Pathophysiology and Therapy: A Translational Perspective. Circ. Res. 2020, 127, 51–72. [Google Scholar] [CrossRef]
- Yang, B.; Lin, H.; Xiao, J.; Lu, Y.; Luo, X.; Li, B.; Zhang, Y.; Xu, C.; Bai, Y.; Wang, H.; et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat. Med. 2007, 13, 486–491. [Google Scholar] [CrossRef]
- Li, Y.D.; Hong, Y.F.; Yusufuaji, Y.; Tang, B.P.; Zhou, X.H.; Xu, G.J.; Li, J.X.; Sun, L.; Zhang, J.H.; Xin, Q.; et al. Altered expression of hyperpolarization-activated cyclic nucleotide-gated channels and microRNA-1 and -133 in patients with age-associated atrial fibrillation. Mol. Med. Rep. 2015, 12, 3243–3248. [Google Scholar] [CrossRef]
- Girmatsion, Z.; Biliczki, P.; Bonauer, A.; Wimmer-Greinecker, G.; Scherer, M.; Moritz, A.; Bukowska, A.; Goette, A.; Nattel, S.; Hohnloser, S.H.; et al. Changes in microRNA-1 expression and IK1 up-regulation in human atrial fibrillation. Heart Rhythm 2009, 6, 1802–1809. [Google Scholar] [CrossRef]
- Chiang, D.Y.; Kongchan, N.; Beavers, D.L.; Alsina, K.M.; Voigt, N.; Neilson, J.R.; Jakob, H.; Martin, J.F.; Dobrev, D.; Wehrens, X.H.; et al. Loss of microRNA-106b-25 Cluster Promotes Atrial Fibrillation by Enhancing Ryanodine Receptor Type-2 Expression and Calcium Release. Circ. Arrhythm. Electrophysiol. 2014, 7, 1214–1222. [Google Scholar] [CrossRef] [Green Version]
- Cañón, S.; Caballero, R.; Herraiz-Martínez, A.; Pérez-Hernández, M.; López, B.; Atienza, F.; Jalife, J.; Hove-Madsen, L.; Delpón, E.; Bernad, A. miR-208b upregulation interferes with calcium handling in HL-1 atrial myocytes: Implications in human chronic atrial fibrillation. J. Mol. Cell. Cardiol. 2016, 99, 162–173. [Google Scholar] [CrossRef]
- Van den Berg, N.W.E.; Kawasaki, M.; Berger, W.R.; Neefs, J.; Meulendijks, E.; Tijsen, A.J.; de Groot, J.R. MicroRNAs in Atrial Fibrillation: From Expression Signatures to Functional Implications. Cardiovasc. Drugs Ther. 2017, 31, 345–365. [Google Scholar]
- Shen, N.N.; Zhang, C.; Li, Z.; Kong, L.C.; Wang, X.H.; Gu, Z.C.; Wang, J.L. MicroRNA expression signatures of atrial fibrillation: The critical systematic review and bioinformatics analysis. Exp. Biol. Med. 2020, 245, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.K.; Kafert-Kasting, S.; Thum, T. Preclinical and Clinical Development of Noncoding RNA Therapeutics for Cardiovascular Disease. Circ. Res. 2020, 126, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Setten, R.L.; Rossi, J.J.; Han, S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef]
- Ishikawa, K.; Weber, T.; Hajjar, R.J. Human Cardiac Gene Therapy. Circ. Res. 2018, 123, 601–613. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Lee, S.S. Therapeutic advances of miRNAs: A preclinical and clinical update. J. Adv. Res. 2020, 28, 127–138. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Protein | Disease Phenotype | References |
|---|---|---|
| RyR2 | CPVT, ARVD/C2, AF | Priori et al., 2001 [12]; Marks et al., 2002 [13]; Yano et al., 2005 [14] |
| SERCA2a | HF | Hasenfuss et al., 1994 [15]; Meyer et al., 2006 [16]; Flesch et al., 1996 [17] |
| PLN | ARVD, DCM | Zwaag et al., 2012 [18]; Jordan et al., 2021 [19] |
| CSQ2 | CPVT | Lahat et al., 2001 [20]; Postma et al., 2002 [21] |
| CaM | CPVT, LQTS | Chazin and Johnson. 2020 [22] |
| Triadin | CPVT | Roux-Buisson et al., 2012 [23]; Rooryck et al., 2015 [24] |
| Junctin | DCM | Gergs et al. 2007 [25] |
| HRC | Arrhythmias, DCM, AF | Arvanitis et al., 2008 [26]; Amioka et al., 2019 [27] |
| microRNA ID | Pathways Related Target Genes | Ca2+ Homeostasis Related Target Genes | Changes in HD | References |
|---|---|---|---|---|
| miR-1 | Cell differentiation, heart development | NCX1, RyR2, MCU | Down in HF, HT, ICM or Up in DCM | Barwari et al., 2016 [67]; Harada et al., 2014 [68]; Watson et al., 2015 [72]; Quan et al., 2018 [73]; Oh et al., 2018 [74] |
| miR-21 | Inflammation | LTCC | Up in HT, DCM, CM | |
| miR-22 | Apoptosis | SERCA2a | Up in HT, HF | |
| miR-25 | Heart development | SERCA2a, MCU | Up in HF | |
| miR-132 | Cell proliferation | NCX1 | Up in HT, DCM | |
| miR-133 | Cardiac hypertrophy | RyR2 | Down in HF, HT, AF, DCM, ICM or Up in CM | |
| miR-145 | Heart development | CaMKIIδ | Up in DCM, AS | |
| miR-146a | Inflammation | SUMO1 | Up in HF & CHD | |
| miR-214 | Cardiac hypertrophy | NCX1, CaMKIIδ | Up in MI, DCM, ICM, AS | |
| miR-328 | Fibrosis | SERCA2a, LTCC | Down in HF or Up in HT | |
| miR-494 | Apoptosis | CaMKIIδ | Down in IHD, MI | |
| miR-574 | Apoptosis | SERCA2a | Up in MI |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.-H.; Kho, C. MicroRNAs and Calcium Signaling in Heart Disease. Int. J. Mol. Sci. 2021, 22, 10582. https://doi.org/10.3390/ijms221910582
Park J-H, Kho C. MicroRNAs and Calcium Signaling in Heart Disease. International Journal of Molecular Sciences. 2021; 22(19):10582. https://doi.org/10.3390/ijms221910582
Chicago/Turabian StylePark, Jae-Ho, and Changwon Kho. 2021. "MicroRNAs and Calcium Signaling in Heart Disease" International Journal of Molecular Sciences 22, no. 19: 10582. https://doi.org/10.3390/ijms221910582
APA StylePark, J.-H., & Kho, C. (2021). MicroRNAs and Calcium Signaling in Heart Disease. International Journal of Molecular Sciences, 22(19), 10582. https://doi.org/10.3390/ijms221910582