
Should We Open Fire on Microglia? Depletion Models as Tools to Elucidate Microglial Role in Health and Alzheimer’s Disease

 , ,
, ,
Abstract
:1. Microglia: Micro in Size but Macro in Functions, Highly Important in Alzheimer’s Disease
2. Pharmacological and Genetic Microglial Depletion Models
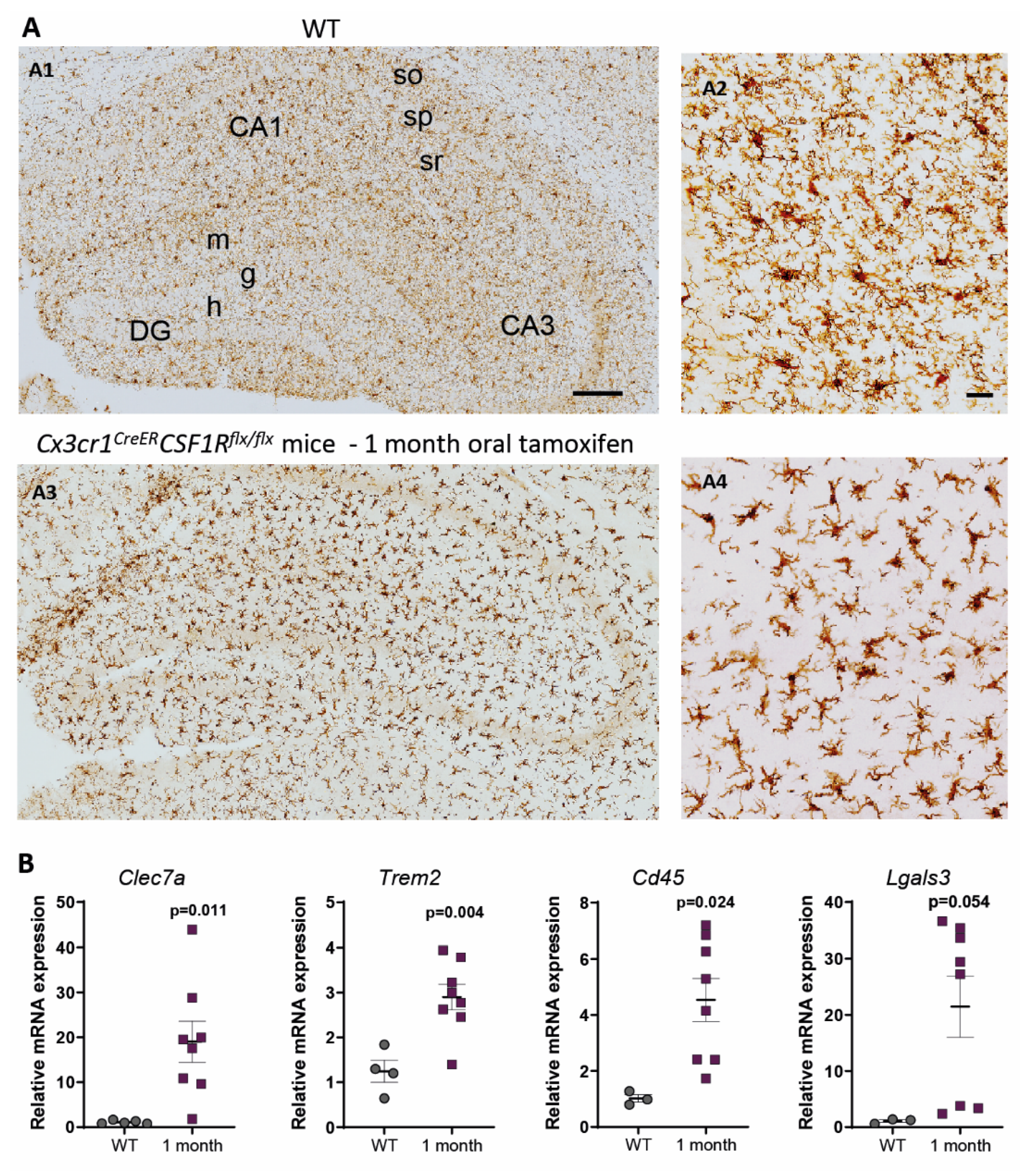
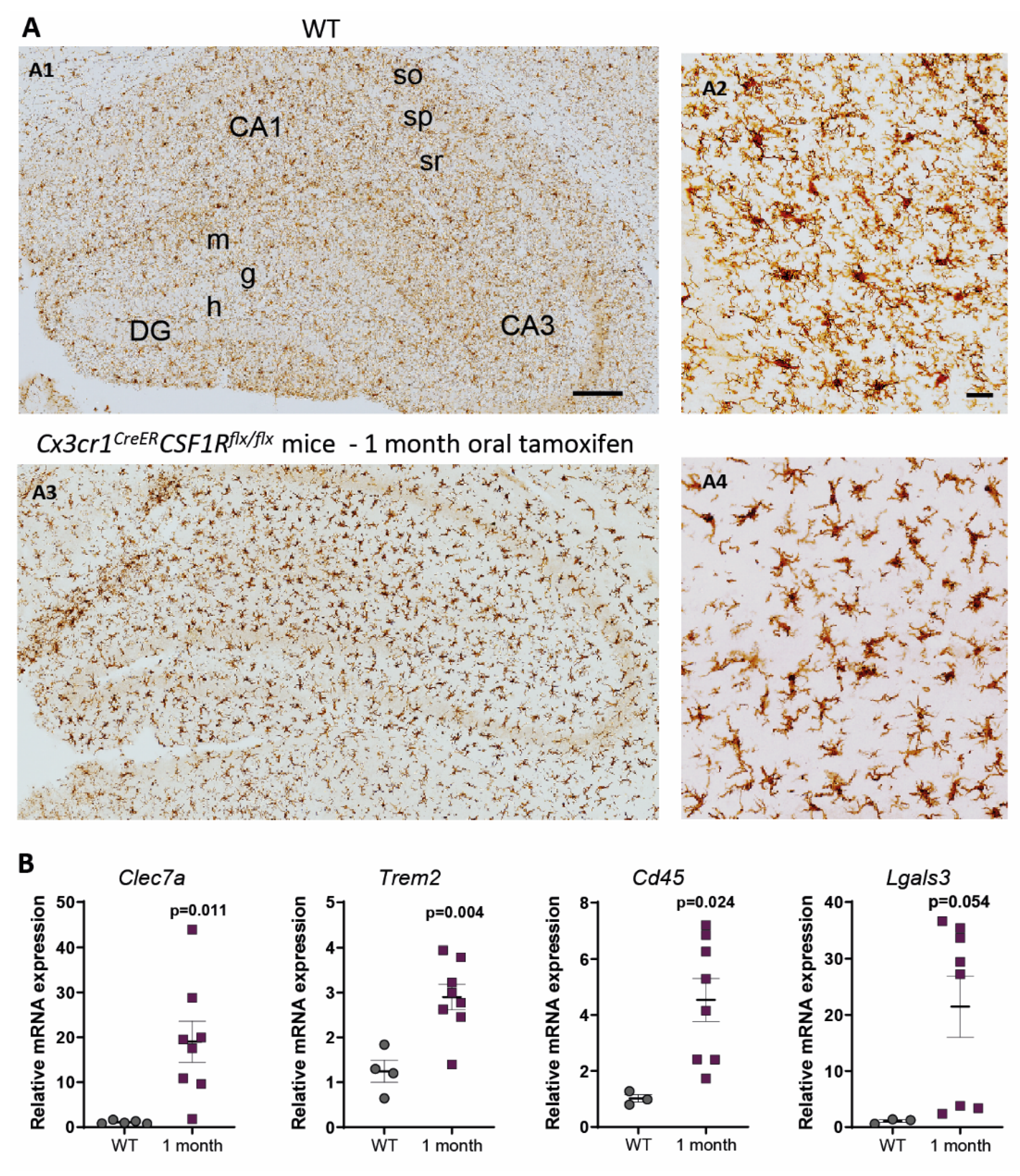
3. Characterization of Cx3cr1CreER/Csf1rflx/flx Mice. Is It a Good Approach to Study Microglial Dysfunction?
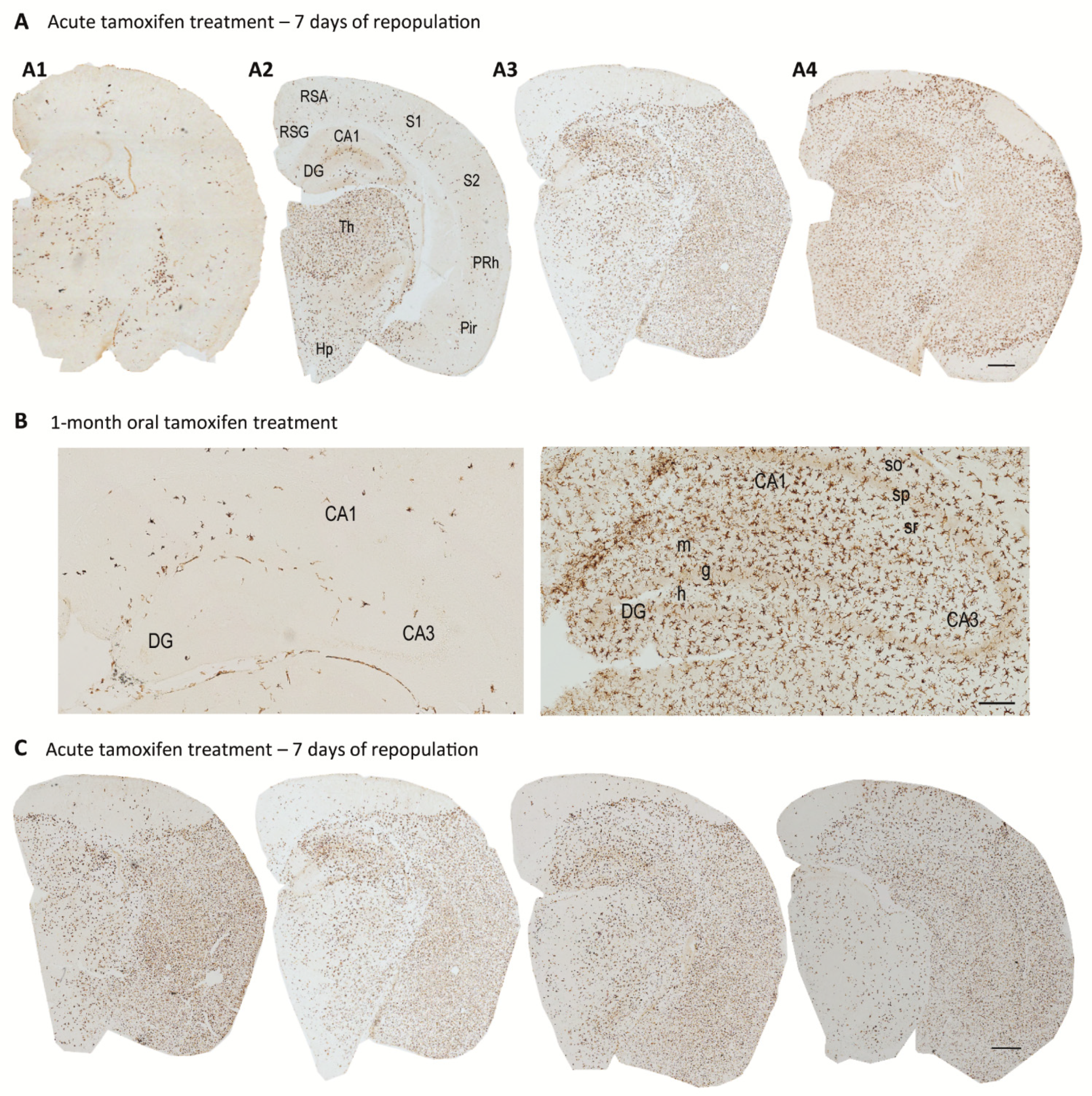
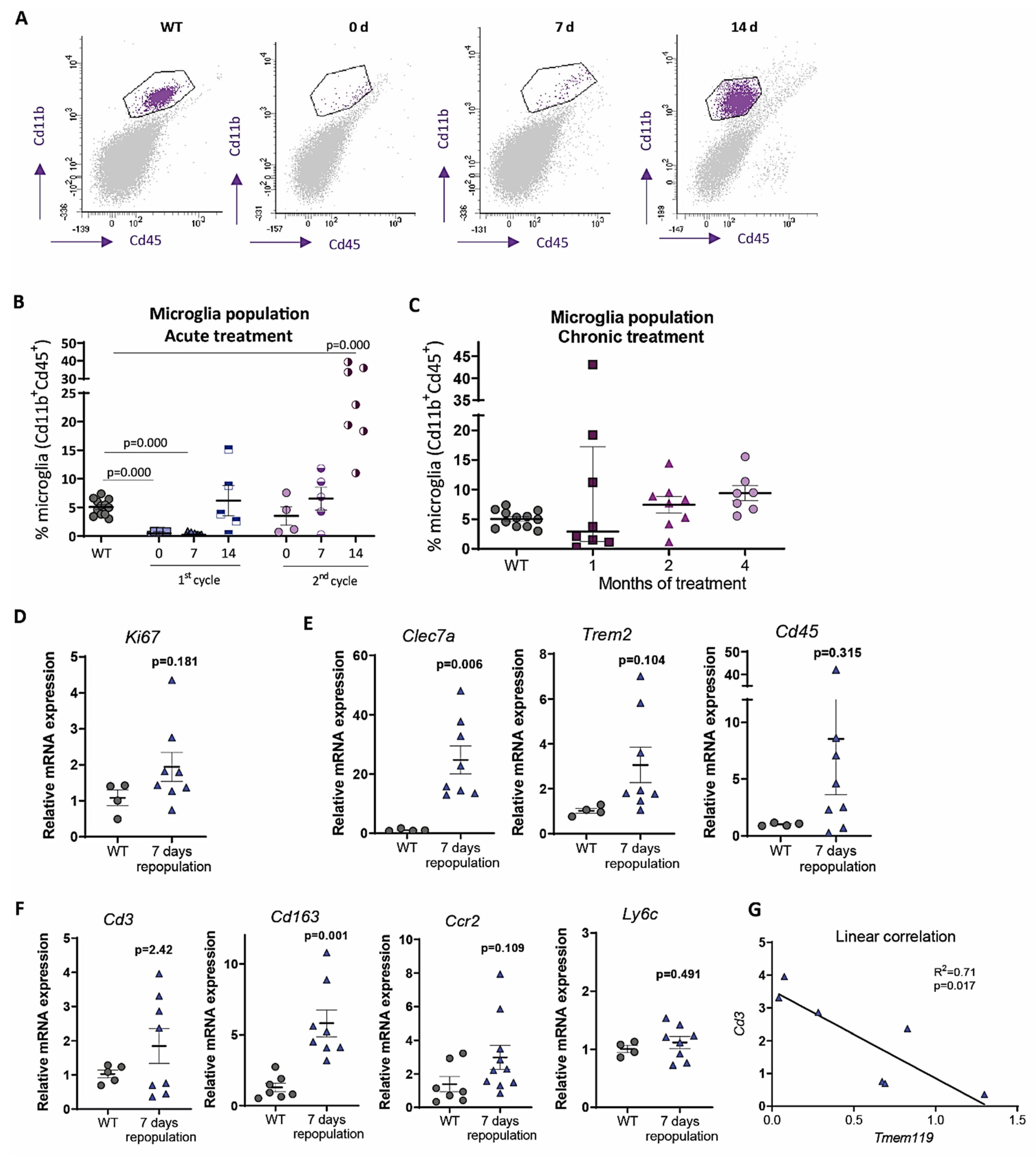
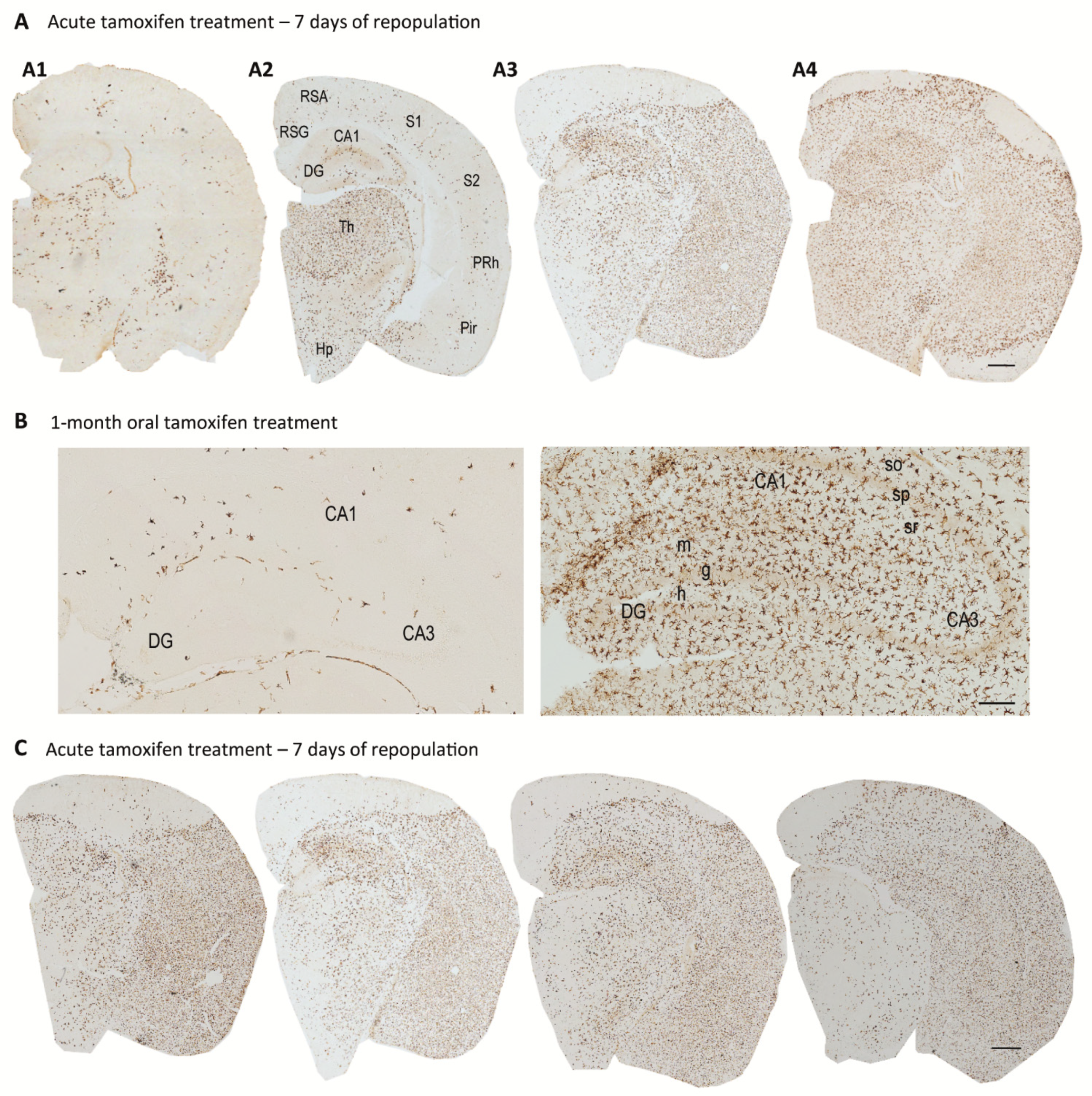
4. Origin and Pattern of Microglial Repopulation
5. Microglial Depletion as a Model of Microglial Replacement
6. Do Microglia Refresh or Poison AD Progression?

{kind=link}
{kind=link}
{kind=link}
| Pathology | Depletion Model | Outcomes | References |
|---|---|---|---|
| Aβ | PLX5622 in 5xFAD mice, from 4- to 5-month-old. | 50% microglia depletion. Reduction of microgliosis and plaque burden, enhancement of neuritic dystrophies. | [62] |
| Aβ | PLX3397 in 5xFAD mice, from 9- to 10-month-old. | Around 50% microglia depletion. Decrease in Aβ deposition and rescue of dopaminergic signaling. | [65] |
| Aβ | PLX5622 in APP/PS1 mice, from 12- to 13-month-old. | Diminution of leukotriene biosynthesis and the neuronal 5-lipoxygenase. | [92] |
| Aβ | PLX5622 in 5xFAD mice from 1.5 to 4- or 7-month-olds. | 97% microglia depletion. Reduction of plaque deposition, but increase of cerebral amyloid angiopathy formation. | [45] |
| Aβ | PLX3397 in 5xFAD mice from 2- to 5-month-old. | 70–80% microglia depletion. Reduction of intraneuronal amyloid, neuritic plaque deposition and improvement in cognitive functions (fear conditioning tests). | [93] |
| Aβ | Diphtheria toxin in 15 months-old Cx3cr1CreER/+:R26DTR/+/APPxPS1 mice, for 1–2 weeks. | 90% depletion. No changes in the number of Aβ plaques, but an increase in size. | [94] |
| Aβ | GWS2580 in APP/PS1 mice from 6- to 9-month-old. | 30% reduction of microglia. No changes in the number of Aβ plaques. Improved performance in memory and behavioral tasks. | [43] |
| Aβ | PLX3397 in 5xFAD from 10- to 11-month-old. | 90% microglia depletion. No alterations in β-amyloid levels or plaque load, but rescue of dendritic spine loss and improvements in contextual memory. | [56] |
| Aβ and Tau | PLX5562 in 3xTg mice for 3 months. | 30% microglia depletion. No changes in total or phosphorylated Tau. Improvements in cognition. | [64] |
| Aβ and Tau | PLX3397 from 5.5- to 7-month-old in 5xFAD/PS19 Tau -injected mice. | 81% microglia depletion. Higher reduction in non-plaque-associated microglia. No changes in Aβ pathology, reduction in Tau pathology and neurodegeneration. | [71] |
| Aβ and Tau | PLX3397 from 6- to 9-month-old in 5xFAD mice injected with AD-Tau. | Improved cognitive and neuronal deficits. Enhancement of Tau seeding and spreading around plaques. | [88] |
| Tau | Cx3cr1CreER/R26DTA/hTAU mice, treated with tamoxifen for 2–3 months at different ages. | 60% microglia depletion. No changes in soluble oligomeric, phosphorylated or total aggregated Tau levels. | [66] |
| Tau | PLX3397 in P301S APOE E4 mice from 6- to 9-month-old. | Total microglia depletion. Protection from brain volume loss and neurodegeneration. Reduction of Tau pathology progression. | [49] |
| Tau | PLX3397 in rTg4510 mice, from 12- to 15-month-old. | 30% microglia depletion. No changes in Tau burden, cortical atrophy, blood vessels or glial activation. | [63] |
| Tau | (a) Clodronate liposomes and PLX3397 in AAV-GFP/Tau injected C57BL/6 mice. (b) PLX3397 in PS19 mice. In both cases, from 3.5- to 4.5-month-old. | 70–80% (a) and 90% (b) microglia depletion. Reduction of phospho-Tau. | [95] |
6.1. Microglial Interplay with Aβ Pathology Progression
6.2. Microglial Contribution to Tau Pathology
6.3. Is Microglia Renewal a Promising Therapeutic Approach for AD?
7. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Nguyen, A.T.; Wang, K.; Hu, G.; Wang, X.; Miao, Z.; Azevedo, J.A.; Suh, E.; Van Deerlin, V.M.; Choi, D.; Roeder, K.; et al. APOE and TREM2 Regulate Amyloid-Responsive Microglia in Alzheimer’s Disease. Acta Neuropathol. 2020. [Google Scholar] [CrossRef]
- Wang, H.; Dey, K.K.; Chen, P.-C.; Li, Y.; Niu, M.; Cho, J.-H.; Wang, X.; Bai, B.; Jiao, Y.; Chepyala, S.R.; et al. Integrated Analysis of Ultra-Deep Proteomes in Cortex, Cerebrospinal Fluid and Serum Reveals a Mitochondrial Signature in Alzheimer’s Disease. Mol. Neurodegener. 2020, 15, 43. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the Distribution and Morphology of Microglia in the Normal Adult Mouse Brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Askew, K.; Li, K.; Olmos-Alonso, A.; Garcia-Moreno, F.; Liang, Y.; Richardson, P.; Tipton, T.; Chapman, M.A.; Riecken, K.; Beccari, S.; et al. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep. 2017, 18, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Réu, P.; Khosravi, A.; Bernard, S.; Mold, J.E.; Salehpour, M.; Alkass, K.; Perl, S.; Tisdale, J.; Possnert, G.; Druid, H.; et al. The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep. 2017, 20, 779–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, C.R., Jr.; Knopman, D.S.; Jagust, W.J.; Shaw, L.M.; Aisen, P.S.; Weiner, M.W.; Petersen, R.C.; Trojanowski, J.Q. Hypothetical Model of Dynamic Biomarkers of the Alzheimer’s Pathological Cascade. Lancet Neurol. 2010, 9, 119–128. [Google Scholar] [CrossRef] [Green Version]
- He, Z.; Guo, J.L.; McBride, J.D.; Narasimhan, S.; Kim, H.; Changolkar, L.; Zhang, B.; Gathagan, R.J.; Yue, C.; Dengler, C.; et al. Amyloid-β Plaques Enhance Alzheimer’s Brain Tau-Seeded Pathologies by Facilitating Neuritic Plaque Tau Aggregation. Nat. Med. 2018, 24, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Clayton, K.; Delpech, J.C.; Herron, S.; Iwahara, N.; Ericsson, M.; Saito, T.; Saido, T.C.; Ikezu, S.; Ikezu, T. Plaque Associated Microglia Hyper-Secrete Extracellular Vesicles and Accelerate Tau Propagation in a Humanized APP Mouse Model. Mol. Neurodegener. 2021, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-Cell Transcriptomic Analysis of Alzheimer’s Disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A Single-Cell Atlas of Entorhinal Cortex from Individuals with Alzheimer’s Disease Reveals Cell-Type-Specific Gene Expression Regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Sagar; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; et al. Spatial and Temporal Heterogeneity of Mouse and Human Microglia at Single-Cell Resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Zhou, Y.; Song, W.M.; Andhey, P.S.; Swain, A.; Levy, T.; Miller, K.R.; Poliani, P.L.; Cominelli, M.; Grover, S.; Gilfillan, S.; et al. Human and Mouse Single-Nucleus Transcriptomics Reveal TREM2-Dependent and—Independent Cellular Responses in Alzheimer’s Disease. Nat. Med. 2020, 26, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Condello, C.; Yuan, P.; Schain, A.; Grutzendler, J. Microglia Constitute a Barrier That Prevents Neurotoxic Protofibrillar Aβ42 Hotspots around Plaques. Nat. Commun. 2015, 6, 6176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- March-Diaz, R.; Lara-Ureña, N.; Romero-Molina, C.; Heras-Garvin, A.; Luis, C.O.S.; Alvarez-Vergara, M.I.; Sanchez-Garcia, M.A.; Sanchez-Mejias, E.; Davila, J.C.; Rosales-Nieves, A.E.; et al. Hypoxia Compromises the Mitochondrial Metabolism of Alzheimer’s Disease Microglia via HIF1. Nat. Aging 2021, 1, 385–399. [Google Scholar] [CrossRef]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.G.; Ngu, H.; et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Molina, C.; Navarro, V.; Sanchez-Varo, R.; Jimenez, S.; Fernandez-Valenzuela, J.J.; Sanchez-Mico, M.V.; Muñoz-Castro, C.; Gutierrez, A.; Vitorica, J.; Vizuete, M. Distinct Microglial Responses in Two Transgenic Murine Models of TAU Pathology. Front. Cell. Neurosci. 2018, 12, 421. [Google Scholar] [CrossRef] [Green Version]
- Leyns, C.E.G.; Gratuze, M.; Narasimhan, S.; Jain, N.; Koscal, L.J.; Jiang, H.; Manis, M.; Colonna, M.; Lee, V.M.Y.; Ulrich, J.D.; et al. TREM2 Function Impedes Tau Seeding in Neuritic Plaques. Nat. Neurosci. 2019, 22, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Gómez-Isla, T.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. A Phenotypic Change but Not Proliferation Underlies Glial Responses in Alzheimer Disease. Am. J. Pathol. 2013, 182, 2332–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and Protective Microglial Activation in Alzheimer’s Disease: A Prospective Study Using 18F-DPA-714 PET Imaging. Brain J. Neurol. 2016, 139, 1252–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An Early and Late Peak in Microglial Activation in Alzheimer’s Disease Trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef]
- Navarro, V.; Sanchez-Mejias, E.; Jimenez, S.; Muñoz-Castro, C.; Sanchez-Varo, R.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Microglia in Alzheimer’s Disease: Activated, Dysfunctional or Degenerative. Front. Aging Neurosci. 2018, 10, 140. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, S.; Baglietto-Vargas, D.; Caballero, C.; Moreno-Gonzalez, I.; Torres, M.; Sanchez-Varo, R.; Ruano, D.; Vizuete, M.; Gutierrez, A.; Vitorica, J. Inflammatory Response in the Hippocampus of PS1M146L/APP751SL Mouse Model of Alzheimer’s Disease: Age-Dependent Switch in the Microglial Phenotype from Alternative to Classic. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 11650–11661. [Google Scholar] [CrossRef]
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic Analysis of Purified Human Cortical Microglia Reveals Age-Associated Changes. Nat. Neurosci. 2017, 20, 1162–1171. [Google Scholar] [CrossRef]
- Sanchez-Mejias, E.; Navarro, V.; Jimenez, S.; Sanchez-Mico, M.; Sanchez-Varo, R.; Nuñez-Diaz, C.; Trujillo-Estrada, L.; Davila, J.C.; Vizuete, M.; Gutierrez, A.; et al. Soluble Phospho-Tau from Alzheimer’s Disease Hippocampus Drives Microglial Degeneration. Acta Neuropathol. 2016, 132, 897–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierksma, A.; Escott-Price, V.; Strooper, B.D. Translating Genetic Risk of Alzheimer’s Disease into Mechanistic Insight and Drug Targets. Science 2020, 370, 61–66. [Google Scholar] [CrossRef]
- Gutierrez, A.; Vitorica, J. Toward a New Concept of Alzheimer’s Disease Models: A Perspective from Neuroinflammation. J. Alzheimers Dis. 2018, 64, S329–S338. [Google Scholar] [CrossRef]
- Bohlen, C.J.; Bennett, F.C.; Tucker, A.F.; Collins, H.Y.; Mulinyawe, S.B.; Barres, B.A. Diverse Requirements for Microglial Survival, Specification, and Function Revealed by Defined-Medium Cultures. Neuron 2017, 94, 759–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, M.R.P.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. CSF1 Receptor Signaling Is Necessary for Microglia Viability, Which Unmasks a Cell That Rapidly Repopulates the Microglia-Depleted Adult Brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Li, Y.; Yu, M.; Yang, F.; Tu, M.; Xu, H. Notch Signaling Regulates Microglial Activation and Inflammatory Reactions in a Rat Model of Temporal Lobe Epilepsy. Neurochem. Res. 2018, 43, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Elegheert, J.; Desfosses, A.; Shkumatov, A.V.; Wu, X.; Bracke, N.; Verstraete, K.; Van Craenenbroeck, K.; Brooks, B.R.; Svergun, D.I.; Vergauwen, B.; et al. Extracellular Complexes of the Hematopoietic Human and Mouse CSF-1 Receptor Are Driven by Common Assembly Principles. Structure 2011, 19, 1762–1772. [Google Scholar] [CrossRef] [Green Version]
- Felix, J.; De Munck, S.; Verstraete, K.; Meuris, L.; Callewaert, N.; Elegheert, J.; Savvides, S.N. Structure and Assembly Mechanism of the Signaling Complex Mediated by Human CSF-1. Structure 2015, 23, 1621–1631. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Lin, W.Y.; Chen, Y.; Stawicki, S.; Mukhyala, K.; Wu, Y.; Martin, F.; Bazan, J.F.; Starovasnik, M.A. Structural Basis for the Dual Recognition of Helical Cytokines IL-34 and CSF-1 by CSF-1R. Structure 2012, 20, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Player, M. Colony-Stimulating Factor-1 Receptor Inhibitors for the Treatment of Cancer and Inflammatory Disease. Curr. Top. Med. Chem. 2009, 9. [Google Scholar] [CrossRef]
- Chitu, V.; Stanley, E.R. Chapter Seven—Regulation of Embryonic and Postnatal Development by the CSF-1 Receptor. In Current Topics in Developmental Biology; Jenny, A., Ed.; Protein Kinases in Development and Disease; Academic Press: Cambridge, MA, USA, 2017; Volume 123, pp. 229–275. [Google Scholar]
- Erblich, B.; Zhu, L.; Etgen, A.M.; Dobrenis, K.; Pollard, J.W. Absence of Colony Stimulation Factor-1 Receptor Results in Loss of Microglia, Disrupted Brain Development and Olfactory Deficits. PLoS ONE 2011, 6, e26317. [Google Scholar] [CrossRef] [Green Version]
- Green, K.N.; Crapser, J.D.; Hohsfield, L.A. To Kill a Microglia: A Case for CSF1R Inhibitors. Trends Immunol. 2020, 41, 771–784. [Google Scholar] [CrossRef]
- Wu, W.; Li, Y.; Wei, Y.; Bosco, D.B.; Xie, M.; Zhao, M.-G.; Richardson, J.R.; Wu, L.-J. Microglial Depletion Aggravates the Severity of Acute and Chronic Seizures in Mice. Brain. Behav. Immun. 2020, 89, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhu, K.; Zhang, X.-M.; Harris, R.A. Enforced Microglial Depletion and Repopulation as a Promising Strategy for the Treatment of Neurological Disorders. Glia 2019, 67, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Olmos-Alonso, A.; Schetters, S.T.T.; Sri, S.; Askew, K.; Mancuso, R.; Vargas-Caballero, M.; Holscher, C.; Perry, V.H.; Gomez-Nicola, D. Pharmacological Targeting of CSF1R Inhibits Microglial Proliferation and Prevents the Progression of Alzheimer’s-like Pathology. Brain J. Neurol. 2016, 139, 891–907. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, R.; Fryatt, G.; Cleal, M.; Obst, J.; Pipi, E.; Monzón-Sandoval, J.; Ribe, E.; Winchester, L.; Webber, C.; Nevado, A.; et al. CSF1R Inhibitor JNJ-40346527 Attenuates Microglial Proliferation and Neurodegeneration in P301S Mice. Brain 2019, 142, 3243–3264. [Google Scholar] [CrossRef]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained Microglial Depletion with CSF1R Inhibitor Impairs Parenchymal Plaque Development in an Alzheimer’s Disease Model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef] [PubMed]
- Lei, F.; Cui, N.; Zhou, C.; Chodosh, J.; Vavvas, D.G.; Paschalis, E.I. CSF1R Inhibition by a Small-Molecule Inhibitor Is Not Microglia Specific; Affecting Hematopoiesis and the Function of Macrophages. Proc. Natl. Acad. Sci. USA 2020, 117, 23336–23338. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.L.; Jimenez-Andrade, J.M.; Chartier, S.; Tsai, J.; Burton, E.A.; Habets, G.; Lin, P.S.; West, B.L.; Mantyh, P.W. Targeting Cells of the Myeloid Lineage Attenuates Pain and Disease Progression in a Prostate Model of Bone Cancer. Pain 2015, 156, 1692–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitu, V.; Gokhan, Ş.; Nandi, S.; Mehler, M.F.; Stanley, E.R. Emerging Roles for CSF-1 Receptor and Its Ligands in the Nervous System. Trends Neurosci. 2016, 39, 378–393. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Manis, M.; Long, J.; Wang, K.; Sullivan, P.M.; Serrano, J.R.; Hoyle, R.; Holtzman, D.M. Microglia Drive APOE-Dependent Neurodegeneration in a Tauopathy Mouse Model. J. Exp. Med. 2019, 216, 2546–2561. [Google Scholar] [CrossRef]
- Jäkel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef] [Green Version]
- Gowing, G.; Vallières, L.; Julien, J.-P. Mouse Model for Ablation of Proliferating Microgliain Acute CNS Injuries. Glia 2006, 53, 331–337. [Google Scholar] [CrossRef]
- Bruttger, J.; Karram, K.; Wörtge, S.; Regen, T.; Marini, F.; Hoppmann, N.; Klein, M.; Blank, T.; Yona, S.; Wolf, Y.; et al. Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity 2015, 43, 92–106. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, M.; Im, S.-K.; Fang, S. Mouse Cre-LoxP System: General Principles to Determine Tissue-Specific Roles of Target Genes. Lab. Anim. Res. 2018, 34, 147–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronk, J.C.; Filiano, A.J.; Louveau, A.; Marin, I.; Marsh, R.; Ji, E.; Goldman, D.H.; Smirnov, I.; Geraci, N.; Acton, S.; et al. Peripherally Derived Macrophages Can Engraft the Brain Independent of Irradiation and Maintain an Identity Distinct from Microglia. J. Exp. Med. 2018, 215, 1627–1647. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, T.; Feng, G. Tmem119-EGFP and Tmem119-CreERT2 Transgenic Mice for Labeling and Manipulating Microglia. eNeuro 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.P.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating Microglia in Alzheimer’s Mice Prevents Neuronal Loss without Modulating Amyloid-β Pathology. Brain J. Neurol. 2016, 139, 1265–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Echeverry, S.; Shi, X.Q.; Yang, M.; Yang, Q.Z.; Wang, G.Y.F.; Chambon, J.; Wu, Y.C.; Fu, K.Y.; De Koninck, Y.; et al. Dynamics of Spinal Microglia Repopulation Following an Acute Depletion. Sci. Rep. 2016, 6, 22839. [Google Scholar] [CrossRef] [Green Version]
- Elmore, M.R.P.; Lee, R.J.; West, B.L.; Green, K.N. Characterizing Newly Repopulated Microglia in the Adult Mouse: Impacts on Animal Behavior, Cell Morphology, and Neuroinflammation. PLoS ONE 2015, 10, e0122912. [Google Scholar] [CrossRef] [PubMed]
- Rice, R.A.; Pham, J.; Lee, R.J.; Najafi, A.R.; West, B.L.; Green, K.N. Microglial Repopulation Resolves Inflammation and Promotes Brain Recovery after Injury. Glia 2017, 65, 931–944. [Google Scholar] [CrossRef]
- Grathwohl, S.A.; Kälin, R.E.; Bolmont, T.; Prokop, S.; Winkelmann, G.; Kaeser, S.A.; Odenthal, J.; Radde, R.; Eldh, T.; Gandy, S.; et al. Formation and Maintenance of Alzheimer’s Disease Beta-Amyloid Plaques in the Absence of Microglia. Nat. Neurosci. 2009, 12, 1361–1363. [Google Scholar] [CrossRef]
- Faustino, J.V.; Wang, X.; Johnson, C.E.; Klibanov, A.; Derugin, N.; Wendland, M.F.; Vexler, Z.S. Microglial Cells Contribute to Endogenous Brain Defenses after Acute Neonatal Focal Stroke. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 12992–13001. [Google Scholar] [CrossRef] [Green Version]
- Casali, B.T.; MacPherson, K.P.; Reed-Geaghan, E.G.; Landreth, G.E. Microglia Depletion Rapidly and Reversibly Alters Amyloid Pathology by Modification of Plaque Compaction and Morphologies. Neurobiol. Dis. 2020, 142, 104956. [Google Scholar] [CrossRef]
- Bennett, R.E.; Bryant, A.; Hu, M.; Robbins, A.B.; Hopp, S.C.; Hyman, B.T. Partial Reduction of Microglia Does Not Affect Tau Pathology in Aged Mice. J. Neuroinflamm. 2018, 15, 311. [Google Scholar] [CrossRef]
- Dagher, N.N.; Najafi, A.R.; Kayala, K.M.N.; Elmore, M.R.P.; White, T.E.; Medeiros, R.; West, B.L.; Green, K.N. Colony-Stimulating Factor 1 Receptor Inhibition Prevents Microglial Plaque Association and Improves Cognition in 3xTg-AD Mice. J. Neuroinflamm. 2015, 12, 139. [Google Scholar] [CrossRef] [Green Version]
- Son, Y.; Jeong, Y.J.; Shin, N.-R.; Oh, S.J.; Nam, K.R.; Choi, H.-D.; Choi, J.Y.; Lee, H.-J. Inhibition of Colony-Stimulating Factor 1 Receptor by PLX3397 Prevents Amyloid Beta Pathology and Rescues Dopaminergic Signaling in Aging 5xFAD Mice. Int. J. Mol. Sci. 2020, 21, 5553. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Pieber, M.; Han, J.; Blomgren, K.; Zhang, X.-M.; Harris, R.A.; Lund, H. Absence of Microglia or Presence of Peripherally-Derived Macrophages Does Not Affect Tau Pathology in Young or Old HTau Mice. Glia 2020, 68, 1466–1478. [Google Scholar] [CrossRef] [Green Version]
- Najafi, A.R.; Crapser, J.; Jiang, S.; Ng, W.; Mortazavi, A.; West, B.L.; Green, K.N. A Limited Capacity for Microglial Repopulation in the Adult Brain. Glia 2018. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Xu, Z.; Xiong, S.; Sun, F.; Qin, G.; Hu, G.; Wang, J.; Zhao, L.; Liang, Y.-X.; Wu, T.; et al. Repopulated Microglia Are Solely Derived from the Proliferation of Residual Microglia after Acute Depletion. Nat. Neurosci. 2018, 21, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Okamoto, T.; Hara, Y.; Komine, O.; Tamada, H.; Maeda, M.; Osako, F.; Kobayashi, M.; Nishiyama, A.; Kataoka, Y.; et al. Astrocytic Phagocytosis Is a Compensatory Mechanism for Microglial Dysfunction. EMBO J. 2020, 39, e104464. [Google Scholar] [CrossRef] [PubMed]
- Zhan, L.; Krabbe, G.; Du, F.; Jones, I.; Reichert, M.C.; Telpoukhovskaia, M.; Kodama, L.; Wang, C.; Cho, S.; Sayed, F.; et al. Proximal Recolonization by Self-Renewing Microglia Re-Establishes Microglial Homeostasis in the Adult Mouse Brain. PLoS Biol. 2019, 17, e3000134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodder, C.; Scheyltjens, I.; Stancu, I.C.; Botella Lucena, P.; Gutiérrez de Ravé, M.; Vanherle, S.; Vanmierlo, T.; Cremers, N.; Vanrusselt, H.; Brône, B.; et al. CSF1R Inhibition Rescues Tau Pathology and Neurodegeneration in an A/T/N Model with Combined AD Pathologies, While Preserving Plaque Associated Microglia. Acta Neuropathol. Commun. 2021, 9, 108. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 Lipid Sensing Sustains the Microglial Response in an Alzheimer’s Disease Model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [Green Version]
- Zhan, L.; Fan, L.; Kodama, L.; Sohn, P.D.; Wong, M.Y.; Mousa, G.A.; Zhou, Y.; Li, Y.; Gan, L. A MAC2-Positive Progenitor-like Microglial Population Is Resistant to CSF1R Inhibition in Adult Mouse Brain. eLife 2020, 9, e51796. [Google Scholar] [CrossRef]
- Shemer, A.; Grozovski, J.; Tay, T.L.; Tao, J.; Volaski, A.; Süß, P.; Ardura-Fabregat, A.; Gross-Vered, M.; Kim, J.-S.; David, E.; et al. Engrafted Parenchymal Brain Macrophages Differ from Microglia in Transcriptome, Chromatin Landscape and Response to Challenge. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment Drives Selection and Function of Enhancers Controlling Tissue-Specific Macrophage Identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef]
- Varvel, N.H.; Grathwohl, S.A.; Baumann, F.; Liebig, C.; Bosch, A.; Brawek, B.; Thal, D.R.; Charo, I.F.; Heppner, F.L.; Aguzzi, A.; et al. Microglial Repopulation Model Reveals a Robust Homeostatic Process for Replacing CNS Myeloid Cells. Proc. Natl. Acad. Sci. USA 2012, 109, 18150–18155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, H.; Pieber, M.; Parsa, R.; Han, J.; Grommisch, D.; Ewing, E.; Kular, L.; Needhamsen, M.; Espinosa, A.; Nilsson, E.; et al. Competitive Repopulation of an Empty Microglial Niche Yields Functionally Distinct Subsets of Microglia-like Cells. Nat. Commun. 2018, 9, 4845. [Google Scholar] [CrossRef]
- Plemel, J.R.; Stratton, J.A.; Michaels, N.J.; Rawji, K.S.; Zhang, E.; Sinha, S.; Baaklini, C.S.; Dong, Y.; Ho, M.; Thorburn, K.; et al. Microglia Response Following Acute Demyelination Is Heterogeneous and Limits Infiltrating Macrophage Dispersion. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, W.D.; Wojtas, B.; Gielniewski, B.; Miró-Mur, F.; Pedragosa, J.; Zawadzka, M.; Pilanc, P.; Planas, A.M.; Kaminska, B. Defining Molecular Identity and Fates of CNS-Border Associated Macrophages after Ischemic Stroke in Rodents and Humans. Neurobiol. Dis. 2020, 137, 104722. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Ishikawa, E.; Matsuda, M.; Sugii, N.; Kohzuki, H.; Akutsu, H.; Sakamoto, N.; Takano, S.; Matsumura, A. Infiltration of CD163-Positive Macrophages in Glioma Tissues after Treatment with Anti-PD-L1 Antibody and Role of PI3Kγ Inhibitor as a Combination Therapy with Anti-PD-L1 Antibody in in Vivo Model Using Temozolomide-Resistant Murine Glioma-Initiating Cells. Brain Tumor Pathol. 2020, 37, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Swanson, M.E.V.; Scotter, E.L.; Smyth, L.C.D.; Murray, H.C.; Ryan, B.; Turner, C.; Faull, R.L.M.; Dragunow, M.; Curtis, M.A. Identification of a Dysfunctional Microglial Population in Human Alzheimer’s Disease Cortex Using Novel Single-Cell Histology Image Analysis. Acta Neuropathol. Commun. 2020, 8, 170. [Google Scholar] [CrossRef]
- Tan, Y.-L.; Yuan, Y.; Tian, L. Microglial Regional Heterogeneity and Its Role in the Brain. Mol. Psychiatry 2020, 25, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Thion, M.S.; Ginhoux, F.; Garel, S. Microglia and Early Brain Development: An Intimate Journey. Science 2018, 362, 185–189. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Fryatt, G.L.; Ghorbani, M.; Obst, J.; Menassa, D.A.; Martin-Estebane, M.; Muntslag, T.A.O.; Olmos-Alonso, A.; Guerrero-Carrasco, M.; Thomas, D.; et al. Replicative Senescence Dictates the Emergence of Disease-Associated Microglia and Contributes to Aβ Pathology. Cell Rep. 2021, 35. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic Regulation of Microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W.; Mochly-Rosen, D. Fragmented Mitochondria Released from Microglia Trigger A1 Astrocytic Response and Propagate Inflammatory Neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, L.; Wang, X.; Ma, W.; Lazere, A.; Qian, H.-H.; Zhang, J.; Abu-Asab, M.; Fariss, R.N.; Roger, J.E.; et al. Repopulating Retinal Microglia Restore Endogenous Organization and Function under CX3CL1-CX3CR1 Regulation. Sci. Adv. 2018, 4, eaap8492. [Google Scholar] [CrossRef] [Green Version]
- Gratuze, M.; Chen, Y.; Parhizkar, S.; Jain, N.; Strickland, M.R.; Serrano, J.R.; Colonna, M.; Ulrich, J.D.; Holtzman, D.M. Activated Microglia Mitigate Aβ-Associated Tau Seeding and Spreading. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mico, M.V.; Jimenez, S.; Gomez-Arboledas, A.; Muñoz-Castro, C.; Romero-Molina, C.; Navarro, V.; Sanchez-Mejias, E.; Nuñez-Diaz, C.; Sanchez-Varo, R.; Galea, E.; et al. Amyloid-β Impairs the Phagocytosis of Dystrophic Synapses by Astrocytes in Alzheimer’s Disease. Glia 2021, 69, 997–1011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cui, D. Evolving Models and Tools for Microglial Studies in the Central Nervous System. Neurosci. Bull. 2021. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef]
- Michael, J.; Unger, M.S.; Poupardin, R.; Schernthaner, P.; Mrowetz, H.; Attems, J.; Aigner, L. Microglia Depletion Diminishes Key Elements of the Leukotriene Pathway in the Brain of Alzheimer’s Disease Mice. Acta Neuropathol. Commun. 2020, 8, 129. [Google Scholar] [CrossRef]
- Sosna, J.; Philipp, S.; Albay, R.; Reyes-Ruiz, J.M.; Baglietto-Vargas, D.; LaFerla, F.M.; Glabe, C.G. Early Long-Term Administration of the CSF1R Inhibitor PLX3397 Ablates Microglia and Reduces Accumulation of Intraneuronal Amyloid, Neuritic Plaque Deposition and Pre-Fibrillar Oligomers in 5XFAD Mouse Model of Alzheimer’s Disease. Mol. Neurodegener. 2018, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Hu, W.; Tsai, J.; Li, W.; Gan, W.-B. Microglia Limit the Expansion of β-Amyloid Plaques in a Mouse Model of Alzheimer’s Disease. Mol. Neurodegener. 2017, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of Microglia and Inhibition of Exosome Synthesis Halt Tau Propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef]
- Reed-Geaghan, E.G.; Croxford, A.L.; Becher, B.; Landreth, G.E. Plaque-Associated Myeloid Cells Derive from Resident Microglia in an Alzheimer’s Disease Model. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.K.; McIntyre, L.L.; Marsh, S.E.; Schneider, C.A.; Hoover, E.M.; Walsh, C.M.; Lodoen, M.B.; Blurton-Jones, M.; Inlay, M.A. CD11a Expression Distinguishes Infiltrating Myeloid Cells from Plaque-Associated Microglia in Alzheimer’s Disease. Glia 2019, 67, 844–856. [Google Scholar] [CrossRef]
- Huang, Y.; Happonen, K.E.; Burrola, P.G.; O’Connor, C.; Hah, N.; Huang, L.; Nimmerjahn, A.; Lemke, G. Microglia Use TAM Receptors to Detect and Engulf Amyloid β Plaques. Nat. Immunol. 2021, 22, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid Toxicity in Alzheimer’s Disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-Derived ASC Specks Cross-Seed Amyloid-β in Alzheimer’s Disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Mendes, M.S.; Le, L.; Atlas, J.; Brehm, Z.; Ladron-de-Guevara, A.; Matei, E.; Lamantia, C.; McCall, M.N.; Majewska, A.K. The Role of P2Y12 in the Kinetics of Microglial Self-Renewal and Maturation in the Adult Visual Cortex in Vivo. eLife 2021, 10, e61173. [Google Scholar] [CrossRef]
- Perea, J.R.; Bolós, M.; Avila, J. Microglia in Alzheimer’s Disease in the Context of Tau Pathology. Biomolecules 2020, 10, 1439. [Google Scholar] [CrossRef] [PubMed]
- Bolós, M.; Perea, J.R.; Avila, J. Alzheimer’s Disease as an Inflammatory Disease. Biomol. Concepts 2017, 8, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, S.; Yamazaki, T.; Makioka, K.; Fujita, Y.; Ikeda, M.; Takatama, M.; Okamoto, K.; Yokoo, H.; Ikeda, Y. Hypersialylation Is a Common Feature of Neurofibrillary Tangles and Granulovacuolar Degenerations in Alzheimer’s Disease and Tauopathy Brains. Neuropathology 2016, 36, 333–345. [Google Scholar] [CrossRef]
- Bolós, M.; Llorens-Martín, M.; Jurado-Arjona, J.; Hernández, F.; Rábano, A.; Avila, J. Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J. Alzheimers Dis. 2015, 50, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Hopp, S.C.; Lin, Y.; Oakley, D.; Roe, A.D.; DeVos, S.L.; Hanlon, D.; Hyman, B.T. The Role of Microglia in Processing and Spreading of Bioactive Tau Seeds in Alzheimer’s Disease. J. Neuroinflamm. 2018, 15, 269. [Google Scholar] [CrossRef] [Green Version]
- Luo, W.; Liu, W.; Hu, X.; Hanna, M.; Caravaca, A.; Paul, S.M. Microglial Internalization and Degradation of Pathological Tau Is Enhanced by an Anti-Tau Monoclonal Antibody. Sci. Rep. 2015, 5, 11161. [Google Scholar] [CrossRef] [Green Version]
- Andersson, C.R.; Falsig, J.; Stavenhagen, J.B.; Christensen, S.; Kartberg, F.; Rosenqvist, N.; Finsen, B.; Pedersen, J.T. Antibody-Mediated Clearance of Tau in Primary Mouse Microglial Cultures Requires Fcγ-Receptor Binding and Functional Lysosomes. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Hu, W.; Tung, Y.C.; Liu, F.; Gong, C.-X.; Iqbal, K. Tau Passive Immunization Blocks Seeding and Spread of Alzheimer Hyperphosphorylated Tau-Induced Pathology in 3 × Tg-AD Mice. Alzheimers Res. Ther. 2018, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acker, C.M.; Forest, S.K.; Zinkowski, R.; Davies, P.; d’Abramo, C. Sensitive Quantitative Assays for Tau and Phospho-Tau in Transgenic Mouse Models. Neurobiol. Aging 2013, 34, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, J.; McGowan, E.; Rockwood, J.; Melrose, H.; Nacharaju, P.; Slegtenhorst, M.V.; Gwinn-Hardy, K.; Murphy, M.P.; Baker, M.; Yu, X.; et al. Neurofibrillary Tangles, Amyotrophy and Progressive Motor Disturbance in Mice Expressing Mutant (P301L) Tau Protein. Nat. Genet. 2000, 25, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef] [Green Version]
- Ramsden, M.; Kotilinek, L.; Forster, C.; Paulson, J.; McGowan, E.; SantaCruz, K.; Guimaraes, A.; Yue, M.; Lewis, J.; Carlson, G.; et al. Age-Dependent Neurofibrillary Tangle Formation, Neuron Loss, and Memory Impairment in a Mouse Model of Human Tauopathy (P301L). J. Neurosci. 2005, 25, 10637–10647. [Google Scholar] [CrossRef]
- Bemiller, S.M.; McCray, T.J.; Allan, K.; Formica, S.V.; Xu, G.; Wilson, G.; Kokiko-Cochran, O.N.; Crish, S.D.; Lasagna-Reeves, C.A.; Ransohoff, R.M.; et al. TREM2 Deficiency Exacerbates Tau Pathology through Dysregulated Kinase Signaling in a Mouse Model of Tauopathy. Mol. Neurodegener. 2017, 12, 74. [Google Scholar] [CrossRef]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of Tau Pathology by the Microglial Fractalkine Receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive Microglia Drive Tau Pathology and Contribute to the Spreading of Pathological Tau in the Brain. Brain J. Neurol. 2015, 138, 1738–1755. [Google Scholar] [CrossRef]
- Acharya, M.M.; Green, K.N.; Allen, B.D.; Najafi, A.R.; Syage, A.; Minasyan, H.; Le, M.T.; Kawashita, T.; Giedzinski, E.; Parihar, V.K.; et al. Elimination of Microglia Improves Cognitive Function Following Cranial Irradiation. Sci. Rep. 2016, 6, 31545. [Google Scholar] [CrossRef]
- Li, M.; Li, Z.; Ren, H.; Jin, W.-N.; Wood, K.; Liu, Q.; Sheth, K.N.; Shi, F.-D. Colony Stimulating Factor 1 Receptor Inhibition Eliminates Microglia and Attenuates Brain Injury after Intracerebral Hemorrhage. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 2383–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W.-N.; Shi, S.X.-Y.; Li, Z.; Li, M.; Wood, K.; Gonzales, R.J.; Liu, Q. Depletion of Microglia Exacerbates Postischemic Inflammation and Brain Injury. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 2224–2236. [Google Scholar] [CrossRef] [Green Version]
- Szalay, G.; Martinecz, B.; Lénárt, N.; Környei, Z.; Orsolits, B.; Judák, L.; Császár, E.; Fekete, R.; West, B.L.; Katona, G.; et al. Microglia Protect against Brain Injury and Their Selective Elimination Dysregulates Neuronal Network Activity after Stroke. Nat. Commun. 2016, 7, 11499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally Administered Colony Stimulating Factor 1 Receptor Inhibitor PLX3397 in Recurrent Glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium Phase II Study. Neuro-Oncology 2016, 18, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Streit, W.J.; Khoshbouei, H.; Bechmann, I. Dystrophic Microglia in Late-Onset Alzheimer’s Disease. Glia 2020, 68, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mustafa, M.; Yuede, C.M.; Salazar, S.V.; Kong, P.; Long, H.; Ward, M.; Siddiqui, O.; Paul, R.; Gilfillan, S.; et al. Anti-Human TREM2 Induces Microglia Proliferation and Reduces Pathology in an Alzheimer’s Disease Model. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Ostrowitzki, S.; Deptula, D.; Thurfjell, L.; Barkhof, F.; Bohrmann, B.; Brooks, D.J.; Klunk, W.E.; Ashford, E.; Yoo, K.; Xu, Z.X.; et al. Mechanism of Amyloid Removal in Patients with Alzheimer Disease Treated with Gantenerumab. Arch. Neurol. 2012, 69, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Bohrmann, B.; Baumann, K.; Benz, J.; Gerber, F.; Huber, W.; Knoflach, F.; Messer, J.; Oroszlan, K.; Rauchenberger, R.; Richter, W.F.; et al. Gantenerumab: A Novel Human Anti-Aβ Antibody Demonstrates Sustained Cerebral Amyloid-β Binding and Elicits Cell-Mediated Removal of Human Amyloid-β. J. Alzheimers Dis. 2012, 28, 49–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novakovic, D.; Feligioni, M.; Scaccianoce, S.; Caruso, A.; Piccinin, S.; Schepisi, C.; Errico, F.; Mercuri, N.B.; Nicoletti, F.; Nisticò, R. Profile of Gantenerumab and Its Potential in the Treatment of Alzheimer’s Disease. Drug Des. Dev. Ther. 2013, 7, 1359–1364. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romero-Molina, C.; Navarro, V.; Jimenez, S.; Muñoz-Castro, C.; Sanchez-Mico, M.V.; Gutierrez, A.; Vitorica, J.; Vizuete, M. Should We Open Fire on Microglia? Depletion Models as Tools to Elucidate Microglial Role in Health and Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 9734. https://doi.org/10.3390/ijms22189734
Romero-Molina C, Navarro V, Jimenez S, Muñoz-Castro C, Sanchez-Mico MV, Gutierrez A, Vitorica J, Vizuete M. Should We Open Fire on Microglia? Depletion Models as Tools to Elucidate Microglial Role in Health and Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(18):9734. https://doi.org/10.3390/ijms22189734
Chicago/Turabian StyleRomero-Molina, Carmen, Victoria Navarro, Sebastian Jimenez, Clara Muñoz-Castro, Maria V. Sanchez-Mico, Antonia Gutierrez, Javier Vitorica, and Marisa Vizuete. 2021. "Should We Open Fire on Microglia? Depletion Models as Tools to Elucidate Microglial Role in Health and Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 18: 9734. https://doi.org/10.3390/ijms22189734
APA StyleRomero-Molina, C., Navarro, V., Jimenez, S., Muñoz-Castro, C., Sanchez-Mico, M. V., Gutierrez, A., Vitorica, J., & Vizuete, M. (2021). Should We Open Fire on Microglia? Depletion Models as Tools to Elucidate Microglial Role in Health and Alzheimer’s Disease. International Journal of Molecular Sciences, 22(18), 9734. https://doi.org/10.3390/ijms22189734