
The Effects of Aβ1-42 Binding to the SARS-CoV-2 Spike Protein S1 Subunit and Angiotensin-Converting Enzyme 2

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
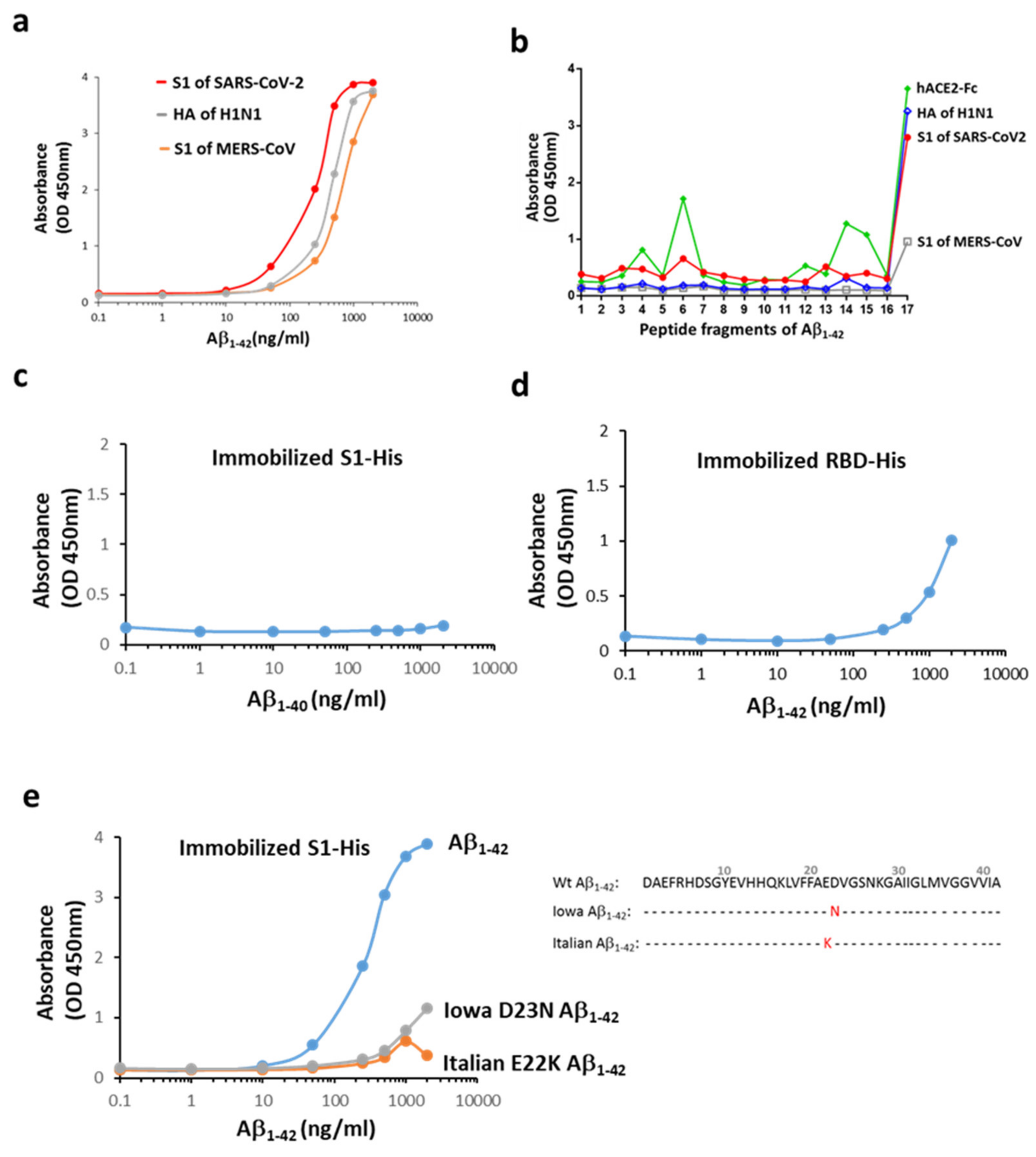
2.1. Aβ1-42 Binds to Viral Proteins with a High Affinity to the S1 of SARS-CoV-2
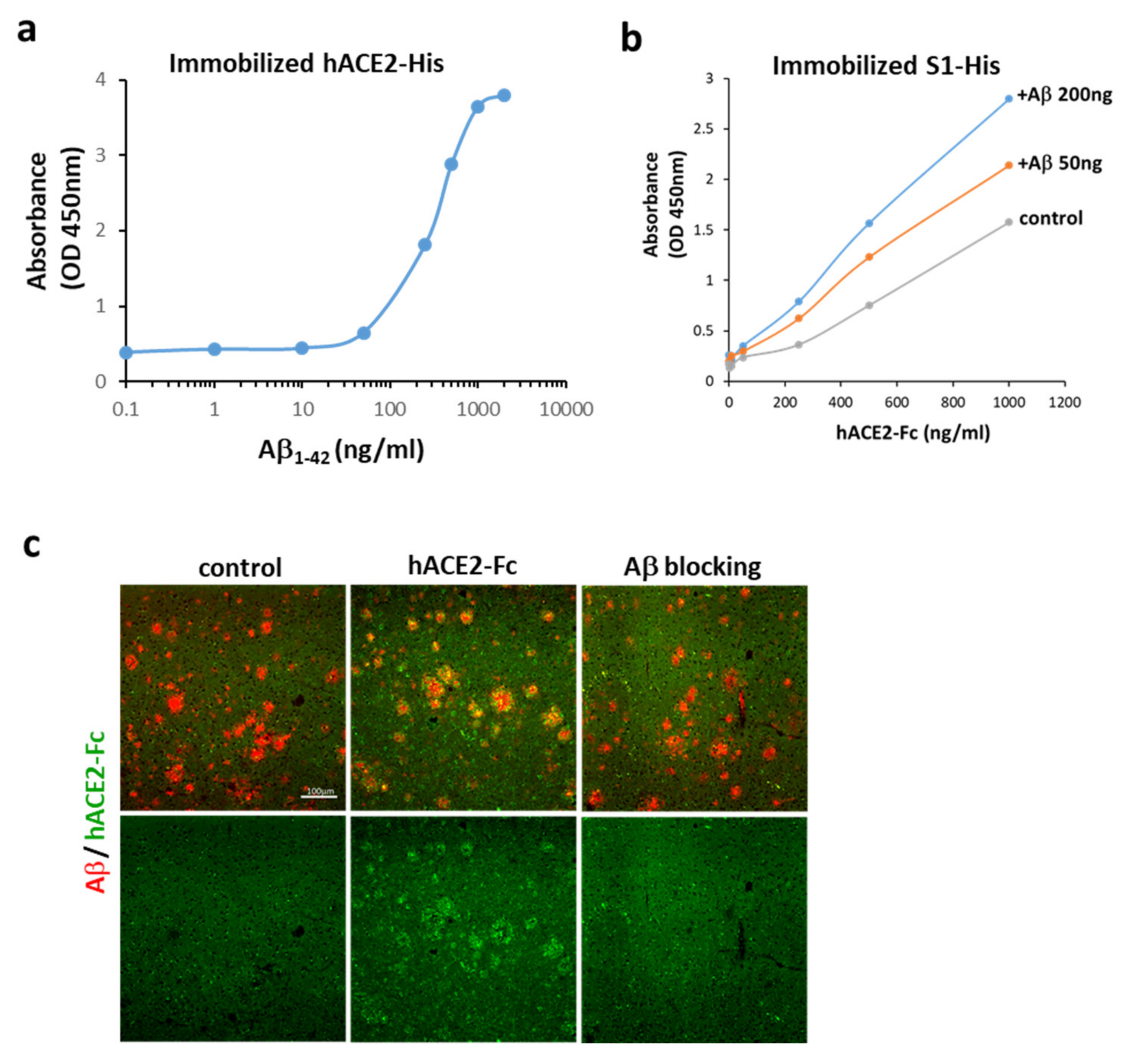
2.2. Aβ1-42 Binds to hACE2 and Strengthens the Binding of the S1 of SARS-CoV-2 to hACE2
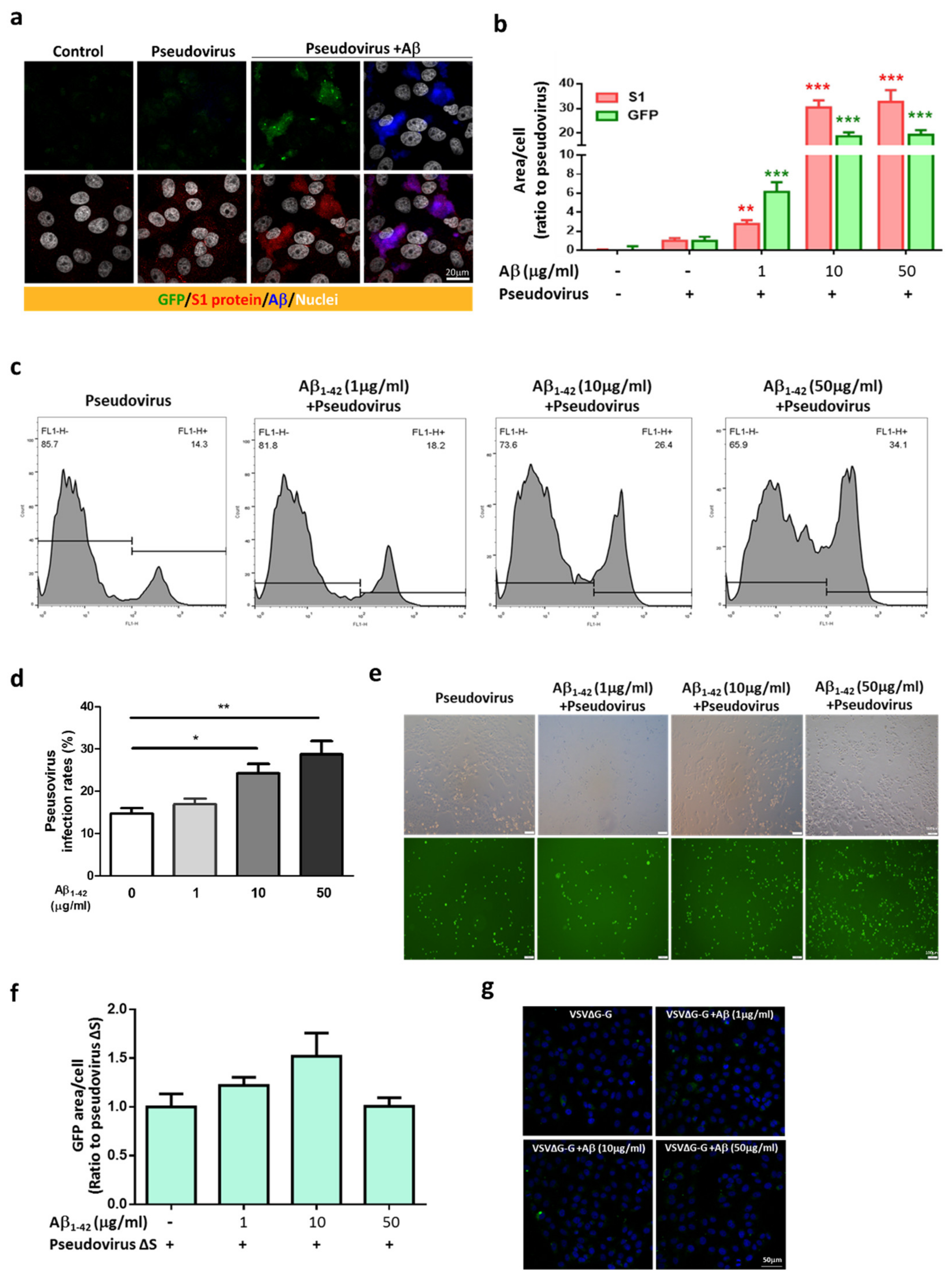
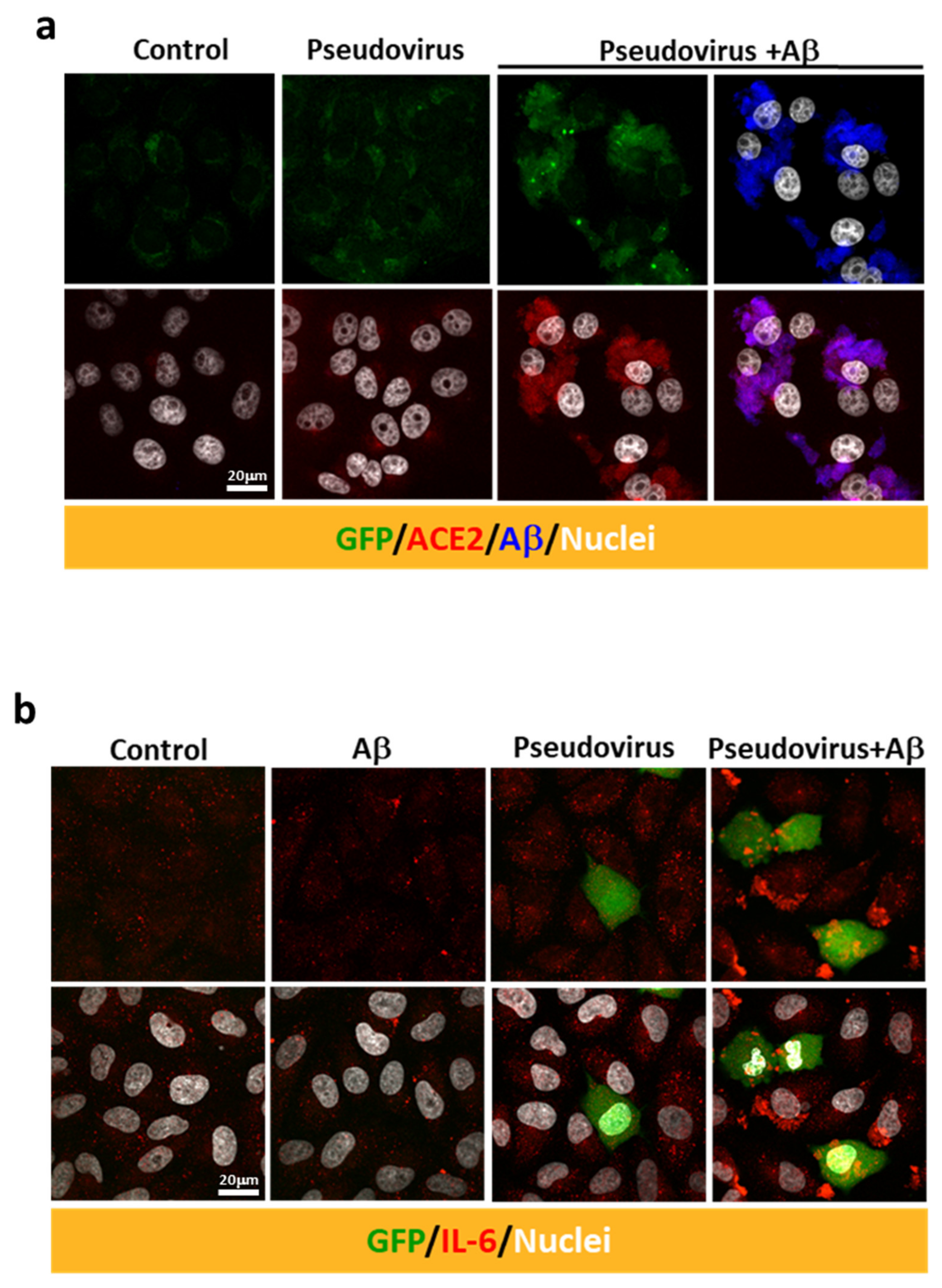
2.3. Aβ1-42 Increases the Infectivity of SARS-CoV-2 Pseudovirus and IL-6 in Host Cells
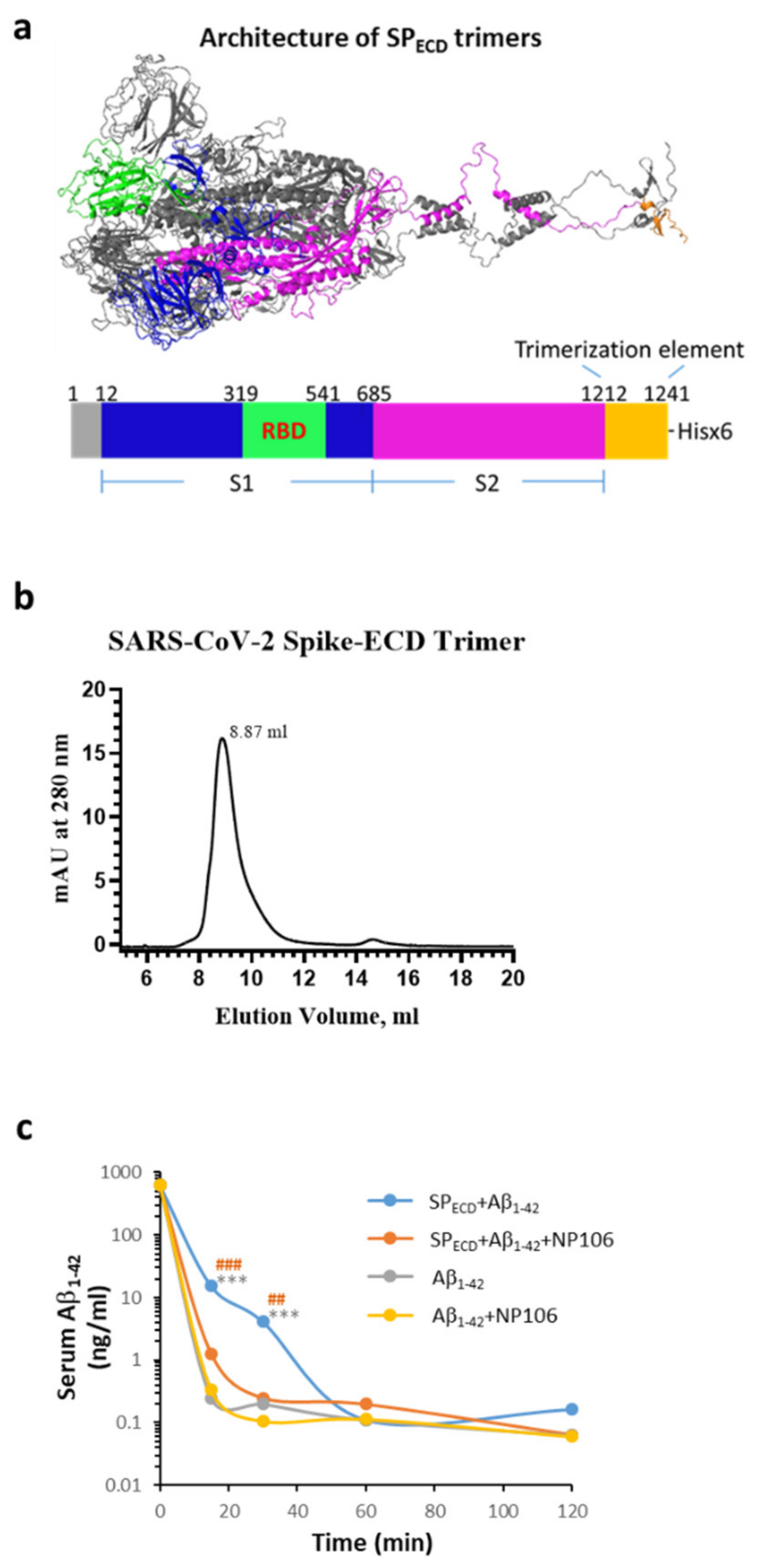
2.4. The Extracellular Domain (ECD) of the S Protein of SARS-CoV-2 (SPECD) Reduces Aβ Clearance in the Blood
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Generation of SARS-CoV-2 Pseudovirus
4.3. Confocal Microscopy and Flow Cytometry for Measuring the Pseudoviral Infection in Vero E6 Cells
4.4. IL-6 Expression in A549 Cells after Infection with SARS-CoV-2 Pseudovirus
4.5. Functional ELISA Binding Assay
4.6. Linear Binding Epitope of Viral Proteins on Aβ1-42
4.7. Interaction between hACE2-Fc and Aβ Plaques Examined by Confocal Microscopy
4.8. Novel Aβ Antibody NP106: Linear Epitope Mapping and Binding Affinity to Aβ1-42
4.9. Measurement of Serum Aβ1-42 Levels in Mice with Intravenous Inoculation of Aβ1-42
4.10. The Effects of SARS-CoV-2 SPECD Trimers on Aβ1-42 Clearance in the Blood
4.11. Statistical Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Mallapaty, S. The coronavirus is most deadly if you are older and male—New data reveal the risks. Nature 2020, 585, 16–17. [Google Scholar] [CrossRef]
- Apicella, M.; Campopiano, M.C.; Mantuano, M.; Mazoni, L.; Coppelli, A.; Del Prato, S. COVID-19 in people with diabetes: Understanding the reasons for worse outcomes. Lancet Diabetes Endocrinol. 2020. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Kremer, S.; Lersy, F.; Anheim, M.; Merdji, H.; Schenck, M.; Oesterlé, H.; Bolognini, F.; Messie, J.; Khalil, A.; Gaudemer, A.; et al. Neurologic and neuroimaging findings in COVID-19 patients: A retrospective multicenter study. Neurology 2020. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef]
- Solomon, I.H.; Normandin, E.; Bhattacharyya, S.; Mukerji, S.S.; Keller, K.; Ali, A.S.; Adams, G.; Hornick, J.L.; Padera, R.F., Jr.; Sabeti, P. Neuropathological Features of Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Atkins, J.L.; Masoli, J.A.H.; Delgado, J.; Pilling, L.C.; Kuo, C.L.; Kuchel, G.A.; Melzer, D. Preexisting Comorbidities Predicting COVID-19 and Mortality in the UK Biobank Community Cohort. J. Gerontol. A Biol. Sci. Med. Sci. 2020. [Google Scholar] [CrossRef]
- Perry, G. Alzheimer’s Disease Patients in the Crosshairs of COVID-19. J. Alzheimers Dis. 2020, 76, 1. [Google Scholar] [CrossRef] [PubMed]
- Azarpazhooh, M.R.; Amiri, A.; Morovatdar, N.; Steinwender, S.; Rezaei Ardani, A.; Yassi, N.; Biller, J.; Stranges, S.; Tokazebani Belasi, M.; Neya, S.K.; et al. Correlations between COVID-19 and burden of dementia: An ecological study and review of literature. J. Neurol. Sci. 2020, 416, 117013. [Google Scholar] [CrossRef]
- Livingston, G.; Rostamipour, H.; Gallagher, P.; Kalafatis, C.; Shastri, A.; Huzzey, L.; Liu, K.; Sommerlad, A.; Marston, L. Prevalence, management, and outcomes of SARS-CoV-2 infections in older people and those with dementia in mental health wards in London, UK: A retrospective observational study. Lancet Psychiatry 2020. [Google Scholar] [CrossRef]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.S.; Cashman, N.R. Passive immunotherapies targeting Aβ and tau in Alzheimer’s disease. Neurobiol. Dis. 2020, 144, 105010. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. Biomarkers for Alzheimer’s disease: Current status and prospects for the future. J. Intern. Med. 2018, 284, 643–663. [Google Scholar] [CrossRef]
- Lin, Y.S.; Pande, V.S. Effects of Familial Mutations on the Monomer Structure of Aβ42. Biophys. J. 2012, 103, L47–L49. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vijaya Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef]
- Eimer, W.A.; Vijaya Kumar, D.K.; Navalpur Shanmugam, N.K.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018, 99, 56–63. [Google Scholar] [CrossRef]
- Allnutt, M.A.; Johnson, K.; Bennett, D.A.; Connor, S.M.; Troncoso, J.C.; Pletnikova, O.; Albert, M.S.; Resnick, S.M.; Scholz, S.W.; De Jager, P.L.; et al. Human Herpesvirus 6 Detection in Alzheimer’s Disease Cases and Controls across Multiple Cohorts. Neuron 2020, 105, 1027–1035. [Google Scholar] [CrossRef]
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018, 14, 591–604. [Google Scholar] [CrossRef]
- Meakin, P.J.; Coull, B.M.; Tuharska, Z.; McCaffery, C.; Akoumianakis, I.; Antoniades, C.; Brown, J.; Griffin, K.J.; Platt, F.; Ozber, C.H.; et al. Elevated circulating amyloid concentrations in obesity and diabetes promote vascular dysfunction. J. Clin. Investig. 2020, 130, 4104–4117. [Google Scholar]
- Brubaker, W.D.; Crane, A.; Johansson, J.U.; Yen, K.; Garfinkel, K.; Mastroeni, D.; Asok, P.; Bradt, B.; Sabbagh, M.; Wallace, T.L.; et al. Peripheral complement interactions with amyloid β peptide (Aβ) in Alzheimer’s disease: 1. Erythrocyte clearance of Aβ. Alzheimers Dement. 2017, 13, 1397–1409. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Ye, Z.; Reixach, N.; Friske, L.; Levy, C.; Das, P.; Golde, T.; Masliah, E.; Roberts, A.R.; Bartfai, T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Abeta toxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 2681–2686. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, L.; Shi, C.; di Lorenzo, D.; Van Baelen, A.C.; Tonali, N. The Positive Side of the Alzheimer’s Disease Amyloid Cross-Interactions: The Case of the Aβ 1-42 Peptide with Tau, TTR, CysC, and ApoA1. Molecules 2020, 25, 2439. [Google Scholar] [CrossRef] [PubMed]
- Poejo, J.; Salazar, J.; Mata, A.M.; Gutierrez-Merino, C. The Relevance of Amyloid β-Calmodulin Complexation in Neurons and Brain Degeneration in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4976. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, X.; Zhao, M.; Gottesdiener, A.; Luo, W.; Paul, S. Tau pathogenesis is promoted by Aβ1-42 but not Aβ1-40. Mol. Neurodegener. 2014, 9, 52. [Google Scholar] [CrossRef]
- Vilella, A.; Daini, E.; De Benedictis, C.A.; Grabrucker, A.M. Targeting Metal Homeostasis as a Therapeutic Strategy for Alzheimer’s Disease. In Alzheimer’s Disease: Drug Discovery [Internet]; Huang, X., Ed.; Exon Publications: Brisbane, Australia, 2020; Chapter 5. [Google Scholar]
- Kehoe, P.G.; Wong, S.; Al Mulhim, N.; Palmer, L.E.; Miners, J.S. Angiotensin-converting enzyme 2 is reduced in Alzheimer’s disease in association with increasing amyloid-β and tau pathology. Alzheimers Res. Ther. 2016, 8, 50. [Google Scholar] [CrossRef]
- Evans, C.E.; Miners, J.S.; Piva, G.; Willis, C.L.; Heard, D.M.; Kidd, E.J.; Good, M.A.; Kehoe, P.G. ACE2 activation protects against cognitive decline and reduces amyloid pathology in the Tg2576 mouse model of Alzheimer’s disease. Acta Neuropathol. 2020, 139, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Shults, N.V.; Harris, B.T.; Suzuki, Y.J. Angiotensin-converting enzyme 2 (ACE2) is upregulated in Alzheimer’s disease brain. bioRxiv 2020. [Google Scholar] [CrossRef]
- Idrees, D.; Kumar, V. SARS-CoV-2 spike protein interactions with amyloidogenic proteins: Potential clues to neurodegeneration. Biochem. Biophys. Res. Commun. 2021, 554, 94–98. [Google Scholar] [CrossRef]
- Whitt, M.A. Generation of VSV pseudotypes using recombinant ΔG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J. Virol. Methods 2010, 169, 365–374. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, J.T.-A.; Tien, C.-F.; Yu, G.-Y.; Shen, S.; Lee, Y.-H.; Hsu, P.-C.; Wang, Y.; Chao, P.-K.; Tsay, H.-J.; Shie, F.-S. The Effects of Aβ1-42 Binding to the SARS-CoV-2 Spike Protein S1 Subunit and Angiotensin-Converting Enzyme 2. Int. J. Mol. Sci. 2021, 22, 8226. https://doi.org/10.3390/ijms22158226
Hsu JT-A, Tien C-F, Yu G-Y, Shen S, Lee Y-H, Hsu P-C, Wang Y, Chao P-K, Tsay H-J, Shie F-S. The Effects of Aβ1-42 Binding to the SARS-CoV-2 Spike Protein S1 Subunit and Angiotensin-Converting Enzyme 2. International Journal of Molecular Sciences. 2021; 22(15):8226. https://doi.org/10.3390/ijms22158226
Chicago/Turabian StyleHsu, John Tsu-An, Chih-Feng Tien, Guann-Yi Yu, Santai Shen, Yi-Hsuan Lee, Pei-Chien Hsu, Yun Wang, Po-Kuan Chao, Huey-Jen Tsay, and Feng-Shiun Shie. 2021. "The Effects of Aβ1-42 Binding to the SARS-CoV-2 Spike Protein S1 Subunit and Angiotensin-Converting Enzyme 2" International Journal of Molecular Sciences 22, no. 15: 8226. https://doi.org/10.3390/ijms22158226
APA StyleHsu, J. T.-A., Tien, C.-F., Yu, G.-Y., Shen, S., Lee, Y.-H., Hsu, P.-C., Wang, Y., Chao, P.-K., Tsay, H.-J., & Shie, F.-S. (2021). The Effects of Aβ1-42 Binding to the SARS-CoV-2 Spike Protein S1 Subunit and Angiotensin-Converting Enzyme 2. International Journal of Molecular Sciences, 22(15), 8226. https://doi.org/10.3390/ijms22158226