
Building Personalized Cancer Therapeutics through Multi-Omics Assays and Bacteriophage-Eukaryotic Cell Interactions


Abstract
:1. Introduction
2. Neoantigen—Personalized Cancer Therapeutic Target
2.1. Accurate Neoantigen Identification Lays the Foundations for Personalized Cancer Treatment
2.1.1. Prediction-Based Neoantigen Identification
2.1.2. Functional Analysis-Based Neoantigen Identification
2.1.3. Directly Detecting and Quantifying Neoantigens
3. Phage-Cell Interactions and Their Therapeutic Effects
3.1. Phage Biology and Its Applications
3.2. Phage Therapy through Phage-Bacteria Interaction
3.3. Phage Display and Phage-Eukaryotic Cell Neoantigen Interaction
4. Off-the-Shelf and Personalized Cancer Drugs Developed through Phage Display
5. Conclusions
Funding
Conflicts of Interest
References
- Rajagopala, S.V.; Vashee, S.; Oldfield, L.M.; Suzuki, Y.; Venter, J.C.; Telenti, A.; Nelson, K.E. The Human Microbiome and Cancer. Cancer Prev. Res. 2017, 10, 226–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, J.; Zhang, X. How human microbiome talks to health and disease. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 1595–1601. [Google Scholar] [CrossRef] [PubMed]
- Levaditi, C. Les Ultravirus: Considérés à Travers le Microscope Electronique; La Press méd: Paris, France, 1942; Volume 17, pp. 203–207. [Google Scholar]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Ebrahimizadeh, W.; Rajabibazl, M. Bacteriophage Vehicles for Phage Display: Biology, Mechanism, and Application. Curr. Microbiol. 2014, 69, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Skora, A.D.; Douglass, J.; Hwang, M.S.; Tam, A.J.; Blosser, R.L.; Gabelli, S.; Cao, J.; Diaz, L.; Papadopoulos, N.; Kinzler, K.W.; et al. Generation of MANAbodies specific to HLA-restricted epitopes encoded by somatically mutated genes. Proc. Natl. Acad. Sci. USA 2015, 112, 9967–9972. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Gallia, G.L.; Zhang, M.; Ning, Y.; Haffner, M.C.; Batista, D.; Binder, Z.A.; Bishop, J.A.; Hann, C.L.; Hruban, R.H.; Ishii, M.; et al. Genomic analysis identifies frequent deletions of Dystrophin in olfactory neuroblastoma. Nat. Commun. 2018, 9, 5410. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Heemskerk, B.; Kvistborg, P.; Schumacher, T.N.M. The cancer antigenome. EMBO J. 2012, 32, 194–203. [Google Scholar] [CrossRef]
- Riaz, N.; Morris, L.; Havel, J.; Makarov, V.; Desrichard, A.; Chan, T.A. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016, 28, 411–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Sileni, V.C.; Gonzalez, R.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, P.A.; Shuqiang, L.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Douglass, J.; Han-Chung Hsiue, E.; Mog, B.J.; Hwang, S.M.; Dinapoli, S.R.; Pearlamn, A.H.; Miller, M.S.; Wright, K.M.; Azurmendi, P.A.; Wang, Q.; et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci. Immunol. 2021, 6, eabd5515. [Google Scholar] [CrossRef] [PubMed]
- Hsiue, E.H.-C.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; Di Napoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, eabc8697. [Google Scholar] [CrossRef]
- Levy, S.E.; Myers, R.M. Advancements in Next-Generation Sequencing. Annu. Rev. Genom. Hum. Genet. 2016, 17, 95–115. [Google Scholar] [CrossRef] [Green Version]
- Levy, S.E.; Boone, B.E. Next-Generation Sequencing Strategies. Cold Spring Harb. Perspect. Med. 2018, 9, a025791. [Google Scholar] [CrossRef]
- Yohe, S.; Thyagarajan, B. Review of Clinical Next-Generation Sequencing. Arch. Pathol. Lab. Med. 2017, 141, 1544–1557. [Google Scholar] [CrossRef] [Green Version]
- Diamond, E.L.; Subbiah, V.; Lockhart, A.C.; Blay, J.-Y.; Puzanov, I.; Chau, I.; Raje, N.S.; Wolf, J.; Erinjeri, J.P.; Torrisin, J.; et al. Vemurafenib for BRAF V600-Mutant Erdheim-Chester Disease and Langerhans Cell Histiocytosis: Analysis of Data from the Histology-Independent, Phase 2, Open-label VE-BASKET Study. JAMA Oncol. 2018, 4, 384–388. [Google Scholar] [CrossRef] [Green Version]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S.; et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef]
- Targeted Cancer Therapies. Available online: https://www.cancer.gov/ (accessed on 31 July 2021).
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, C.D.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Li, L.; Goedegebuure, S.; Gillanders, W. Preclinical and clinical development of neoantigen vaccines. Ann. Oncol. 2017, 28, xii11–xii17. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Neoantigen quality, not quantity. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bassani-Sternberg, M.; Pletscher-Frankild, S.; Jensen, L.J.; Mann, M. Mass Spectrometry of Human Leukocyte Antigen Class I Peptidomes Reveals Strong Effects of Protein Abundance and Turnover on Antigen Presentation. Mol. Cell. Proteom. 2015, 14, 658–673. [Google Scholar] [CrossRef] [Green Version]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2015, 32, 511–517. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lundegaard, C.; Nielsen, M. Pan-specific MHC class I predictors: A benchmark of HLA class I pan-specific prediction methods. Bioinformatics 2008, 25, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Kenter, G.G.; Welters, M.J.P.; Valentijn, A.R.P.M.; Lowik, M.J.G.; Der Meer, D.M.A.B.-V.; Vloon, A.P.G.; Essahsah, F.; Fathers, L.M.; Offringa, R.; Drijfhout, J.W.; et al. Vaccination against HPV-16 Oncoproteins for Vulvar Intraepithelial Neoplasia. N. Engl. J. Med. 2009, 361, 1838–1847. [Google Scholar] [CrossRef] [Green Version]
- Van Poelgeest, M.I.; Welters, M.J.P.; Vermeji, R.; Stynenbisch, L.F.M.; Loof, N.M.; Berends-van der Meer, D.M.A.; Löwik, M.J.G.; Hamming, I.L.E.; van Esch, E.M.G.; Hellebrekers, B.W.J.; et al. Vaccination against Oncoproteins of HPV16 for Noninvasive Vulvar/Vaginal Lesions: Lesion Clearance Is Related to the Strength of the T-Cell Response. Clin. Cancer Res. 2016, 22, 2342–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandran, V.P.; Initiative, A.P.C.G.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef]
- Kim, S.; Kim, H.S.; Kim, E.; Lee, M.G.; Shin, E.C.; Paik, S. Neopepsee: Accurate genome-level prediction of neoantigens by harnessing sequence and amino acid immuno-genicity information. Ann. Oncol. 2018, 29, 1030–1036. [Google Scholar] [CrossRef]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2017, 359, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Hundal, J.; Carreno, B.M.; Petti, A.A.; Linette, G.P.; Griffith, O.L.; Mardis, E.R.; Griffith, M. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Stevanović, S.; Pasetto, A.; Helman, S.R.; Gartner, J.J.; Prickett, T.D.; Howie, B.; Robins, H.S.; Robbins, P.F.; Klebanoff, C.; Rosenberg, S.A.; et al. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 2017, 356, 200–205. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Olsson, N.; Wagar, L.; Haabeth, O.A.W.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 2017, 543, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Liepe, J.; Marino, F.; Sidney, J.; Jeko, A.; Bunting, D.E.; Sette, A.; Kloetzel, P.M.; Stumpf, M.P.H.; Heck, A.J.R.; Mishto, M. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science 2016, 354, 354–358. [Google Scholar] [CrossRef] [Green Version]
- Laumont, C.M.; Daouda, T.; Laverdure, J.-P.; Bonneil, É.; Caron-Lizotte, O.; Hardy, M.-P.; Granados, D.P.; Durette, C.; Lemieux, S.; Thibault, P.; et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat. Commun. 2016, 7, 10238. [Google Scholar] [CrossRef]
- Abelin, J.; Keskin, D.B.; Sarkizova, S.; Hartigan, C.R.; Zhang, W.; Sidney, J.; Stevens, J.; Lane, W.; Zhang, G.L.; Eisenhaure, T.M.; et al. Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity 2017, 46, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Pearson, H.; Daouda, T.; Granados, D.P.; Durette, C.; Bonneil, E.; Courcelles, M.; Rodenbrock, A.; Laverdure, J.-P.; Côté, C.; Mader, S.; et al. MHC class I–associated peptides derive from selective regions of the human genome. J. Clin. Investig. 2016, 126, 4690–4701. [Google Scholar] [CrossRef]
- Jørgensen, K.W.; Rasmussen, M.; Buus, S.; Nielsen, M. NetMHCstab- predicting stability of peptide-MHC-I complexes; impacts for cytotoxic T lymphocyte epitope discovery. Immunology 2013, 141, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Brunak, S.; Lund, O.; Nielsen, M. An integrative approach to CTL epitope prediction: A combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur. J. Immunol. 2005, 35, 2295–2303. [Google Scholar] [CrossRef]
- Singh-Jasuja, H.; Emmerich, N.P.N.; Rammensee, H.-G. The Tüingen approach: Identification, selection, and validation of tumor-associated HLA peptides for cancer therapy. Cancer Immunol. Immunother. 2004, 53, 187–195. [Google Scholar] [CrossRef]
- Wu, J.; Wang, W.; Zhang, J.; Zhou, B.; Zhao, W.; Su, Z.; Gu, X.; Wu, J.; Zhou, Z.; Chen, S.; et al. DeepHLApan: A Deep Learning Approach for Neoantigen Prediction Considering Both HLA-Peptide Binding and Immunogenicity. Front. Immunol. 2019, 10, 2559. [Google Scholar] [CrossRef] [Green Version]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.J.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Wang, Q.; Douglass, J.; Hwang, M.S.; Hsiue, E.H.-C.; Mog, B.J.; Zhang, M.; Papadopoulos, N.; Kinzler, K.W.; Zhou, S.; Vogelstein, B. Direct Detection and Quantification of Neoantigens. Cancer Immunol. Res. 2019, 7, 1748–1754. [Google Scholar] [CrossRef]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.-C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Anonymous. The problem with neoantigen prediction. Nat. Biotechnol. 2017, 35, 97. [Google Scholar]
- Vitiello, A.; Zanetti, M. Neoantigen prediction and the need for validation. Nat. Biotechnol. 2017, 35, 815–817. [Google Scholar] [CrossRef] [PubMed]
- Hadrup, S.R.; Bakker, A.; Shu, C.J.; Andersen, R.S.; Van Veluw, J.; Hombrink, P.; Castermans, E.; Straten, P.T.; Blank, C.; Haanen, J.B.; et al. Parallel detection of antigen-specific T-cell responses by multidimensional encoding of MHC multimers. Nat. Methods 2009, 6, 520–526. [Google Scholar] [CrossRef]
- Bentzen, A.K. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA bar-codes. Nat. Biotechnol. 2016, 34, 1037–1045. [Google Scholar] [CrossRef]
- Bentzen, A.K.; Hadrup, S.R. Evolution of MHC-based technologies used for detection of antigen-responsive T cells. Cancer Immunol. Immunother. 2017, 66, 657–666. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.-C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient Identification of Mutated Cancer Antigens Recognized by T Cells Associated with Durable Tumor Regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef] [Green Version]
- Danilova, L.; Anagnostou, V.; Caushi, J.X.; Sidhom, J.-W.; Guo, H.; Chan, H.Y.; Suri, P.; Tam, A.J.; Zhang, J.; El Asmar, M.; et al. The Mutation-Associated Neoantigen Functional Expansion of Specific T Cells (MANAFEST) Assay: A Sensitive Platform for Monitoring Antitumor Immunity. Cancer Immunol. Res. 2018, 6, 888–899. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Matsuda, T.; Ikeda, Y.; Park, J.-H.; Leisegang, M.; Yoshimura, S.; Hikichi, T.; Harada, M.; Zewde, M.G.; Sato, S.; et al. Effective screening of T cells recognizing neoantigens and construction of T-cell receptor-engineered T cells. Oncotarget 2018, 9, 11009–11019. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef]
- Caron, E.; Aebersold, R.; Banaei-Esfahani, A.; Chong, C.; Bassani-Sternberg, M. A Case for a Human Immuno-Peptidome Project Consortium. Immunity 2017, 47, 203–208. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Csordas, A.; Sun, Z.; Jarnuczak, A.; Perez-Riverol, Y.; Ternent, T.; Campbell, D.S.; Llinares, M.B.; Okuda, S.; Kawano, S.; et al. The ProteomeXchange consortium in 2017: Supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2016, 45, D1100–D1106. [Google Scholar] [CrossRef] [PubMed]
- Spraggins, J.M.; Djambazova, K.V.; Rivera, E.; Migas, L.G.; Neumann, E.K.; Fuetterer, A.; Suetering, J.; Gödecke, N.; Ly, A.; Van De Plas, R.; et al. High-Performance Molecular Imaging with MALDI Trapped Ion-Mobility Time-of-Flight (timsTOF) Mass Spectrometry. Anal. Chem. 2019, 91, 14552–14560. [Google Scholar] [CrossRef]
- Zubarev, R.A.; Makarov, A. Orbitrap mass spectrometry. Anal. Chem. 2013, 85, 5288–5296. [Google Scholar] [CrossRef] [PubMed]
- Kote, S.; Pirog, A.; Bedran, G.; Alfaro, J.; Dapic, I. Mass Spectrometry-Based Identification of MHC-Associated Peptides. Cancers 2020, 12, 535. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.; Gfeller, D.; Coukos, G.; Bassani-Sternberg, M. ‘Hotspots’ of Antigen Presentation Revealed by Human Leukocyte Antigen Ligandomics for Neoantigen Prioritization. Front. Immunol. 2017, 8, 1367. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Absolute abundance for the masses. Nat. Biotechnol. 2009, 27, 825–826. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.J.; Dexter, A.; Bunch, J. Exploring Ion Suppression in Mass Spectrometry Imaging of a Heterogeneous Tissue. Anal. Chem. 2018, 90, 5637–5645. [Google Scholar] [CrossRef] [PubMed]
- Annesley, T.M. Ion Suppression in Mass Spectrometry. Clin. Chem. 2003, 49, 1041–1044. [Google Scholar] [CrossRef] [Green Version]
- de Verteuil, D. Deletion of immunoproteasome subunits imprints on the transcriptome and has a broad impact on peptides presented by major histocompatibility complex I molecules. Mol. Cell Proteomics. 2010, 9, 2034–2047. [Google Scholar] [CrossRef] [Green Version]
- Hebert, A.S.; Prasad, S.; Belford, M.W.; Bailey, D.J.; McAlister, G.C.; Abbatiello, S.E.; Huguet, R.; Wouters, E.R.; Dunyach, J.-J.; Brademan, D.R.; et al. Comprehensive Single-Shot Proteomics with FAIMS on a Hybrid Orbitrap Mass Spectrometer. Anal. Chem. 2018, 90, 9529–9537. [Google Scholar] [CrossRef] [PubMed]
- Garimella, S.V.; Ibrahim, Y.M.; Wbb, I.K.; Ipsen, A.B.; Chen, T.-C.; Molmachev, A.V.; Baker, E.S.; Anderson, G.A.; Smith, R.D. Ion manipulations in structures for lossless ion manipulations (SLIM): Computational evaluation of a 90 degrees turn and a switch. Analyst 2015, 140, 6845–6852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaminathan, J.; Boulgakov, A.; Hernandez, E.T.; Bardo, A.M.; Bachman, J.L.; Marotta, J.; Johnson, A.M.; Anslyn, E.V.; Marcotte, E.M. Highly parallel single-molecule identification of proteins in zeptomole-scale mixtures. Nat. Biotechnol. 2018, 36, 1076–1082. [Google Scholar] [CrossRef]
- Picotti, P.; Aebersold, R. Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nat. Methods 2012, 9, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Gallien, S.; Duriez, E.; Domon, B. Selected reaction monitoring applied to proteomics. J. Mass Spectrom. 2011, 46, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Hopfgartner, G. Triple quadrupole linear ion trap mass spectrometer for the analysis of small molecules and macro-molecules. J. Mass Spectrom. 2004, 39, 845–855. [Google Scholar] [CrossRef]
- Haag, A.M. Mass Analyzers and Mass Spectrometers. Adv. Exp. Med. Biol. 2016, 919, 157–169. [Google Scholar] [CrossRef]
- Loos, G.; Van Schepdael, A.; Cabooter, D. Quantitative mass spectrometry methods for pharmaceutical analysis. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150366. [Google Scholar] [CrossRef] [Green Version]
- Calvo, E.; Camafeita, E.; Fernandez-Gutierrez, B.; Lopez, J.A. Applying selected reaction monitoring to targeted proteomics. Expert Rev. Proteom. 2011, 8, 165–173. [Google Scholar] [CrossRef]
- Kuhn, E.; Wu, J.; Karl, J.; Liao, H.; Zolg, W.; Guild, B. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and13C-labeled peptide standards. Proteomics 2004, 4, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Picotti, P.; Bodenmiller, B.; Mueller, L.N.; Domon, B.; Aebersold, R. Full Dynamic Range Proteome Analysis of S. cerevisiae by Targeted Proteomics. Cell 2009, 138, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Song, E.; Nie, S.; Rodland, K.D.; Liu, T.; Qian, W.-J.; Smith, R.D. Advances in targeted proteomics and applications to biomedical research. Proteomics 2016, 16, 2160–2182. [Google Scholar] [CrossRef] [Green Version]
- Bourmaud, A.; Gallien, S.; Domon, B. Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: Prin-ciple and applications. Proteomics 2016, 16, 2146–2159. [Google Scholar] [CrossRef] [PubMed]
- Peterson, A.C.; Russell, J.D.; Bailey, D.J.; Westphall, M.S.; Coon, J.J. Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Mol. Cell. Proteom. 2012, 11, 1475–1488. [Google Scholar] [CrossRef] [Green Version]
- Ronsein, G.E.; Pamir, N.; von Haller, P.D.; Kim, D.S.; Oda, M.N.; Jarvik, G.P.; Vaisar, T.; Heinecke, J.W. Parallel reaction monitoring (PRM) and selected reaction monitoring (SRM) exhibit comparable linearity, dynamic range and precision for targeted quantitative HDL proteomics. J. Proteom. 2014, 113, 388–399. [Google Scholar] [CrossRef] [Green Version]
- Zweigenbaum, J.; Henion, J. Bioanalytical high-throughput selected reaction monitoring-LC/MS determination of selected estrogen receptor modulators in human plasma: 2000 samples/day. Anal. Chem. 2000, 72, 2446–2454. [Google Scholar] [CrossRef] [PubMed]
- Pretty, J.R.; Deng, H.; Goeringer, D.E.; Van Berkel, G.J. Electrochemically modulated preconcentration and matrix elimination for organic analytes coupled on-line with electrospray mass spectrometry. Anal. Chem. 2000, 72, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Jensen, O.N.; Larsen, M.R.; Roepstorff, P. Mass spectrometric identification and microcharacterization of proteins from electrophoretic gels: Strategies and applications. Proteins 1998, 2, 74–89. [Google Scholar] [CrossRef]
- Whitelegge, J.P.; Gundersen, C.B.; Faull, K.F. Electrospray-ionization mass spectrometry of intact intrinsic membrane proteins. Protein Sci. 1998, 7, 1423–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Chaerkady, R.; Wu, J.; Hwang, H.J.; Papadopoulos, N.; Kopelovich, L.; Maitra, A.; Matthaei, H.; Eshleman, J.R.; Hruban, R.H.; et al. Mutant proteins as cancer-specific biomarkers. Proc. Natl. Acad. Sci. USA 2011, 108, 2444–2449. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhang, M.; Tomita, T.; Vogelstein, J.T.; Zhou, S.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Selected reaction monitoring approach for validating peptide biomarkers. Proc. Natl. Acad. Sci. USA 2017, 114, 13519–13524. [Google Scholar] [CrossRef] [Green Version]
- Gallien, S.; Bourmaud, A.; Kim, S.Y.; Domon, B. Technical considerations for large-scale parallel reaction monitoring analysis. J. Proteom. 2014, 100, 147–159. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, A.M.; Buchanan, A.H.; Kinde, I.; Warren, A.; Honushefsky, A.; Cohain, A.T.; Ledbetter, D.H.; Sanfilippo, F.; Sheridan, K.; Rosica, D.; et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 2020, 369, eabb9601. [Google Scholar] [CrossRef] [PubMed]
- Shasha, S.M.; Sharon, N.; Inbar, M. [Bacteriophages as antibacterial agents]. Harefuah 2004, 143, 121–125. [Google Scholar]
- Altamirano, F.L.G.; Barr, J.J. Phage Therapy in the Postantibiotic Era. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Tian, T.; Liu, W.; Zhu, Z.; Yang, C.J. Advance in phage display technology for bioanalysis. Biotechnol. J. 2016, 11, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [Green Version]
- Ventola, C.L. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.A.; Siddiqui, R. War on terror cells: Killing the host that harbours ‘superbugs’ is an infection control strategy in our fight against infectious diseases. Pathog. Glob. Health 2014, 108, 4–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, S.M. Role of efflux pumps in the antibiotic resistance of bacteria embedded in a biofilm. Virulence 2013, 4, 223–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, D.; Wang-Kan, X.; Neuberger, A.; Van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Genet. 2018, 16, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Summers, W.C. The strange history of phage therapy. Bacteriophage 2012, 2, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Labrie, S.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Genet. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Liu, C.G.; Green, S.I.; Min, L.; Clark, J.R.; Salazar, K.; Terwilliger, A.L.; Kaplan, H.B.; Trautner, B.W.; Ramig, R.F.; Maresso, A.W. Phage-Antibiotic Synergy Is Driven by a Unique Combination of Antibacterial Mechanism of Action and Stoichiometry. MBio 2020, 11. [Google Scholar] [CrossRef]
- Gurney, J.; Pradier, L.; Griffin, J.S.; Gougat-Barbera, C.; Chan, B.K.; Turner, E.P.; Kaltz, O.; Hochberg, E.M. Phage steering of antibiotic-resistance evolution in the bacterial pathogen, Pseudomonas aeruginosa. Evol. Med. Public Health 2020, 2020, 148–157. [Google Scholar] [CrossRef]
- Moreira, G.M.S.G.; Fühner, V.; Hust, M. Epitope Mapping by Phage Display. J. Immunol. Methods 2017, 1701, 497–518. [Google Scholar] [CrossRef]
- Hartley, O. The use of phage display in the study of receptors and their ligands. J. Recept. Signal Transduct. 2002, 22, 373–392. [Google Scholar] [CrossRef]
- Kushwaha, R.; Payne, C.M.; Downie, A.B. Uses of phage display in agriculture: A review of food-related pro-tein-protein interactions discovered by biopanning over diverse baits. Comput. Math. Methods Med. 2013, 2013, 653759. [Google Scholar] [CrossRef] [Green Version]
- Rojas, G.; Carmenate, T.; Santo-Tomás, J.F.; Valiente, P.A.; Becker, M.; Riverón, A.P.; Tundidor, Y.; Ortiz, Y.; De Cossio-Diaz, J.F.; Graca, L.; et al. Directed evolution of super-secreted variants from phage-displayed human Interleukin-2. Sci. Rep. 2019, 9, 800. [Google Scholar] [CrossRef]
- Mimmi, S.; Maisano, D.; Quinto, I.; Iaccino, E. Phage Display: An Overview in Context to Drug Discovery. Trends Pharmacol. Sci. 2019, 40, 87–91. [Google Scholar] [CrossRef]
- Jara-Acevedo, R.; Díez, P.; González-González, M.; Dégano, R.M.; Ibarrola, N.; Góngora, R.; Orfao, A.; Fuentes, M. Screening Phage-Display Antibody Libraries Using Protein Arrays. Methods Mol. Biol. 2017, 1701, 365–380. [Google Scholar] [CrossRef]
- Vandormael, P.; Verschueren, P.; De Winter, L.; Somers, V. cDNA phage display for the discovery of theranostic autoantibodies in rheumatoid arthritis. Immunol. Res. 2016, 65, 307–325. [Google Scholar] [CrossRef]
- Omidfar, K.; Daneshpour, M. Advances in phage display technology for drug discovery. Expert Opin. Drug Discov. 2015, 10, 651–669. [Google Scholar] [CrossRef]
- Portes, L.D.S. Subtractive phage display selection for screening and identification of peptide sequences with po-tential use in serodiagnosis of paracoccidioidomycosis caused by Paracoccidioides brasiliensis. Lett. Appl. Microbiol. 2017, 65, 346–353. [Google Scholar] [CrossRef]
- Rouet, R.; Jackson, K.J.L.; Langley, D.B.; Christ, D. Next-Generation Sequencing of Antibody Display Repertoires. Front. Immunol. 2018, 9, 118. [Google Scholar] [CrossRef] [Green Version]
- Brockmann, E.-C. Selection of Stable scFv Antibodies by Phage Display. Methods Mol. Biol. 2012, 907, 123–144. [Google Scholar] [CrossRef]
- Ranganathan, R. Putting Evolution to Work. Cell 2018, 175, 1449–1451. [Google Scholar] [CrossRef] [Green Version]
- Mullard, A. FDA approves 100th monoclonal antibody product. Nat. Rev. Drug Discov. 2021. [Google Scholar] [CrossRef]
- Yuri Laguna Terai, C.H.; Wang, B.; Kang, X.; Han, J.; Douglass, J.A.; Han-Chuang Hsiue, E.; Purohit, R.; de Silva, T.; Wang, Q. Valid-NEO: An Integrated System for Neoantigen Validation from Limited Clinical Samples. Anal. Chem. 2021. Under Revision. [Google Scholar]
- Zhao, W.; Wu, J.; Chen, S.; Zhou, Z. Shared neoantigens: Ideal targets for off-the-shelf cancer immunotherapy. Pharmacogenomics 2020, 21, 637–645. [Google Scholar] [CrossRef]
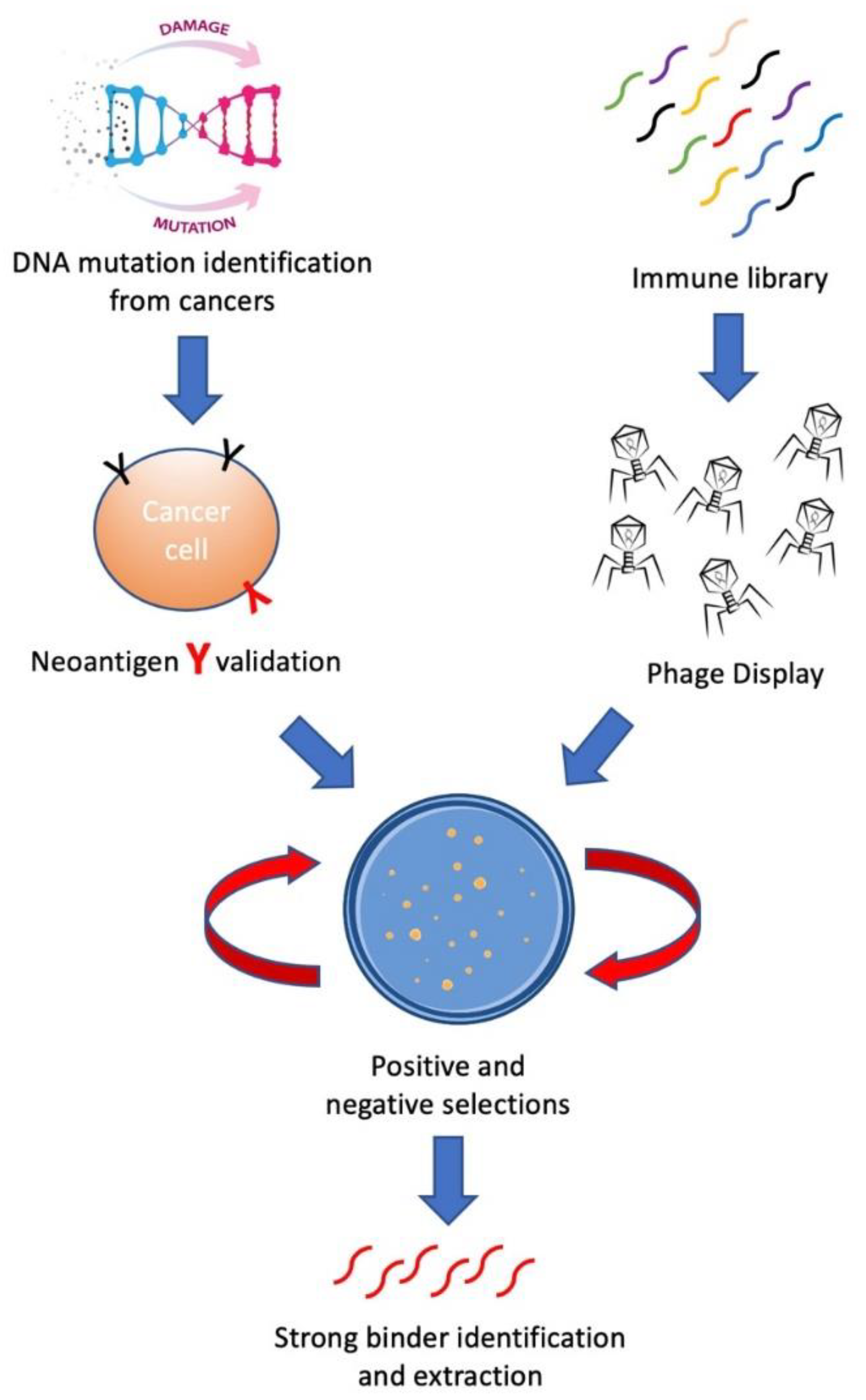

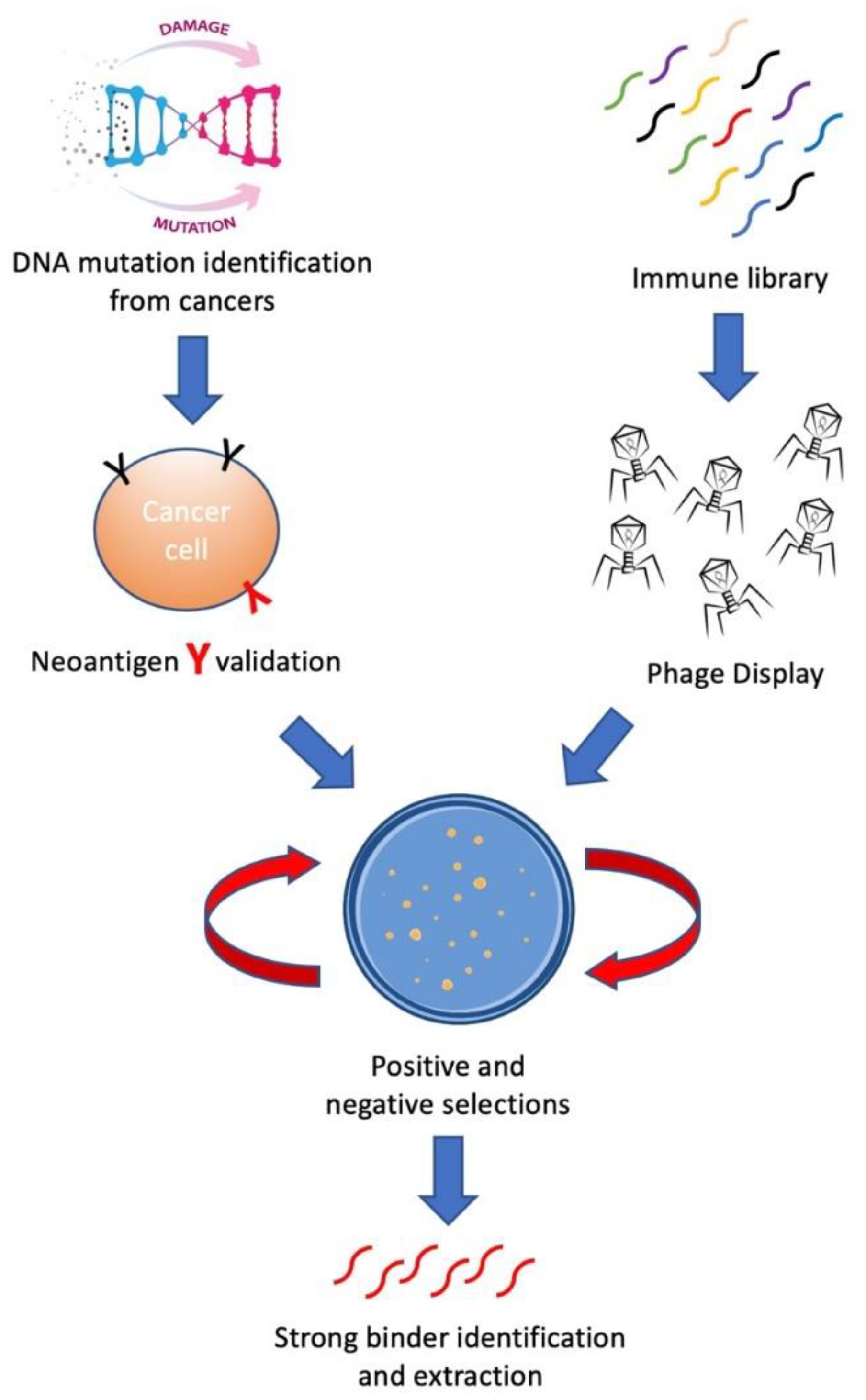{kind=link}
| DNA Change | Type | Consequences | Percentage in Cancer Patients |
|---|---|---|---|
| chr7:g.140753336A>T | Substitution | Missense BRAF V600E | 4.93% |
| chr2:g.208248388C>T | Substitution | Missense IDH1 R132H | 3.15% |
| chr12:g.25245350C>T | Substitution | Missense KRAS G12D | 2.61% |
| chr3:g.179218303G>A | Substitution | Missense PIK3CA E545K | 2.34% |
| chr3:g.179234297A>G | Substitution | Missense PIK3CA H1047R | 2.22% |
| chr12:g.25245350C>A | Substitution | Missense KRAS G12V | 2.06% |
| chr3:g.179218294G>A | Substitution | Missense PIK3CA E542K | 1.50% |
| chr17:g.7675088C>T | Substitution | Missense TP53 R175H | 1.49% |
| chr1:g.114713908T>C | Substitution | Missense NRAS Q61R | 1.44% |
| chr17:g.7674220C>T | Substitution | Missense TP53 R248Q | 1.16% |
| chr17:g.7673803G>A | Substitution | Missense TP53 R273C | 1.13% |
| chr1:g.114713909G>T | Substitution | Missense NRAS Q61K | 0.97% |
| chr12:g.25245351C>A | Substitution | Missense KRAS G12C | 0.97% |
| chr12:g.25245347C>T | Substitution | Missense KRAS G13D | 0.95% |
| chr1:g.6197725delT | Deletion | Frameshift RPL22 K15Rfs*5 | 0.93% |
| chr17:g.7673802C>T | Substitution | Missense TP53 R273H | 0.91% |
| chr17:g.7674221G>A | Substitution | Missense TP53 R248W | 0.89% |
| chr17:g.58357800delC | Deletion | Frameshift RNF43 G659Vfs*41 | 0.89% |
| chr17:g.7673776G>A | Substitution | Missense TP53 R282W | 0.83% |
| chr17:g.7674894G>A | Substitution | Stop Gained TP53 R213* | 0.72% |
| chr6:g.167003333delT | Deletion | Intron FGFR1OP | 0.69% |
| chr12:g.25227341T>G | Substitution | Missense KRAS Q61H | 0.66% |
| chr17:g.7674872T>C | Substitution | Missense TP53 Y220C | 0.65% |
| chr10:g.87965537delT | Deletion | 3 Prime UTR PTEN | 0.64% |
| chr3:g.179199088G>A | Substitution | Missense PIK3CA R88Q | 0.62% |
| chr1:g.64841314delT | Deletion | Frameshift JAK1 K860Nfs*16 | 0.57% |
| chr12:g.25245351C>G | Substitution | Missense KRAS G12R | 0.54% |
| chr17:g.7674945G>A | Substitution | Stop Gained TP53 R196* | 0.53% |
| chr17:g.20204950delA | Deletion | Frameshift SPECC1 N303Tfs*63 | 0.50% |
| chr2:g.208248389G>A | Substitution | Missense IDH1 R132C | 0.49% |
| chr10:g.87933148G>A | Substitution | Missense PTEN R130Q | 0.47% |
| chr14:g.104780214C>T | Substitution | Missense AKT1 E17K | 0.47% |
| chr9:g.21971121G>A | Substitution | Stop Gained CDKN2A R80* | 0.45% |
| chr12:g.25245350C>G | Substitution | Missense KRAS G12A | 0.45% |
| chr17:g.7674230C>T | Substitution | Missense TP53 G245S | 0.42% |
| chr10:g.87933147C>G | Substitution | Missense PTEN R130G | 0.41% |
| chr5:g.112839942C>T | Substitution | Stop Gained APC R1450* | 0.41% |
| chr17:g.7675076T>C | Substitution | Missense TP53 H179R | 0.41% |
| chr5:g.159099589delT | Deletion | 5 Prime UTR EBF1 | 0.39% |
| chr10:g.87957915C>T | Substitution | Stop Gained PTEN R233* | 0.37% |
| chr3:g.179234297A>T | Substitution | Missense PIK3CA H1047L | 0.34% |
| chr4:g.152328233G>A | Substitution | Missense FBXW7 R465C | 0.33% |
| chr7:g.140753337C>T | Substitution | Missense BRAF V600M | 0.33% |
| chr8:g.102277121delT | Deletion | Frameshift UBR5 E2121Kfs*28 | 0.33% |
| chr17:g.7670685G>A | Substitution | Stop Gained TP53 R342* | 0.33% |
| chr16:g.67611435_67611436insA | Insertion | Frameshift CTCF T204Nfs*26 | 0.33% |
| chr14:g.55684263delA | Deletion | 3 Prime UTR KTN1 | 0.32% |
| chr12:g.25245351C>T | Substitution | Missense KRAS G12S | 0.32% |
| chr17:g.7673704G>A | Substitution | Stop Gained TP53 R306* | 0.32% |
| chr4:g.1801841C>G | Substitution | Missense FGFR3 S249C | 0.32% |
| chr3:g.179203765T>A | Substitution | Missense PIK3CA N345K | 0.31% |
| chr17:g.7674953T>C | Substitution | Missense TP53 H193R | 0.30% |
| chr17:g.7675143C>A | Substitution | Missense TP53 V157F | 0.30% |
| chrX:g.77508202delA | Deletion | 3 Prime UTR ATRX | 0.30% |
| chr7:g.55191822T>G | Substitution | Missense EGFR L858R | 0.30% |
| chr17:g.39711955C>T | Substitution | Missense ERBB2 S310F | 0.30% |
| chr1:g.114716124C>G | Substitution | Missense NRAS G13R | 0.30% |
| chr19:g.3118944A>T | Substitution | Missense GNA11 Q209L | 0.30% |
| chr1:g.26779440delG | Deletion | Frameshift ARID1A D1850Tfs*33 | 0.29% |
| chr1:g.26779863C>T | Substitution | Stop Gained ARID1A R1989* | 0.29% |
| chr5:g.112840254_112840255insA | Insertion | Frameshift APC T1556Nfs*3 | 0.29% |
| chr19:g.52212718C>G | Substitution | Missense PPP2R1A P179R | 0.28% |
| chr2:g.222201320delT | Deletion | Intron PAX3 | 0.28% |
| chrX:g.40062191T>C | Substitution | Missense BCOR N1459S | 0.28% |
| chr1:g.114716126C>T | Substitution | Missense NRAS G12D | 0.27% |
| chr4:g.152328232C>T | Substitution | Missense FBXW7 R465H | 0.27% |
| chr10:g.87933147C>T | Substitution | Stop Gained PTEN R130* | 0.26% |
| chr17:g.7675994C>A | Substitution | Splice Region TP53 T125T | 0.26% |
| chr3:g.179221146G>A | Substitution | Missense PIK3CA E726K | 0.26% |
| chr17:g.7675124T>C | Substitution | Missense TP53 Y163C | 0.26% |
| chr5:g.158698822delA | Deletion | 3 Prime UTR EBF1 | 0.26% |
| chr12:g.132676598G>C | Substitution | Missense POLE P286R | 0.26% |
| chr1:g.114713908T>A | Substitution | Missense NRAS Q61L | 0.26% |
| chr12:g.4301917delT | Deletion | 3 Prime UTR CCND2 | 0.25% |
| chr12:g.49040709delG | Deletion | Frameshift KMT2D P2354Lfs*30 | 0.25% |
| chr10:g.87958013delA | Deletion | Frameshift PTEN K267Rfs*9 | 0.25% |
| chr10:g.87961042delTACT | Deletion | Frameshift PTEN T319* | 0.24% |
| chr14:g.65076348delA | Deletion | 3 Prime UTR MAX | 0.24% |
| chr9:g.77794572T>G | Substitution | Missense GNAQ Q209P | 0.24% |
| chr11:g.533874T>C | Substitution | Missense HRAS Q61R | 0.24% |
| chr3:g.179199690G>A | Substitution | Missense PIK3CA G118D | 0.24% |
| chr13:g.39343745delT | Deletion | 3 Prime UTR LHFP | 0.24% |
| chr17:g.7673802C>A | Substitution | Missense TP53 R273L | 0.24% |
| chr17:g.7675085C>A | Substitution | Missense TP53 C176F | 0.23% |
| chr10:g.121520163G>C | Substitution | Missense FGFR2 S252W | 0.23% |
| chr9:g.21971187G>A | Substitution | Stop Gained CDKN2A R58* | 0.23% |
| chr7:g.91973771delA | Deletion | Frameshift AKAP9 K39Rfs*17 | 0.23% |
| chr2:g.60458275delT | Deletion | 3 Prime UTR BCL11A | 0.23% |
| chr4:g.152326137G>C | Substitution | Missense FBXW7 R505G | 0.23% |
| chr12:g.25225628C>T | Substitution | Missense KRAS A146T | 0.23% |
| chr17:g.7674947A>G | Substitution | Missense TP53 I195T | 0.23% |
| chr5:g.112838220C>T | Substitution | Stop Gained APC R876* | 0.22% |
| chr17:g.7674957G>A | Substitution | Stop Gained TP53 Q192* | 0.22% |
| chr4:g.105240988delT | Deletion | Intron TET2 | 0.22% |
| chr17:g.7674216C>A | Substitution | Missense TP53 R249S | 0.22% |
| chr3:g.181713439delA | Deletion | 3 Prime UTR SOX2 | 0.22% |
| chr3:g.41224622C>T | Substitution | Missense CTNNB1 S37F | 0.22% |
| chr17:g.7673767C>T | Substitution | Missense TP53 E285K | 0.22% |
| chr5:g.112838934C>T | Substitution | Stop Gained APC R1114* | 0.22% |
| chr17:g.7675085C>T | Substitution | Missense TP53 C176Y | 0.22% |
| Total | 58.23% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q. Building Personalized Cancer Therapeutics through Multi-Omics Assays and Bacteriophage-Eukaryotic Cell Interactions. Int. J. Mol. Sci. 2021, 22, 9712. https://doi.org/10.3390/ijms22189712
Wang Q. Building Personalized Cancer Therapeutics through Multi-Omics Assays and Bacteriophage-Eukaryotic Cell Interactions. International Journal of Molecular Sciences. 2021; 22(18):9712. https://doi.org/10.3390/ijms22189712
Chicago/Turabian StyleWang, Qing. 2021. "Building Personalized Cancer Therapeutics through Multi-Omics Assays and Bacteriophage-Eukaryotic Cell Interactions" International Journal of Molecular Sciences 22, no. 18: 9712. https://doi.org/10.3390/ijms22189712
APA StyleWang, Q. (2021). Building Personalized Cancer Therapeutics through Multi-Omics Assays and Bacteriophage-Eukaryotic Cell Interactions. International Journal of Molecular Sciences, 22(18), 9712. https://doi.org/10.3390/ijms22189712