
High Mutational Heterogeneity, and New Mutations in the Human Coagulation Factor V Gene. Future Perspectives for Factor V Deficiency Using Recombinant and Advanced Therapies

 , , ,
, , , 
Abstract
1. Introduction
2. Results
2.1. Clinical Characteristics of the Two Patients with Factor V Deficiency
2.1.1. Case A History
2.1.2. Case B History
2.2. Hematological and Coagulation Analysis
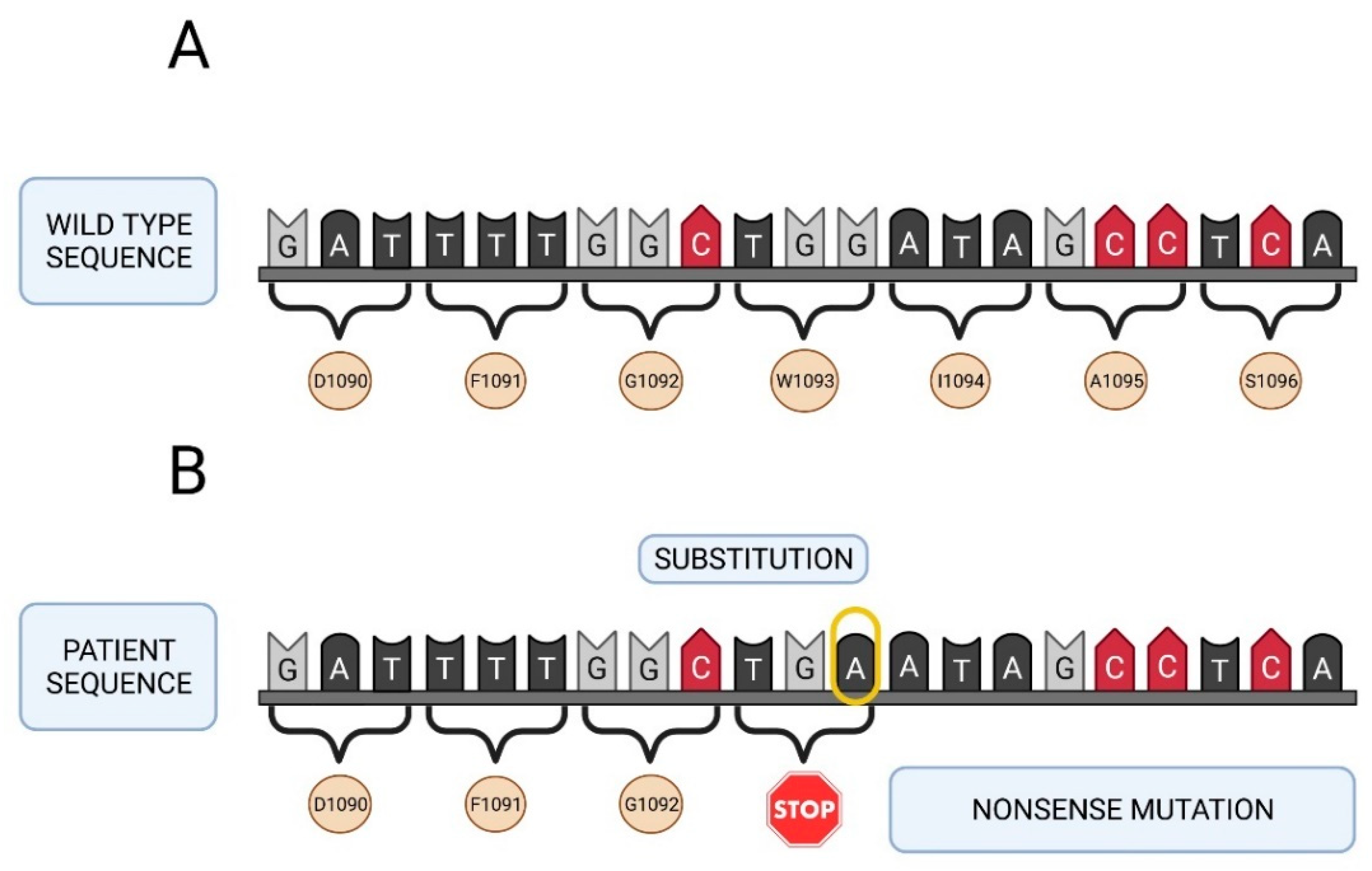
2.3. Molecular Study of the F5 Gene in the Two Patients and Their Parents
3. Discussion
4. Materials and Methods
4.1. Blood Collection
4.2. Hematological and Coagulation Tests
4.3. DNA Extraction
4.4. Mutational Analysis
4.5. Sequence Alignment
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Tabibian, S.; Shiravand, Y.; Shams, M.; Safa, M.; Gholami, M.S.; Heydari, F.; Ahmadi, A.; Rashidpanah, J.; Dorgalaleh, A. A Comprehensive Overview of Coagulation Factor V and Congenital Factor V Deficiency. Semin. Thromb. Hemost. 2019, 45, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, S.; Reglińska-Matveyev, N.; Gierula, M.; Camire, R.M.; Crawley, J.T.B.; Lane, D.A.; Ahnström, J. Factor V has an anticoagulant cofactor activity that targets the early phase of coagulation. J. Biol. Chem. 2017, 292, 9335–9344. [Google Scholar] [CrossRef]
- Van Doorn, P.; Rosing, J.; Duckers, C.; Hackeng, T.; Simioni, P.; Castoldi, E. Factor V Has Anticoagulant Activity in Plasma in the Presence of TFPIα: Difference between FV1 and FV2. Thromb. Haemost. 2018, 118, 1194–1202. [Google Scholar] [CrossRef]
- Segers, K.; Dahlbäck, B.; Nicolaes, G. Coagulation Factor V and Thrombophilia: Background and Mechanisms. Thromb. Haemost. 2007, 98, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Hirbawi, J.; Vaughn, J.L.; Bukys, M.A.; Vos, H.L.; Kalafatis, M. Contribution of Amino Acid Region 659-663 of Factor Va Heavy Chain to the Activity of Factor Xa within Prothrombinase. Biochemistry 2010, 49, 8520–8534. [Google Scholar] [CrossRef]
- Camire, R.M. A New Look at Blood Coagulation Factor V. Curr. Opin. Hematol. 2011, 18, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Mazurkiewicz-Pisarek, A.; Płucienniczak, G.; Ciach, T.; Płucienniczak, A. The Factor VIII Protein and Its Function. Acta Biochim. Pol. 2016, 63, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Vadivel, K.; Kumar, Y.; Bunce, M.W.; Camire, R.M.; Bajaj, M.S.; Bajaj, S.P. Interaction of Factor V B-Domain Acidic Region with Its Basic Region and with TFPI/TFPI2: Structural Insights from Molecular Modeling Studies. Int. Biol. Rev. 2017, 1. Available online: http://journals.ke-i.org/index.php/ibr/article/view/1334/975 (accessed on 2 September 2021).
- Bos, M.H.A.; Camire, R.M. Blood Coagulation Factors V and VIII: Molecular Mechanisms of Procofactor Activation. J. Coagul. Disord. 2010, 2, 19–27. [Google Scholar]
- Van Doorn, P.; Rosing, J.; Wielders, S.J.; Hackeng, T.M.; Castoldi, E. The C-Terminus of Tissue Factor Pathway Inhibitor-α Inhibits Factor V Activation by Protecting the Arg 1545 Cleavage Site. J. Thromb. Haemost. 2017, 15, 140–149. [Google Scholar] [CrossRef]
- Favaloro, E.J. Genetic Testing for Thrombophilia-Related Genes: Observations of Testing Patterns for Factor V Leiden (G1691A) and Prothrombin Gene “Mutation” (G20210A). Semin. Thromb. Hemost. 2019, 45, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Ruben, E.A.; Rau, M.J.; Fitzpatrick, J.; Di Cera, E. Cryo-EM structures of human coagulation factors V and Va. Blood 2021, 137, 3137–3144. [Google Scholar] [CrossRef]
- Hoffman, M.; Monroe, D.A. Cell-Based Model of Hemostasis. Thromb. Haemost. 2001, 85, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A. The Cell-Based Model of Coagulation. J. Vet. Emerg. Crit. Care 2009, 19, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Mann, K.G. Prothrombinase: The Paradigm for Membrane Bound Enzyme Complexes; a Memoir. J. Thromb. Thrombolysis 2021. [Google Scholar] [CrossRef] [PubMed]
- Stoilova-McPhie, S. Factor VIII and Factor V Membrane Bound Complexes. In Macromolecular Protein Complexes III: Structure and Function; Harris, J.R., Marles-Wright, J., Eds.; Subcellular Biochemistry; Springer International Publishing: Cham, Switzerland, 2021; Volume 96, pp. 153–175. [Google Scholar] [CrossRef]
- Kane, W.H.; Majerus, P.W. Purification and Characterization of Human Coagulation Factor V. J. Biol. Chem. 1981, 256, 1002–1007. [Google Scholar] [CrossRef]
- Gould, W.R.; Silveira, J.R.; Tracy, P.B. Unique in Vivo Modifications of Coagulation Factor V Produce a Physically and Functionally Distinct Platelet-Derived Cofactor. J. Biol. Chem. 2004, 279, 2383–2393. [Google Scholar] [CrossRef]
- Von Drygalski, A.; Bhat, V.; Gale, A.J.; Burnier, L.; Cramer, T.J.; Griffin, J.H.; Mosnier, L.O. An Engineered Factor Va Prevents Bleeding Induced by Anticoagulant Wt Activated Protein C. PLoS ONE 2014, 9, e104304. [Google Scholar] [CrossRef]
- Gale, A.J.; Bhat, V.; Pellequer, J.L.; Griffin, J.H.; Mosnier, L.O.; Von Drygalski, A. Safety, Stability and Pharmacokinetic Properties of SuperFactor Va, a Novel Engineered Coagulation Factor V for Treatment of Severe Bleeding. Pharm. Res. 2016, 33, 1517–1526. [Google Scholar] [CrossRef]
- Bhat, V.; von Drygalski, A.; Gale, A.J.; Griffin, J.H.; Mosnier, L.O. Improved Coagulation and Haemostasis in Haemophilia with Inhibitors by Combinations of SuperFactor Va and Factor VIIa. Thromb. Haemost. 2016, 115, 551–561. [Google Scholar] [CrossRef]
- Cui, J.; O’Shea, K.S.; Purkayastha, A.; Saunders, T.L.; Ginsburg, D. Fatal haemorrhage and incomplete block to embryogenesis in mice lacking coagulation factor V. Nature 1996, 384, 66–68. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Cui, J.; Taylor, J.; Yang, A.; Gruber, S.; Ginsburg, D. Rescue of Fatal Neonatal Hemorrhage in Factor V Deficient Mice by Low Level Transgene Expression. Thromb. Haemost. 2000, 83, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Weyand, A.C.; Grzegorski, S.J.; Rost, M.S.; Lavik, K.I.; Ferguson, A.C.; Menegatti, M.; Richter, C.E.; Asselta, R.; Duga, S.; Peyvandi, F.; et al. Analysis of Factor V in Zebrafish Demonstrates Minimal Levels Needed for Early Hemostasis. Blood Adv. 2019, 3, 1670–1680. [Google Scholar] [CrossRef]
- Cripe, L.D.; Moore., K.D.; Kane, W.H. Structure of the gene for human coagulation factor V. Biochemistry 1992, 31, 3777–3785. [Google Scholar] [CrossRef]
- Dall’Osso, C.; Guella, I.; Duga, S.; Locatelli, N.; Paraboschi, E.M.; Spreafico, M.; Afrasiabi, A.; Pechlaner, C.; Peyvandi, F.; Tenchini, M.L.; et al. Molecular Characterization of Three Novel Splicing Mutations Causing Factor V Deficiency and Analysis of the F5 Gene Splicing Pattern. Haematologica 2008, 93, 1505–1513. [Google Scholar] [CrossRef]
- Guasch, J.F.; Cannegieter, S.; Reitsma, P.H.; Van ‘t Veer-Korthof, E.T.; Bertina, R.M. Severe Coagulation Factor V Deficiency Caused by a 4 Bp Deletion in the Factor V Gene: Genetic Defect in Severe Factor V Deficiency. Br. J. Haematol. 1998, 101, 32–39. [Google Scholar] [CrossRef]
- The Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff (HGMD). Available online: http://www.hgmd.cf.ac.uk/ac/index.php (accessed on 2 September 2021).
- Caudill, J.S.; Sood, R.; Zehnder, J.L.; Pruthi, R.K.; Steensma, D.P. Severe Coagulation Factor V Deficiency Associated with an Interstitial Deletion of Chromosome 1q. J. Thromb. Haemost. 2007, 5, 626–628. [Google Scholar] [CrossRef]
- Guella, I.; Paraboschi, E.M.; van Schalkwyk, W.A.; Asselta, R.; Duga, S. Identification of the First Alu-Mediated Large Deletion Involving the F5 Gene in a Compound Heterozygous Patient with Severe Factor V Deficiency. Thromb. Haemost. 2011, 106, 296–303. [Google Scholar] [CrossRef]
- Lippi, G.; Favaloro, E.J.; Montagnana, M.; Manzato, F.; Guidi, G.C.; Franchini, M. Inherited and Acquired Factor V Deficiency. Blood Coagul. Fibrinolysis 2011, 22, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Thalji, N.; Camire, R. Parahemophilia: New Insights into Factor V Deficiency. Semin. Thromb. Hemost. 2013, 39, 607–612. [Google Scholar] [CrossRef]
- Congenital Factor V Deficiency. The Portal for Rare Diseases and Orphan Drugs Orphanet. Available online: https://www.orpha.net/consor/cgi-bin/index.php?lng=EN (accessed on 2 September 2021).
- Mannucci, P.M.; Duga, S.; Peyvandi, F. Recessively Inherited Coagulation Disorders. Blood 2004, 104, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Jenny, R.J.; Pittman, D.D.; Toole, J.J.; Kriz, R.W.; Aldape, R.A.; Hewick, R.M.; Kaufman, R.J.; Mann, K.G. Complete cDNA and Derived Amino Acid Sequence of Human Factor V. Proc. Natl. Acad. Sci. USA 1987, 84, 4846–4850. [Google Scholar] [CrossRef] [PubMed]
- Owren, P. Parahaemophilia. Lancet 1947, 249, 446–448. [Google Scholar] [CrossRef]
- Miletich, J.P.; Majerus, D.W.; Majerus, P.W. Patients with Congenital Factor V Deficiency Have Decreased Factor Xa Binding Sites on Their Platelets. J. Clin. Investig. 1978, 62, 824–831. [Google Scholar] [CrossRef]
- Duckers, C.; Simioni, P.; Spiezia, L.; Radu, C.; Dabrilli, P.; Gavasso, S.; Rosing, J.; Castoldi, E. Residual Platelet Factor V Ensures Thrombin Generation in Patients with Severe Congenital Factor V Deficiency and Mild Bleeding Symptoms. Blood 2010, 115, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, E.; Duckers, C.; Radu, C.; Spiezia, L.; Rossetto, V.; Tagariello, G.; Rosing, J.; Simioni, P. Homozygous F5 Deep-Intronic Splicing Mutation Resulting in Severe Factor V Deficiency and Undetectable Thrombin Generation in Platelet-Rich Plasma: Deep-Intronic Mutation Causing Severe FV Deficiency. J. Thromb. Haemost. 2011, 9, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Duckers, C.; Simioni, P.; Spiezia, L.; Radu, C.; Gavasso, S.; Rosing, J.; Castoldi, E. Low Plasma Levels of Tissue Factor Pathway Inhibitor in Patients with Congenital Factor V Deficiency. Blood 2008, 112, 3615–3623. [Google Scholar] [CrossRef]
- Jain, S.; Acharya, S.S. Management of Rare Coagulation Disorders in 2018. Transfus. Apher. Sci. 2018, 57, 705–712. [Google Scholar] [CrossRef]
- Peyvandi, F.; Di Michele, D.; Bolton-Maggs, P.H.B.; Lee, C.A.; Tripodi, A.; Srivastava, A. Classification of Rare Bleeding Disorders (RBDs) Based on the Association between Coagulant Factor Activity and Clinical Bleeding Severity: Classification of Rare Bleeding Disorders. J. Thromb. Haemost. 2012, 10, 1938–1943. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.K.; Hendrickson, J.E.; Bahel, P.; Siddon, A.J.; Rinder, H.M.; Tormey, C.A. Factor V activity in apheresis platelets: Implications for management of FV deficiency. Transfusion 2021, 61, 405–409. [Google Scholar] [CrossRef]
- Drzymalski, D.M.; Elsayes, A.H.; Ward, K.R.; House, M.; Manica, V.S. Platelet transfusion as treatment for factor V deficiency in the parturient: A case report. Transfusion 2019, 59, 2234–2237. [Google Scholar] [CrossRef]
- Heger, A.; Svae, T.E.; Neisser-Svae, A.; Jordan, S.; Behizad, M.; Römisch, J. Biochemical Quality of the Pharmaceutically Licensed Plasma OctaplasLG® after Implementation of a Novel Prion Protein (PrPSc) Removal Technology and Reduction of the Solvent/Detergent (S/D) Process Time. Vox Sang. 2009, 97, 219–225. [Google Scholar] [CrossRef]
- Cushing, M.M.; Asmis, L.; Calabia, C.; Rand, J.H.; Haas, T. Efficacy of Solvent/Detergent Plasma after Storage at 2–8 °C for 5 Days in Comparison to Other Plasma Products to Improve Factor V Levels in Factor V Deficient Plasma. Transfus. Apher. Sci. 2016, 55, 114–119. [Google Scholar] [CrossRef]
- Spinella, P.C.; Borasino, S.; Alten, J. Solvent/Detergent-Treated Plasma in the Management of Pediatric Patients Who Require Replacement of Multiple Coagulation Factors: An Open-Label, Multicenter, Post-Marketing Study. Front. Pediatr. 2020, 8, 572. [Google Scholar] [CrossRef]
- Goulenok, T.; Vasco, C.; Faille, D.; Ajzenberg, N.; De Raucourt, E.; Dupont, A.; Frere, C.; James, C.; Rabut, E.; Rugeri, L.; et al. RAVI study group. Acquired factor V inhibitor: A nation-wide study of 38 patients. Br. J. Haematol. 2021, 192, 892–899. [Google Scholar] [CrossRef]
- Lunghi, B.; Castoldi, E.; Mingozzi, F.; Bernardi, F.; Castaman, G. A Novel Factor V Null Mutation Detected in a Thrombophilic Patient with Pseudo-Homozygous APC Resistance and in an Asymptomatic Unrelated Subject. Blood 1998, 92, 1463–1464. [Google Scholar] [CrossRef]
- Patent “In Vitro Method to Recover the Expression of the F5 Gene Encoding Coagulation Factor V”. Invention Patent with Examination. Patent recognition. Office for the Transfer of Research Results. 2021. Available online: https://consultas2.oepm.es/pdf/ES/0000/000/02/78/53/ES-2785323_B2.pdf (accessed on 2 September 2021).
- In Vitro Method to Recover the Expression of the F5 Gene Encoding Coagulation Factor V. Transfer catalog. Office for the Transfer of Research Results. 2021. Available online: https://www.ucm.es/otri/complutransfer-metodo-in-vitro-para-recuperar-la-expresion-del-gen-f5-que-codifica-el-factor-v-de-la-coagulacion-1 (accessed on 2 September 2021).
- Ajzner, E.; Balogh, I.; Szabó, T.; Marosi, A.; Haramura, G.; Muszbek, L. Severe Coagulation Factor V Deficiency Caused by 2 Novel Frameshift Mutations: 2952delT in Exon 13 and 5493insG in Exon 16 of Factor 5 Gene. Blood 2002, 99, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Maheswari, K.; Wadhwa, L. Role of consanguinity in paediatric neurological disorders. Int. J. Contemp. Pediatrics 2016, 3, 939–942. [Google Scholar] [CrossRef][Green Version]
- Bhinder, M.A.; Sadia, H.; Mahmood, N.; Qasim, M.; Hussain, Z.; Rashid, M.M.; Zahoor, M.Y.; Bhatti, R.; Shehzad, W.; Waryah, A.M.; et al. Consanguinity: A Blessing or Menace at Population Level? Ann. Hum. Genet. 2019, 83, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Oniya, O.; Neves, K.; Ahmed, B.; Konje, J.C. A Review of the Reproductive Consequences of Consanguinity. Eur. J. Obstet. Gynecol. Reprod. Biol. 2019, 232, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Naderi, M.; Tabibian, S.; Alizadeh, S.; Hosseini, S.; Zaker, F.; Bamedi, T.; Shamsizadeh, M.; Dorgalaleh, A. Congenital Factor V Deficiency: Comparison of the Severity of Clinical Presentations among Patients with Rare Bleeding Disorders. Acta Haematol. 2015, 133, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Bhopal, R.S.; Petherick, E.S.; Wright, J.; Small, N. Potential Social, Economic and General Health Benefits of Consanguineous Marriage: Results from the Born in Bradford Cohort Study. Eur. J. Public Health 2014, 24, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Castoldi, E.; Lunghi, B.; Mingozzi, F.; Muleo, G.; Redaelli, R.; Mariani, G.; Bernardi, F. A Missense Mutation (Y1702C) in the Coagulation Factor V Gene Is a Frequent Cause of Factor V Deficiency in the Italian Population. Haematologica 2001, 86, 629–633. [Google Scholar] [PubMed]
- Kong, R.X.; Xie, Y.S.; Xie, H.X.; Luo, S.S.; Wang, M.S. Analysis of Phenotype and Genotype of a Family with Hereditary Coagulation Factor V Deficiency Caused by A Compound Heterozygous Mutation. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2020, 28, 2033–2038. [Google Scholar] [CrossRef]
- Asselta, R.; Tenchini, M.L.; Duga, S. Inherited Defects of Coagulation Factor V: The Hemorrhagic Side: Factor V and Bleeding Disorders. J. Thromb. Haemost. 2006, 4, 26–34. [Google Scholar] [CrossRef]
- Duga, S.; Montefusco, M.C.; Asselta, R.; Malcovati, M.; Peyvandi, F.; Santagostino, E.; Mannucci, P.M.; Tenchini, M.L. Arg2074Cys Missense Mutation in the C2 Domain of Factor V Causing Moderately Severe Factor V Deficiency: Molecular Characterization by Expression of the Recombinant Protein. Blood 2003, 101, 173–177. [Google Scholar] [CrossRef]
- Nuzzo, F.; Bulato, C.; Nielsen, B.I.; Lee, K.; Wielders, S.J.; Simioni, P.; Key, N.S.; Castoldi, E. Characterization of an Apparently Synonymous F5 Mutation Causing Aberrant Splicing and Factor V Deficiency. Haemophilia 2015, 21, 241–248. [Google Scholar] [CrossRef]
- Cunha, M.L.R.; Bakhtiari, K.; Peter, J.; Marquart, J.A.; Meijers, J.C.M.; Middeldorp, S. A Novel Mutation in the F5 Gene (Factor V Amsterdam) Associated with Bleeding Independent of Factor V Procoagulant Function. Blood 2015, 125, 1822–1825. [Google Scholar] [CrossRef]
- Yamazaki, T.; Nicolaes, G.A.; Sørensen, K.W.; Dahlbäck, B. Molecular basis of quantitative factor V deficiency associated with factor V R2 haplotype. Blood 2002, 100, 2515–2521. [Google Scholar] [CrossRef]
- Montefusco, M.C.; Duga, S.; Asselta, R.; Malcovati, M.; Peyvandi, F.; Santagostino, E.; Mannucci, P.M.; Tenchini, M.L. Clinical and Molecular Characterization of 6 Patients Affected by Severe Deficiency of Coagulation Factor V: Broadening of the Mutational Spectrum of Factor V Gene and in Vitro Analysis of the Newly Identified Missense Mutations. Blood 2003, 102, 3210–3216. [Google Scholar] [CrossRef][Green Version]
- Chen, T.Y.; Lin, T.M.; Chen, H.Y.; Wu, C.L.; Tsao, C.J. Gly392Cys Missense Mutation in the A2 Domain of Factor V Causing Severe Factor V Deficiency: Molecular Characterization by Expression of the Recombinant Protein. Thromb. Haemost. 2005, 93, 614–615. [Google Scholar] [CrossRef]
- Liu, H.C.; Shen, M.C.; Eng, H.L.; Wang, C.H.; Lin, T.M. Asp68His Mutation in the A1 Domain of Human Factor V Causes Impaired Secretion and Ineffective Translocation. Haemophilia 2014, 20, e318–e326. [Google Scholar] [CrossRef]
- Friedmann, A.P.; Koutychenko, A.; Wu, C.; Fredenburgh, J.C.; Weitz, J.I.; Gross, P.L.; Xu, P.; Ni, F.; Kim, P.Y. Identification and Characterization of a Factor Va-Binding Site on Human Prothrombin Fragment 2. Sci. Rep. 2019, 9, 2436. [Google Scholar] [CrossRef]
- Peyvandi, F.; Garagiola, I.; Young, G. The Past and Future of Haemophilia: Diagnosis, Treatments, and Its Complications. Lancet 2016, 388, 187–197. [Google Scholar] [CrossRef]
- Zhang, B.; Zheng, C.; Zhu, M.; Tao, J.; Vasievich, M.P.; Baines, A.; Kim, J.; Schekman, R.; Kaufman, R.J.; Ginsburg, D. Mice Deficient in LMAN1 Exhibit FV and FVIII Deficiencies and Liver Accumulation of A1-Antitrypsin. Blood 2011, 118, 3384–3391. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, P.V.; Dill, J.L.; Zhou, Q.; Fay, P.J. Contribution of Factor VIIIa A2 and A3-C1-C2 Subunits to the Affinity for Factor IXa in Factor Xase. Biochemistry 2004, 43, 5094–5101. [Google Scholar] [CrossRef]
- DeAngelis, J.P.; Wakabayashi, H.; Fay, P.J. Sequences Flanking Arg336 in Factor VIIIa Modulate Factor Xa-Catalyzed Cleavage Rates at This Site and Cofactor Function. J. Biol. Chem. 2012, 287, 15409–15417. [Google Scholar] [CrossRef] [PubMed]
- Williamson, D.; Brown, K.; Luddington, R.; Baglin, C.; Baglin, T. Factor V Cambridge: A New Mutation (Arg306→Thr) Associated With Resistance to Activated Protein C. Blood 1998, 91, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zhang, B. Combined Deficiency of Coagulation Factors V and VIII: An Update. Semin. Thromb. Hemost. 2013, 39, 613–620. [Google Scholar] [CrossRef]
- Everett, L.A.; Khoriaty, R.N.; Zhang, B.; Ginsburg, D. Altered Phenotype in LMAN1-Deficient Mice with Low Levels of Residual LMAN1 Expression. Blood Adv. 2020, 4, 5635–5643. [Google Scholar] [CrossRef] [PubMed]
- Altenhoff, A.M.; Glover, N.M.; Dessimoz, C. Inferring Orthology and Paralogy. In Evolutionary Genomics; Anisimova, M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; Volume 1910, pp. 149–175. [Google Scholar] [CrossRef]
- Yang, T.L.; Cui, J.; Rehumtulla, A.; Yang, A.; Moussalli, M.; Kaufman, R.J.; Ginsburg, D. The Structure and Function of Murine Factor V and Its Inactivation by Protein C. Blood 1998, 91, 4593–4599. [Google Scholar] [CrossRef]
- Rallapalli, P.M.; Orengo, C.A.; Studer, R.A.; Perkins, S.J. Positive Selection during the Evolution of the Blood Coagulation Factors in the Context of Their Disease-Causing Mutations. Mol. Biol. Evol. 2014, 31, 3040–3056. [Google Scholar] [CrossRef] [PubMed]
- Mariz, J.P.V.; Nery, M.F. Unraveling the Molecular Evolution of Blood Coagulation Genes in Fishes and Cetaceans. Front. Mar. Sci. 2020, 7, 592383. [Google Scholar] [CrossRef]
- Kretz, C.A.; Weyand, A.C.; Shavit, J.A. Modeling Disorders of Blood Coagulation in the Zebrafish. Curr. Pathobiol. Rep. 2015, 3, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Fish, R.J.; Freire, C.; Di Sanza, C.; Neerman-Arbez, M. Venous Thrombosis and Thrombocyte Activity in Zebrafish Models of Quantitative and Qualitative Fibrinogen Disorders. Int. J. Mol. Sci. 2021, 22, 655. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.; Jagadeeswaran, P. Microkinetic Coagulation Assays for Human and Zebrafish Plasma. Blood. Coagul. Fibrinolysis 2021, 32, 50–56. [Google Scholar] [CrossRef]
- Kuta, P.; Melling, N.; Zimmermann, R.; Achenbach, S.; Eckstein, R.; Strobel, J. Clotting Factor Activity in Fresh Frozen Plasma after Thawing with a New Radio Wave Thawing Device. Transfusion 2019, 59, 1857–1861. [Google Scholar] [CrossRef]
- Marchesini, E.; Morfini, M.; Valentino, L. Recent Advances in the Treatment of Hemophilia: A Review. BTT 2021, 15, 221–235. [Google Scholar] [CrossRef]
- Sankar, A.D.; Weyand, A.C.; Pipe, S.W. The Evolution of Recombinant Factor Replacement for Hemophilia. Transfus. Apher. Sci. 2019, 58, 596–600. [Google Scholar] [CrossRef] [PubMed]
- High, K.A.; Roncarolo, M.G. Gene Therapy. N. Engl. J. Med. 2019, 381, 455–464. [Google Scholar] [CrossRef]
- Anguela, X.M.; High, K.A. Entering the Modern Era of Gene Therapy. Annu. Rev. Med. 2019, 70, 273–288. [Google Scholar] [CrossRef]
- Kamiyama, Y.; Naritomi, Y.; Moriya, Y.; Yamamoto, S.; Kitahashi, T.; Maekawa, T.; Yahata, M.; Hanada, T.; Uchiyama, A.; Noumaru, A.; et al. Biodistribution Studies for Cell Therapy Products: Current Status and Issues. Regen. Ther. 2021, 18, 202–216. [Google Scholar] [CrossRef]
- Buzhor, E.; Leshansky, L.; Blumenthal, J.; Barash, H.; Warshawsky, D.; Mazor, Y.; Shtrichman, R. Cell-Based Therapy Approaches: The Hope for Incurable Diseases. Regen. Med. 2014, 9, 649–672. [Google Scholar] [CrossRef] [PubMed]
- Weinshilboum, R.M.; Wang, L. Pharmacogenomics: Precision Medicine and Drug Response. Mayo Clin. Proc. 2017, 92, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- De Haan, P.; Van Diemen, F.R.; Toscano, M.G. Viral Gene Delivery Vectors: The next Generation Medicines for Immune-Related Diseases. Hum. Vaccin. Immunother. 2021, 17, 14–21. [Google Scholar] [CrossRef]
- Athanasopoulos, T.; Munye, M.M.; Yáñez-Muñoz, R.J. Nonintegrating Gene Therapy Vectors. Hematol. Oncol. Clin. North Am. 2017, 31, 753–770. [Google Scholar] [CrossRef]
- Zu, H.; Gao, D. Non-Viral Vectors in Gene Therapy: Recent Development, Challenges, and Prospects. AAPS J. 2021, 23, 78. [Google Scholar] [CrossRef]
- Gähwiler, E.K.N.; Motta, S.E.; Martin, M.; Nugraha, B.; Hoerstrup, S.P.; Emmert, M.Y. Human iPSCs and Genome Editing Technologies for Precision Cardiovascular Tissue Engineering. Front. Cell Dev. Biol. 2021, 9, 639699. [Google Scholar] [CrossRef]
- García-Bernal, D.; García-Arranz, M.; Yáñez, R.M.; Hervás-Salcedo, R.; Cortés, A.; Fernández-García, M.; Hernando-Rodríguez, M.; Quintana-Bustamante, O.; Bueren, J.A.; García-Olmo, D.; et al. The Current Status of Mesenchymal Stromal Cells: Controversies, Unresolved Issues and Some Promising Solutions to Improve Their Therapeutic Efficacy. Front. Cell Dev. Biol. 2021, 9, 650664. [Google Scholar] [CrossRef] [PubMed]
- Fomin, M.E.; Togarrati, P.P.; Muench, M.O. Progress and Challenges in the Development of a Cell-Based Therapy for Hemophilia A. J. Thromb. Haemost. 2014, 12, 1954–1965. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rodríguez-Merchán, E.C.; De Pablo-Moreno, J.A.; Liras, A. Gene Therapy in Hemophilia: Recent Advances. Int. J. Mol. Sci. 2021, 22, 7647. [Google Scholar] [CrossRef] [PubMed]
- Parri, M.S.; Gianetti, J.; Dushpanova, A.; Della Pina, F.; Saracini, C.; Marcucci, R.; Giusti, B.; Berti, S. Pantoprazole Significantly Interferes with Antiplatelet Effect of Clopidogrel: Results of a Pilot Randomized Trial. Int. J. Cardiol. 2013, 167, 2177–2181. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Arbini, A.; Chantarangkul, V.; Mannucci, P.M. Recombinant Tissue Factor as Substitute for Conventional Thromboplastin in the Prothrombin Time Test. Thromb. Haemost. 1992, 67, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Van den Besselaar, A.M.; Neuteboom, J.; Bertina, R.M. Effect of Synthetic Phospholipids on the Response of the Activated Partial Thromboplastin Time to Heparin. Blood Coagul. Fibrinolysis 1993, 4, 895–903. [Google Scholar] [CrossRef] [PubMed]
- Ray, M.J.; Hawson, G.A.T. A Comparison of Two APTT Reagents Which Use Silica Activators. Clin. Lab. Haematol. 1989, 11, 221–232. [Google Scholar] [CrossRef]
- Den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M.; et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.M.; Taly, J.F.; Notredame, C. T-Coffee: A Web Server for the Multiple Sequence Alignment of Protein and RNA Sequences Using Structural Information and Homology Extension. Nucleic Acids Res. 2011, 39, W13–W17. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PT | PA | aPTT | F | INR | FVa | |
|---|---|---|---|---|---|---|
| Patient A * | 49.6 | 16.0 | 146.5 | 4.4 | 4.6 | <1 |
| Patient B ** | 50.1 | 10.7 | 129.5 | 3.7 | 5.8 | <1 |
| Father A | 12.2 | 83.0 | 37.8 | 3.2 | 1.1 | 21.0 |
| Mother A | 11.0 | 96.0 | 34.4 | 3.6 | 1.0 | 62.9 |
| Father B | 11.5 | 86.8 | 30.3 | 3.3 | 1.1 | 45.9 |
| Mother B | 12.0 | 92.3 | 26.8 | 3.7 | 1.0 | 46.4 |
| F5 Gene † | Exon | Intron | FV Protein | SNP | MAF § | Polyphen | M-Taster | SIFT | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | c.237A>G | 2 | p.Gln79Gln | rs6028 | 23 | ** | ** | ** | |
| 2 | c.952+76C>T | 6 | * | rs2239854 | 27 | ** | ** | ** | |
| 3 | c.1238T>C | 8 | p.Met413Thr | rs6033 | 5 | Benign | Polymorphism | Permissive | |
| 4 | c.1242A>G | 8 | p.Lys414Lys | rs6035 | 7 | ** | ** | ** | |
| 5 | c.1297-121T>C | 8 | * | rsl0800456 | 43 | ** | ** | ** | |
| 6 | c.1380C>T | 9 | p.Asn460Asn | rs6015 | 5 | ** | ** | ** | |
| 7 | c.1397-164C>T | 9 | * | rs7537742 | 50 | ** | ** | ** | |
| 8 | c.1716G>A | 11 | p.Glu572Glu | rs6036 | 5 | ** | ** | ** | |
| 9 | c.1926C>A | 12 | p.Thr642Thr | rs6037 | 5 | ** | ** | ** | |
| 10 | c.2218C>T | 13 | p.Arg740 * | ** | ** | ** | ** | ** | |
| 11 | c.2289A>G | 13 | p.Glu763Glu | rs6024 | 5 | ** | ** | ** | |
| 12 | c.2450A>C | 13 | p.Asn817Thr | rs6018 | 5 | Benign | Polymorphism | Permissive | |
| 13 | c.2758A>G | 13 | p.Arg920Gly | ** | ** | Benign | Polymorphism | Deleterious | |
| 14 | c.3279G>A | 13 | p.Trp1093 * | ** | ** | ** | ** | ** | |
| 15 | c.3865T>C | 13 | p.Phe1289Leu | ** | ** | Benign | Polymorphism | Permissive | |
| 16 | c.3939C>T | 13 | p.Ser1313Ser | ** | ** | ** | ** | ** | |
| 17 | c.3980A>G | 13 | p.His1327Arg | rs1800595 | 5 | Benign | Polymorphism | Permissive | |
| 18 | c.4095C>T | 13 | p.Thr1365Thr | rs9332607 | 26 | ** | ** | ** | |
| 19 | c.4796+50A>C | 13 | * | ** | ** | ** | ** | ** | |
| 20 | c.5209-134T>C | 15 | * | ** | ** | ** | ** | ** | |
| 21 | c.5290A>G | 16 | p.Met1764Val | rs6030 | 29 | Benign | Polymorphism | Permissive | |
| 22 | c.6194-20C>A | 22 | * | rs6013 | 5 | ** | ** | ** | |
| 23 | c.6529-65A>C | 24 | * | rs2227243 | 5 | ** | ** | ** | |
| 24 | c.6665A>G | 25 | p.Asp2222Gly | rs6027 | 5 | Benign | Polymorphism | Permissive |
| Exon * | Primer | Sequence 5′ → 3′ | Size (bp) |
|---|---|---|---|
| 1 | F5_1F | CACCTGCAGTAA AACAGTCAC | 532 |
| F5_1R | AGCCATGACATTGCAAAGGG | ||
| 2 | F5_2F | ACAGTTTGGGTTTCTACTGTG | 461 |
| F5_2R | GCATGTGAATGCCAAATTACCC | ||
| 3 | F5_3F | AAGTGAGTCAGCCTCAGGAC | 451 |
| F5_3R | AATGCAGGTCTAGAGGACTC | ||
| 4 | F5_4F | TACATGAGCATAGAAATGGGC | 528 |
| F5_4R | TCAAACAATGATCTGGTCTCC | ||
| 5 | F5_5F | CCCCAAAGCAAGAAGGTATC | 488 |
| F5_5R | CCTTCTTGATAGGGAGTTGC | ||
| 6 | F5_6F | AGGGCACAAACTACAACTGG | 494 |
| F5_6R | TGAGGAAAGTTTGTCTGCGG | ||
| 7 | F5_7F | TCTTGCCTTTTCTGGATGCC | 463 |
| F5_7R | CCAATACATGTGTCCCCTTG | ||
| 8 | F5_8F | ATGCAGGAGACAAATCAGAAG | 572 |
| F5_8R | TTGAGAAACTGTCTCAGATCC | ||
| 9 | F5_9F | AATGCTCCTGCCAAGTGATG | 442 |
| F5_9R | AACTCCTGAAGTGAGAAGGG | ||
| 10 | F5_10F | GCAATATTAATTGGTTCCAGCG | 413 |
| F5_10R | TCTCTTGAAGGAAATGCCCC | ||
| 11 | F5_11F | GGAATAGAGAATCCTTTCCC | 433 |
| F5_11R | AAGTCTTTGGACTGGAAGTG | ||
| 12 | F5_12F | AATCACTGCTTTGACACAACC | 459 |
| F5_12R | TTGAAAGAAAAGCCTGCAGGG | ||
| 13 | F5_13AF | TAGGTCACAGACAAGCAGTG | 562 |
| F5_13AR | CTTTCTGAGGTTCTGCAAGG | ||
| F5_13BF | TGGCTGCAGCATTAGGAATC | 549 | |
| F5_13BR | CCAGTGTCTTGGCTAGGAAGG | ||
| F5_13CF | TAAGCATAAGGGACCCAAGG | 568 | |
| F5_13CR | TCTTAGAGGGTGAAAGGTCC | ||
| F5_13DF | AACAAGCCTGGAAAGCAGAG | 501 | |
| F5_13DR | TTCACTGAGCTCTGGAGAAG | ||
| F5_13EF | GACACTGGTCAGGCAAGCTG | 651 | |
| F5_13ER | TGGAGAAATGGGCATCTGAC | ||
| F5_13FF | AGATGCCCATTTCTCCAGAC | 577 | |
| F5_13FR | AGATCTGTCTCACCAAGGTC | ||
| F5_13GF | CAACCCTTTCTCTAGACCTC | 520 | |
| F5_13GR | AGTCATCTTCACTGCTCTGG | ||
| F5_13HF | CTTATCCAGACCTTGGTCAG | 469 | |
| F5_13HR | ATAGGGGAACCAGACTGTTC | ||
| 14 | F5_14F | AGGTCATAGGAAGACTTACC | 480 |
| F5_14R | TCACCTATAGCTCTCTTGCC | ||
| 15 | F5_15F | ACTTGGGCCATATCTCACAG | 502 |
| F5_15R | GAAATAACCCCGACTCTTCC | ||
| 16 | F5_16F | GATCAATCAGAGGAAGGAGG | 506 |
| F5_16R | GTCTCAGAAGCATCTCATGTC | ||
| 17 | F5_17F | GGGAATGCAGAATCATGAGG | 449 |
| F5_17R | TTTGGGTCTATGGGTTTGCC | ||
| 18 | F5_18F | GAAAGCCTCTTGTGAAGCAG | 365 |
| F5_18R | CAATGCAATCAGACCATGGG | ||
| 19 | F5_19F | TTAAGTCAGGGCCACACAAG | 357 |
| F5_19R | CCCAAATGGAGCTGCTTCAC | ||
| 20 | F5_20F | TACAACACAGGTCCTCCAAG | 339 |
| F5_20R | GCCTCACACTTAGTACTTGC | ||
| 21 | F5_21F | AGGCAGTGTGTGACTTGTTG | 355 |
| F5_21R | CCATATGACCCTTAGAAAGCC | ||
| 22 | F5_22F | CTGGAACTGGAATTATCCCC | 425 |
| F5_22R | CAAAGGTTTTCCTAGGAGCC | ||
| 23 | F5_23F | GCCTGAGAACAGTATTTGGC | 414 |
| F5_23R | ATACTCCTGCTTCCCAGATC | ||
| 24 | F5_24F | GAGACTGTGAATCCTAAGGG | 420 |
| F5_24R | AGAGGTGGTACATGTCACTG | ||
| 25 | F5_25F | GTTTAAGGCTGCAGTGAGCC | 473 |
| F5_25R | CTTACTTACTGGTAGCAAGGAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernal, S.; Pelaez, I.; Alias, L.; Baena, M.; De Pablo-Moreno, J.A.; Serrano, L.J.; Camero, M.D.; Tizzano, E.F.; Berrueco, R.; Liras, A. High Mutational Heterogeneity, and New Mutations in the Human Coagulation Factor V Gene. Future Perspectives for Factor V Deficiency Using Recombinant and Advanced Therapies. Int. J. Mol. Sci. 2021, 22, 9705. https://doi.org/10.3390/ijms22189705
Bernal S, Pelaez I, Alias L, Baena M, De Pablo-Moreno JA, Serrano LJ, Camero MD, Tizzano EF, Berrueco R, Liras A. High Mutational Heterogeneity, and New Mutations in the Human Coagulation Factor V Gene. Future Perspectives for Factor V Deficiency Using Recombinant and Advanced Therapies. International Journal of Molecular Sciences. 2021; 22(18):9705. https://doi.org/10.3390/ijms22189705
Chicago/Turabian StyleBernal, Sara, Irene Pelaez, Laura Alias, Manel Baena, Juan A. De Pablo-Moreno, Luis J. Serrano, M. Dolores Camero, Eduardo F. Tizzano, Ruben Berrueco, and Antonio Liras. 2021. "High Mutational Heterogeneity, and New Mutations in the Human Coagulation Factor V Gene. Future Perspectives for Factor V Deficiency Using Recombinant and Advanced Therapies" International Journal of Molecular Sciences 22, no. 18: 9705. https://doi.org/10.3390/ijms22189705
APA StyleBernal, S., Pelaez, I., Alias, L., Baena, M., De Pablo-Moreno, J. A., Serrano, L. J., Camero, M. D., Tizzano, E. F., Berrueco, R., & Liras, A. (2021). High Mutational Heterogeneity, and New Mutations in the Human Coagulation Factor V Gene. Future Perspectives for Factor V Deficiency Using Recombinant and Advanced Therapies. International Journal of Molecular Sciences, 22(18), 9705. https://doi.org/10.3390/ijms22189705