
Inhibition of BMI-1 Induces Apoptosis through Downregulation of DUB3-Mediated Mcl-1 Stabilization



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
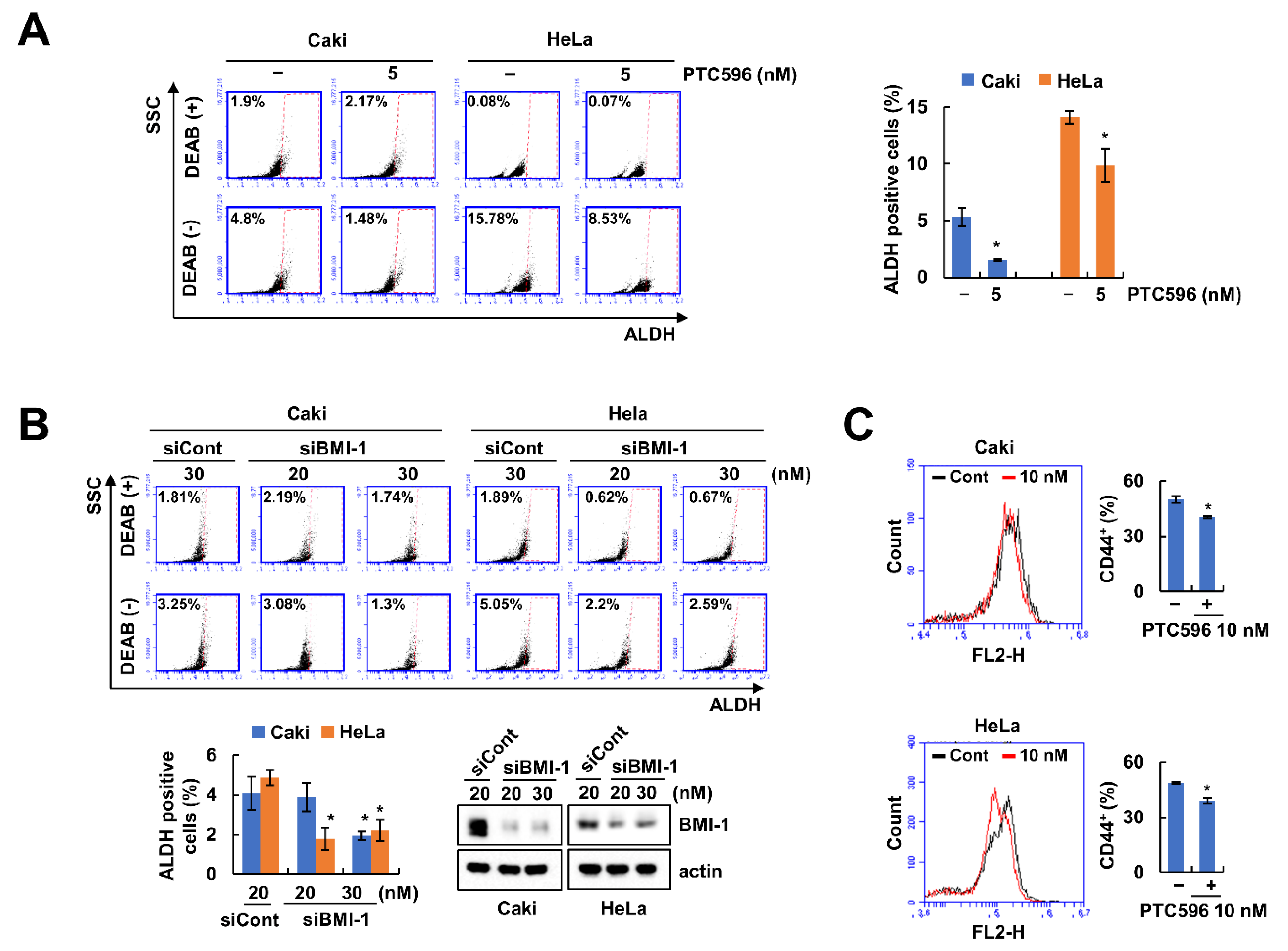
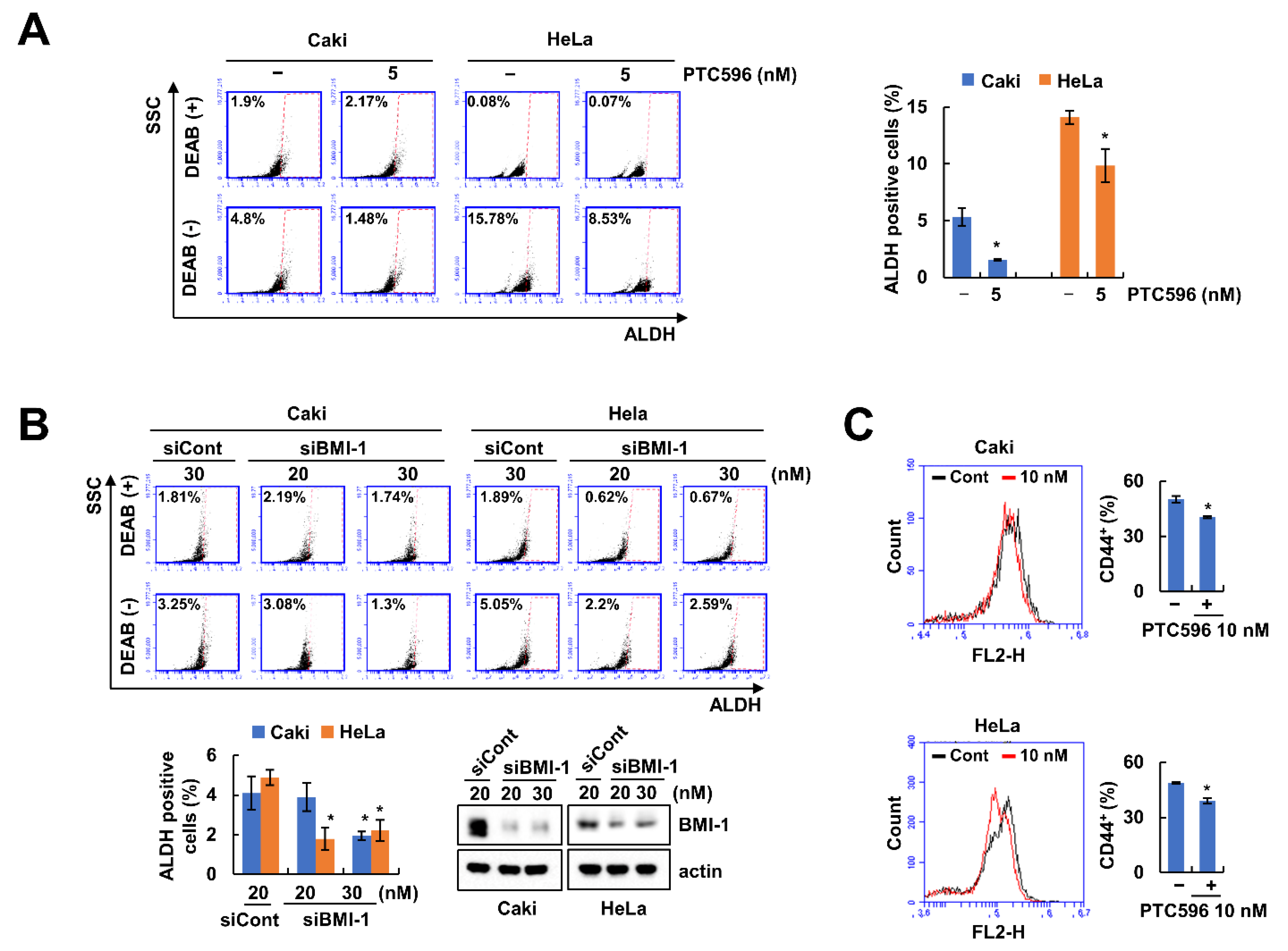
2.1. BMI-1 Inhibitor PTC596 Decreases the Cancer Stem-Like Cells (CSCs) Population
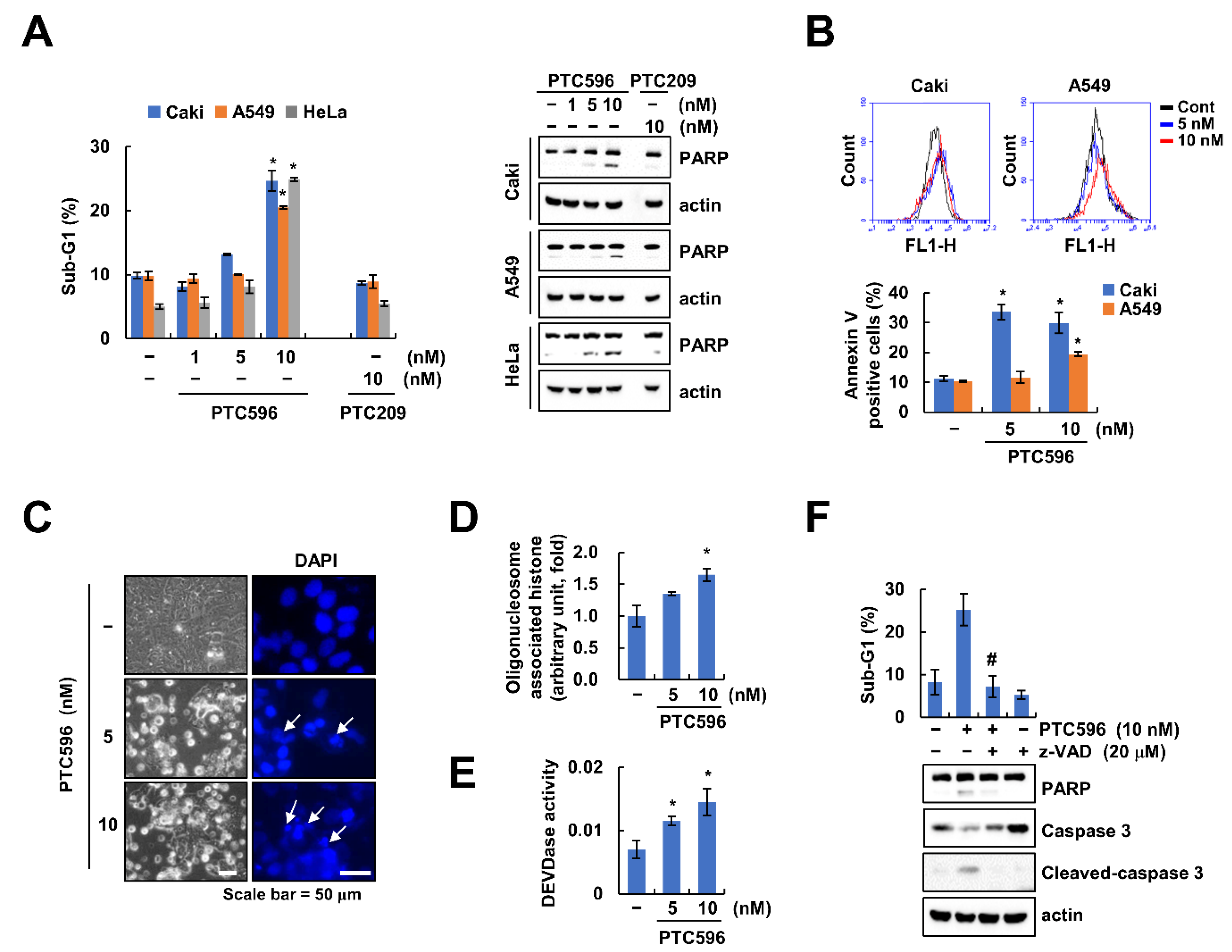
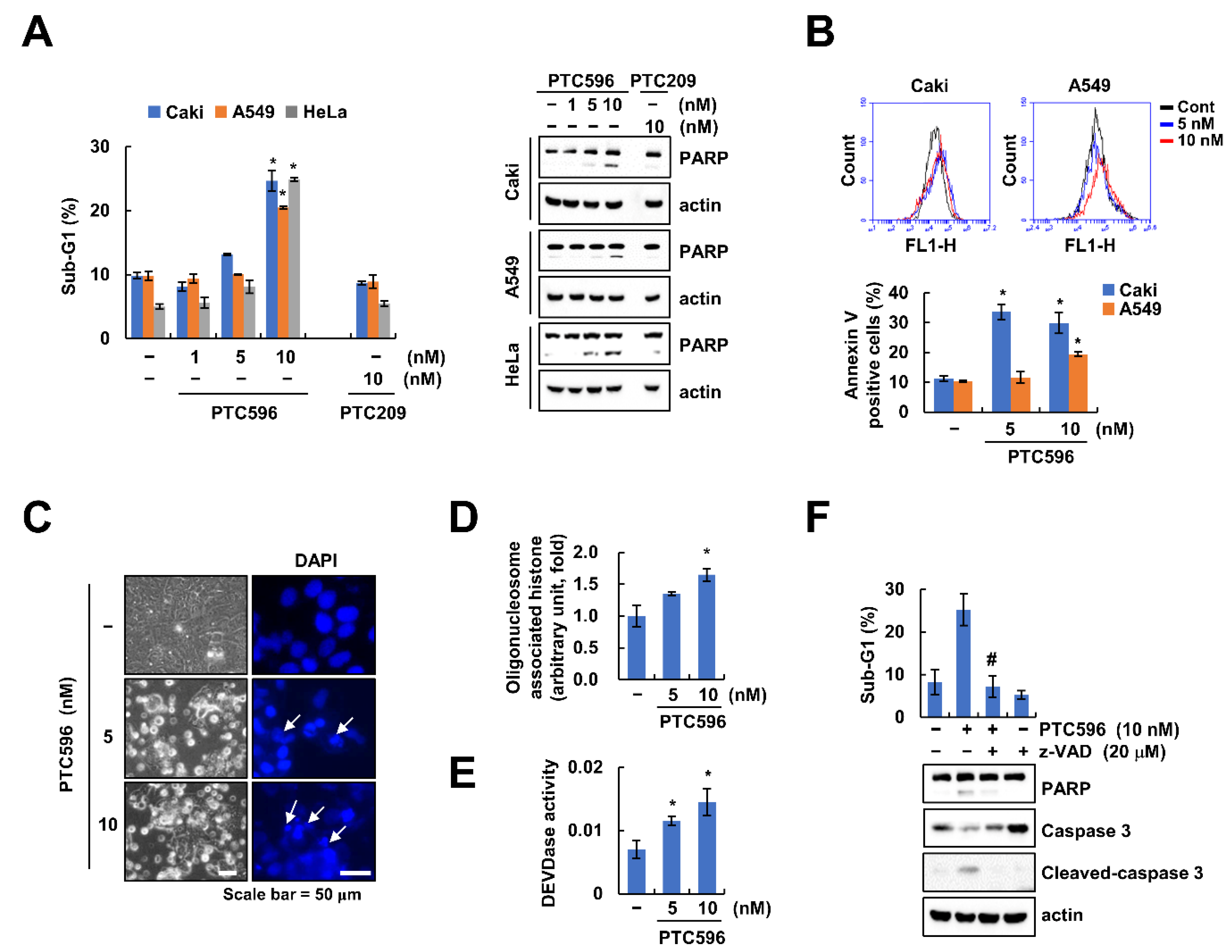
2.2. PTC596 Induces Apoptosis through Caspase-3 Activation
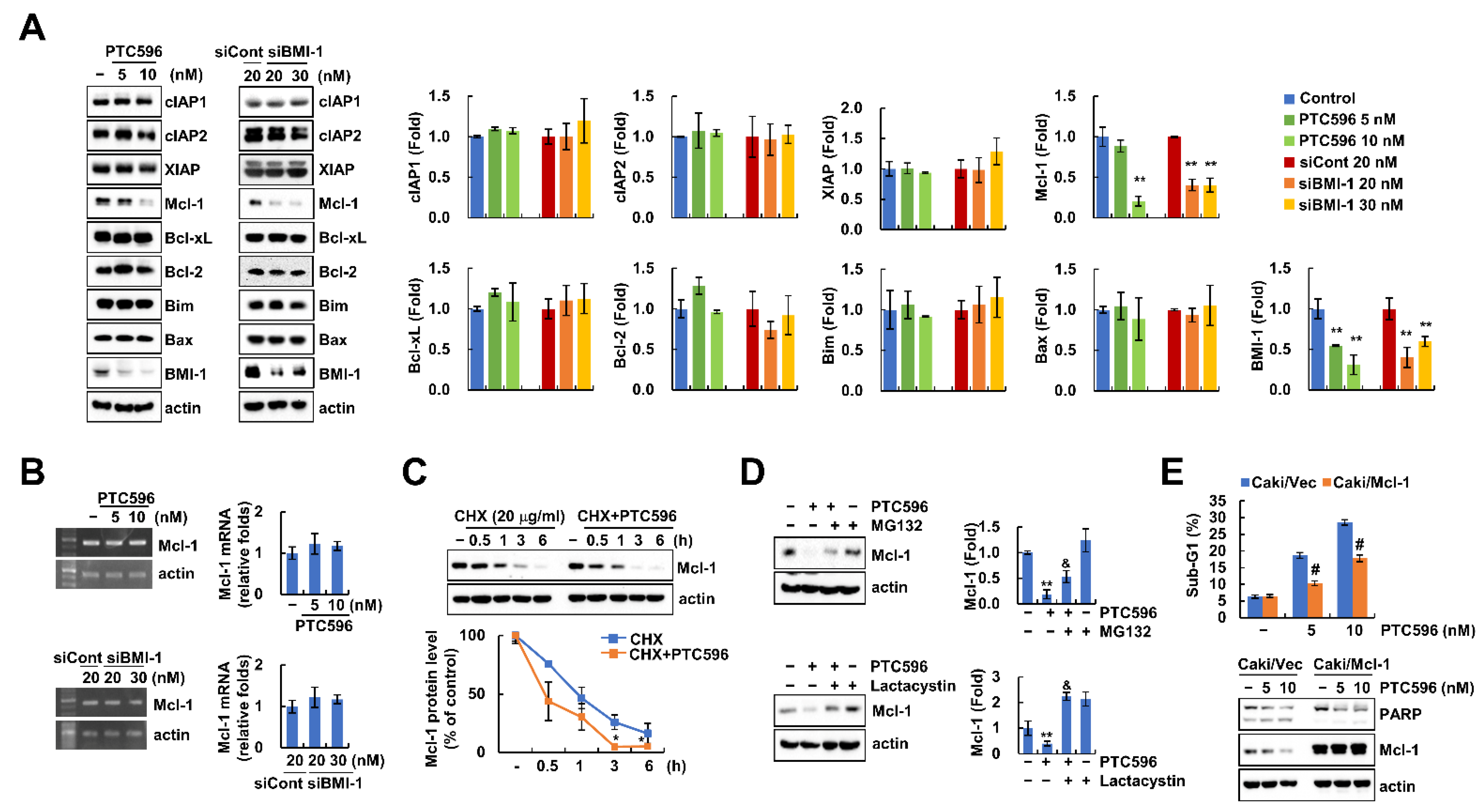
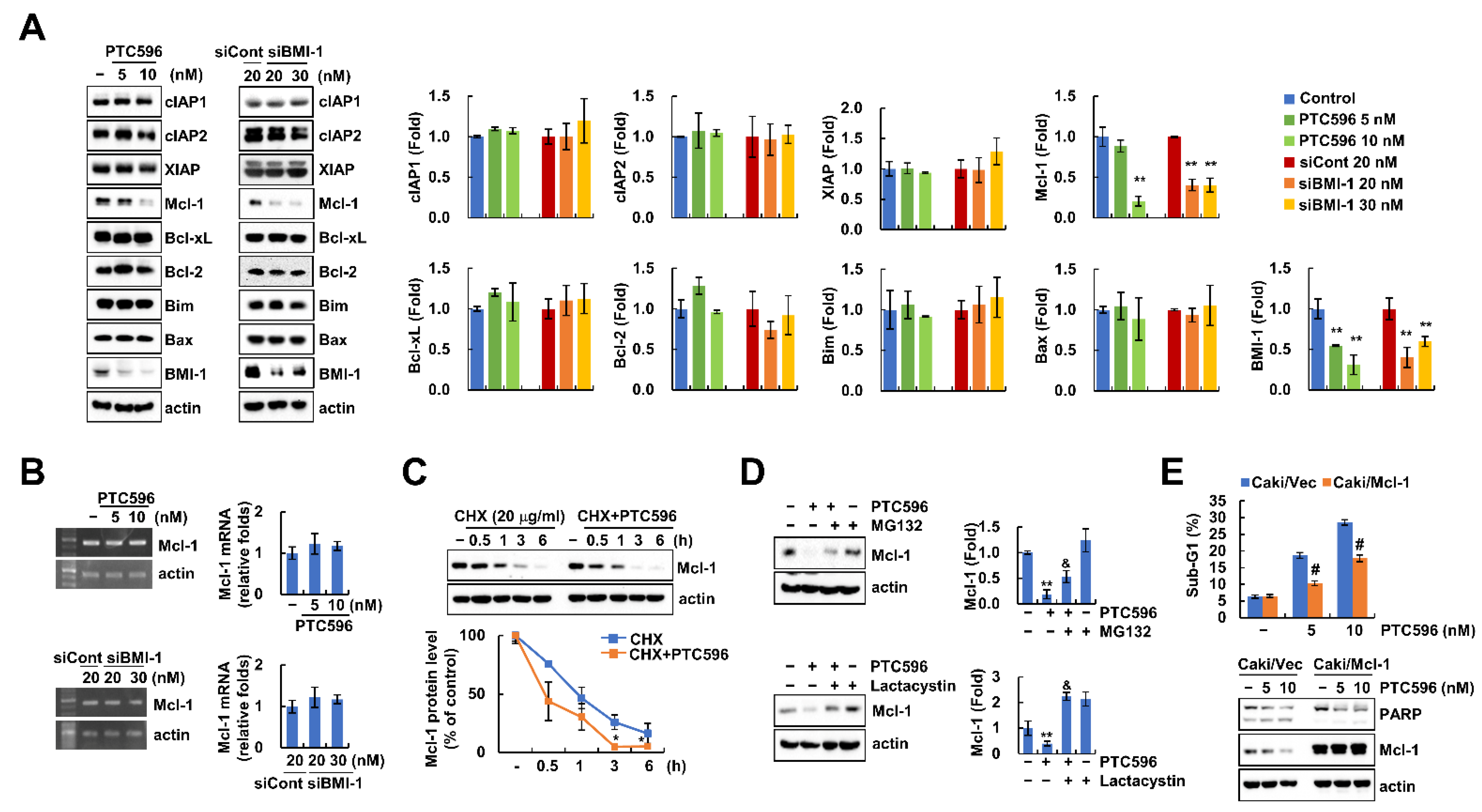
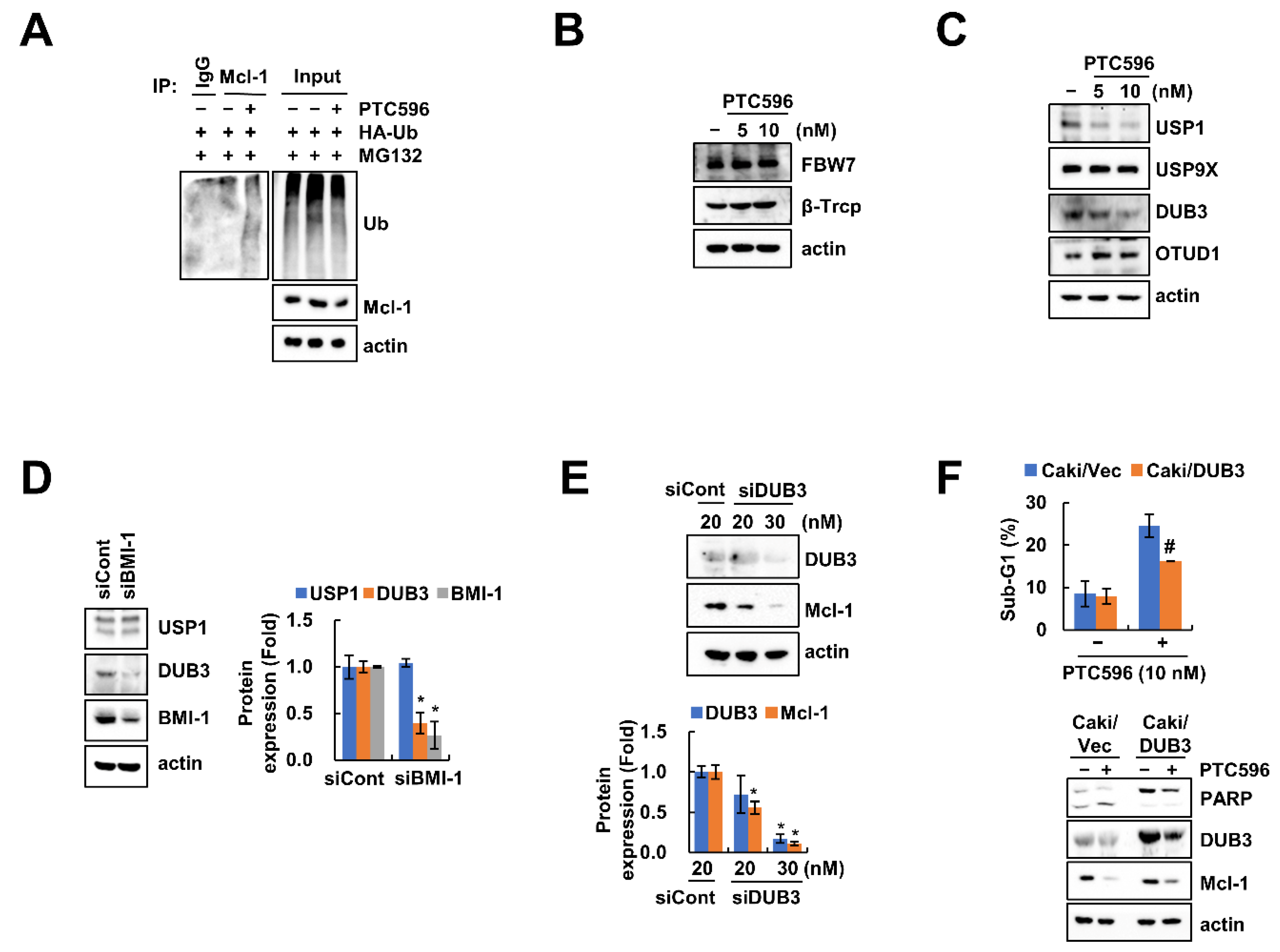
2.3. PTC596 Downregulates Mcl-1 Expression at the Post-Translational Level
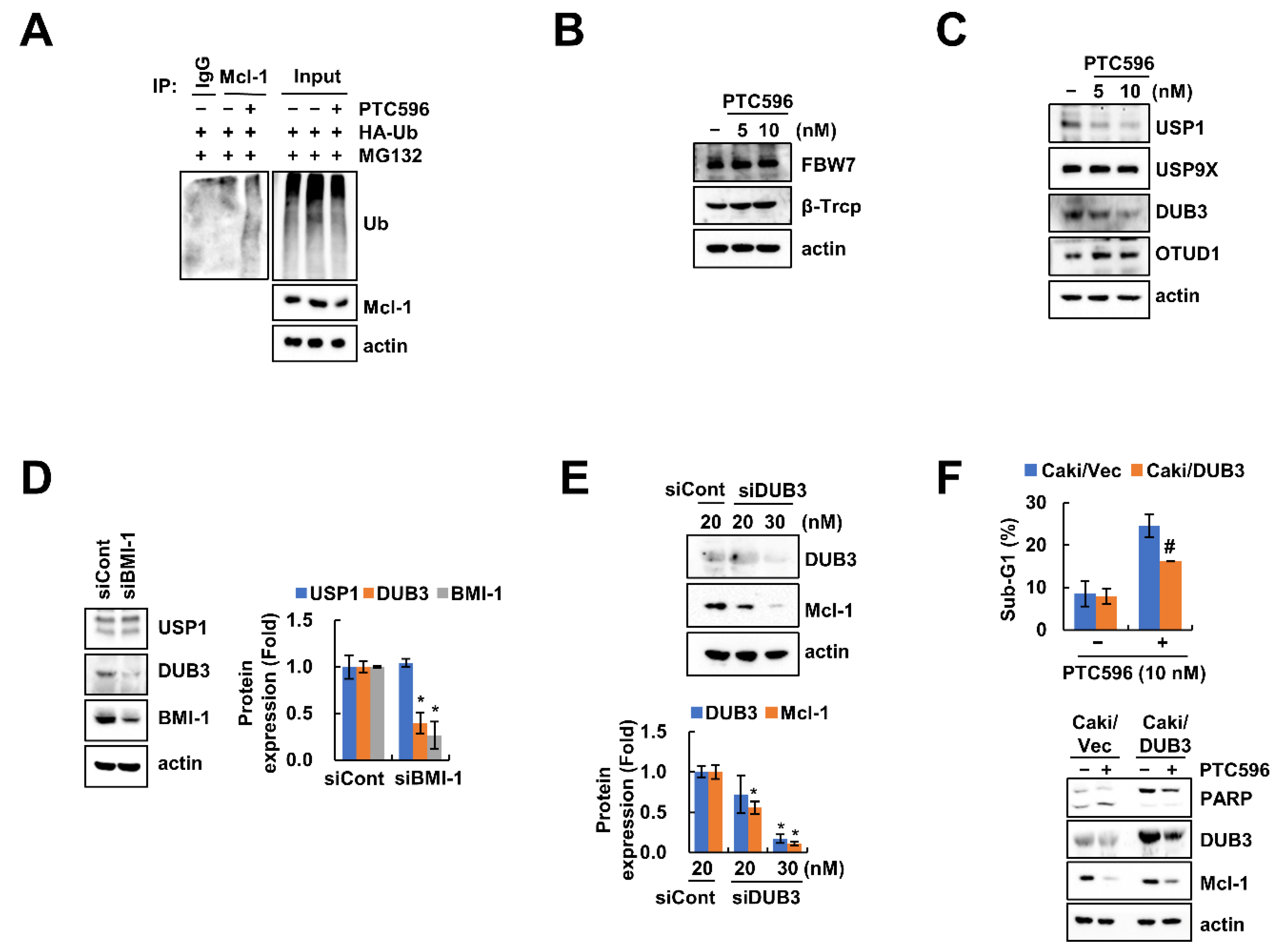
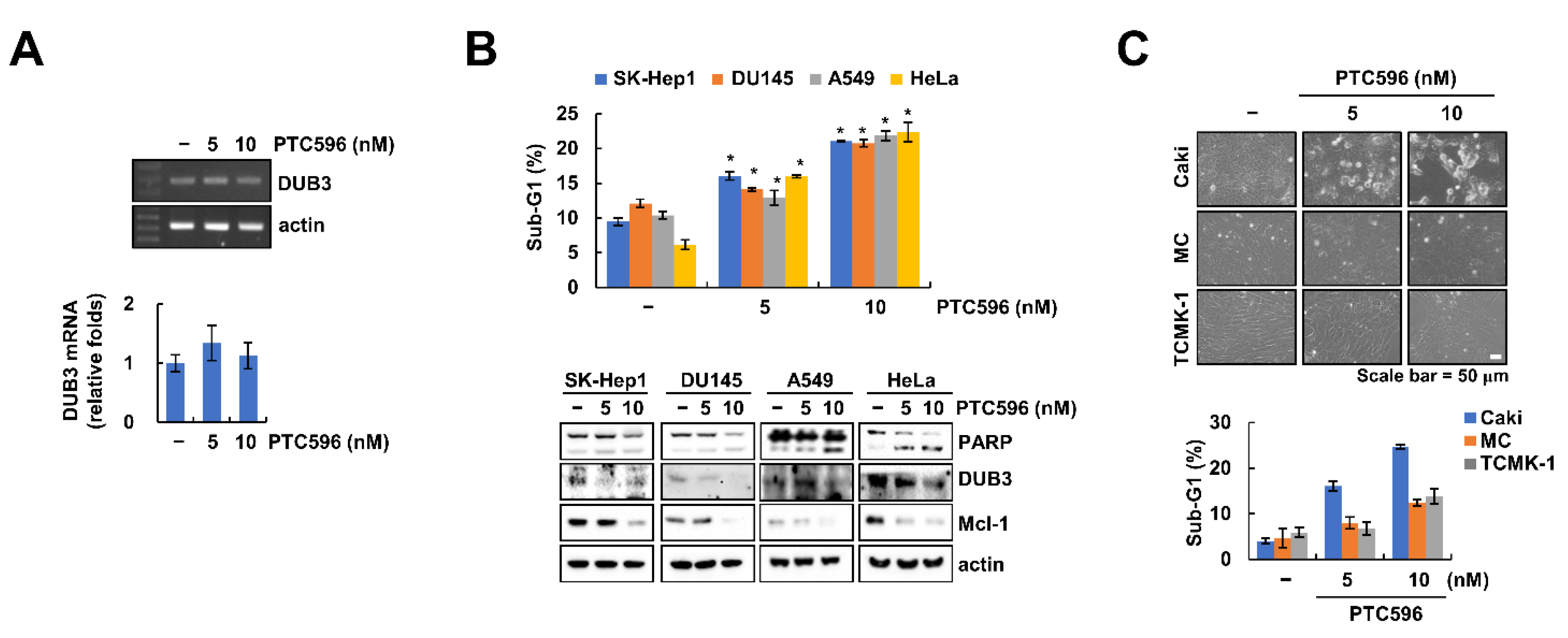
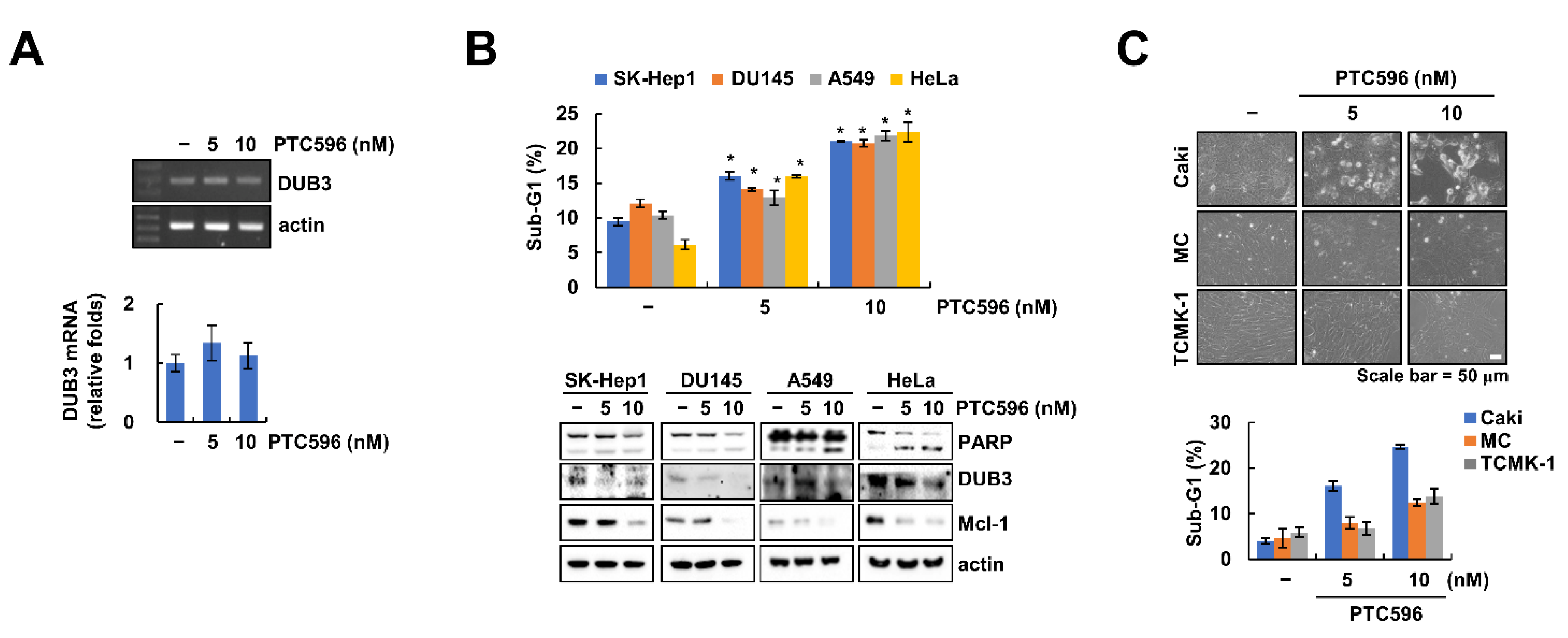
2.4. Downregulation of DUB3 Is Associated with PTC596-Mediated Mcl-1 Degradation and Apoptosis
2.5. The Apoptotic Effects of PTC596 in Various Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Chemicals
4.2. Cancer Cell Stem-Like Cell Population Analysis
4.3. Flow Cytometry and Western Blotting Analysis
4.4. Annexin V staining
4.5. DNA Fragmentation and DAPI Staining
4.6. Asp-Glu-Val-Aspase Activity Assay
4.7. Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and Quantitative Polymerase Chain Reaction (qPCR)
4.8. Transfection
4.9. Deubiquitination Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, R.; Rofstad, E.K. Cancer stem cells (CSCs), cervical CSCs and targeted therapies. Oncotarget 2017, 8, 35351–35367. [Google Scholar] [CrossRef] [Green Version]
- Nunes, T.; Hamdan, D.; Leboeuf, C.; El Bouchtaoui, M.; Gapihan, G.; Nguyen, T.T.; Meles, S.; Angeli, E.; Ratajczak, P.; Lu, H.; et al. Targeting Cancer Stem Cells to Overcome Chemoresistance. Int. J. Mol. Sci. 2018, 19, 4036. [Google Scholar] [CrossRef] [Green Version]
- Wicha, M.S. Targeting self-renewal, an Achilles’ heel of cancer stem cells. Nat. Med. 2014, 20, 14–15. [Google Scholar] [CrossRef]
- Alkema, M.J.; Wiegant, J.; Raap, A.K.; Berns, A.; van Lohuizen, M. Characterization and chromosomal localization of the human proto-oncogene BMI-1. Hum. Mol. Genet. 1993, 2, 1597–1603. [Google Scholar] [CrossRef]
- Park, I.K.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003, 423, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Raaphorst, F.M. Self-renewal of hematopoietic and leukemic stem cells: A central role for the Polycomb-group gene Bmi-1. Trends Immunol. 2003, 24, 522–524. [Google Scholar] [CrossRef]
- Abdouh, M.; Facchino, S.; Chatoo, W.; Balasingam, V.; Ferreira, J.; Bernier, G. BMI1 sustains human glioblastoma multiforme stem cell renewal. J. Neurosci. 2009, 29, 8884–8896. [Google Scholar] [CrossRef]
- Lukacs, R.U.; Memarzadeh, S.; Wu, H.; Witte, O.N. Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell 2010, 7, 682–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biehs, B.; Hu, J.K.; Strauli, N.B.; Sangiorgi, E.; Jung, H.; Heber, R.P.; Ho, S.; Goodwin, A.F.; Dasen, J.S.; Capecchi, M.R.; et al. BMI1 represses Ink4a/Arf and Hox genes to regulate stem cells in the rodent incisor. Nat. Cell Biol. 2013, 15, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Choy, B.; Bandla, S.; Xia, Y.; Tan, D.; Pennathur, A.; Luketich, J.D.; Godfrey, T.E.; Peters, J.H.; Sun, J.; Zhou, Z. Clinicopathologic characteristics of high expression of Bmi-1 in esophageal adenocarcinoma and squamous cell carcinoma. BMC Gastroenterol. 2012, 12, 146. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Feng, J.Q.; Shen, X.M.; Wang, H.Y.; Liu, Y.; Zhou, Z.T. Two stem cell markers, ATP-binding cassette, G2 subfamily (ABCG2) and BMI-1, predict the transformation of oral leukoplakia to cancer: A long-term follow-up study. Cancer 2012, 118, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.X.; Liu, X.D.; Luo, Z.Y.; Zhang, X.H.; Luo, X.Q.; Chen, X.; Jiang, H.; Xu, L. Upregulation of the proto-oncogene Bmi-1 predicts a poor prognosis in pediatric acute lymphoblastic leukemia. BMC Cancer 2017, 17, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Chen, L.; Bao, Z.; Liu, Y.; Liu, G.; Li, F.; Li, L. BMI1 promotes invasion and metastasis in endometrial adenocarcinoma and is a poor prognostic factor. Oncol. Rep. 2020, 43, 1630–1640. [Google Scholar] [PubMed]
- Yang, D.; Liu, H.Q.; Yang, Z.; Fan, D.; Tang, Q.Z. BMI1 in the heart: Novel functions beyond tumorigenesis. EBioMedicine 2021, 63, 103193. [Google Scholar] [CrossRef]
- Bolomsky, A.; Schlangen, K.; Schreiner, W.; Zojer, N.; Ludwig, H. Targeting of BMI-1 with PTC-209 shows potent anti-myeloma activity and impairs the tumour microenvironment. J. Hematol. Oncol. 2016, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Li, Z.; Wu, Y.; Huang, R.; Zhu, Y.; Zhang, W.; Wang, Y.; Cheng, J. Pharmacological inhibition of Bmi1 by PTC-209 impaired tumor growth in head neck squamous cell carcinoma. Cancer Cell Int. 2017, 17, 107. [Google Scholar] [CrossRef] [Green Version]
- Dey, A.; Xiong, X.; Crim, A.; Dwivedi, S.K.D.; Mustafi, S.B.; Mukherjee, P.; Cao, L.; Sydorenko, N.; Baiazitov, R.; Moon, Y.C.; et al. Evaluating the Mechanism and Therapeutic Potential of PTC-028, a Novel Inhibitor of BMI-1 Function in Ovarian Cancer. Mol. Cancer Ther. 2018, 17, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolomsky, A.; Muller, J.; Stangelberger, K.; Lejeune, M.; Duray, E.; Breid, H.; Vrancken, L.; Pfeiffer, C.; Hubl, W.; Willheim, M.; et al. The anti-mitotic agents PTC-028 and PTC596 display potent activity in pre-clinical models of multiple myeloma but challenge the role of BMI-1 as an essential tumour gene. Br. J. Haematol. 2020, 190, 877–890. [Google Scholar] [CrossRef]
- Nishida, Y.; Maeda, A.; Kim, M.J.; Cao, L.; Kubota, Y.; Ishizawa, J.; AlRawi, A.; Kato, Y.; Iwama, A.; Fujisawa, M.; et al. The novel BMI-1 inhibitor PTC596 downregulates MCL-1 and induces p53-independent mitochondrial apoptosis in acute myeloid leukemia progenitor cells. Blood Cancer J. 2017, 7, e527. [Google Scholar] [CrossRef]
- Maeda, A.; Nishida, Y.; Weetall, M.; Cao, L.; Branstrom, A.; Ishizawa, J.; Nii, T.; Schober, W.D.; Abe, Y.; Matsue, K.; et al. Targeting of BMI-1 expression by the novel small molecule PTC596 in mantle cell lymphoma. Oncotarget 2018, 9, 28547–28560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seipel, K.; Kopp, B.; Bacher, U.; Pabst, T. BMI1-Inhibitor PTC596 in Combination with MCL1 Inhibitor S63845 or MEK Inhibitor Trametinib in the Treatment of Acute Leukemia. Cancers 2021, 13, 581. [Google Scholar] [CrossRef] [PubMed]
- Mojsa, B.; Lassot, I.; Desagher, S. Mcl-1 ubiquitination: Unique regulation of an essential survival protein. Cells 2014, 3, 418–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreso, A.; van Galen, P.; Pedley, N.M.; Lima-Fernandes, E.; Frelin, C.; Davis, T.; Cao, L.; Baiazitov, R.; Du, W.; Sydorenko, N.; et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat. Med. 2014, 20, 29–36. [Google Scholar] [CrossRef]
- Dey, A.; Mustafi, S.B.; Saha, S.; Kumar Dhar Dwivedi, S.; Mukherjee, P.; Bhattacharya, R. Inhibition of BMI1 induces autophagy-mediated necroptosis. Autophagy 2016, 12, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Eberle-Singh, J.A.; Sagalovskiy, I.; Maurer, H.C.; Sastra, S.A.; Palermo, C.F.; Decker, A.R.; Kim, M.J.; Sheedy, J.; Mollin, A.; Cao, L.; et al. Effective Delivery of a Microtubule Polymerization Inhibitor Synergizes with Standard Regimens in Models of Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2019, 25, 5548–5560. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I.; O’Mara, E.; Laskin, O.L.; Gao, L.; Baird, J.D.; Spiegel, R.J.; Kaushik, D.; Weetall, M.; Colacino, J.; O’Keefe, K.; et al. Pharmacokinetics and Safety of PTC596, a Novel Tubulin-Binding Agent, in Subjects With Advanced Solid Tumors. Clin. Pharmacol. Drug Dev. 2021, 10, 940–949. [Google Scholar] [CrossRef]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Koo, J.; Guan, B.; Yue, P.; Deng, X.; Chen, M.; Khuri, F.R.; Sun, S.Y. The E3 ubiquitin ligases beta-TrCP and FBXW7 cooperatively mediates GSK3-dependent Mcl-1 degradation induced by the Akt inhibitor API-1, resulting in apoptosis. Mol. Cancer 2013, 12, 146. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Li, S.; Cui, X.; Han, K.; Wang, J.; Hou, X.; Cui, L.; He, S.; Xiao, J.; Yang, Y. Inhibition of Ubiquitin Specific Protease 1 Sensitizes Colorectal Cancer Cells to DNA-Damaging Chemotherapeutics. Front. Oncol. 2019, 9, 1406. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Zhong, N.; Liu, G.; Chen, K.; Liu, X.; Su, L.; Singhal, S. Usp9x- and Noxa-mediated Mcl-1 downregulation contributes to pemetrexed-induced apoptosis in human non-small-cell lung cancer cells. Cell Death Dis. 2014, 5, e1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Luo, Q.; Zhao, P.; Chang, W.; Wang, Y.; Shu, T.; Ding, F.; Li, B.; Liu, Z. MGMT-activated DUB3 stabilizes MCL1 and drives chemoresistance in ovarian cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 2961–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Lin, Y.; Feng, J.; Qi, Y.; Wang, X.; Lin, Q.; Shi, W.; Zheng, E.; Wang, W.; Hou, Z.; et al. The deubiquitinating enzyme OTUD1 antagonizes BH3-mimetic inhibitor induced cell death through regulating the stability of the MCL1 protein. Cancer Cell Int. 2019, 19, 222. [Google Scholar] [CrossRef] [Green Version]
- Min, K.J.; Shahriyar, S.A.; Kwon, T.K. Arylquin 1, a potent Par-4 secretagogue, induces lysosomal membrane permeabilization-mediated non-apoptotic cell death in cancer cells. Toxicol. Res. 2020, 36, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Woo, S.M.; Kwon, T.K. The Histone Lysine-specific Demethylase 1 Inhibitor, SP2509 Exerts Cytotoxic Effects against Renal Cancer Cells through Downregulation of Bcl-2 and Mcl-1. J. Cancer Prev. 2020, 25, 79–86. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, K.; Woo, S.-M.; Seo, S.-U.; Kwon, T.-K. Inhibition of BMI-1 Induces Apoptosis through Downregulation of DUB3-Mediated Mcl-1 Stabilization. Int. J. Mol. Sci. 2021, 22, 10107. https://doi.org/10.3390/ijms221810107
Wu K, Woo S-M, Seo S-U, Kwon T-K. Inhibition of BMI-1 Induces Apoptosis through Downregulation of DUB3-Mediated Mcl-1 Stabilization. International Journal of Molecular Sciences. 2021; 22(18):10107. https://doi.org/10.3390/ijms221810107
Chicago/Turabian StyleWu, Kaixin, Seon-Min Woo, Seung-Un Seo, and Taeg-Kyu Kwon. 2021. "Inhibition of BMI-1 Induces Apoptosis through Downregulation of DUB3-Mediated Mcl-1 Stabilization" International Journal of Molecular Sciences 22, no. 18: 10107. https://doi.org/10.3390/ijms221810107
APA StyleWu, K., Woo, S.-M., Seo, S.-U., & Kwon, T.-K. (2021). Inhibition of BMI-1 Induces Apoptosis through Downregulation of DUB3-Mediated Mcl-1 Stabilization. International Journal of Molecular Sciences, 22(18), 10107. https://doi.org/10.3390/ijms221810107