
Nanoparticles Targeting Innate Immune Cells in Tumor Microenvironment


Abstract
:1. Introduction
2. Nanoparticles for Targeting TAMs
2.1. TAMs Depletion
2.2. Inhibiting Monocyte Recruitment
2.3. TAM Reprogramming
2.4. Blocking CD47-Sirpα Signaling
3. Nanoparticles for Targeting DCs
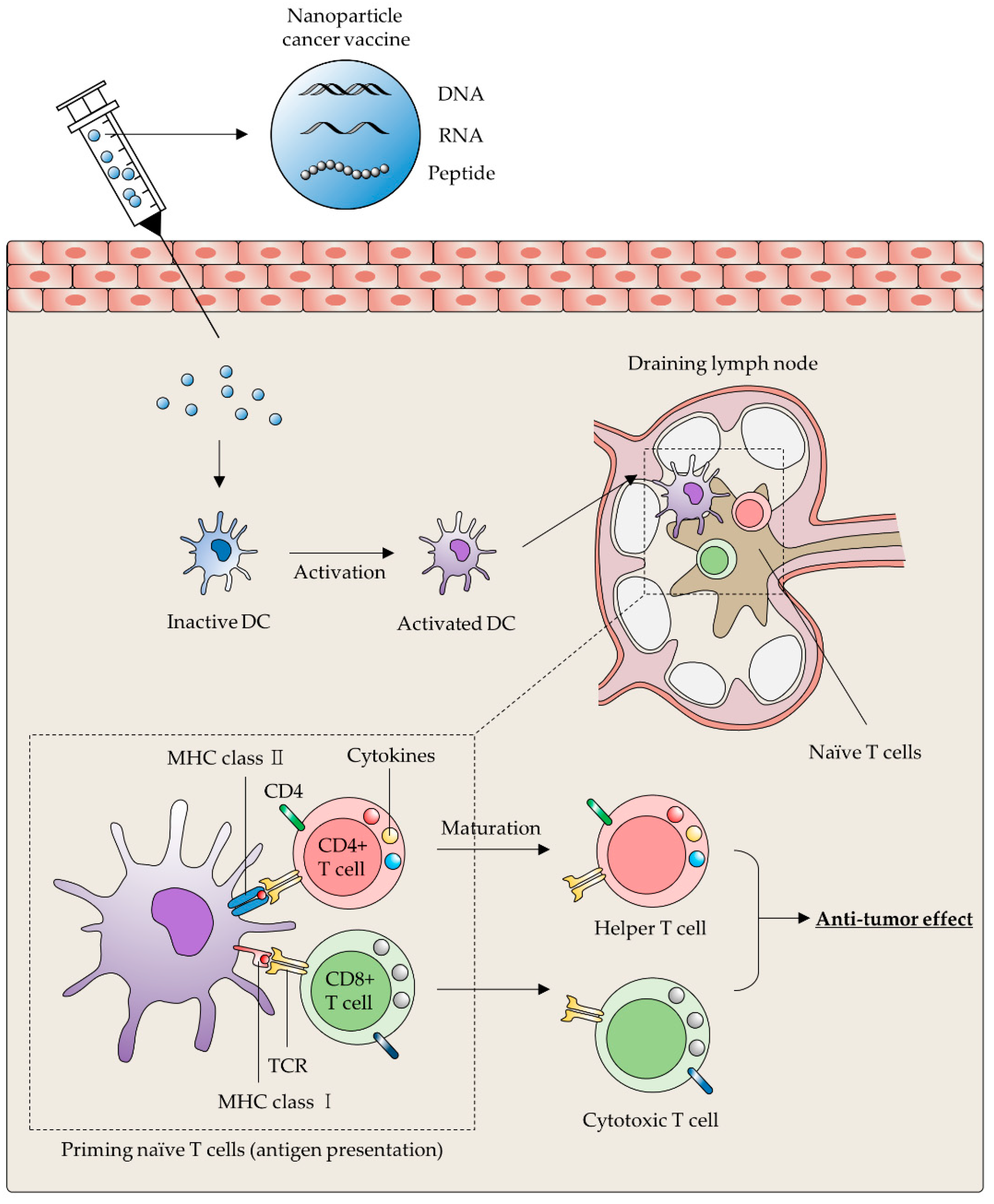
3.1. Cancer Vaccines

{kind=link}
{kind=link}
| Categories | Types | Features | References |
|---|---|---|---|
| Lipid-based 1 NPs | Liposome |
| [41,48,72,73,74,75] |
| 2 LNPs | |||
| Inorganic NPs | 3 AuNPs |
| [37,38,47,56,76,77,78,79,80] |
| Silica NPs | |||
| Selenium NPs | |||
| 4 AgNPs | |||
| Calcium bisphosphonate | |||
| Iron NPs | |||
| Polymer-based NPs | Polymeric micelle |
| [36,43,50,81,82,83,84] |
| Dendrimer | |||
| Cationic polymeric NPs | |||
| Poly-β-amino ester | |||
| Extracellular vesicle | Extracellular vesicle |
| [51,52,57,85,86] |
| Nanoparticle | Ligand/Target | Payload | Purpose | Reference |
|---|---|---|---|---|
| Lipid-based NPs | Mannose/Mannose receptor | - | TAM reprogramming | [48] |
| α-1 M2pep/2 SR-B1 | Anti-CSF-1R siRNA | Inhibiting monocyte recruitment | [41] | |
| - | 3 OVA, Poly(I:C), 4 gp100, 5 TRP2 | DC-based cancer vaccine | [72,74] | |
| T1 DNA aptamer | 6 Dox | MDSC depletion | [75] | |
| Inorganic NPs | 8 FORL2 | 9 MTX | TAM depletion | [36] |
| CD163 antibody/CD163 | - | [38] | ||
| - | CCR2 siRNA | Inhibiting monocyte recruitment | [43] | |
| - | RFP, CpG-ODN, OVA | DC-based cancer vaccine | [76] | |
| Sirpα/CD47 | - | Blockade CD47-Sirpα signaling | [56] | |
| - | Dox, 10 ATRA, IL-2 | MDSC depletion | [87] | |
| α-EGFR, α-4-1BB, and α-CD16 | 7 EPI | NK cell activation | [84] | |
| Polymer-based NPs | Mannose/Mannose receptor | OVA, CCR7 pDNA | DC-based cancer vaccine | [82] |
| - | OVA, CpG | [83] | ||
| - | 11 IRF5 mRNA | TAM reprogramming | [50] | |
| Extracellular vesicle | - | STAT6 inhibitor, IKKβ siRNA | TAM reprogramming | [53] |
| Sirpα variants/CD47 | - | Blockade CD47-Sirpα signaling | [57] | |
| CD40L/CD40 | - | DC-based cancer vaccine | [85] | |
| 12 NKG2D ligand and IL-15Rα | - | NK cell activation | [86] |
3.1.1. Lipid-Based Nanoparticle
3.1.2. Inorganic Nanoparticles
3.1.3. Polymer-Based Nanoparticles
3.1.4. Extracellular Vesicles
3.2. DC Activation
4. Nanoparticles for MDSCs Depletion
5. Nanoparticles for Activating NK Cells
6. Nanoparticles for Targeting Neutrophils
7. Challenges and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TME | Tumor Microenvironment |
| ECM | Extracellular Matrix |
| TAMs | Tumor-Associated Macrophages |
| DCs | Dendritic Cells |
| NKs | Natural Killer Cells |
| ADCC | Antibody-Dependent Cellular Cytotoxicity |
| ADCP | Antibody-Dependent Cellular Phagocytosis |
| CAR-T | Chimeric Antigen Receptor-T |
| MDSCs | Myeloid-Derived Suppressor Cells |
| IFN-γ | Interferon-γ |
| FOLR2 | Folate Receptor-2 |
| CaBP | Calcium Bisphosphonate |
| CSF-1 | Colony-Stimulating Factor-1 |
| MCSF | Macrophage Colony-Stimulating Factor |
| siRNA | Small Interfering RNA |
| CCL2 | Chemokine Ligand 2 |
| APCs | Antigen Presenting Cells |
| PBAE | Poly-Beta-Amino-Esters |
| IRF5 | Interferon Regulatory Factor 5 |
| BMDMs | Bone Marrow-Derived Macrophages |
| miRNAs | MicroRNAs |
| SIRPα | Signal-Regulatory Protein Alpha |
| MHC I | Major Histocompatibility Complex I |
| Th | Helper T Cell |
| Tc | Cytotoxic T Cell |
| TAA | Tumor-Associated Antigen |
| NPs | Nanoparticles |
| AuNPs | Gold Nanoparticles |
| LN | Lymph Nodes |
| CT | Computed Tomography |
| MSNs | Mesoporous Silica Nanoparticles |
| SA | Steric Acid |
| M-COSA | Mannose-engineered COSA |
| BMDCs | Bone Marrow-Drived Dendritic Cells |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| CCR5 | Chemokine Receptor type 5 |
| Tregs | Regulatory T Cells |
| iNOS | Inducible Nitric Oxide Synthase |
| ROS | Reactive Oxygen Species |
| RNS | Reactive Nitrogen Species |
| ATRA | All-Trans Retinoic Acid |
| EGFR | Epidermal Growth Factor Receptor |
| Dex | Dendritic cells-derived exosomes |
| cNPs | Cationic Nanoparticles |
| NKT | Natural Killer T cells |
| PTT | Photothermal Therapy |
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Sormendi, S.; Wielockx, B. Hypoxia Pathway proteins as central mediators of metabolism in the tumor cells and their microenvironment. Front. Immunol. 2018, 9, 40. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, V.; Turdo, A.; Di Franco, S.; Dieli, F.; Todaro, M.; Stassi, G. Tumor and its microenvironment: A synergistic interplay. Semin. Cancer Biol. 2013, 23, 522–532. [Google Scholar] [CrossRef]
- Chen, F.; Zhuang, X.; Lin, L.; Yu, P.; Wang, Y.; Shi, Y.; Hu, G.; Sun, Y. New horizons in tumor microenvironment biology: Challenges and opportunities. BMC Med. 2015, 13, 45. [Google Scholar] [CrossRef] [Green Version]
- Huai, Y.; Hossen, M.N.; Wilhelm, S.; Bhattacharya, R.; Mukherjee, P. Nanoparticle interactions with the tumor microenvironment. Bioconjug. Chem. 2019, 30, 2247–2263. [Google Scholar] [CrossRef]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14 (Suppl. S2), 49. [Google Scholar] [CrossRef] [Green Version]
- Ginefra, P.; Lorusso, G.; Vannini, N. Innate immune cells and their contribution to T-Cell-based immunotherapy. Int. J. Mol. Sci. 2020, 21, 4441. [Google Scholar] [CrossRef] [PubMed]
- Shihab, I.; Khalil, B.A.; Elemam, N.M.; Hachim, I.Y.; Hachim, M.Y.; Hamoudi, R.A.; Maghazachi, A.A. Understanding the Role of innate immune cells and identifying genes in breast cancer microenvironment. Cancers 2020, 12, 2226. [Google Scholar] [CrossRef] [PubMed]
- Anfray, C.; Ummarino, A.; Andon, F.T.; Allavena, P. Current strategies to target tumor-associated-macrophages to improve anti-tumor immune responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moynihan, K.D.; Irvine, D.J. Roles for Innate Immunity in Combination Immunotherapies. Cancer Res. 2017, 77, 5215–5221. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.R.; Corrales, L.; Gajewski, T.F. Innate immune recognition of cancer. Annu. Rev. Immunol. 2015, 33, 445–474. [Google Scholar] [CrossRef]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Narayana, A. Applications of nanotechnology in cancer: A literature review of imaging and treatment. J. Nucl. Med. Radiat. Ther. 2014, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Lungu, I.I.; Grumezescu, A.M.; Volceanov, A.; Andronescu, E. Nanobiomaterials used in cancer therapy: An up-to-date overview. Molecules 2019, 24, 3547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Wu, T.; Bao, Y.; Zhang, Z. Nanotechnology based therapeutic modality to boost anti-tumor immunity and collapse tumor defense. J. Control. Release 2017, 256, 26–45. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering precision nanoparticles for drug delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Kim, H.; Kim, E.H.; Kwak, G.; Chi, S.G.; Kim, S.H.; Yang, Y. Exosomes: Cell-derived nanoplatforms for the delivery of cancer therapeutics. Int. J. Mol. Sci. 2020, 22, 14. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhang, Y.S.; Hobson, D.; Hydbring, P. Nanoparticles for immune system targeting. Drug Discov. Today 2017, 22, 1295–1301. [Google Scholar] [CrossRef]
- Yuan, A.; Hsiao, Y.J.; Chen, H.Y.; Chen, H.W.; Ho, C.C.; Chen, Y.Y.; Liu, Y.C.; Hong, T.H.; Yu, S.L.; Chen, J.J.; et al. Opposite effects of M1 and M2 macrophage subtypes on lung cancer progression. Sci. Rep. 2015, 5, 14273. [Google Scholar] [CrossRef] [Green Version]
- Mehla, K.; Singh, P.K. Metabolic regulation of macrophage polarization in cancer. Trends Cancer 2019, 5, 822–834. [Google Scholar] [CrossRef]
- Li, X.; Liu, R.; Su, X.; Pan, Y.; Han, X.; Shao, C.; Shi, Y. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol. Cancer 2019, 18, 177. [Google Scholar] [CrossRef] [Green Version]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sanchez-Rodriguez, R.; Scolaro, T.; Castegna, A. The metabolic signature of macrophage responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Jeong, H.; Bae, Y.; Shin, K.; Kang, S.; Kim, H.; Oh, J.; Bae, H. Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. J. Immunother. Cancer 2019, 7, 147. [Google Scholar] [CrossRef] [Green Version]
- Rubio, C.; Munera-Maravilla, E.; Lodewijk, I.; Suarez-Cabrera, C.; Karaivanova, V.; Ruiz-Palomares, R.; Paramio, J.M.; Duenas, M. Macrophage polarization as a novel weapon in conditioning tumor microenvironment for bladder cancer: Can we turn demons into gods? Clin. Transl. Oncol. 2019, 21, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Penn, C.A.; Yang, K.; Zong, H.; Lim, J.Y.; Cole, A.; Yang, D.; Baker, J.; Goonewardena, S.N.; Buckanovich, R.J. Therapeutic impact of nanoparticle therapy targeting tumor-associated macrophages. Mol. Cancer Ther. 2018, 17, 96–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Yi, X.; Dong, Z.; Xu, J.; Liang, C.; Chao, Y.; Wang, Y.; Yang, K.; Liu, Z. Calcium Bisphosphonate nanoparticles with chelator-free radiolabeling to deplete tumor-associated macrophages for enhanced cancer radioisotope therapy. ACS Nano 2018, 12, 11541–11551. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lee, J.S.; Kim, J.E.; Kim, J.-W.; Bok, S.; Keum, K.C.; Koh, W.-G.; Koom, W.S. Enhancement of antitumor effect of radiotherapy via combination with Au@SiO2 nanoparticles targeted to tumor-associated macrophages. J. Ind. Eng. Chem. 2020, 84, 349–357. [Google Scholar] [CrossRef]
- Xu, F.; Wei, Y.; Tang, Z.; Liu, B.; Dong, J. Tumorassociated macrophages in lung cancer: Friend or foe? (Review). Mol. Med. Rep. 2020, 22, 4107–4115. [Google Scholar]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Qiao, S.; Dai, Y.; Xu, G.; Dai, B.; Lu, L.; Yu, X.; Luo, Q.; Zhang, Z. Molecular-targeted immunotherapeutic strategy for melanoma via dual-targeting nanoparticles delivering small interfering RNA to tumor-associated macrophages. ACS Nano 2017, 11, 9536–9549. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Vadgama, J.V.; Wang, P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun. Signal. 2020, 18, 82. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, Y.; Chen, K.G.; Luo, Y.L.; Wang, J. Cationic polymeric nanoparticle delivering CCR2 siRNA to inflammatory monocytes for tumor microenvironment modification and cancer therapy. Mol. Pharm. 2018, 15, 3642–3653. [Google Scholar] [CrossRef] [PubMed]
- Pudlarz, A.; Szemraj, J. Nanoparticles as carriers of proteins, peptides and other therapeutic molecules. Open Life Sci. 2018, 13, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Riaz Ahmed, K.B.; Nagy, A.M.; Brown, R.P.; Zhang, Q.; Malghan, S.G.; Goering, P.L. Silver nanoparticles: Significance of physicochemical properties and assay interference on the interpretation of in vitro cytotoxicity studies. Toxicol. In Vitro 2017, 38, 179–192. [Google Scholar] [CrossRef]
- Zhang, X.F.; Liu, Z.G.; Shen, W.; Gurunathan, S. Silver nanoparticles: Synthesis, characterization, properties, applications, and therapeutic approaches. Int. J. Mol. Sci. 2016, 17, 1534. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tang, H.; Liu, J.H.; Wang, H.; Liu, Y. Evaluation of the adjuvant effect of silver nanoparticles both in vitro and in vivo. Toxicol. Lett. 2013, 219, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Yang, Y.; Dong, W.; Gao, Y.; Meng, Y.; Wang, H.; Li, L.; Jin, J.; Ji, M.; Xia, X.; et al. Drug-free mannosylated liposomes inhibit tumor growth by promoting the polarization of tumor-associated macrophages. Int. J. Nanomed. 2019, 14, 3203–3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Yin, W.; Wu, A.; Tang, Y.; Wang, J.; Pan, Z.; Lin, T.; Zhang, M.; Chen, B.; Duan, Y.; et al. Dual-targeting to cancer cells and M2 macrophages via biomimetic delivery of mannosylated albumin nanoparticles for drug-resistant cancer therapy. Adv. Funct. Mater. 2017, 27, 1700403. [Google Scholar] [CrossRef]
- Zhang, F.; Parayath, N.N.; Ene, C.I.; Stephan, S.B.; Koehne, A.L.; Coon, M.E.; Holland, E.C.; Stephan, M.T. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 2019, 10, 3974. [Google Scholar] [CrossRef]
- Choo, Y.W.; Kang, M.; Kim, H.Y.; Han, J.; Kang, S.; Lee, J.R.; Jeong, G.J.; Kwon, S.P.; Song, S.Y.; Go, S.; et al. M1 macrophage-derived nanovesicles potentiate the anticancer efficacy of immune checkpoint inhibitors. ACS Nano 2018, 12, 8977–8993. [Google Scholar] [CrossRef]
- Neupane, K.R.; McCorkle, J.R.; Kopper, T.J.; Lakes, J.E.; Aryal, S.P.; Abdullah, M.; Snell, A.A.; Gensel, J.C.; Kolesar, J.; Richards, C.I. Macrophage-engineered vesicles for therapeutic delivery and bidirectional reprogramming of immune cell polarization. ACS Omega 2021, 6, 3847–3857. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Guo, Y.; Li, B.; Li, X.; Wang, Y.; Han, S.; Cheng, D.; Shuai, X. M2-Like tumor-associated macrophage-targeted codelivery of STAT6 Inhibitor and IKKbeta siRNA induces M2-to-M1 repolarization for cancer immunotherapy with low immune side effects. ACS Cent. Sci. 2020, 6, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Luo, X.; Chen, C.; Tang, Y.; Li, L.; Mo, B.; Liang, H.; Yu, S. The Ap-2alpha/Elk-1 axis regulates Sirpalpha-dependent tumor phagocytosis by tumor-associated macrophages in colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrissey, M.A.; Vale, R.D. CD47 suppresses phagocytosis by repositioning SIRPA and preventing integrin activation. bioRxiv 2019. [Google Scholar]
- Rao, L.; Zhao, S.K.; Wen, C.; Tian, R.; Lin, L.; Cai, B.; Sun, Y.; Kang, F.; Yang, Z.; He, L.; et al. Activating Macrophage-mediated cancer immunotherapy by genetically edited nanoparticles. Adv. Mater. 2020, 32, e2004853. [Google Scholar] [CrossRef]
- Koh, E.; Lee, E.J.; Nam, G.H.; Hong, Y.; Cho, E.; Yang, Y.; Kim, I.S. Exosome-SIRPalpha, a CD47 blockade increases cancer cell phagocytosis. Biomaterials 2017, 121, 121–129. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Diebold, S.S. Determination of T-cell fate by dendritic cells. Immunol. Cell Biol. 2008, 86, 389–397. [Google Scholar] [CrossRef]
- Eisenbarth, S.C. Dendritic cell subsets in T cell programming: Location dictates function. Nat. Rev. Immunol. 2019, 19, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.R.; De Palma, M. Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat. Commun. 2019, 10, 5408. [Google Scholar] [CrossRef] [PubMed]
- De Vries, I.J.; Krooshoop, D.J.; Scharenborg, N.M.; Lesterhuis, W.J.; Diepstra, J.H.; Van Muijen, G.N.; Strijk, S.P.; Ruers, T.J.; Boerman, O.C.; Oyen, W.J.; et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003, 63, 12–17. [Google Scholar] [PubMed]
- Anguille, S.; Smits, E.L.; Lion, E.; van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Ledford, H. Therapeutic cancer vaccine survives biotech bust. Nature 2015, 519, 17–18. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.H.; Tran, T.T.P.; Nguyen, H.T.; Phung, C.D.; Jeong, J.H.; Stenzel, M.H.; Jin, S.G.; Yong, C.S.; Truong, D.H.; Kim, J.O. Nanoparticles for dendritic cell-based immunotherapy. Int. J. Pharm. 2018, 542, 253–265. [Google Scholar] [CrossRef]
- Mukalel, A.J.; Riley, R.S.; Zhang, R.; Mitchell, M.J. Nanoparticles for nucleic acid delivery: Applications in cancer immunotherapy. Cancer Lett. 2019, 458, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.J.; Xu, S.; Wang, H.M.; Ling, Y.; Dong, J.; Xia, R.D.; Sun, X.H. Nanoparticles: Oral delivery for protein and peptide drugs. AAPS PharmSciTech 2019, 20, 190. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.G. Small-molecule delivery by nanoparticles for anticancer therapy. Trends Mol. Med. 2010, 16, 594–602. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Megia, E.; Novoa-Carballal, R.; Quinoa, E.; Riguera, R. Conjugation of bioactive ligands to PEG-grafted chitosan at the distal end of PEG. Biomacromolecules 2007, 8, 833–842. [Google Scholar] [CrossRef]
- Van den Berg, J.H.; Oosterhuis, K.; Hennink, W.E.; Storm, G.; van der Aa, L.J.; Engbersen, J.F.; Haanen, J.B.; Beijnen, J.H.; Schumacher, T.N.; Nuijen, B. Shielding the cationic charge of nanoparticle-formulated dermal DNA vaccines is essential for antigen expression and immunogenicity. J. Control. Release 2010, 141, 234–240. [Google Scholar] [CrossRef]
- Varypataki, E.M.; van der Maaden, K.; Bouwstra, J.; Ossendorp, F.; Jiskoot, W. Cationic liposomes loaded with a synthetic long peptide and poly(I:C): A defined adjuvanted vaccine for induction of antigen-specific T cell cytotoxicity. AAPS J. 2015, 17, 216–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, P.L.; Lin, H.J.; Wang, H.W.; Tsai, W.Y.; Lin, S.F.; Chien, M.Y.; Liang, P.H.; Huang, Y.Y.; Liu, D.Z. Galactosylated liposome as a dendritic cell-targeted mucosal vaccine for inducing protective anti-tumor immunity. Acta Biomater. 2015, 11, 356–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef]
- Liu, H.; Mai, J.; Shen, J.; Wolfram, J.; Li, Z.; Zhang, G.; Xu, R.; Li, Y.; Mu, C.; Zu, Y.; et al. A novel DNA aptamer for dual targeting of polymorphonuclear myeloid-derived suppressor cells and tumor cells. Theranostics 2018, 8, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.H.; Kwon, H.K.; An, S.; Kim, D.; Kim, S.; Yu, M.K.; Lee, J.H.; Lee, T.S.; Im, S.H.; Jon, S. Imageable antigen-presenting gold nanoparticle vaccines for effective cancer immunotherapy in vivo. Angew. Chem. Int. Ed. Engl. 2012, 51, 8800–8805. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, X.; Cheng, Y.; Guo, X.; Yuan, W. Iron oxide nanoparticles-based vaccine delivery for cancer treatment. Mol. Pharm. 2018, 15, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Zhong, X.; Du, G.; Hou, Y.; Zhang, Y.; Zhang, Z.; Gong, T.; Zhang, L.; Sun, X. The pore size of mesoporous silica nanoparticles regulates their antigen delivery efficiency. Sci. Adv. 2020, 6, eaaz4462. [Google Scholar] [CrossRef]
- Orlowski, P.; Tomaszewska, E.; Ranoszek-Soliwoda, K.; Gniadek, M.; Labedz, O.; Malewski, T.; Nowakowska, J.; Chodaczek, G.; Celichowski, G.; Grobelny, J.; et al. Tannic Acid-modified silver and gold nanoparticles as novel stimulators of dendritic cells activation. Front. Immunol. 2018, 9, 1115. [Google Scholar] [CrossRef]
- Song, Z.; Luo, W.; Zheng, H.; Zeng, Y.; Wang, J.; Chen, T. Translational Nanotherapeutics reprograms immune microenvironment in malignant pleural effusion of lung adenocarcinoma. Adv. Healthc. Mater. 2021, 10, e2100149. [Google Scholar] [CrossRef]
- Kim, K.S.; Han, J.H.; Choi, S.H.; Jung, H.Y.; Park, J.D.; An, H.J.; Kim, S.E.; Kim, D.H.; Doh, J.; Han, D.K.; et al. Cationic Nanoparticle-Mediated Activation of Natural Killer Cells for Effective Cancer Immunotherapy. ACS Appl. Mater. Interfaces 2020, 12, 56731–56740. [Google Scholar] [CrossRef]
- Yang, X.; Lian, K.; Meng, T.; Liu, X.; Miao, J.; Tan, Y.; Yuan, H.; Hu, F. Immune adjuvant targeting micelles allow efficient dendritic cell migration to lymph nodes for enhanced cellular immunity. ACS Appl. Mater. Interfaces 2018, 10, 33532–33544. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, H.; Xu, L.; Chao, Y.; Wang, C.; Han, X.; Dong, Z.; Chang, H.; Peng, R.; Cheng, Y.; et al. Nanovaccine based on a protein-delivering dendrimer for effective antigen cross-presentation and cancer immunotherapy. Biomaterials 2019, 207, 1–9. [Google Scholar] [CrossRef]
- Au, K.M.; Park, S.I.; Wang, A.Z. Trispecific natural killer cell nanoengagers for targeted chemoimmunotherapy. Sci. Adv. 2020, 6, eaba8564. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L.; Lin, Z.; Tao, L.; Chen, M. More efficient induction of antitumor T cell immunity by exosomes from CD40L gene-modified lung tumor cells. Mol. Med. Rep. 2014, 9, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Reiners, K.S.; Dassler, J.; Coch, C.; Pogge von Strandmann, E. Role of Exosomes released by dendritic cells and/or by tumor targets: Regulation of NK cell plasticity. Front. Immunol. 2014, 5, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, M.; Tang, J.; Qiao, Q.; Wu, T.; Qi, Y.; Tan, S.; Gao, X.; Zhang, Z. Biodegradable hollow mesoporous silica nanoparticles for regulating tumor microenvironment and enhancing antitumor efficiency. Theranostics 2017, 7, 3276–3292. [Google Scholar] [CrossRef]
- Ding, B.S.; Dziubla, T.; Shuvaev, V.V.; Muro, S.; Muzykantov, V.R. Advanced drug delivery systems that target the vascular endothelium. Mol. Interv. 2006, 6, 98–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, S.; Wu, S.Y. The use of lipid-based nanocarriers for targeted pain therapies. Front. Pharmacol 2013, 4, 143. [Google Scholar] [CrossRef] [Green Version]
- Koning, G.A.; Storm, G. Targeted drug delivery systems for the intracellular delivery of macromolecular drugs. Drug Discov. Today 2003, 8, 482–483. [Google Scholar] [CrossRef]
- Metselaar, J.M.; Storm, G. Liposomes in the treatment of inflammatory disorders. Expert Opin. Drug Deliv. 2005, 2, 465–476. [Google Scholar] [CrossRef]
- Leung, A.K.; Tam, Y.Y.; Chen, S.; Hafez, I.M.; Cullis, P.R. Microfluidic mixing: A general method for encapsulating macromolecules in lipid nanoparticle systems. J. Phys. Chem. B 2015, 119, 8698–8706. [Google Scholar] [CrossRef]
- Patel, S.; Ryals, R.C.; Weller, K.K.; Pennesi, M.E.; Sahay, G. Lipid nanoparticles for delivery of messenger RNA to the back of the eye. J. Control. Release 2019, 303, 91–100. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater 2021, 1–17. [Google Scholar]
- Love, K.T.; Mahon, K.P.; Levins, C.G.; Whitehead, K.A.; Querbes, W.; Dorkin, J.R.; Qin, J.; Cantley, W.; Qin, L.L.; Racie, T.; et al. Lipid-like materials for low-dose, in vivo gene silencing. Proc. Natl. Acad. Sci. USA 2010, 107, 1864–1869. [Google Scholar] [CrossRef] [Green Version]
- Vhora, I.; Lalani, R.; Bhatt, P.; Patil, S.; Misra, A. Lipid-nucleic acid nanoparticles of novel ionizable lipids for systemic BMP-9 gene delivery to bone-marrow mesenchymal stem cells for osteoinduction. Int. J. Pharm. 2019, 563, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Dings, R.P.M.; Cannon, M.; Vang, K.B. Design of gold nanoparticles in dendritic cell-based vaccines. Part. Part. Syst. Charact. 2018, 35, 1800109. [Google Scholar] [CrossRef]
- Yildiz, A.; Kaya, Y.; Tanriverdi, O. Effect of the interaction between selenium and zinc on DNA repair in association with cancer prevention. J. Cancer Prev. 2019, 24, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Neve, J. Selenium as a risk factor for cardiovascular diseases. J. Cardiovasc. Risk 1996, 3, 42–47. [Google Scholar] [CrossRef]
- Sun, W.; Zhu, J.; Li, S.; Tang, C.; Zhao, Q.; Zhang, J. Selenium supplementation protects against oxidative stress-induced cardiomyocyte cell cycle arrest through activation of PI3K/AKT. Metallomics 2020, 12, 1965–1978. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Liu, T.; Chen, T. Overcoming blood-brain barrier by HER2-targeted nanosystem to suppress glioblastoma cell migration, invasion and tumor growth. J. Mater. Chem. B 2018, 6, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Nabavinia, M.; Beltran-Huarac, J. Recent progress in iron oxide nanoparticles as therapeutic magnetic agents for cancer treatment and tissue engineering. ACS Appl. Bio Mater. 2020, 3, 8172–8187. [Google Scholar] [CrossRef]
- Schwenk, M.H. Ferumoxytol: A new intravenous iron preparation for the treatment of iron deficiency anemia in patients with chronic kidney disease. Pharmacotherapy 2010, 30, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Corot, C.; Robert, P.; Idee, J.M.; Port, M. Recent advances in iron oxide nanocrystal technology for medical imaging. Adv. Drug. Deliv. Rev. 2006, 58, 1471–1504. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dong, W.F.; Sun, H.B. Multifunctional superparamagnetic iron oxide nanoparticles: Design, synthesis and biomedical photonic applications. Nanoscale 2013, 5, 7664–7684. [Google Scholar] [CrossRef] [PubMed]
- Kievit, F.M.; Zhang, M. Surface engineering of iron oxide nanoparticles for targeted cancer therapy. Acc. Chem. Res. 2011, 44, 853–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manjili, H.K.; Malvandi, H.; Mousavi, M.S.; Attari, E.; Danafar, H. In vitro and in vivo delivery of artemisinin loaded PCL-PEG-PCL micelles and its pharmacokinetic study. Artif. Cells Nanomed. Biotechnol. 2018, 46, 926–936. [Google Scholar] [CrossRef] [Green Version]
- Yao, Q.; Liu, Y.; Kou, L.; Tu, Y.; Tang, X.; Zhu, L. Tumor-targeted drug delivery and sensitization by MMP2-responsive polymeric micelles. Nanomedicine 2019, 19, 71–80. [Google Scholar] [CrossRef]
- Namazi, H.; Adeli, M. Dendrimers of citric acid and poly (ethylene glycol) as the new drug-delivery agents. Biomaterials 2005, 26, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Saluja, V.; Mankoo, A.; Saraogi, G.K.; Tambuwala, M.M.; Mishra, V. Smart dendrimers: Synergizing the targeting of anticancer bioactives. J. Drug Deliv. Sci. Technol. 2019, 52, 15–26. [Google Scholar] [CrossRef]
- Adams, M.L.; Lavasanifar, A.; Kwon, G.S. Amphiphilic block copolymers for drug delivery. J. Pharm. Sci. 2003, 92, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A.; Safaiyan, S.; Rafati, S. Improvement of different vaccine delivery systems for cancer therapy. Mol. Cancer 2011, 10, 3. [Google Scholar] [CrossRef]
- Chang, H.; Lv, J.; Gao, X.; Wang, X.; Wang, H.; Chen, H.; He, X.; Li, L.; Cheng, Y. Rational design of a polymer with robust efficacy for intracellular protein and peptide delivery. Nano Lett. 2017, 17, 1678–1684. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.S.; Liu, F.; Huang, L. Implications of pharmacokinetic behavior of lipoplex for its inflammatory toxicity. Adv. Drug Deliv. Rev. 2005, 57, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, P.R.; Mandel, R.J.; Xiong, W.D.; Kroeger, K.; Castro, M.G. Immune responses to adenovirus and adeno-associated vectors used for gene therapy of brain diseases: The role of immunological synapses in understanding the cell biology of neuroimmune interactions. Curr. Gene Ther. 2007, 7, 347–360. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Ichihara, M.; Wang, X.; Yamamoto, K.; Kimura, J.; Majima, E.; Kiwada, H. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J. Control. Release 2006, 112, 15–25. [Google Scholar] [CrossRef]
- Yang, Y.; Hong, Y.; Cho, E.; Kim, G.B.; Kim, I.S. Extracellular vesicles as a platform for membrane-associated therapeutic protein delivery. J. Extracell. Vesicles 2018, 7, 1440131. [Google Scholar] [CrossRef] [Green Version]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human dendritic cells: Their heterogeneity and clinical application potential in cancer immunotherapy. Front. Immunol. 2018, 9, 3176. [Google Scholar] [CrossRef]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.J.; Pulendran, B.; Palucka, K. Immunobiology of dendritic cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef]
- Mbongue, J.C.; Nieves, H.A.; Torrez, T.W.; Langridge, W.H. The role of dendritic cell maturation in the induction of insulin-dependent diabetes mellitus. Front. Immunol. 2017, 8, 327. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Zhao, Y.; Guo, H.; Li, Y.; Tewary, P.; Xing, G.; Hou, W.; Oppenheim, J.J.; Zhang, N. [Gd@C(82)(OH)(22)](n) nanoparticles induce dendritic cell maturation and activate Th1 immune responses. ACS Nano 2010, 4, 1178–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikawa, M.; Kato, H.; Okumura, M.; Narazaki, M.; Kanazawa, Y.; Miwa, N.; Shinohara, H. Paramagnetic water-soluble metallofullerenes having the highest relaxivity for MRI contrast agents. Bioconjug. Chem. 2001, 12, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Sozzani, S.; Luini, W.; Borsatti, A.; Polentarutti, N.; Zhou, D.; Piemonti, L.; D’Amico, G.; Power, C.A.; Wells, T.N.; Gobbi, M.; et al. Receptor expression and responsiveness of human dendritic cells to a defined set of CC and CXC chemokines. J. Immunol. 1997, 159, 1993–2000. [Google Scholar] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Bronte, V.; Zanovello, P. Regulation of immune responses by L-arginine metabolism. Nat. Rev. Immunol. 2005, 5, 641–654. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Hernandez, C.P.; Quiceno, D.; Dubinett, S.M.; Zabaleta, J.; Ochoa, J.B.; Gilbert, J.; Ochoa, A.C. Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J. Exp. Med. 2005, 202, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Ochoa, A.C. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: Mechanisms and therapeutic perspectives. Immunol. Rev. 2008, 222, 180–191. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Zea, A.H.; Culotta, K.S.; Zabaleta, J.; Ochoa, J.B.; Ochoa, A.C. Regulation of T cell receptor CD3zeta chain expression by L-arginine. J. Biol. Chem. 2002, 277, 21123–21129. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Nagaraj, S.; Gabrilovich, D.I. Tumor-associated CD8+ T cell tolerance induced by bone marrow-derived immature myeloid cells. J. Immunol. 2005, 175, 4583–4592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10(+) and IL-35(+) Treg cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019, 20, 724–735. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.A.; Tomita, Y.; Jankowska-Gan, E.; Lema, D.A.; Arvedson, M.P.; Nair, A.; Bracamonte-Baran, W.; Zhou, Y.; Meyer, K.K.; Zhong, W.; et al. Treg-cell-derived IL-35-coated extracellular vesicles promote infectious tolerance. Cell Rep. 2020, 30, 1039–1051.e5. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Krelin, Y.; Dvorkin, T.; Bjorkdahl, O.; Segal, S.; Dinarello, C.A.; Voronov, E.; Apte, R.N. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J. Immunol. 2005, 175, 8200–8208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunt, S.K.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J. Immunol. 2006, 176, 284–290. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Chen, J.; Zhang, W.; Zhang, R.; Ye, Y.; Liu, P.; Yu, W.; Wei, F.; Ren, X.; Yu, J. Interleukin-6 trans-signaling pathway promotes immunosuppressive myeloid-derived suppressor cells via suppression of suppressor of cytokine signaling 3 in breast cancer. Front. Immunol. 2017, 8, 1840. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007, 67, 4507–4513. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Le, Q.V.; Yang, G.; Wu, Y.; Jang, H.W.; Shokouhimehr, M.; Oh, Y.K. Nanomaterials for modulating innate immune cells in cancer immunotherapy. Asian J. Pharm. Sci. 2019, 14, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Terme, M.; Flament, C.; Taieb, J.; Andre, F.; Novault, S.; Escudier, B.; Robert, C.; Caillat-Zucman, S.; Tursz, T.; et al. Dendritic cell-derived exosomes promote natural killer cell activation and proliferation: A role for NKG2D ligands and IL-15Ralpha. PLoS ONE 2009, 4, e4942. [Google Scholar] [CrossRef]
- Bandyopadhyay, K.; Marrero, I.; Kumar, V. NKT cell subsets as key participants in liver physiology and pathology. Cell Mol. Immunol. 2016, 13, 337–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, D.; Zhao, Q.; Yu, J.; Zhang, F.; Zhang, H.; Wang, Z. Nanoparticle Targeting of Neutrophils for Improved Cancer Immunotherapy. Adv. Healthc. Mater. 2016, 5, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Li, S.; Zhou, H.; Tang, X.; Wu, Y.; Jiang, W.; Tian, Z.; Zhou, X.; Yang, X.; Wang, Y. Chemotaxis-driven delivery of nano-pathogenoids for complete eradication of tumors post-phototherapy. Nat. Commun. 2020, 11, 1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Zheng, L.; Chen, W.; Weng, W.; Song, J.; Ji, J. Delivery strategies of cancer immunotherapy: Recent advances and future perspectives. J. Hematol. Oncol. 2019, 12, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvistborg, P.; Philips, D.; Kelderman, S.; Hageman, L.; Ottensmeier, C.; Joseph-Pietras, D.; Welters, M.J.; van der Burg, S.; Kapiteijn, E.; Michielin, O.; et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci. Transl. Med. 2014, 6, 254ra128. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Kamphorst, A.O.; Im, S.J.; Kissick, H.T.; Pillai, R.N.; Ramalingam, S.S.; Araki, K.; Ahmed, R. CD8 T Cell exhaustion in chronic infection and cancer: Opportunities for interventions. Annu. Rev. Med. 2018, 69, 301–318. [Google Scholar] [CrossRef]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 2017, 170, 1120–1133 e17. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.S.; Sansom, D.M. Confusing signals: Recent progress in CTLA-4 biology. Trends Immunol. 2015, 36, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef]
- Mohanty, R.; Chowdhury, C.R.; Arega, S.; Sen, P.; Ganguly, P.; Ganguly, N. CAR T cell therapy: A new era for cancer treatment (Review). Oncol. Rep. 2019, 42, 2183–2195. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hodi, F.S.; Robert, C. CTLA-4 and PD-1/PD-L1 blockade: New immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin. Cancer Res. 2013, 19, 5300–5309. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, H.; Kim, E.H.; Chi, S.-G.; Kim, S.H.; Yang, Y. Nanoparticles Targeting Innate Immune Cells in Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 10009. https://doi.org/10.3390/ijms221810009
Jang H, Kim EH, Chi S-G, Kim SH, Yang Y. Nanoparticles Targeting Innate Immune Cells in Tumor Microenvironment. International Journal of Molecular Sciences. 2021; 22(18):10009. https://doi.org/10.3390/ijms221810009
Chicago/Turabian StyleJang, Hochung, Eun Hye Kim, Sung-Gil Chi, Sun Hwa Kim, and Yoosoo Yang. 2021. "Nanoparticles Targeting Innate Immune Cells in Tumor Microenvironment" International Journal of Molecular Sciences 22, no. 18: 10009. https://doi.org/10.3390/ijms221810009
APA StyleJang, H., Kim, E. H., Chi, S.-G., Kim, S. H., & Yang, Y. (2021). Nanoparticles Targeting Innate Immune Cells in Tumor Microenvironment. International Journal of Molecular Sciences, 22(18), 10009. https://doi.org/10.3390/ijms221810009