
Development of Effective Therapeutic Molecule from Natural Sources against Coronavirus Protease

 ,
,  ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. ADME/Tox Prediction
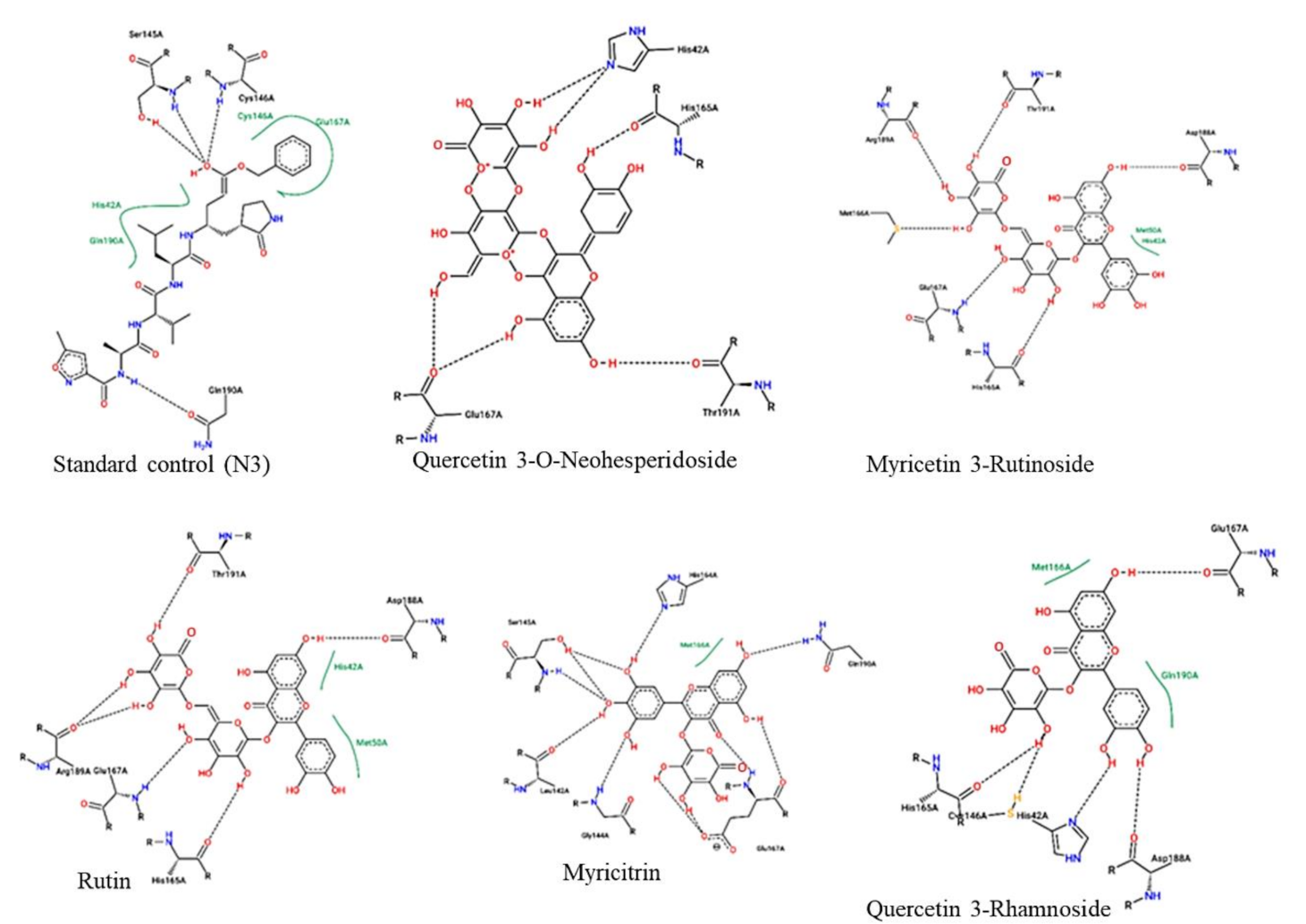
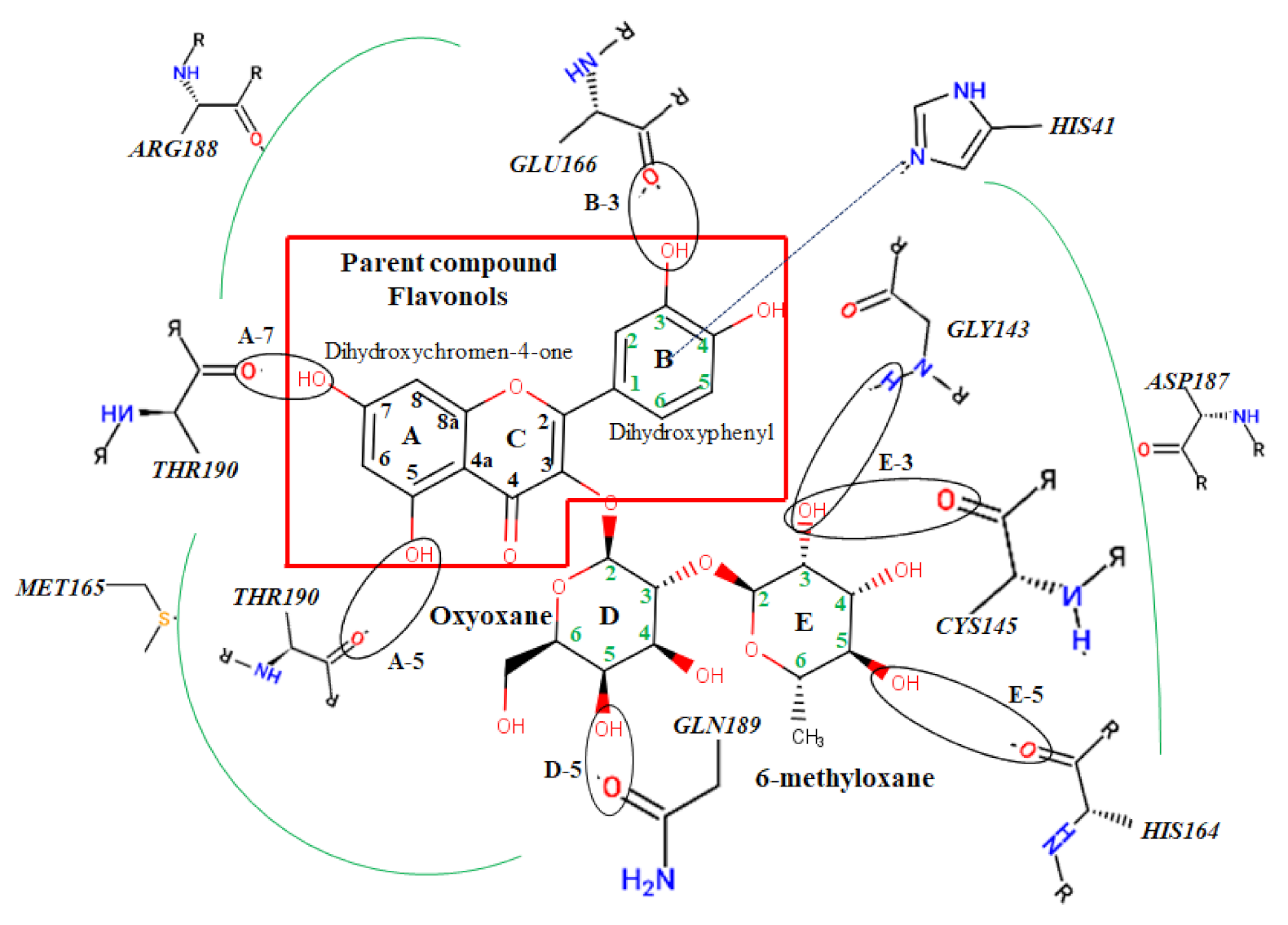
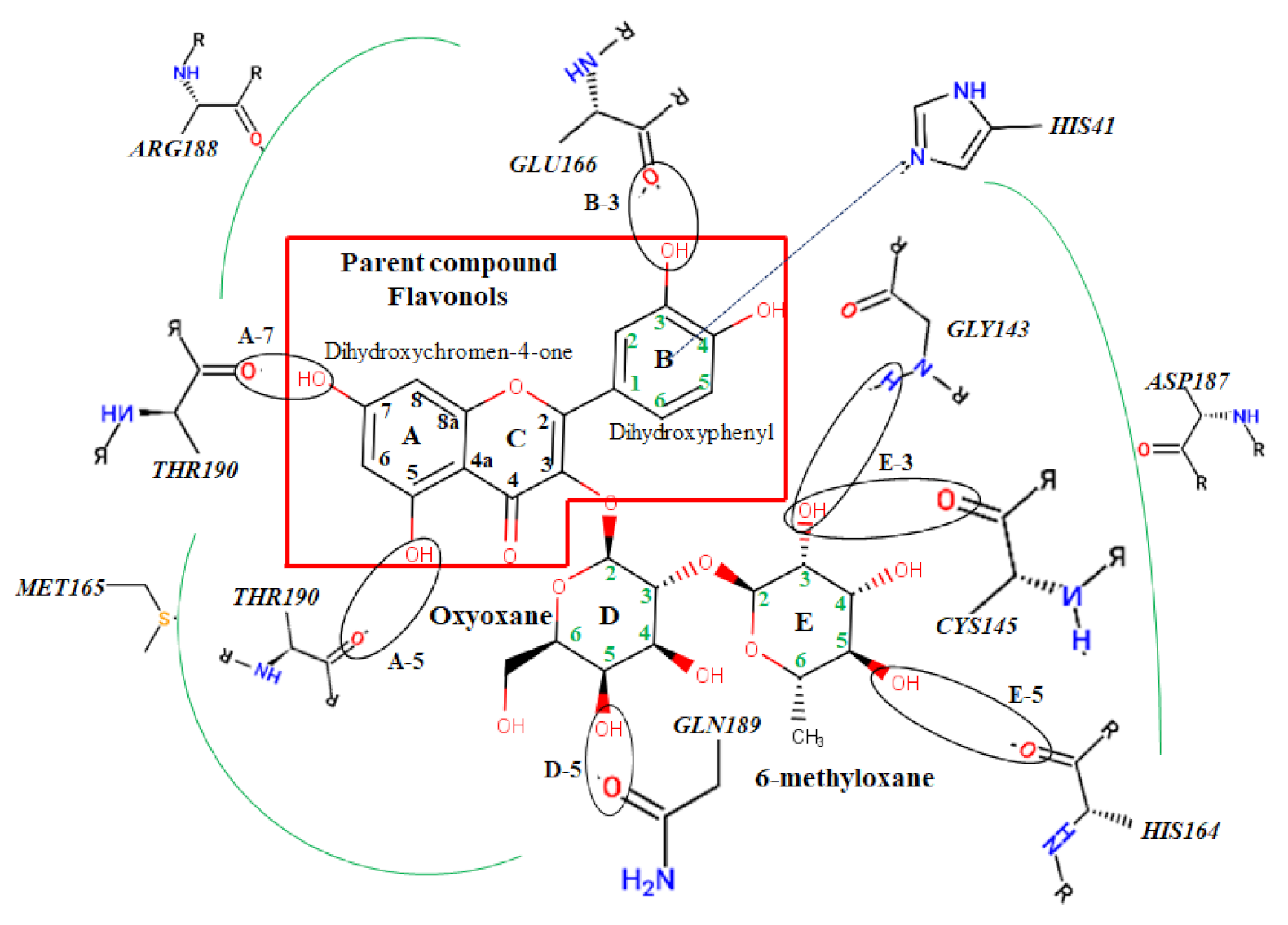
2.2. Docking Calculations
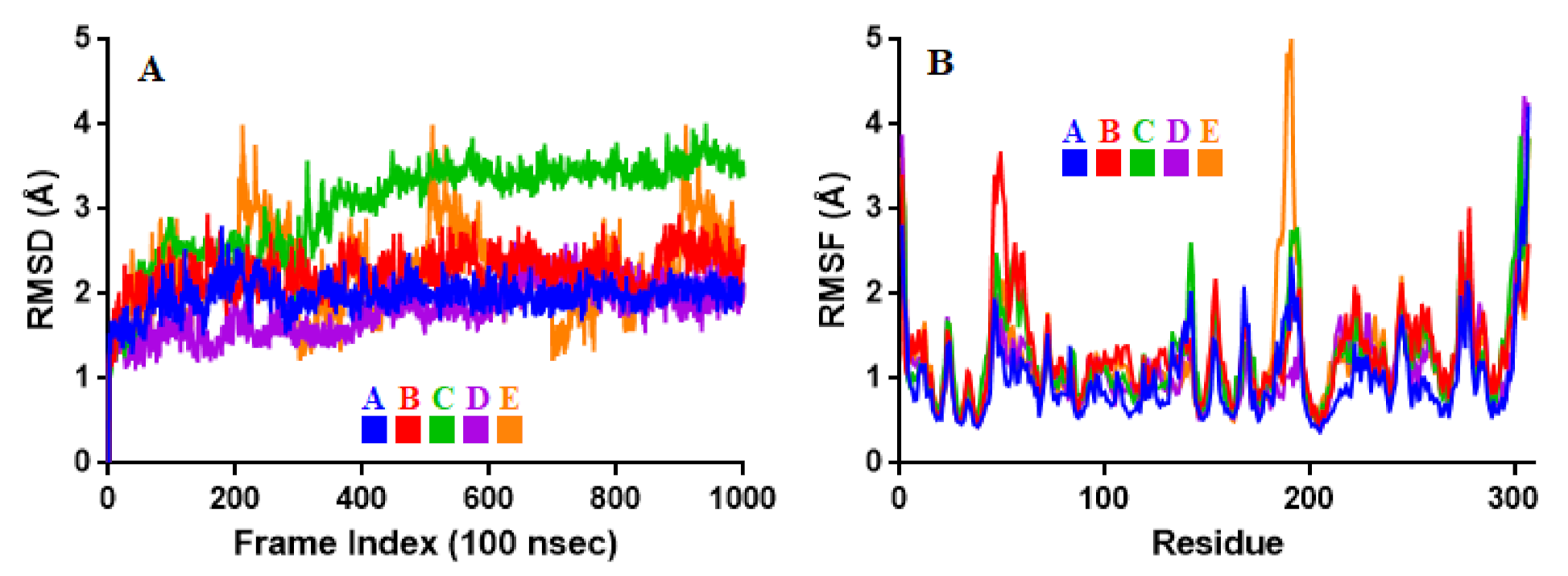
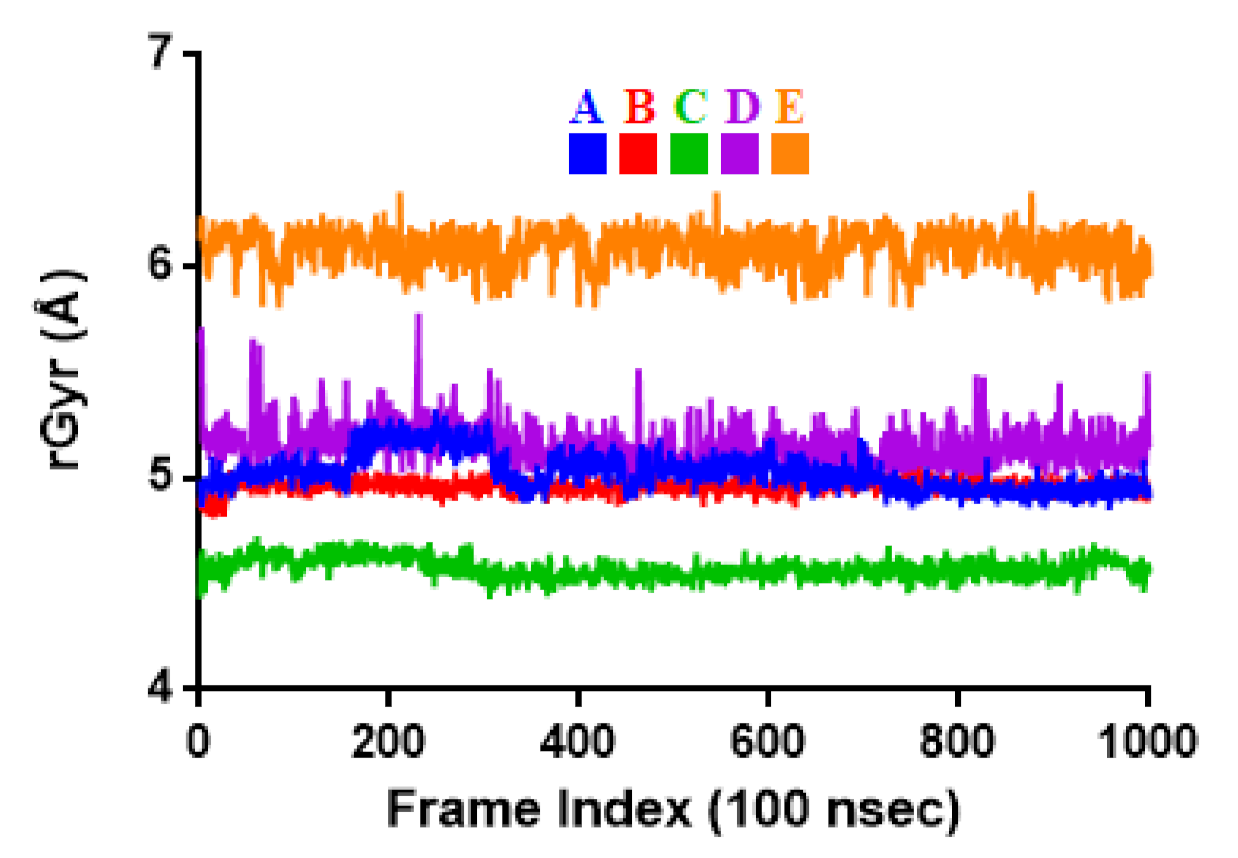
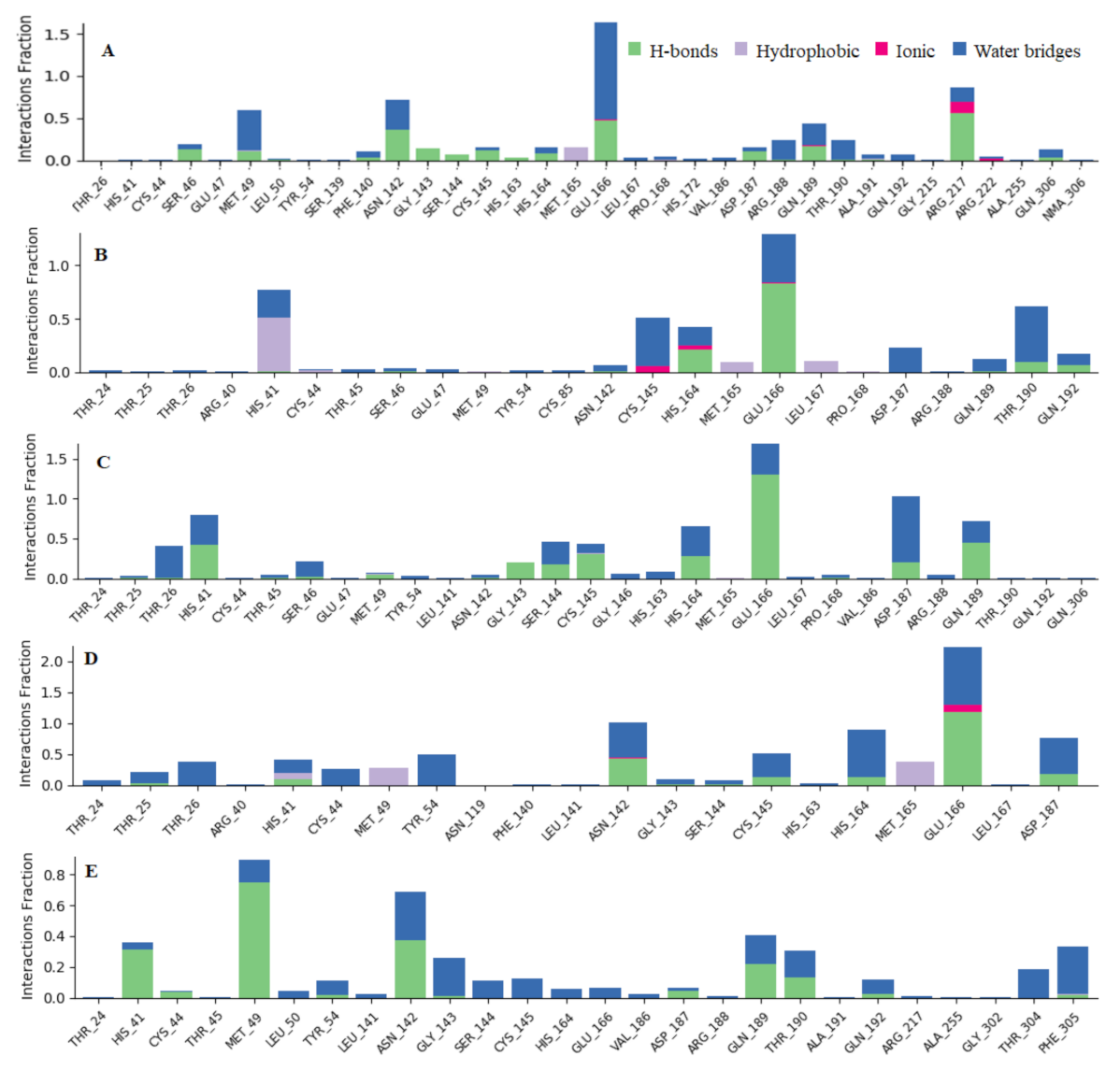
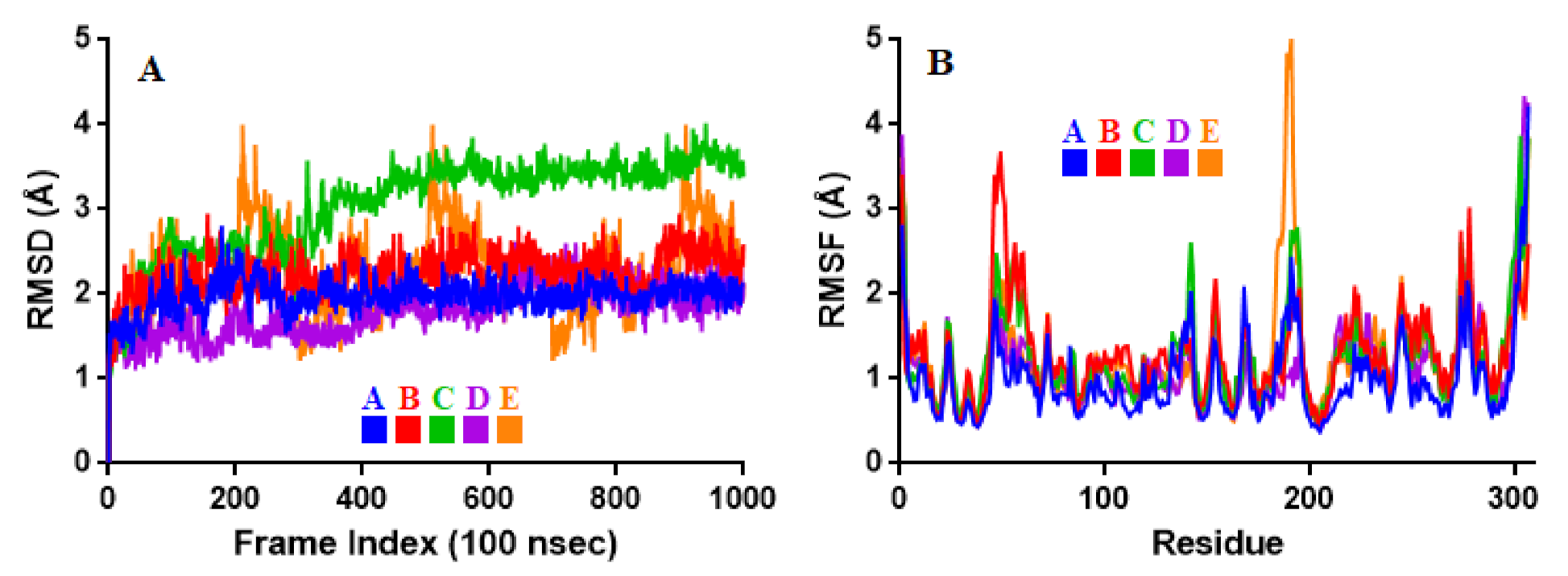
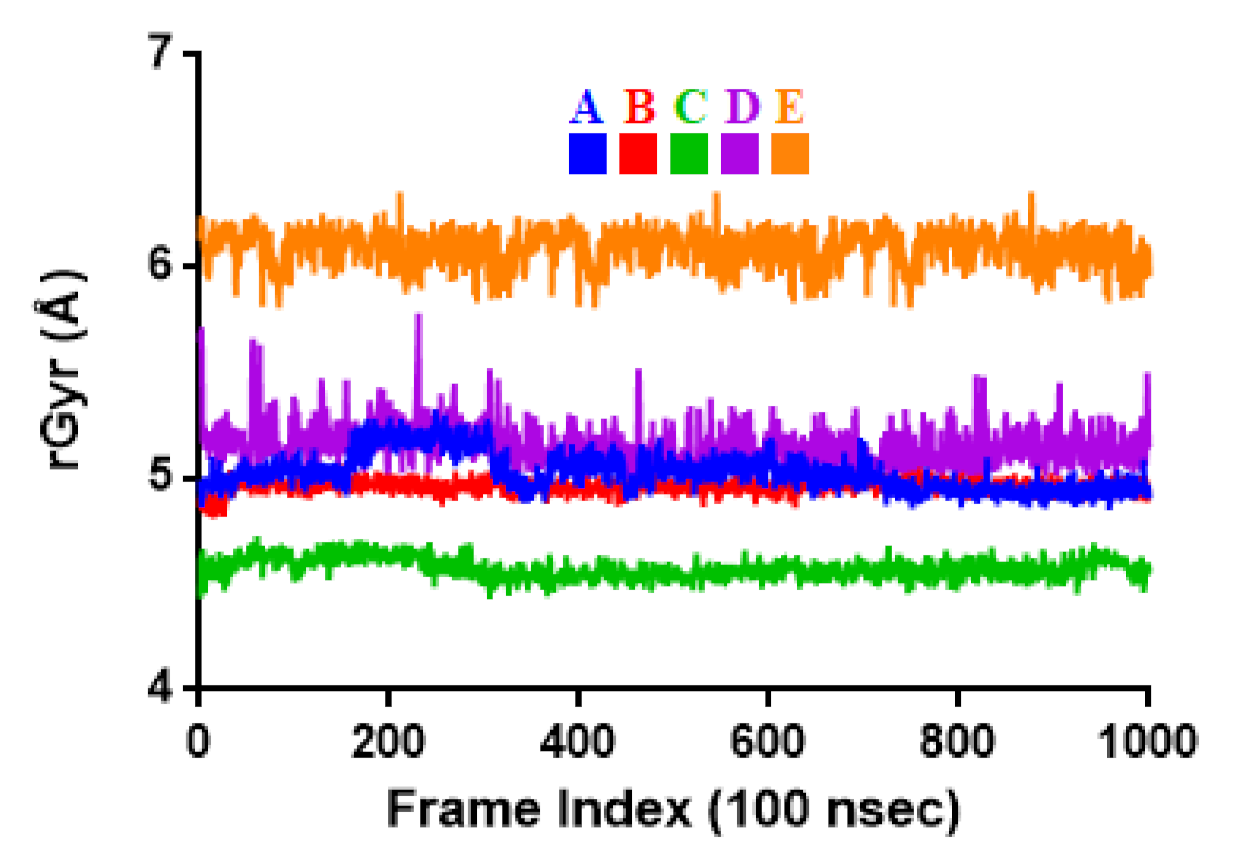
2.3. Molecular Dynamic Simulation
3. Discussion
3.1. Drug-Likeness (ADME/Tox) Analysis
3.2. Docking Analysis
3.3. Molecular Dynamic Simulation
4. Materials and Methods
4.1. ADME/Tox Analysis (Drug-Likeness)
4.2. Protein Preparation
4.3. Ligand Selection and Preparation
4.4. Molecular Docking Calculation
4.5. ∆G Energy Calculation of the Docked Complexes
4.6. Molecular Dynamics Simulation (MDs)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muhammed, Y. Molecular targets for COVID-19 drug development: Enlightening Nigerians about the pandemic and future treatment. Biosaf. Health 2020, 2, 210–216. [Google Scholar] [CrossRef]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8, 15092. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Sun, J.; Mao, Z.; Wang, L.; Lu, Y.; Li, J. A guideline for homology modeling of the proteins from newly discovered betacoronavirus, 2019 novel coronavirus (2019-nCoV). J. Med. Virol. 2020, 92, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S. Research and Development on Therapeutic Agents and Vaccines for Covid-19 and Related Human Coronavirus Diseases; ACS Publications: Washington, DC, USA, 2020. [Google Scholar]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadaka, A.O.; Sibuyi, N.R.S.; Adewale, F.; Bakare, O.O.; Akanbi, M.O.; Klein, A.; Madiehe, A.M.; Meyer, M. Understanding the epidemiology, pathophysiology, diagnosis and management of SARS-CoV-2. J. Int. Med. Res. 2020, 48, 0300060520949077. [Google Scholar] [CrossRef]
- Needle, D.; Lountos, G.T.; Waugh, D.S. Structures of theMiddle East respiratory syndrome coronavirus3C-like protease reveal insights into substrate specificity. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015, 71, 1102–1111. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-chymotrypsin like protease (3CLPro) inhibitors as potential anti-SARS-CoV-2 agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- Qamar, M.T.U.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef]
- Gyebi, G.A.; Ogunro, O.B.; Adegunloye, A.P.; Ogunyemi, O.M.; Afolabi, S.O. Potential inhibitors of coronavirus 3-chymotrypsin-like protease (3CLpro): An in silico screening of alkaloids and terpenoids from African medicinal plants. J. Biomol. Struct. Dyn. 2020, 39, 3396–3408. [Google Scholar] [CrossRef]
- Roviello, V.; Musumeci, D.; Mokhir, A.; Roviello, G.N. Evidence of protein binding by a nucleopeptide based on a thymine-decorated L-diaminopropanoic acid through CD and in silico studies. Curr. Med. Chem. 2021, 28, 1. [Google Scholar] [CrossRef]
- Vicidomini, C.; Roviello, V.; Roviello, G.N. In Silico Investigation on the Interaction of Chiral Phytochemicals from Opuntia ficusindica with SARS-CoV-2 Mpro. Symmetry 2021, 13, 1041. [Google Scholar] [CrossRef]
- Upadhyay, S.; Dixit, M. Role of Polyphenols and Other Phytochemicals on Molecular Signaling. Oxid. Med. Cell. Longev. 2015, 2015, 504253. [Google Scholar] [CrossRef]
- Graf, B.L.; Raskin, I.; Cefalu, W.T.; Ribnicky, D.M. Plant-derived therapeutics for the treatment of metabolic syndrome. Curr. Opin. Investig. Drugs 2010, 11, 1107. [Google Scholar] [CrossRef]
- García-Lafuente, A.; Guillamón, E.; Villares, A.; Rostagno, M.A.; Martínez, J.A. Flavonoids as an-ti-inflammatory agents: Implications in cancer and cardiovascular disease. Inflamm. Res. 2009, 58, 537–552. [Google Scholar] [CrossRef]
- Ichikawa, D.; Matsui, A.; Imai, M.; Sonoda, Y.; Kasahara, T. Effect of various catechins on the IL-12p40 pro-duction by murine peritoneal macrophages and a macrophage cell line, J774. 1. Biol. Pharm. Bull. 2004, 27, 1353–1358. [Google Scholar] [CrossRef]
- Loo, G. Redox-sensitive mechanisms of phytochemical-mediated inhibition of cancer cell proliferation (review). J. Nutr. Biochem. 2003, 14, 64–73. [Google Scholar] [CrossRef]
- Tosetti, F.; Noonan, D.M.; Albini, A. Metabolic regulation and redox activity as mechanisms for angiopre-vention by dietary phytochemicals. Int. J. Cancer 2009, 125, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Kowshik, J.; Baba, A.B.; Giri, H.; Reddy, G.D.; Dixit, M.; Nagini, S. Astaxanthin Inhibits JAK/STAT-3 Signaling to Abrogate Cell Proliferation, Invasion and Angiogenesis in a Hamster Model of Oral Cancer. PLoS ONE 2014, 9, e109114. [Google Scholar] [CrossRef]
- Yarmolinsky, L.; Huleihel, M.; Zaccai, M.; Ben-Shabat, S. Potent antiviral flavone glycosides from Ficus benjamina leaves. Fitoterapia 2012, 83, 362–367. [Google Scholar] [CrossRef]
- Szlávik, L.; Gyuris, Á.; Minárovits, J.; Forgo, P.; Molnár, J.; Hohmann, J. Alkaloids from Leucojum vernum and antiretroviral activity of Amaryllidaceae alkaloids. Planta Med. Nat. Prod. Med. Plant Res. 2004, 70, 871–873. [Google Scholar]
- Ibrahim, A.K.; Youssef, A.I.; Arafa, A.; Ahmed, S.A. Anti-H5N1 virus flavonoids from Capparis sinaica Veill. Nat. Prod. Res. 2013, 27, 2149–2153. [Google Scholar] [CrossRef]
- Orhan, D.D.; Özçelik, B.; Özgen, S.; Ergun, F. Antibacterial, antifungal, and antiviral activities of some flavonoids. Microbiol. Res. 2010, 165, 496–504. [Google Scholar] [CrossRef]
- Ekor, M. The growing use of herbal medicines: Issues relating to adverse reactions and challenges in monitoring safety. Front. Pharmacol. 2014, 4, 177. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.-T.; Hsu, W.-C.; Lin, C.-C. Antiviral Natural Products and Herbal Medicines. J. Tradit. Complement. Med. 2014, 4, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Pour, P.M.; Fakhri, S.; Asgary, S.; Farzaei, M.H.; Echeverria, J. The signaling pathways, and ther-apeutic targets of antiviral agents: Focusing on the antiviral approaches and clinical perspectives of antho-cyanins in the management of viral diseases. Front. Pharmacol. 2019, 10, 1207. [Google Scholar] [CrossRef] [Green Version]
- Kurapati, K.R.V.; Atluri, V.S.; Samikkannu, T.; Garcia, G.; Nair, M.P.N. Natural Products as Anti-HIV Agents and Role in HIV-Associated Neurocognitive Disorders (HAND): A Brief Overview. Front. Microbiol. 2016, 6, 1444. [Google Scholar] [CrossRef]
- Ninfali, P.; Antonelli, A.; Magnani, M.; Scarpa, E.S. Antiviral Properties of Flavonoids and Delivery Strategies. Nutrition 2020, 12, 2534. [Google Scholar] [CrossRef]
- Chuanasa, T.; Phromjai, J.; Lipipun, V.; Likhitwitayawuid, K.; Suzuki, M.; Pramyothin, P.; Hattori, M.; Shiraki, K. Anti-herpes simplex virus (HSV-1) activity of oxyresveratrol derived from Thai medicinal plant: Mechanism of action and therapeutic efficacy on cutaneous HSV-1 infection in mice. Antivir. Res. 2008, 80, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Kuo, P. Flora Reipublicae Popularis Sinicae; Science Press: Beijing, China, 1987; Volume 9, p. 19. [Google Scholar]
- Zhou, X.; Peng, J.; Fan, G.; Wu, Y. Isolation and purification of flavonoid glycosides from Trollius ledebouri using high-speed counter-current chromatography by stepwise increasing the flow-rate of the mobile phase. J. Chromatogr. A 2005, 1092, 216–221. [Google Scholar] [CrossRef]
- Da Silva, B.P.; Bernardo, R.R.; Parente, J.P. Flavonol glycosides from Costus spicatus. Phytochemistry 2000, 53, 87–92. [Google Scholar] [CrossRef]
- Lau-Cam, C.A.; Chan, H. Flavonoids from Comptonia peregrina. Phytochemistry 1973, 12, 1829. [Google Scholar] [CrossRef]
- Semwal, D.K.; Semwal, R.B.; Combrinck, S.; Viljoen, A. Myricetin: A Dietary Molecule with Diverse Biological Activities. Nutrition 2016, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Wu, C.D. Compounds from Syzygium aromaticum possessing growth inhibitory activity against oral pathogens. J. Nat. Prod. 1996, 59, 987–990. [Google Scholar] [CrossRef]
- Jo, S.; Kim, S.; Shin, D.H.; Kim, M.-S. Inhibition of African swine fever virus protease by myricetin and myricitrin. J. Enzym. Inhib. Med. Chem. 2020, 35, 1045–1049. [Google Scholar] [CrossRef]
- Almutairi, M.M.; Alanazi, W.; Alshammari, M.A.; Alotaibi, M.R.; Alhoshani, A.R.; Al-Rejaie, S.S.; Hafez, M.M.; Al-Shabanah, O.A. Neuro-protective effect of rutin against Cisplatin-induced neurotoxic rat model. BMC Complement. Altern. Med. 2017, 17, 472. [Google Scholar] [CrossRef] [Green Version]
- Elsayed, H.E.; Ebrahim, H.Y.; Mohyeldin, M.M.; Siddique, A.B.; Kamal, A.M.; Haggag, E.; El Sayed, K.A. Rutin as A Novel c-Met Inhibitory Lead for the Control of Triple Negative Breast Malignancies. Nutr. Cancer 2017, 69, 1256–1271. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, C.N.; Setzer, W.N. An in-silico investigation of phytochemicals as antiviral agents against dengue fever. Comb. Chem. High Throughput Screen. 2016, 19, 516–536. [Google Scholar] [CrossRef] [Green Version]
- Ojo, O.A.; Aruleba, R.T.; Adekiya, T.A.; Sibuyi, N.R.S.; Ojo, A.B.; Ajiboye, B.O.; Oyinloye, B.E.; Adeola, H.A.; Fadaka, A.O. Deciphering the interaction of puerarin with cancer macromolecules: An in silico investigation. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Aruleba, R.T.; Sibuyi, N.R.S.; Klein, A.; Madiehe, A.M.; Meyer, M. Inhibitory potential of re-purposed drugs against the SARS-CoV-2 main protease: A computational-aided approach. J. Biomol. Struct. Dyn. 2020. [Google Scholar] [CrossRef]
- Ben-Shabat, S.; Yarmolinsky, L.; Porat, D.; Dahan, A. Antiviral effect of phytochemicals from medicinal plants: Applications and drug delivery strategies. Drug Deliv. Transl. Res. 2020, 10, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Mani, J.S.; Johnson, J.B.; Steel, J.C.; Broszczak, D.A.; Neilsen, P.M.; Walsh, K.B.; Naiker, M. Natural product-derived phytochemicals as potential agents against coronaviruses: A review. Virus Res. 2020, 284, 197989. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Adekiya, T.A.; Aruleba, R.T.; Klein, A.; Fadaka, A.O. In silico inhibition of SGTP4 as a therapeutic target for the treatment of schistosomiasis. J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Cherrak, S.A.; Merzouk, H.; Mokhtari-Soulimane, N. Potential bioactive glycosylated flavonoids as SARS-CoV-2 main protease inhibitors: A molecular docking and simulation studies. PLoS ONE 2020, 15, e0240653. [Google Scholar] [CrossRef]
- Pantsar, T.; Poso, A. Binding Affinity via Docking: Fact and Fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef] [Green Version]
- Rastelli, G.; Del Rio, A.; Degliesposti, G.; Sgobba, M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Beveridge, D.L.; Di Capua, F. Free energy via molecular simulation: Applications to chemical and biomolecular systems. Ann. Rev. Biophys. Biophys. Chem. 1989, 18, 431–492. [Google Scholar] [CrossRef]
- Efridlender, M.; Ekapulnik, Y.; Ekoltai, H. Plant derived substances with anti-cancer activity: From folklore to practice. Front. Plant Sci. 2015, 6, 799. [Google Scholar] [CrossRef]
- Brylinski, M. Aromatic interactions at the ligand-protein interface: Implications for the development of docking scoring functions. Chem. Biol. Drug Des. 2017, 91, 380–390. [Google Scholar] [CrossRef]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Omondi, R.O.; Sibuyi, N.R.; Fadaka, A.O.; Meyer, M.; Jaganyi, D.; Ojwach, S.O. Role of π-conjugation on the coordination behaviour, substitution kinetics, DNA/BSA interactions, and in vitro cytotoxicity of carbox-amide palladium (ii) complexes. Dalton Trans. 2021, 50, 8127–8143. [Google Scholar] [CrossRef]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Arnittali, M.; Rissanou, A.N.; Harmandaris, V. Structure of biomolecules through molecular dynamics simulations. Proc. Comput. Sci. 2019, 156, 69–78. [Google Scholar] [CrossRef]
- Ylilauri, M.; Pentikäinen, O.T. MMGBSA as a Tool to Understand the Binding Affinities of Filamin–Peptide Interactions. J. Chem. Inf. Model. 2013, 53, 2626–2633. [Google Scholar] [CrossRef]
- Fadaka, A.O.; Sibuyi, N.R.S.; Madiehe, A.M.; Meyer, M. Computational insight of dexamethasone against potential targets of SARS-CoV-2. J. Biomol. Struct. Dyn. 2020, 1–11. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
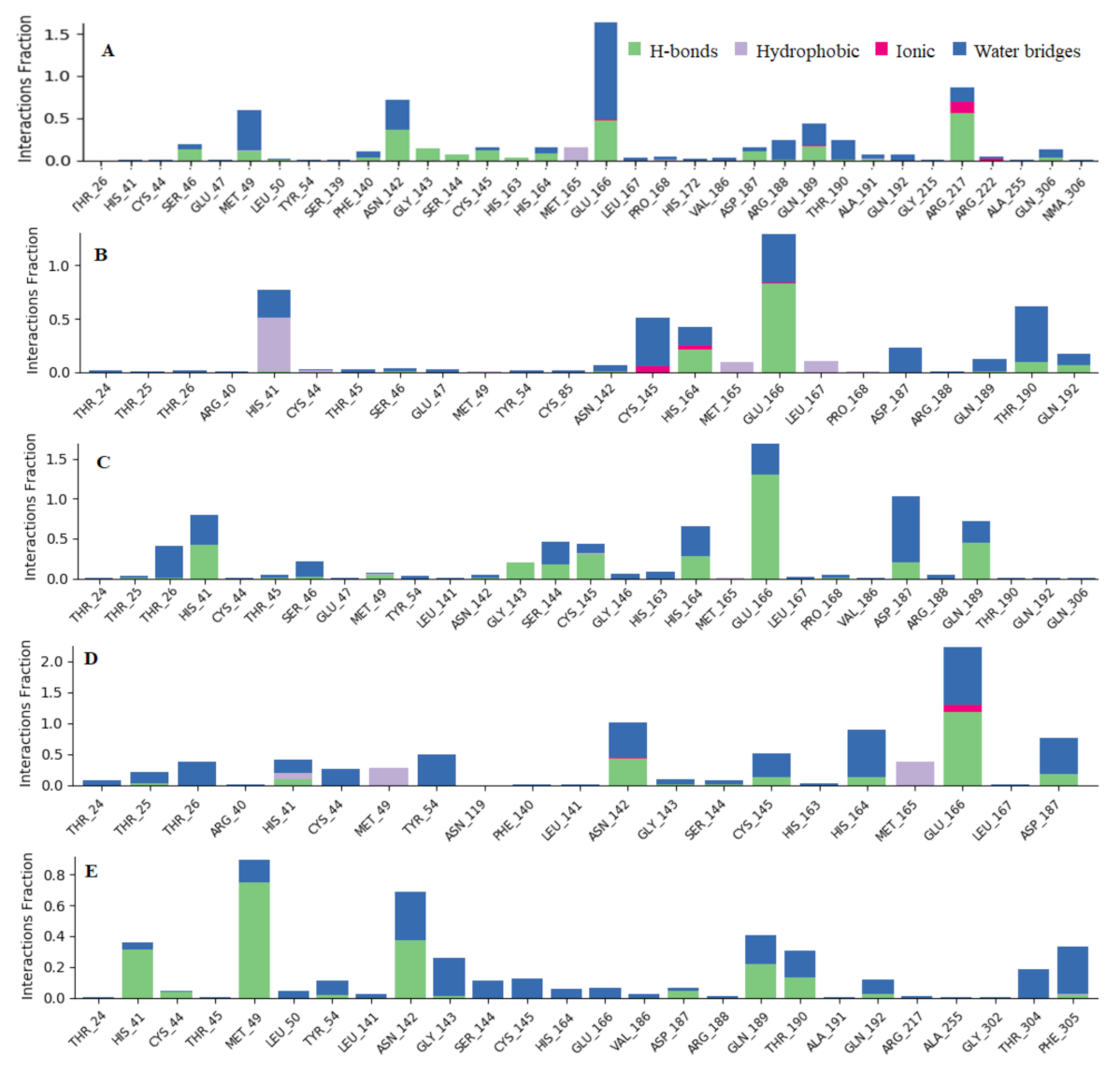{kind=link}
| Compound | ID a | M.W b | ROF c | QplogHERG d | QplogPoW e | QplogKP f | Donor HB | Acceptor HB | QplogS g | QplogBB h |
|---|---|---|---|---|---|---|---|---|---|---|
| Quercetin 3-O-Neohesperidoside | 5748416 | 610.5 | 2 | −6.449 | −1.998 | −6.423 | 9 | 20.55 | −2.932 | −4.728 |
| Myricetin 3-Rutinoside | 44259428 | 626.5 | 2 | −6.394 | −2.455 | −5.583 | 10 | 21.3 | −2.341 | −4.306 |
| Quercetin 3-Rhamnoside | 5353915 | 448.3 | 2 | −5.451 | −0.55 | −6.101 | 6 | 12.05 | −3.196 | −3.312 |
| Rutin | 5280805 | 610.5 | 2 | −5.238 | −2.495 | −7.251 | 9 | 20.55 | −2.175 | −4.503 |
| Myricitrin | 5281673 | 464.3 | 2 | −5.463 | −1.045 | −6.589 | 7 | 12.8 | −2.779 | −3.48 |
| Compound | ID | Ames Toxicity | Carcinogens | Acute Oral Toxicity | Rat Acute Toxicity |
|---|---|---|---|---|---|
| Quercetin 3-O-Neohesperidoside | 5748416 | AT | NC | III | 2.2619 |
| Myricetin 3-Rutinoside | 44259428 | NAT | NC | III | 2.4984 |
| Quercetin 3-Rhamnoside | 5353915 | NAT | NC | III | 2.5458 |
| Rutin | 5280805 | NAT | NC | III | 2.4984 |
| Myricitrin | 5281673 | NAT | NC | III | 2.5458 |
| Name | Dock Score | ∆G Bind | H-Bond | Residues (Å) | Other Bond (Å) |
|---|---|---|---|---|---|
| Standard | −16.5 | −80.88 | 6 | CYS145 (2.13), LEU141 (2.76), PHE140 (2.02), GLU166 (1.68,1.83, 2.05) | Salt bridges (2) |
| Quercetin 3-O-Neohesperidoside | −16.8 | −87.60 | 5 | GLY143 (2.76), CYS145 (2.11), GLN189 (2.11), THR190 (1.76), HIS41 (2.30) | π-π stacking HIS41 (1.49) |
| Myricetin 3-Rutinoside | −12.9 | −87.50 | 7 | CYS145 (2.08), ASN142 (1.75), GLU166 (1.98), THR190 (2.21), ARG188 (1.97), HIS164 (1.90,1.98) | |
| Quercetin 3-Rhamnoside | −10.3 | −80.17 | 4 | LEU141 (1.49), THR190 (1.78), GLU166 (2.01), HIS164 (1.81) | |
| Rutin | −10.0 | −58.95 | 6 | THR190 (1.83), HIS41 (2.08), GLY143 (2.39,1.89), ASN142 (1.92), LEU141 (2.10) | - |
| Myricitrin | −9.1 | −49.22 | 3 | CYS145 (2.51), ASN142 (2.04), THR190 (1.83) | π-π stacking HIS41 (5.37) |
| Properties | A | B | C | D | E |
|---|---|---|---|---|---|
| RMSD | 1.98 ± 0.19 | 2.25 ± 0.26 | 3.05 ± 0.57 | 1.81 ± 0.30 | 2.26 ± 0.51 |
| RMSF | 1.00 ± 0.51 | 1.31 ± 0.55 | 1.27 ± 0.58 | 1.15 ± 0.58 | 1.25 ± 0.65 |
| rGyr | 5.03 ± 0.09 | 4.96 ± 0.03 | 4.57 ± 0.05 | 5.15 ± 0.10 | 6.09 ± 0.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fadaka, A.O.; Sibuyi, N.R.S.; Martin, D.R.; Klein, A.; Madiehe, A.; Meyer, M. Development of Effective Therapeutic Molecule from Natural Sources against Coronavirus Protease. Int. J. Mol. Sci. 2021, 22, 9431. https://doi.org/10.3390/ijms22179431
Fadaka AO, Sibuyi NRS, Martin DR, Klein A, Madiehe A, Meyer M. Development of Effective Therapeutic Molecule from Natural Sources against Coronavirus Protease. International Journal of Molecular Sciences. 2021; 22(17):9431. https://doi.org/10.3390/ijms22179431
Chicago/Turabian StyleFadaka, Adewale Oluwaseun, Nicole Remaliah Samantha Sibuyi, Darius Riziki Martin, Ashwil Klein, Abram Madiehe, and Mervin Meyer. 2021. "Development of Effective Therapeutic Molecule from Natural Sources against Coronavirus Protease" International Journal of Molecular Sciences 22, no. 17: 9431. https://doi.org/10.3390/ijms22179431
APA StyleFadaka, A. O., Sibuyi, N. R. S., Martin, D. R., Klein, A., Madiehe, A., & Meyer, M. (2021). Development of Effective Therapeutic Molecule from Natural Sources against Coronavirus Protease. International Journal of Molecular Sciences, 22(17), 9431. https://doi.org/10.3390/ijms22179431