
A Dansyl-Modified Sphingosine Kinase Inhibitor DPF-543 Enhanced De Novo Ceramide Generation

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
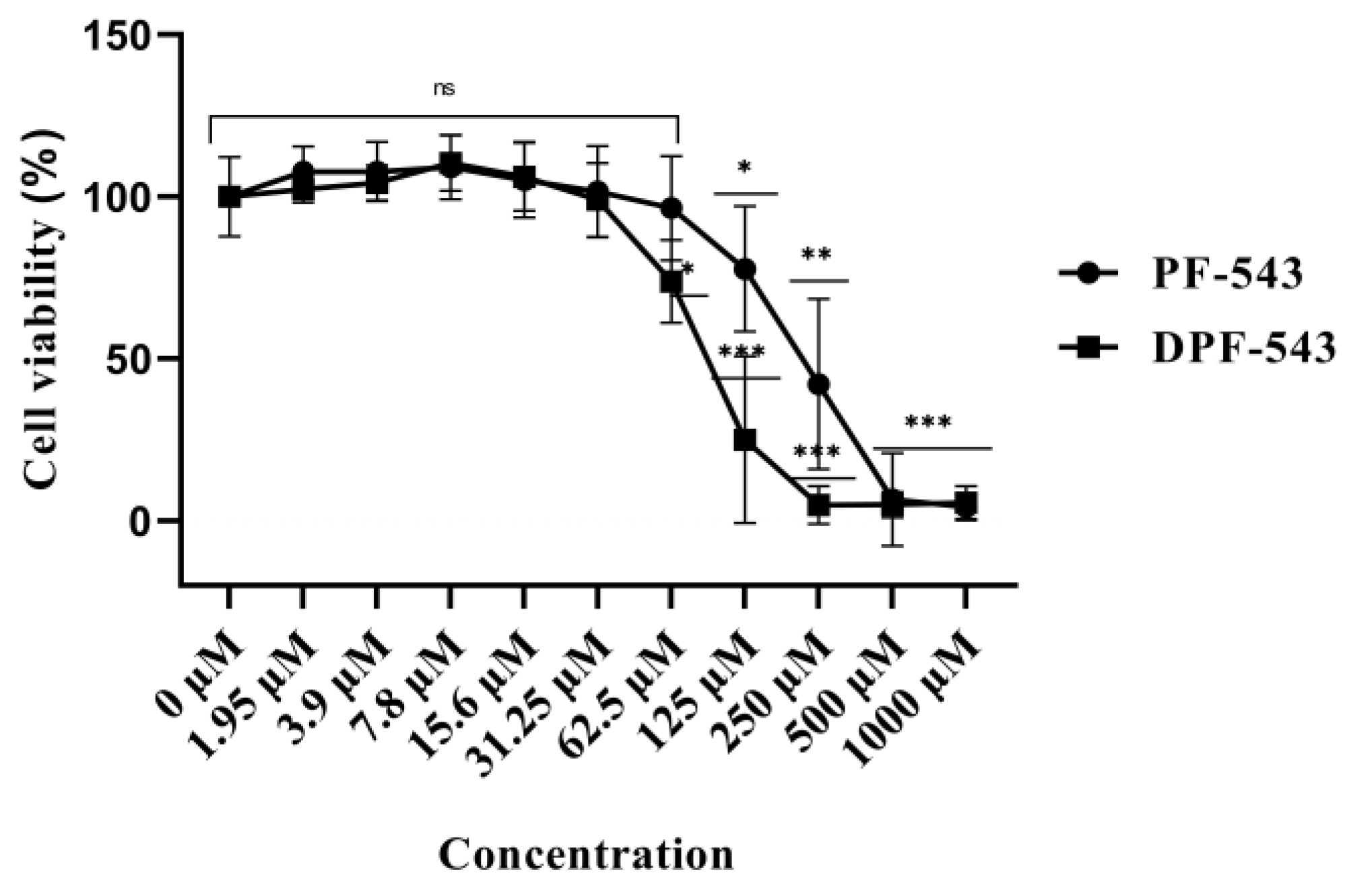
2.1. Cell Viability Evaluation following DPF-543 Treatment
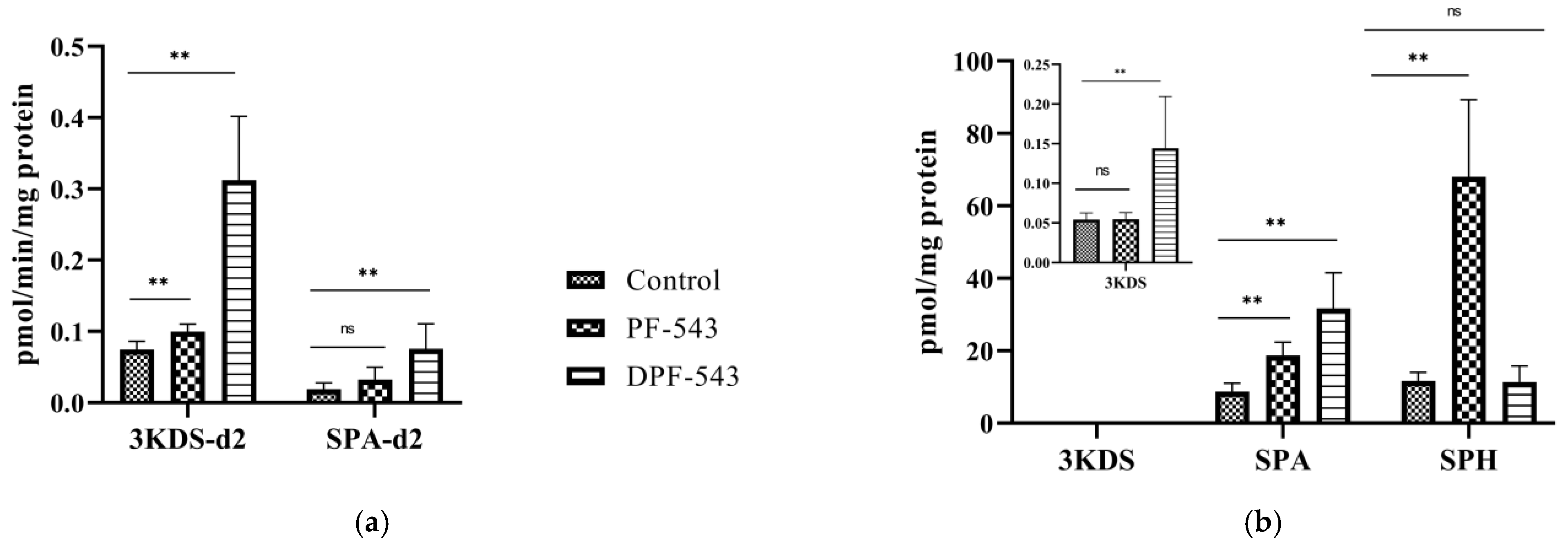
2.2. DPF-543 Activates SPT In Vitro
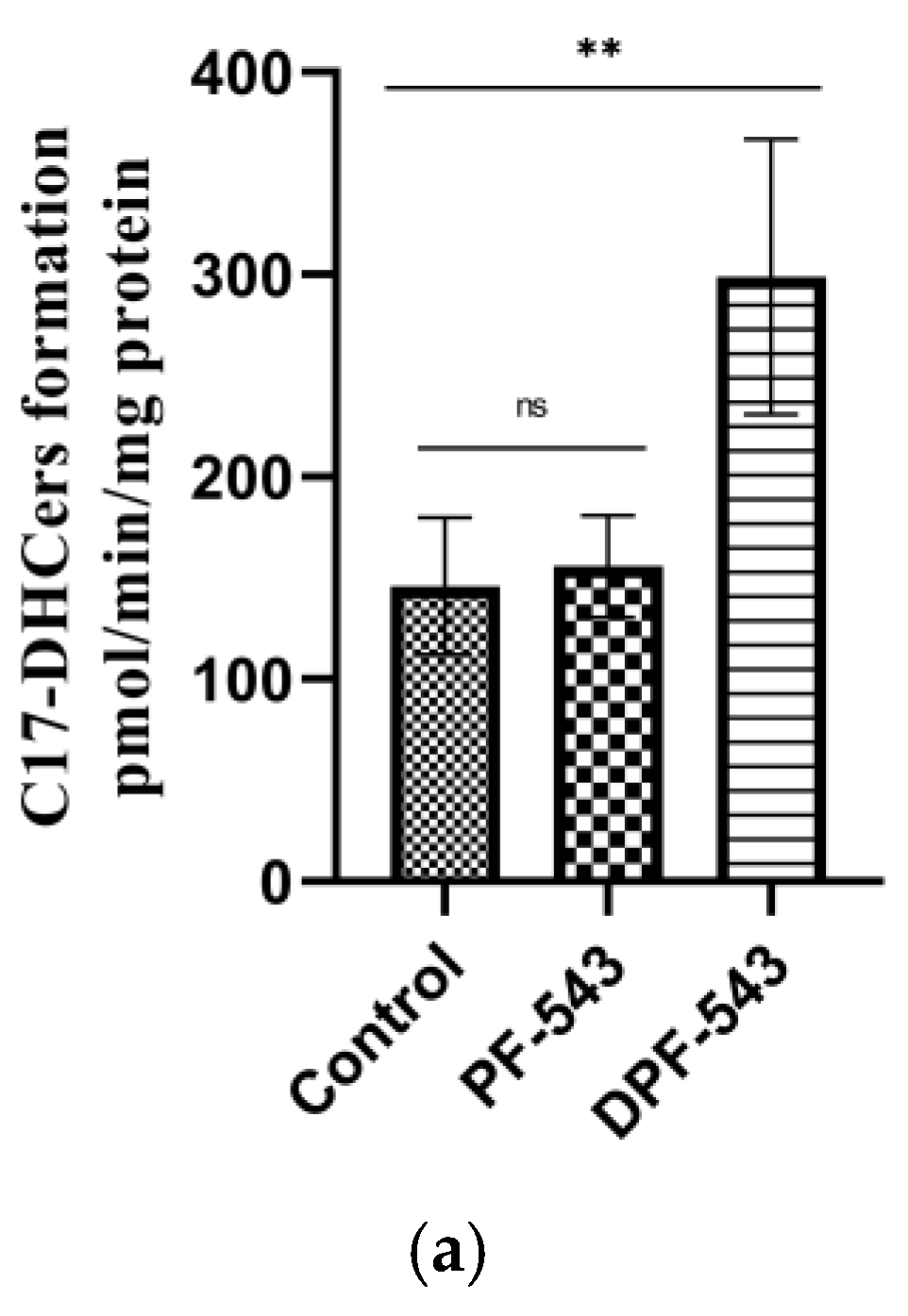
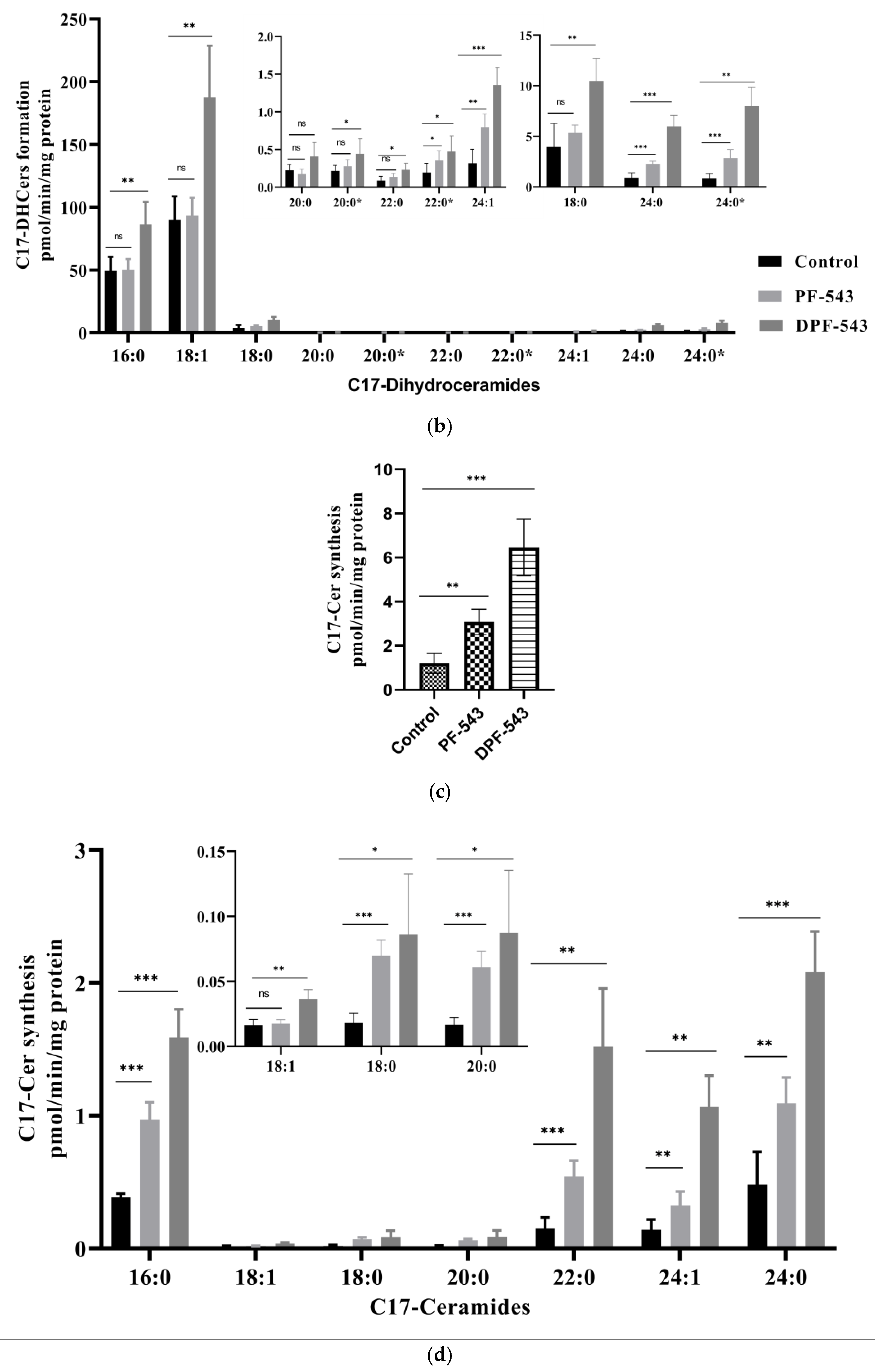
2.3. DPF-543 Strongly Activates CerSs
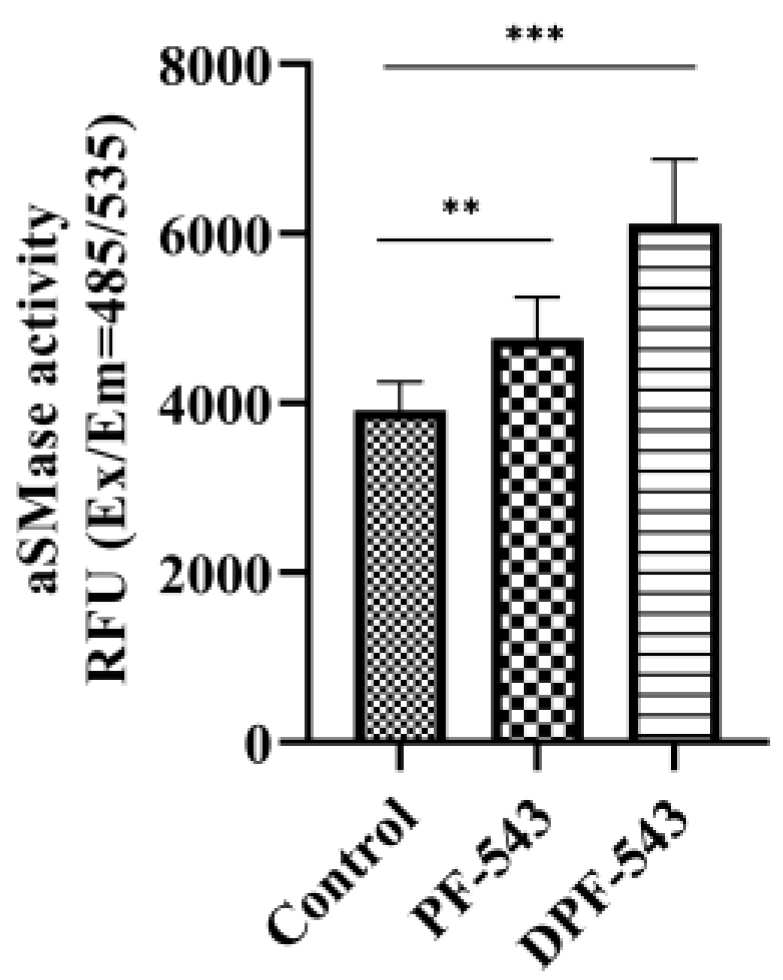
2.4. DPF-543 Activates aSMase
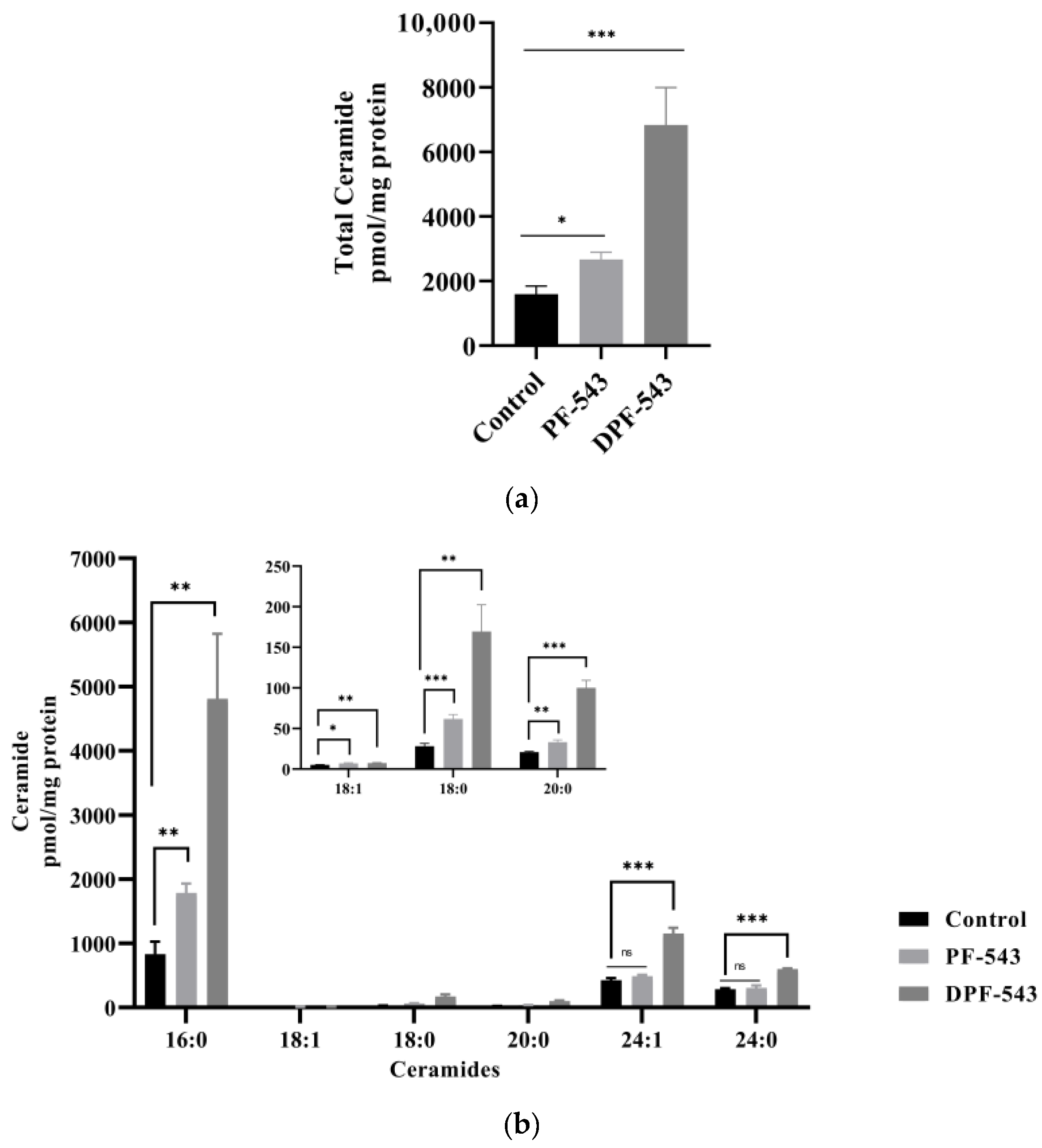
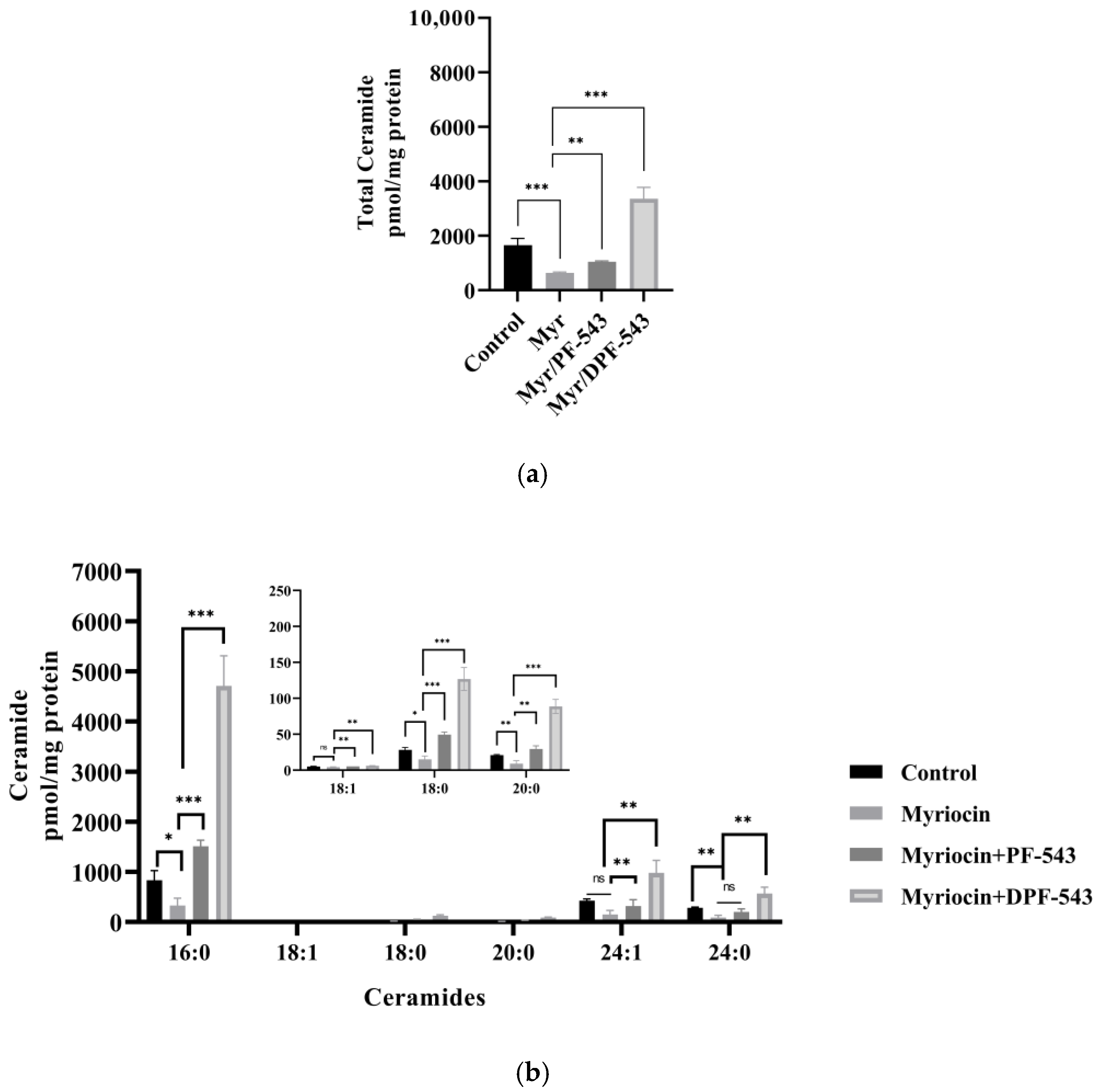
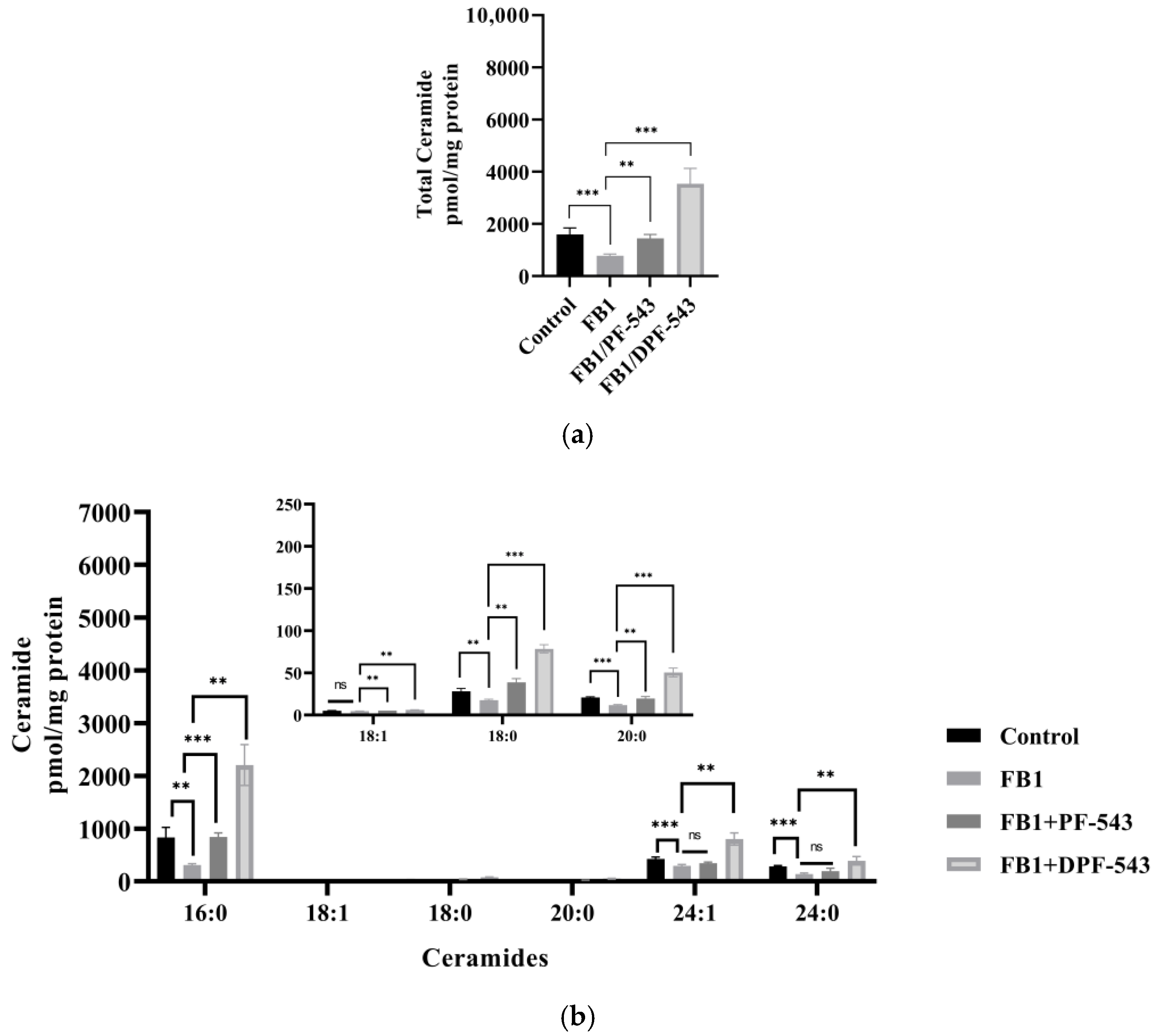
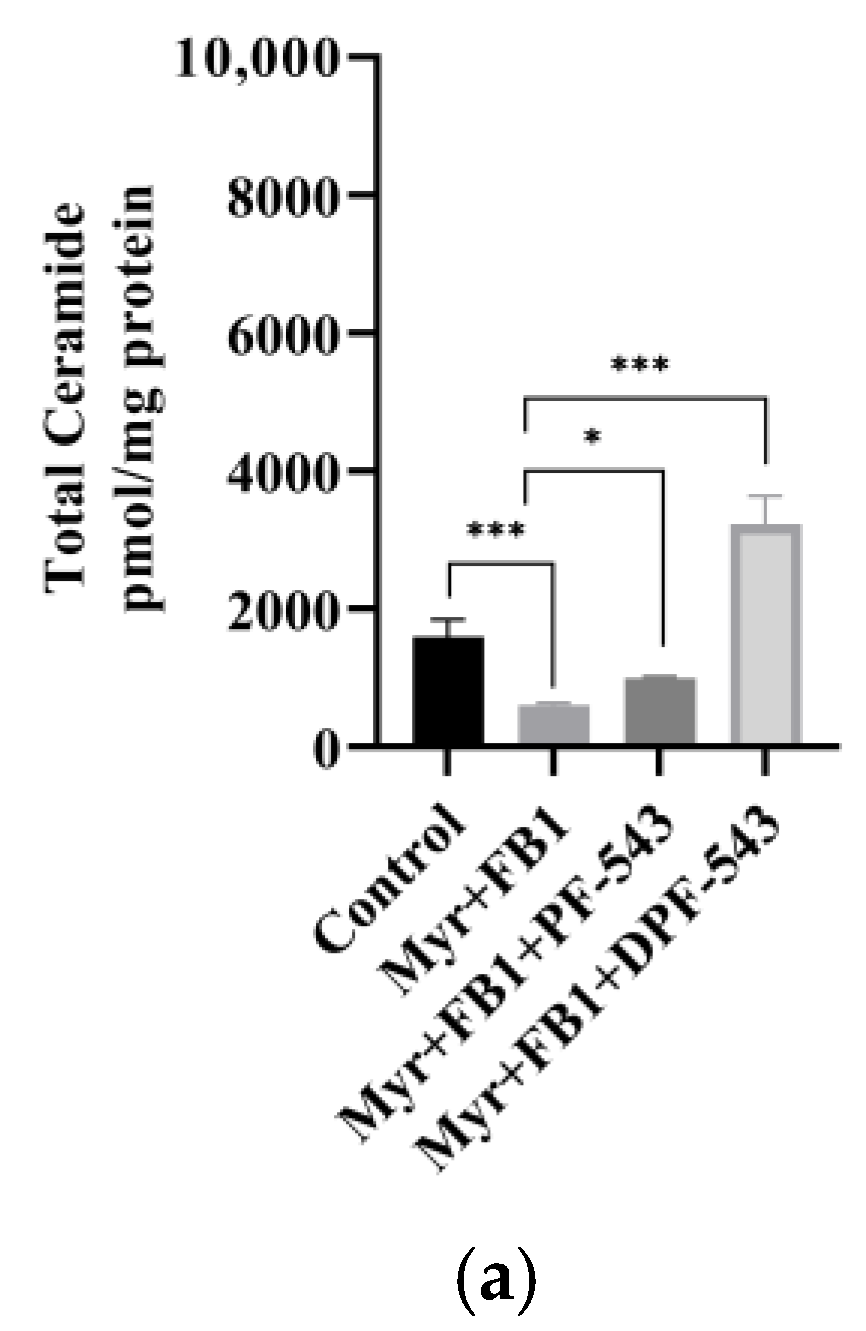
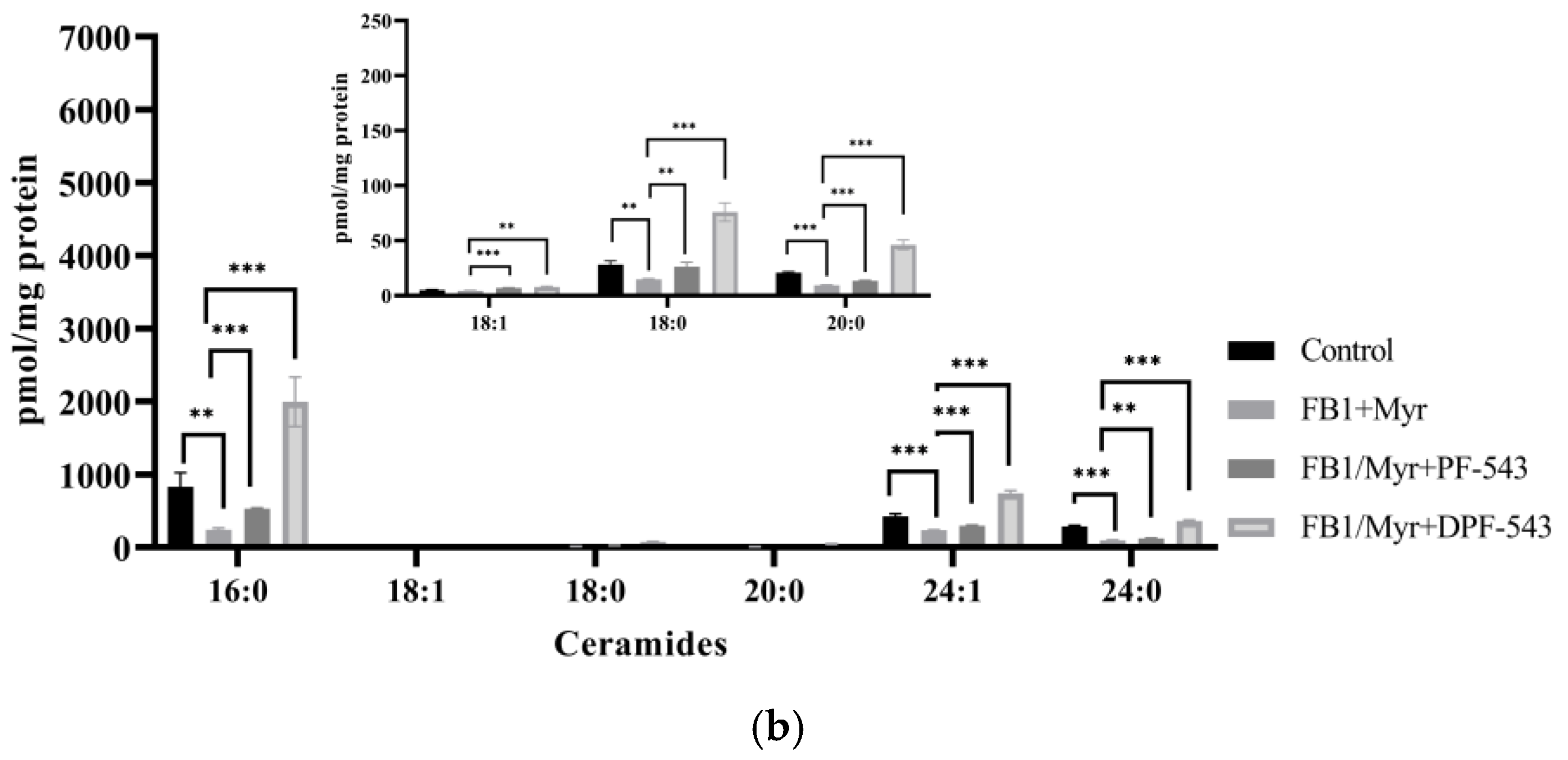
2.5. DPF-543 Activates Cer Synthesis via De Novo Pathway
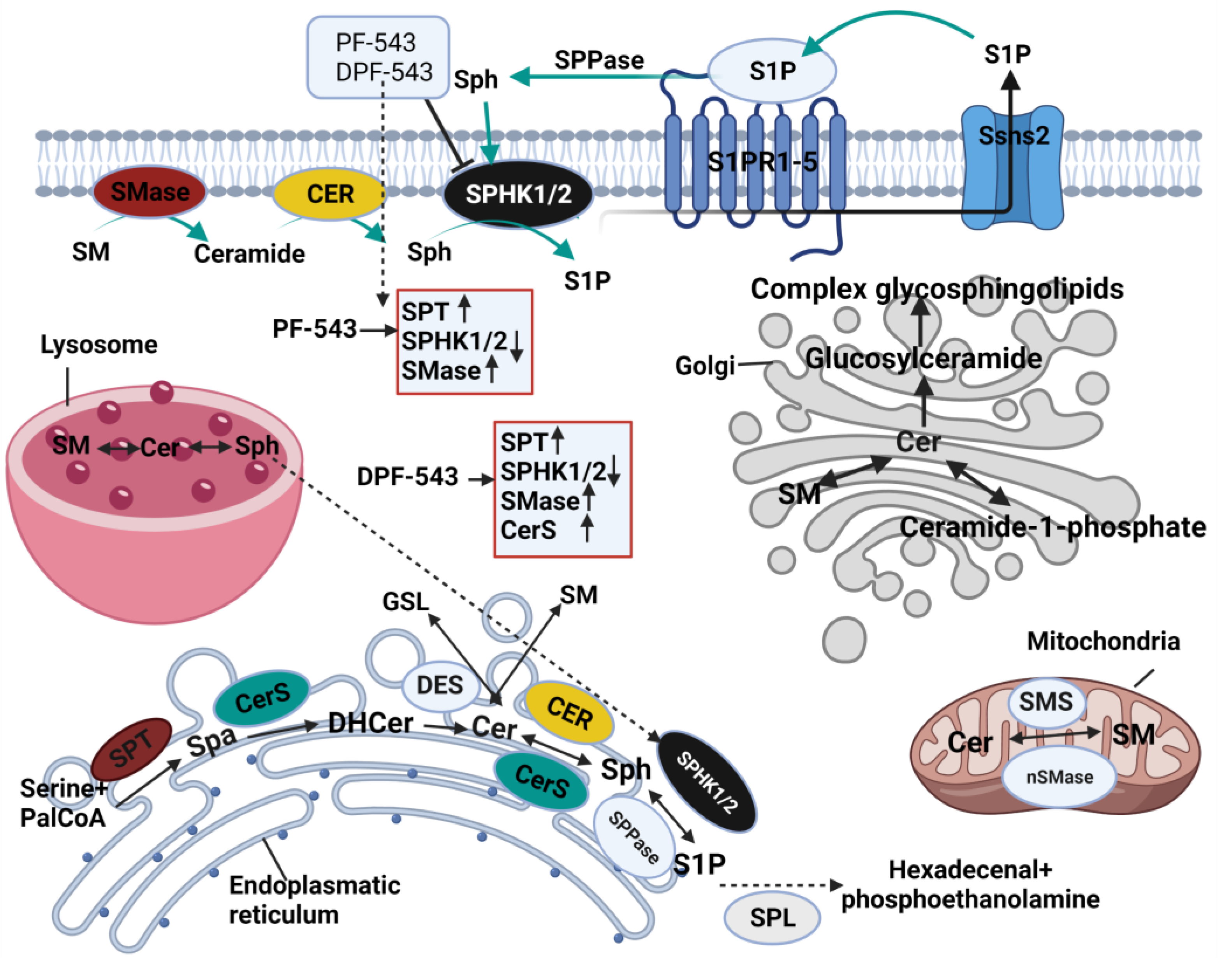
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Viability Test
4.4. Regulation of Sphingolipid Metabolism
4.5. Lipid Extraction
4.6. SPT Activity
4.7. CerS Activity
4.8. aSMase Activity
4.9. Immunoblotting
4.10. LC-MS/MS Conditions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| aSMase | Acid sphingomyelinase |
| Cers | Ceramides |
| CerS | Ceramide synthase |
| CERT | Ceramide transport protein |
| DES1/2 | Desaturase ½ |
| DHCers | Dihydroceramides |
| DHS | Dihydrosphingosine |
| DPF-543 | Dansylated PF-543 |
| ER | Endoplasmatic reticulum |
| FA | Fatty acid |
| FB1 | Fumonisin B1 |
| KDS | 3-ketodihydrosphingosine |
| Myr | Myriocin |
| PalCoA | Palmitoyl Coenzyme-A |
| PLP | Pyridoxal phosphate |
| SPA | Sphinganine |
| SPHK | Sphingosine kinase |
| SM | Sphingomyelin |
| SPN | Sphingosine |
| SPT | Serine palmitoyl transferase |
| S1P | Sphingosine-1-phosphate |
References
- Mandon, E.C.; Ehses, I.; Rother, J.; van Echten, G.; Sandhoff, K. Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. J. Biol. Chem. 1992, 267, 11144–11148. [Google Scholar] [CrossRef]
- Beeler, T.; Bacikova, D.; Gable, K.; Hopkins, L.; Johnson, C.; Slife, H.; Dunn, T. The Saccharomyces cerevisiae TSC10/YBR265w gene encoding 3-ketosphinganine reductase is identified in a screen for temperature-sensitive suppressors of the Ca2+-sensitive csg2Delta mutant. J. Biol. Chem. 1998, 273, 30688–30694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pewzner-Jung, Y.; Ben-Dor, S.; Futerman, A.H. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: Insights into the regulation of ceramide synthesis. J. Biol. Chem. 2006, 281, 25001–25005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahiri, S.; Lee, H.; Mesicek, J.; Fuks, Z.; Haimovitz-Friedman, A.; Kolesnick, R.N.; Futerman, A.H. Kinetic characterization of mammalian ceramide synthases: Determination of K(m) values towards sphinganine. FEBS Lett. 2007, 581, 5289–5294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkataraman, K.; Riebeling, C.; Bodennec, J.; Riezman, H.; Allegood, J.C.; Sullards, M.C.; Merrill, A.H., Jr.; Futerman, A.H. Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast longevity assurance gene 1 (LAG1), regulates N-stearoyl-sphinganine (C18-(dihydro)ceramide) synthesis in a fumonisin B1-independent manner in mammalian cells. J. Biol. Chem. 2002, 277, 35642–35649. [Google Scholar] [CrossRef] [Green Version]
- Teufel, A.; Maass, T.; Galle, P.R.; Malik, N. The longevity assurance homologue of yeast lag1 (Lass) gene family (review). Int. J. Mol. Med. 2009, 23, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Laviad, E.L.; Albee, L.; Pankova-Kholmyansky, I.; Epstein, S.; Park, H.; Merrill, A.H., Jr.; Futerman, A.H. Characterization of ceramide synthase 2: Tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J. Biol. Chem. 2008, 283, 5677–5684. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, Y.; Kihara, A.; Igarashi, Y. LASS3 (longevity assurance homologue 3) is a mainly testis-specific (dihydro)ceramide synthase with relatively broad substrate specificity. Biochem. J. 2006, 398, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, E.A.; Ronald, J.; Rothstein, J.; Rajagopalan, R.; Ranchalis, J.; Wolfbauer, G.; Albers, J.J.; Brunzell, J.D.; Motulsky, A.G.; Rieder, M.J.; et al. Linkage and association of phospholipid transfer protein activity to LASS4. J. Lipid. Res. 2011, 52, 1837–1846. [Google Scholar] [CrossRef] [Green Version]
- Peters, F.; Tellkamp, F.; Brodesser, S.; Wachsmuth, E.; Tosetti, B.; Karow, U.; Bloch, W.; Utermohlen, O.; Kronke, M.; Niessen, C.M. Murine Epidermal Ceramide Synthase 4 Is a Key Regulator of Skin Barrier Homeostasis. J. Investig. Dermatol. 2020, 140, 1927–1937. [Google Scholar] [CrossRef]
- Ito, S.; Ishikawa, J.; Naoe, A.; Yoshida, H.; Hachiya, A.; Fujimura, T.; Kitahara, T.; Takema, Y. Ceramide synthase 4 is highly expressed in involved skin of patients with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Riebeling, C.; Allegood, J.C.; Wang, E.; Merrill, A.H., Jr.; Futerman, A.H. Two mammalian longevity assurance gene (LAG1) family members, trh1 and trh4, regulate dihydroceramide synthesis using different fatty acyl-CoA donors. J. Biol. Chem. 2003, 278, 43452–43459. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, Y.; Kihara, A.; Igarashi, Y. Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem. J. 2005, 390, 263–271. [Google Scholar] [CrossRef]
- Cadena, D.L.; Kurten, R.C.; Gill, G.N. The product of the MLD gene is a member of the membrane fatty acid desaturase family: Overexpression of MLD inhibits EGF receptor biosynthesis. Biochemistry 1997, 36, 6960–6967. [Google Scholar] [CrossRef]
- Funato, K.; Riezman, H. Vesicular and nonvesicular transport of ceramide from ER to the Golgi apparatus in yeast. J. Cell Biol. 2001, 155, 949–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, A.; Schlarmann, P.; Kurokawa, K.; Nakano, A.; Riezman, H.; Funato, K. Tricalbins Are Required for Non-vesicular Ceramide Transport at ER-Golgi Contacts and Modulate Lipid Droplet Biogenesis. iScience 2020, 23, 101603. [Google Scholar] [CrossRef] [PubMed]
- Hanada, K.; Kumagai, K.; Tomishige, N.; Kawano, M. CERT and intracellular trafficking of ceramide. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2007, 1771, 644–653. [Google Scholar] [CrossRef]
- Kitatani, K.; Idkowiak-Baldys, J.; Hannun, Y.A. The sphingolipid salvage pathway in ceramide metabolism and signaling. Cell Signal. 2008, 20, 1010–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coant, N.; Sakamoto, W.; Mao, C.; Hannun, Y.A. Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv. Biol. Regul. 2017, 63, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Hatoum, D.; Haddadi, N.; Lin, Y.; Nassif, N.T.; McGowan, E.M. Mammalian sphingosine kinase (SphK) isoenzymes and isoform expression: Challenges for SphK as an oncotarget. Oncotarget 2017, 8, 36898–36929. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Taiyab, A.; Hussain, A.; Alajmi, M.F.; Islam, A.; Hassan, M.I. Targeting the Sphingosine Kinase/Sphingosine-1-Phosphate Signaling Axis in Drug Discovery for Cancer Therapy. Cancers 2021, 13, 1898. [Google Scholar] [CrossRef]
- Guitton, J.; Bandet, C.L.; Mariko, M.L.; Tan-Chen, S.; Bourron, O.; Benomar, Y.; Hajduch, E.; Le Stunff, H. Sphingosine-1-Phosphate Metabolism in the Regulation of Obesity/Type 2 Diabetes. Cells 2020, 9, 1682. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Alshaker, H.; Cooper, C.; Winkler, M.; Pchejetski, D. The emerging role of FTY720 (Fingolimod) in cancer treatment. Oncotarget 2016, 7, 23106–23127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnute, M.E.; McReynolds, M.D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J.W.; Hall, T.; Chrencik, J.; Kraus, M.; Cronin, C.N.; et al. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 2012, 444, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, M.; Kameyama, H.; Iwai, S.; Yura, Y. Induction of autophagy by sphingosine kinase 1 inhibitor PF-543 in head and neck squamous cell carcinoma cells. Cell Death Discov. 2017, 3, 17047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Zhou, Y.; Shi, Y.; Liu, B. Effect of the Sphingosine Kinase 1 Selective Inhibitor, PF543 on Dextran Sodium Sulfate-Induced Colitis in Mice. DNA Cell Biol. 2019, 38, 1338–1345. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.J.; Huang, L.S.; Watanabe, S.; Joshi, N.; Williams, K.J.N.; Chi, M.; Lu, Z.; Harijith, A.; Yeldandi, A.; et al. The Sphingosine Kinase 1 Inhibitor, PF543, Mitigates Pulmonary Fibrosis by Reducing Lung Epithelial Cell mtDNA Damage and Recruitment of Fibrogenic Monocytes. Int. J. Mol. Sci. 2020, 21, 5595. [Google Scholar] [CrossRef]
- Huang, L.S.; Sudhadevi, T.; Fu, P.; Punathil-Kannan, P.K.; Ebenezer, D.L.; Ramchandran, R.; Putherickal, V.; Cheresh, P.; Zhou, G.; Ha, A.W.; et al. Sphingosine Kinase 1/S1P Signaling Contributes to Pulmonary Fibrosis by Activating Hippo/YAP Pathway and Mitochondrial Reactive Oxygen Species in Lung Fibroblasts. Int. J. Mol. Sci. 2020, 21, 2064. [Google Scholar] [CrossRef] [Green Version]
- Bougault, C.; El Jamal, A.; Briolay, A.; Mebarek, S.; Boutet, M.A.; Garraud, T.; Le Goff, B.; Blanchard, F.; Magne, D.; Brizuela, L. Involvement of sphingosine kinase/sphingosine 1-phosphate metabolic pathway in spondyloarthritis. Bone 2017, 103, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Jozefczuk, E.; Nosalski, R.; Saju, B.; Crespo, E.; Szczepaniak, P.; Guzik, T.J.; Siedlinski, M. Cardiovascular Effects of Pharmacological Targeting of Sphingosine Kinase 1. Hypertension 2020, 75, 383–392. [Google Scholar] [CrossRef]
- Chen, L.; Li, L.; Song, Y.; Lv, T. Blocking SphK1/S1P/S1PR1 Signaling Pathway Alleviates Lung Injury Caused by Sepsis in Acute Ethanol Intoxication Mice. Inflammation 2021, 9, 1–10. [Google Scholar] [CrossRef]
- Shrestha, J.; Hwang, G.T.; Lee, T.; Kim, S.W.; Oh, Y.S.; Kwon, Y.; Hong, S.W.; Kim, S.; Seop Moon, H.; Baek, D.J.; et al. Synthesis and Biological Evaluation of BODIPY-PF-543. Molecules 2019, 24, 4408. [Google Scholar] [CrossRef] [Green Version]
- Park, E.Y.; Lee, T.; Oh, Y.S.; Lee, J.Y.; Shrestha, J.; Hong, S.W.; Jin, Y.J.; Jo, G.; Kim, S.; Hwang, G.T.; et al. Synthesis of dansyl labeled sphingosine kinase 1 inhibitor. Chem. Phys. Lipids. 2018, 215, 29–33. [Google Scholar] [CrossRef]
- Hart, P.C.; Chiyoda, T.; Liu, X.; Weigert, M.; Curtis, M.; Chiang, C.Y.; Loth, R.; Lastra, R.; McGregor, S.M.; Locasale, J.W.; et al. SPHK1 Is a Novel Target of Metformin in Ovarian Cancer. Mol. Cancer Res. 2019, 17, 870–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, M.; Ji, C.; Zhou, Y.; Huang, W.; Ni, W.; Tong, X.; Wei, J.F. Sphingosine kinase inhibitors: A patent review. Int. J. Mol. Med. 2018, 41, 2450–2460. [Google Scholar] [CrossRef]
- Wang, J.; Knapp, S.; Pyne, N.J.; Pyne, S.; Elkins, J.M. Crystal Structure of Sphingosine Kinase 1 with PF-543. ACS Med. Chem. Lett. 2014, 5, 1329–1333. [Google Scholar] [CrossRef] [Green Version]
- Ju, T.; Gao, D.; Fang, Z.Y. Targeting colorectal cancer cells by a novel sphingosine kinase 1 inhibitor PF-543. Biochem. Biophys. Res. Commun. 2016, 470, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Haimovitz-Friedman, A.; Kolesnick, R.N.; Fuks, Z. Ceramide signaling in apoptosis. Br. Med. Bull. 1997, 53, 539–553. [Google Scholar] [CrossRef]
- Ishiyama, M.; Tominaga, H.; Shiga, M.; Sasamoto, K.; Ohkura, Y.; Ueno, K. A combined assay of cell viability and in vitro cytotoxicity with a highly water-soluble tetrazolium salt, neutral red and crystal violet. Biol. Pharm. Bull. 1996, 19, 1518–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockert, J.C.; Horobin, R.W.; Colombo, L.L.; Blazquez-Castro, A. Tetrazolium salts and formazan products in Cell Biology: Viability assessment, fluorescence imaging, and labeling perspectives. Acta. Histochem. 2018, 120, 159–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Ruan, F.; Hakomori, S.; Igarashi, Y. Cooperative inhibitory effect of n,n,n-trimethylsphingosine and sphingosine-1-phosphate, co-incorporated in liposomes, on b16 melanoma cell metastasis-cell-membrane signaling as a target in cancer-therapy.4. Int. J. Oncol. 1995, 7, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Ohta, H.; Yatomi, Y.; Sweeney, E.A.; Hakomori, S.; Igarashi, Y. A possible role of sphingosine in induction of apoptosis by tumor necrosis factor-alpha in human neutrophils. FEBS Lett. 1994, 355, 267–270. [Google Scholar] [CrossRef] [Green Version]
- Bonhoure, E.; Pchejetski, D.; Aouali, N.; Morjani, H.; Levade, T.; Kohama, T.; Cuvillier, O. Overcoming MDR-associated chemoresistance in HL-60 acute myeloid leukemia cells by targeting sphingosine kinase-1. Leukemia 2006, 20, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Peterson, Y.K.; Smith, R.A.; Smith, C.D. Characterization of isoenzyme-selective inhibitors of human sphingosine kinases. PLoS ONE 2012, 7, e44543. [Google Scholar] [CrossRef]
- Venant, H.; Rahmaniyan, M.; Jones, E.E.; Lu, P.; Lilly, M.B.; Garrett-Mayer, E.; Drake, R.R.; Kraveka, J.M.; Smith, C.D.; Voelkel-Johnson, C. The Sphingosine Kinase 2 Inhibitor ABC294640 Reduces the Growth of Prostate Cancer Cells and Results in Accumulation of Dihydroceramides In Vitro and In Vivo. Mol. Cancer Ther. 2015, 14, 2744–2752. [Google Scholar] [CrossRef] [Green Version]
- Paugh, S.W.; Paugh, B.S.; Rahmani, M.; Kapitonov, D.; Almenara, J.A.; Kordula, T.; Milstien, S.; Adams, J.K.; Zipkin, R.E.; Grant, S.; et al. A selective sphingosine kinase 1 inhibitor integrates multiple molecular therapeutic targets in human leukemia. Blood 2008, 112, 1382–1391. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Guo, T.L.; Hait, N.C.; Allegood, J.; Parikh, H.I.; Xu, W.; Kellogg, G.E.; Grant, S.; Spiegel, S.; Zhang, S. Biological characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-phenyl)-propylidene]-thiazolidine-2,4-dione (K145) as a selective sphingosine kinase-2 inhibitor and anticancer agent. PLoS ONE 2013, 8, e56471. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, F.; Casasampere, M.; Sanllehi, P.; Casas, J.; Bujons, J.; Fabrias, G. Inhibition of dihydroceramide desaturase activity by the sphingosine kinase inhibitor SKI II. J. Lipid Res. 2014, 55, 1711–1720. [Google Scholar] [CrossRef] [Green Version]
- Velasco, G.; Galve-Roperh, I.; Sanchez, C.; Blazquez, C.; Haro, A.; Guzman, M. Cannabinoids and ceramide: Two lipids acting hand-by-hand. Life Sci. 2005, 77, 1723–1731. [Google Scholar] [CrossRef]
- Galve-Roperh, I.; Sanchez, C.; Cortes, M.L.; Gomez del Pulgar, T.; Izquierdo, M.; Guzman, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef]
- Yang, Y.H.; Mao, J.W.; Tan, X.L. Research progress on the source, production, and anti-cancer mechanisms of paclitaxel. Chin. J. Nat. Med. 2020, 18, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Charles, A.G.; Han, T.Y.; Liu, Y.Y.; Hansen, N.; Giuliano, A.E.; Cabot, M.C. Taxol-induced ceramide generation and apoptosis in human breast cancer cells. Cancer Chemother. Pharm. 2001, 47, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Devalapally, H.; Duan, Z.; Seiden, M.V.; Amiji, M.M. Paclitaxel and ceramide co-administration in biodegradable polymeric nanoparticulate delivery system to overcome drug resistance in ovarian cancer. Int. J. Cancer 2007, 121, 1830–1838. [Google Scholar] [CrossRef]
- Abdel Hadi, L.; Di Vito, C.; Marfia, G.; Ferraretto, A.; Tringali, C.; Viani, P.; Riboni, L. Sphingosine Kinase 2 and Ceramide Transport as Key Targets of the Natural Flavonoid Luteolin to Induce Apoptosis in Colon Cancer Cells. PLoS ONE 2015, 10, e0143384. [Google Scholar] [CrossRef] [Green Version]
- Kroll, A.; Cho, H.E.; Kang, M.H. Antineoplastic Agents Targeting Sphingolipid Pathways. Front. Oncol. 2020, 10, 833. [Google Scholar] [CrossRef]
- Rutti, M.F.; Richard, S.; Penno, A.; von Eckardstein, A.; Hornemann, T. An improved method to determine serine palmitoyltransferase activity. J. Lipid Res. 2009, 50, 1237–1244. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Qiao, Q.; Toop, H.D.; Morris, J.C.; Don, A.S. A fluorescent assay for ceramide synthase activity. J. Lipid Res. 2012, 53, 1701–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shamshiddinova, M.; Gulyamov, S.; Kim, H.-J.; Jung, S.-H.; Baek, D.-J.; Lee, Y.-M. A Dansyl-Modified Sphingosine Kinase Inhibitor DPF-543 Enhanced De Novo Ceramide Generation. Int. J. Mol. Sci. 2021, 22, 9190. https://doi.org/10.3390/ijms22179190
Shamshiddinova M, Gulyamov S, Kim H-J, Jung S-H, Baek D-J, Lee Y-M. A Dansyl-Modified Sphingosine Kinase Inhibitor DPF-543 Enhanced De Novo Ceramide Generation. International Journal of Molecular Sciences. 2021; 22(17):9190. https://doi.org/10.3390/ijms22179190
Chicago/Turabian StyleShamshiddinova, Maftuna, Shokhid Gulyamov, Hee-Jung Kim, Seo-Hyeon Jung, Dong-Jae Baek, and Yong-Moon Lee. 2021. "A Dansyl-Modified Sphingosine Kinase Inhibitor DPF-543 Enhanced De Novo Ceramide Generation" International Journal of Molecular Sciences 22, no. 17: 9190. https://doi.org/10.3390/ijms22179190
APA StyleShamshiddinova, M., Gulyamov, S., Kim, H.-J., Jung, S.-H., Baek, D.-J., & Lee, Y.-M. (2021). A Dansyl-Modified Sphingosine Kinase Inhibitor DPF-543 Enhanced De Novo Ceramide Generation. International Journal of Molecular Sciences, 22(17), 9190. https://doi.org/10.3390/ijms22179190